

Abstract
This review presents our current understanding of the pathophysiology and potential treatment strategies with respect to mitochondrial disease in children. We focus on pathologies due to mutations in nuclear DNA‐encoded structural and assembly factors of the mitochondrial oxidative phosphorylation (OXPHOS) system, with a particular emphasis on isolated mitochondrial complex I deficiency. Following a brief introduction into mitochondrial disease and OXPHOS function, an overview is provided of the diagnostic process in children with mitochondrial disorders. This includes the impact of whole‐exome sequencing and relevance of cellular complementation studies. Next, we briefly present how OXPHOS mutations can affect cellular parameters, primarily based on studies in patient‐derived fibroblasts, and how this information can be used for the rational design of small‐molecule treatment strategies. Finally, we discuss clinical trial design and provide an overview of small molecules that are currently being developed for treatment of mitochondrial disease.
Keywords: children, clinical trial, drug development, mitochondria, outcome measures
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Metabolism
Glossary
- Blue Native‐PAGE
A polyacrylamide gel‐based electrophoresis technique that allows separation of protein complexes in their native state. It is often used for diagnostic and research purposes regarding the mitochondrial oxidative phosphorylation system.
- Complementation study
A test to study and confirm the pathogenicity of a mutation based on a phenotypic screen. Often such a study is carried out by introducing the wild‐type gene in a patient (‐derived) cell line and assessing the reversal of the biological consequences of the mutated gene. The latter could involve normalization of a depolarized mitochondrial membrane potential, reversal of aberrant mitochondrial structure, or restoration of an enzymatic deficiency.
- Clinical trial
A crucial part of the drug development process consisting of 4 phases. During a clinical trial (phase 1 to 3), the safety, pharmacokinetics, pharmacodynamics, and effectiveness of a compound are assessed in healthy volunteers and patient cohorts. Phase 4 consists of post‐market surveillance.
- Leigh syndrome
A severe pediatric syndrome, first described by Dennis Leigh in 1951, which is caused by either mutations in mitochondrial or nuclear DNA.
- LHON
Leber's hereditary optic neuropathy. A disorder that causes maternally inherited blindness. LHON is frequently caused by mutations in subunits of mitochondrial complex I, encoded by the mitochondrial DNA.
- MELAS
Mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes. A classical mitochondrial disorder primarily caused by the m.3243A>G mutation in the mitochondrial DNA.
- MRI
Magnetic resonance imaging, a non‐invasive technique that uses magnetic fields and radio waves to visualize different parts of the human body. It is often applied during the diagnostic phase when a mitochondrial disorder is suspected for analysis of anatomical and physiological changes.
- Outcome measures
Measures to the study the outcome of an intervention strategy such as the effect of small‐molecule treatment during a clinical trial.
- Whole‐exome sequencing
A technique that allows sequencing of all protein‐coding genes within the genome. The latter is known as the exome and constitutes only a small part (∼1%) of the total genome.
Introduction
Mitochondria are semi‐autonomous organelles that are present in the cytosol of virtually all cells. They consist of a double‐membrane system that envelops the mitochondrial matrix compartment. Functionally, mitochondria are key players in cellular ATP production, fatty acid oxidation, heme biosynthesis, apoptosis induction, heat generation, and calcium homeostasis. Mitochondrial diseases are an expanding group of disorders of which the first signs and symptoms can become apparent from prenatal development to late adulthood (Koopman et al, 2012, 2013; Vafai & Mootha, 2012; Chinnery, 2015; Lightowlers et al, 2015). These disorders can be defined as “primary” (i.e., arising from a mutation in one of the genes encoding a mitochondria‐localized protein) or “secondary” (i.e., arising from an external influence on mitochondria). The latter include off‐target effects of cholesterol‐lowering statin drugs (e.g., Schirris et al, 2015) and defects in the normal breakdown of branched‐chain amino acids as in propionic acidurias, which cause severe combined respiratory chain deficiencies (e.g., Schwab et al, 2006). Here, we primarily focus on mitochondrial diseases that (i) clinically manifest themselves before the age of 18 years and (ii) arise from mutations in nuclear DNA (nDNA)‐encoded structural proteins or assembly factors of the mitochondrial oxidative phosphorylation (OXPHOS) system. The mitochondrial OXPHOS system is embedded in the mitochondrial inner membrane (MIM) and represents the final step in the conversion of nutrients to energy by catalyzing the generation of ATP (Fig 1A). This process is carried out by the combined action of the mitochondrial electron transport chain (ETC) complexes (Fig 1B) and the ATP‐producing FoF1‐ATPase (complex V; Fig 1C). The ETC consists of four multiprotein complexes (complex I to complex IV). Complex I and complex II abstract electrons from reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2), respectively. Subsequently, these electrons are donated to the electron carrier coenzyme Q10, which transports them to complex III. From thereon, electrons are transferred to the electron carrier cytochrome c and transported to complex IV. At the latter complex, the electrons are donated to molecular oxygen (O2) to form water. The energy released by the electron transport is used to drive trans‐MIM proton (H+) efflux from the mitochondrial matrix by complexes I, III, and IV, thereby creating an inward‐directed proton‐motive force (PMF). The latter consists of a chemical (ΔpH) and an electrical component (Δψ; Fig 1C), and is used by complex V to generate ATP by chemiosmotic coupling (Mitchell, 1961). In addition, ΔpH and/or Δψ are essential in sustaining virtually all other mitochondrial functions like the import of pre‐proteins from the cytosol and ion/metabolite exchange (Fig 1C). In normal healthy cells (Fig 1A; red), cellular ATP is predominantly generated through subsequent metabolic reactions of the glycolysis pathway (cytosol), the pyruvate dehydrogenase complex (PDHC, mitochondrial matrix), the tricarboxylic acid (TCA) cycle (mitochondrial matrix), and the OXPHOS system (MIM). This ATP is used to fuel energy‐consuming cellular processes.
Figure 1. Glycolytic and mitochondrial ATP production, the electron transport chain, and oxidative phosphorylation.
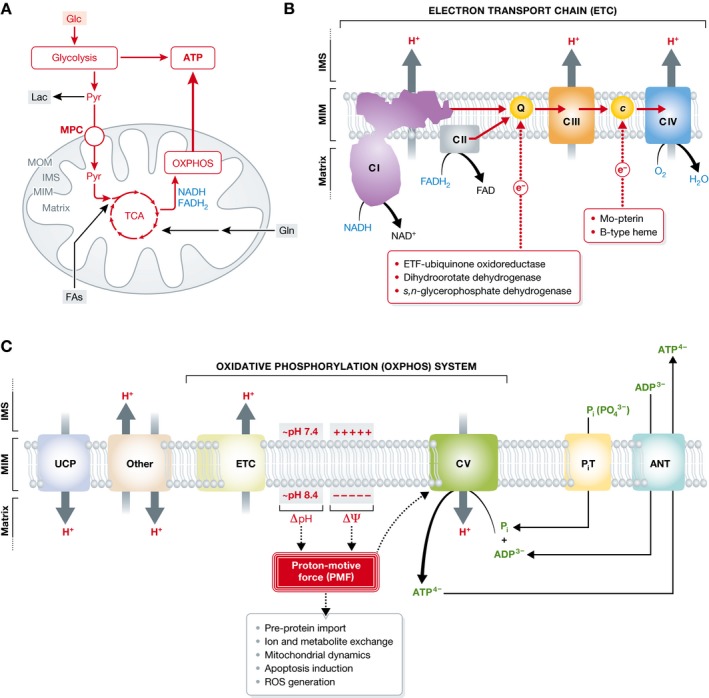
(A) Glucose (Glc) and glutamine (Gln) enter the cell via dedicated transporters. In the cytosol, Glc is converted in the glycolysis pathway into pyruvate (Pyr), which is transported to the mitochondrial matrix by the mitochondrial Pyr carrier (MPC). Alternatively, Pyr can be converted into lactate (Lac) by the action of lactate dehydrogenase (LDH). Inside the mitochondrial matrix, Pyr is converted in to acetyl coenzyme A (acetyl‐CoA; not shown) by pyruvate dehydrogenase (PDH) to enter the tricarboxylic acid (TCA) cycle. The latter supplies the oxidative phosphorylation system (OXPHOS) with substrates in the form of reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH 2). In addition, Gln can enter the mitochondrial matrix where it is converted by glutaminase into glutamate (not shown), a TCA cycle substrate. Also fatty acids can enter the mitochondrial matrix and enter the TCA cycle following their conversion into acetyl‐CoA (not shown). In healthy cells, the conversion of Glc into Pyr and its further metabolic conversion by the TCA and OXPHOS system constitute the major pathway for ATP generation (marked in red). (B) The mitochondrial electron transport chain (ETC) consists of 4 multisubunit protein complexes (complex I to IV) that are embedded in the mitochondrial inner membrane (MIM). Electrons are donated by NADH (at complex I) and FADH 2 (at complex II) to coenzyme Q10 (Q), which transports them to complex III. From thereon, electrons are transported to complex IV by cytochrome c (c) where they are donated to molecular oxygen (O2). Although not discussed here, in addition to the ETC complexes also other proteins can provide coenzyme Q10 and cytochrome c with electrons (red boxes) in a tissue‐dependent manner. During electron transport, energy is liberated and used expel protons (H+) from the mitochondrial matrix intro the inter‐membrane space (IMS) between the MIM and mitochondrial outer membrane (MOM). As a consequence, the mitochondrial matrix displays an increased pH and the MIM has a highly negative‐inside membrane potential (Δψ). (C) Together, the pH (ΔpH) and potential difference (Δψ) across the MIM determine the magnitude of the proton‐motive force (PMF), which is used by the FoF1‐ATPase (complex V) to drive mitochondrial ATP production from inorganic phosphate (Pi) and ADP. In addition to ATP generation, virtually all other mitochondrial processes including ion exchange and pre‐protein import require a proper ΔpH and/or Δψ. The magnitude of the PMF not only depends on the combined action of the ETC and complex V (i.e., the oxidative phosphorylation system; OXPHOS) but also is affected by other electrogenic systems. These include uncoupling proteins (UCPs), the Pi transporter (PiT), and the adenine nucleotide translocator (ANT). This figure was compiled based on (Koopman et al, 2010; Valsecchi et al, 2010; Liemburg‐Apers et al, 2011; Koopman et al, 2012, 2013 and Liemburg‐Apers et al, 2015).
Diagnosing OXPHOS disease in children
The five complexes of the mitochondrial OXPHOS system are assembled from 92 distinct proteins, requiring the assistance of at least 37 nDNA‐encoded assembly factors (Nouws et al, 2012; Koopman et al, 2013). Each OXPHOS complex except complex II is of bi‐genomic origin and contains subunits encoded by mitochondrial DNA (mtDNA) and nDNA (Koopman et al, 2013). Complex I is built up from 44 subunits (7 mtDNA‐ and 37 nDNA‐encoded), complex II of 4 subunits, complex III of 11 subunits (1 mtDNA, 10 nDNA), complex IV of 14 subunits (3 mtDNA, 11 nDNA), and complex V of 19 subunits (2 mtDNA, 17 nDNA). As mentioned in the introduction, we here primarily focus on mitochondrial diseases arising from mutations in nDNA‐encoded structural OXPHOS subunits and assembly factors (Fig 2). Since the first description by Luft of a patient with exercise intolerance and hyperthermia (Luft et al, 1962), the clinical spectrum of diseases associated with OXPHOS malfunction has tremendously broadened. In our opinion, a mitochondrial disease should be considered in any patient presenting with an unexplained mono‐ or multisystemic disease, whether progressive or not. Applying this relatively broad definition has important consequences for the diagnostic approach in children, which is truly challenging given the general lack of awareness within the broader medical community, the relatively low incidence of mitochondrial disease, and the complex genotype–phenotype relationship typical of these inborn errors (Smeitink, 2003). It is also important to increase awareness for mitochondrial disorders to shorten the potentially long delays between appearance of the first symptoms and diagnostic analysis and confirmation of the defect. Regarding the various clinical presentations of mitochondrial OXPHOS disease, detailed reviews have been presented elsewhere (Kisler et al, 2010; Koene & Smeitink, 2011; Lake et al, 2016). The majority of children suffering from mitochondrial OXPHOS disease lack a specific syndromic appearance and some do not display increased blood lactate levels (Triepels et al, 1999). In case of neurodegenerative diseases, mutations in OXPHOS subunits (mtDNA‐ or nDNA‐encoded) and assembly factors have, for instance, been associated with (Koopman et al, 2013): Leigh (‐like) syndrome, leukoencephalopathy, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes) syndrome, NARP (neuropathy, ataxia, and retinitis pigmentosa), Parkinsonism/MELAS, and (susceptibility) modification of PD (Parkinson disease), and AD (Alzheimer's disease).
Figure 2. Currently identified mutations in nDNA‐encoded OXPHOS subunits and assembly factors causing mitochondrial disease in children.
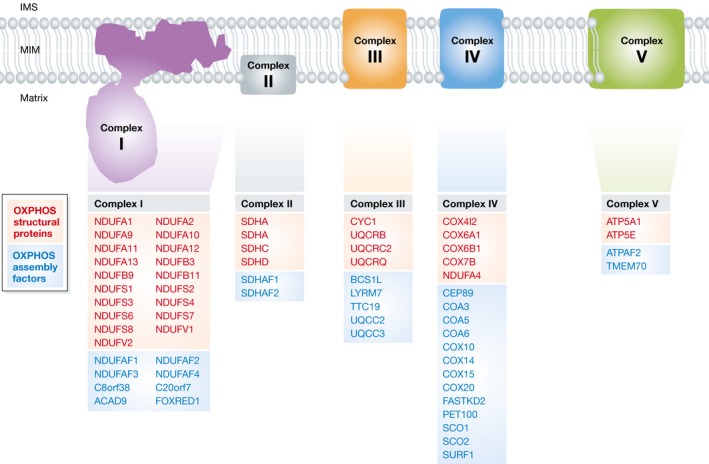
OXPHOS structural subunits in which a pathological mutation was reported are depicted in red for each complex. Assembly factors are indicated (in blue). The data in this figure were compiled from (Koopman et al, 2012, 2013; Mayr et al, 2015; Ostergaard et al, 2015; van Rahden et al, 2015).
The diagnostic process
When clinical signs and symptoms suggest a mitochondrial disorder, the following general strategy is carried out: (i) a detailed analysis of the medical/family history to discriminate between maternal (mtDNA) and Mendelian (nDNA) inheritance, (ii) evaluation of mitochondrial biomarkers like lactate, pyruvate, alanine, fibroblast growth factor 21 (FGF21; Suomalainen et al, 2011) and growth and differentiation factor 15 in blood (GDF15; Yatsuga et al, 2015) or the excretion of TCA cycle intermediates in urine, and (iii) detailed investigations of affected organs and tissues by medical specialists like neurologists, ophthalmologists, cardiologists, and/or by imaging techniques such as echocardiography or magnetic resonance imaging (MRI) of brain, heart, and skeletal muscle, and by neurophysiological studies (such as obtaining an electroencephalogram; EEG). The decision to perform a skeletal muscle biopsy for histopathological, immunohistochemical, and/or enzymological analysis is taken based on the “Bernier” or “Wolf/Smeitink” mitochondrial disease criteria (MDC; Bernier et al, 2002; Wolf & Smeitink, 2002), which might include biochemical (e.g., ATP production rate) and general criteria (e.g., clinical presentation) to establish the probability of a mitochondrial disease. Biochemical analysis usually includes spectrophotometric activity analysis of individual OXPHOS complexes. Blue Native‐PAGE, which can be used to assess the relative amount of fully assembled OXPHOS complexes, can also be a useful tool (e.g., van Rahden et al, 2015). In addition to muscle tissue, enzymes are often examined in cultured skin fibroblasts. In combination with a muscle biopsy, both positive and negative fibroblast results are informative with respect to predicting the underlying genetic defect. Genetic analysis can be carried out either by traditional candidate gene sequencing, or by more comprehensive approaches including direct (whole) exome sequencing (in which all coding regions in the genome are sequenced) or whole‐genome sequencing, often followed by functional complementation analysis in patient‐derived cells.
The impact of whole‐exome sequencing
The implementation of whole‐exome sequencing as a routine molecular diagnostic test for mitochondrial disorders has resulted in the identification of numerous variants in many different genes (DaRe et al, 2013; Lieber et al, 2013; Ohtake et al, 2014; Taylor et al, 2014; Wortmann et al, 2015). Genetic variants include known pathogenic mutations, the clinical/biochemical phenotype of which matches those described in the literature and public databases. In this case, the diagnosis is clear and no further functional tests are necessary. When an unknown gene variant is encountered, there are several possibilities: (i) the variant occurs in a gene that is associated with a known disease and interferes with the function of the gene product; again, this means that no additional functional tests are necessary, (ii) the variant results in an amino acid change at the protein level or possibly interferes with the expression of a disease‐associated gene and/or the splicing of its mRNA; this means that the consequences of the variant are not unambiguously predictable and a functional test can help to establish the diagnosis, (iii) the variant occurs in a gene of unknown function or a gene not previously associated with disease; this means that the diagnosis requires a functional confirmation of the defect. In general, mitochondrial disorders display a poor correlation between genotype and phenotype. This further emphasizes the importance of functional validation of genetic variants (Daud et al, 2015). For genes known to be mutated in OXPHOS disease, the possibility to perform relatively low‐cost diagnostic exome sequencing using DNA extracted from blood samples has certainly sped up and facilitated the diagnostic process (Wortmann et al, 2015). However, it is important to realize that when a negative result is obtained, this does not conclusively rule out the presence of a mitochondrial disease. This is caused by the fact that: (i) the exome sequencing technique displays non‐optimal coverage meaning that mutations in non‐covered parts of the exome cannot be excluded, (ii) even a whole exome covers only approximately 1% of the whole genome, (iii) the pathophysiological effects of many polymorphisms are unknown, and (iv) the number of mutated genes demonstrated to be involved in mitochondrial disease is continuously increasing. As an example of the latter, we demonstrated that a patient with intellectual disability and multisystemic problems displayed an isolated enzymatic complex IV deficiency caused by a pathological mutation in the centrosomal protein of 89 kDa (CEP89), previously not associated with mitochondrial dysfunction (van Bon et al, 2013). As a consequence, it becomes increasingly difficult to classify mitochondrial diseases in children based upon their phenotypic presentation. Therefore, the number of patients that will be correctly diagnosed with a mitochondrial (OXPHOS) disease will be underestimated. A possible solution to this problem could be that patients for whom an obvious explanation for their symptomatology cannot be established are referred to specialized (inter)national centers of excellence (e.g., the Radboud Center for Mitochondrial Medicine in the Netherlands), which can decide on an appropriate cost‐ and time‐effective diagnostic strategy.
The relevance of complementation studies
Genetic complementation studies are carried out by introducing the wild‐type DNA of the mutated gene into patient‐derived cells (usually skin fibroblasts). DNA transduction by lentiviral transmission is an efficient way to achieve genetic complementation (e.g., Jonckheere et al, 2011). Although it is relatively time‐consuming, this procedure yields a population of stably complemented cells. Transient transfections can also be used, for instance, using an insect virus (baculovirus) system (e.g., Kirby et al, 2004; Hoefs et al, 2008). In this approach, the protein to‐be‐overexpressed can be tagged with a fluorescent protein (e.g., Hoefs et al, 2008), or be part of an IRES (internal ribosome entry site)‐containing construct (e.g., Bouabe et al, 2008). Using the IRES strategy allows simultaneous expression of the protein and a fluorescent marker protein without them being physically attached to each other. Although with a transient transfection protocol, not all cells are necessarily transfected, it can be advantageous when a microscopic readout is used since it allows side‐by‐side comparison of transfected (complemented) and non‐transfected (non‐complemented) patient cells. Depending on the nature of the mutation, functional confirmation of pathogenicity can obtained using a relatively simple readout like enzymatic activity (Haack et al, 2012; Ngu et al, 2012; Jonckheere et al, 2013; Szklarczyk et al, 2013) or protein expression levels (assessed by Western blotting; Hoefs et al, 2008). More advanced strategies include comparative analysis of control, patient, and complemented patient primary fibroblasts with respect to mitochondrial morphology (Koopman et al, 2005; Jonckheere et al, 2011) and Δψ (Hoefs et al, 2008). Unfortunately, functional studies of the genetic defect are often lacking in published studies, especially when a large number of patients have been analyzed. Although large‐scale complementation and other functional studies may be practically challenging, they should be carried out to prevent detected variants prematurely or erroneously ending up in pathogenic mutation databases. For example, a recent study described two novel mutations in SCO2 (a complex IV assembly gene) in patients with early‐onset myopia (Jiang et al, 2014). Although the authors did state that additional studies are needed to prove the pathogenicity of these (and other) mutations, the latter are nevertheless listed in the Human Gene Mutation Database (HGMD) as “disease‐causing” ( www.hgmd.cf.ac.uk). This is not to criticize mutation databases as these are extremely valuable to clinical practice, but to illustrate that clinicians and laboratory specialists should remain critical about the information provided by these databases. Similarly, in publications dealing with genetic variants, incorrect or incomplete information can have important clinical implications for individual patients.
Cellular pathophysiology of OXPHOS dysfunction
During the last 15 years, we have strongly advocated the use of living cells (in addition to cell homogenates, isolated mitochondria, and fixed tissue samples), to study the pathophysiology of mitochondrial dysfunction in general and OXPHOS mutations in particular (e.g., Smeitink et al, 2004; Koopman et al, 2010, 2012, 2013; Willems et al, 2015). Live‐cell analysis is crucial since mitochondrial and cellular functions are tightly interconnected. This is illustrated by the following observations: (i) mitochondrial morphology and function are altered by isolation procedures, (ii) the cellular environment is required to provide substrates to mitochondria and the OXPHOS system, (iii) mitochondria exchange ions and metabolites (e.g., ADP, Pi and ATP) with the cytosol, and (iv) mitochondrial and cellular functioning are linked and controlled by (retrograde) signaling pathways (e.g., Palmieri, 2008; Koopman et al, 2010; Picard et al, 2011; Szabo & Zoratti, 2014). In principle, a mutation in an OXPHOS protein‐encoding gene can reduce the catalytic activity of the OXPHOS complex, its protein levels, or both. In case of isolated complex I deficiency, the reduction in the levels of fully assembled complex I in primary patient fibroblasts was linked to a proportional decrease in complex I residual activity (e.g., Valsecchi et al, 2010). This suggests that mutations in nDNA‐encoded complex I subunits reduce the biosynthesis of fully active complex I and/or induce complex I destabilization (i.e., an “expression defect”). Only in a single case (i.e., a patient carrying an Asp446Asn mutation in the NDUFS2 gene of complex I), a reduced complex I activity was not paralleled by a lower level of fully assembled complex I (i.e., a “catalytic defect”; Ngu et al, 2012). Quantitative live‐cell microscopy of primary fibroblasts from 14 control subjects and 24 children with isolated complex I deficiency revealed that patient cells displayed a less negative Δψ (i.e., “depolarization”), increased mitochondrial NADH levels, elevated reactive oxygen (ROS) levels, aberrations in mitochondrial morphology, and disturbed cytosolic and mitochondrial Ca2+ and ATP handling (Koopman et al, 2005, 2007, 2008; Verkaart et al, 2007a; Golubitzky et al, 2011; Valsecchi et al, 2010; Roestenberg et al, 2012; Leman et al, 2015). Interestingly, several of the above aberrations were also observed in fibroblasts carrying mutations in other OXPHOS complexes and assembly factors (e.g., Szklarczyk et al, 2013), in mouse cells with genetic complex I deficiency (e.g., Kruse et al, 2008; Valsecchi et al, 2012, 2013), in cells treated with OXPHOS inhibitors (e.g., Forkink et al, 2014, 2015; Distelmaier et al, 2015; Schöckel et al, 2015), and in cells with mutations in non‐OXPHOS mitochondrial proteins (e.g., Mortiboys et al, 2008; Heeman et al, 2011; Hoffmann et al, 2012). This suggests that, depending on the cellular context and experimental conditions, the observed aberrations in primary skin fibroblasts of patients with inherited complex I deficiency reflect the “general” consequences typical of mitochondrial dysfunction. Correlation analysis suggested that NADH and ROS levels increased as a direct consequence of the reduced expression of fully active complex I (Verkaart et al, 2007a,b). Moreover, patient fibroblasts with a moderate reduction in complex I activity displayed a much smaller increase in ROS levels than fibroblasts with a large reduction in complex I activity (Koopman et al, 2007). In addition, mitochondrial morphology was not affected and appeared fragmented when complex I activity was moderately and greatly reduced, respectively (Koopman et al, 2007). Integrated analysis of cellular and clinical data revealed that fibroblasts from patients with a relatively early age of disease onset/death displayed a large reduction in complex I activity. Similarly, patients with a later age of disease onset/death displayed a much smaller reduction in complex I activity (Blanchet et al, 2011). The same study revealed that the extent of complex I deficiency was proportional to the cellular level of OXPHOS proteins and inversely related to cellular NADH/ROS levels and degree of mitochondrial fragmentation.
Taken together, the above results suggest that complex I activity in patient fibroblasts might be improved by reducing the increased ROS levels and normalizing mitochondrial morphology. Indeed, treating patient fibroblasts with the water‐soluble vitamin E‐derived antioxidant Trolox (6‐hydroxy‐2,5,7,8‐tetramethylchroman‐2‐carboxylic acid) reduced ROS levels and increased the expression of fully assembled complex I and thereby complex I activity (Koopman et al, 2008). A follow‐up study demonstrated that Trolox treatment reversed Δψ depolarization and normalized Ca2+‐stimulated ATP production in patient fibroblasts (Distelmaier et al, 2009). Further mechanistic analysis revealed that ROS and thiol redox state co‐control mitochondrial morphology and function in human skin fibroblasts (Distelmaier et al, 2012; Willems et al, 2013), compatible with the apparent link between ROS levels and regulation of mitochondrial morphology and function (Willems et al, 2015). This suggests that the reduced complex I activity, increased ROS level, and altered mitochondrial morphology, and possibly also other aberrations, might constitute linked therapeutic targets (Koopman et al, 2007; Blanchet et al, 2011, 2015). We thus pursued antioxidant treatment as a potential strategy to mitigate the cellular aberrations in fibroblasts from patients with isolated complex I deficiency. This is discussed further in the section on the development of the novel antioxidant KH176 (see below).
Development of treatments
Overall strategy
A “lead compound” is a chemical molecule displaying pharmacological or biological activity likely to be therapeutically useful, but with a suboptimal chemical structure that requires modification to acquire better drug‐like properties. Such properties would include efficacy, potency, safety, synthesis route, cost, chemical stability, metabolic stability, and, last but not least, patentability (Pritchard et al, 2003). During optimization, the primary selection criterion is usually based on compound efficacy and potency. However, compounds active in one assay may perform poorly in others, indicating that their selection needs to be based on multiple properties. Lead optimization and selection mainly depend on the properties of the molecule in studies involving its absorption, distribution, metabolism, and excretion (ADME properties), its pharmacokinetics (PK) profile, and in vitro safety. Unfortunately, there are no predictive rules for identifying a potentially successful compound. During the selection process, certain additional constraints can be applied depending on the desired in vivo properties of the small molecule. These include the preferred route of administration (e.g., oral bioavailability), the targeted organs (e.g., blood–brain permeability), or potential drug–drug interactions in the studied disease group. Importantly, a certain amount of specific preclinical information is necessary to fulfill the requirements of the ethical/regulatory authorities involved and allow proceeding to the next development phase. To illustrate this process, we here present some of our own experiences in further developing an active pharmaceutical ingredient (API) following lead optimization. This consists of the following steps (Table 1):
Table 1.
Steps in the development of active pharmaceutical ingredients (APIs) toward clinical use
| Phase | Subphase | Activities | Outcomes |
|---|---|---|---|
| Lead optimization (non‐GLP) | In vitro ADME | Chemical stability, solubility, metabolic stability, stability in gastric environment, CYPs interaction, PPB, BBB permeability | Information required for the final selection of the lead compound; it is usually performed on a group of the most potent hit analogs developed during the hit‐to‐lead screen |
| In vitro toxicology | Cytotoxicity, hepatotoxicity, cardiotoxicity, mutagenicity | ||
| In vivo ADME | Pharmacokinetics: (oral) bioavailability, plasma exposure, tissue distribution, metabolism, excretion | ||
| Metabolite profiling | N.a. | Profiling and identification of metabolites formed in hepatocytes of humans and animal species used for non‐clinical safety | Allows the selection of animal species for further in vivo toxicology studies |
| Synthesis scale‐up (GMP) | N.a. | Synthesis of a large batch of API in a GMP fashion | A GMP batch of API produced in quantity sufficient to cover the toxicology and First‐in‐Human studies |
| CMC of the API | N.a. | Validation of API chemical structure; determination of physicochemical parameters | Mandatory information for IB/IMPD or IND filling and for formulation development |
| API stability | N.a. | Stability study of the API under ICH conditions | Expiry date of the API in different storage conditions |
| Pre‐formulation and stability of drug product | N.a. | Selection of the formulation based on physicochemical properties of the API and desired route of administration; determining API stability in the formulation | Optimal formulation and stability data to support GLP toxicology and First‐in‐Human studies |
| GLP‐compliant API detection methods | N.a. | Validation of a GLP‐compliant method to quantify the API in dose formulation studies and biological fluids | GLP‐compliant and validated method to detect and quantify API during toxicology and First‐in‐Human trials |
| Non‐clinical safety | Pilot toxicology study | Dose range finding and 14‐day toxicology study in a single animal species | First information on the dose range and specific toxicity to be monitored during the next phases |
| GLP‐compliant regulatory toxicology study | In vitro: GLP‐compliant in vitro toxicology study | Genetic toxicology, safety pharmacology | |
| In vivo: GLP‐compliant in vivo toxicology | General toxicology | ||
| In vivo: reproductive toxicology study | Adverse effects on embryonic development | ||
| In vivo: juvenile toxicology study | Specific adverse effect on juvenile population | ||
| Drug–drug interaction study | N.a. | Inhibition and substrate potential of API toward CYPs, drug transporters, and UGTs | Prediction of potential adverse effect arising from polypharmacology (i.e., how others drugs and API will impact on each other's ADME properties leading to modified PK/PD profile) |
| Investigational brochure (IB) | N.a. | Compile all preclinical data relevant to study the API in humans under ICH conditions | The IB is required for the clinical investigator to design the clinical trial protocol |
| Investigational medical product dossier (IMPD) | N.a. | Complete the IMPD form with CMC information about drug substance and drug product | The IMPD is required for authorization to enter First‐in‐Human trials |
| Clinical trial application (CTA) form | N.a. | Complete CTA with information about drug product and trial design, site and investigator | The CTA is required to obtain authorization to enter First‐in‐Human trials |
This table summarizes the overall preclinical experimental and administrative steps required for a novel API to entering First‐in‐Human trials. It is based upon experiences within our SME Khondrion and is not intended to be exhaustive. Each API will have a specific development path, and API developers should strictly follow the guidance and requirements from the official agencies involved.
ADME, absorption, distribution, metabolism, and excretion; API, active pharmacological ingredient; BBB, blood–brain barrier; CMC, chemistry and manufacturing controls; CTA, clinical trial application; GLP, good laboratory practices; GMP, good manufacturing practices; CYP, cytochrome P450; IB, investigational brochure; ICH, The International Conference on Harmonisation (of Technical Requirements for Registration of Pharmaceuticals for Human Use); IMPD, investigational medical product dossier; IND, investigational new drug; N.a., not appropriate; PD, pharmacodynamics; PK, pharmacokinetics; PPB, plasma protein binding; UGT, UDP glucuronosyltransferase.
Metabolic profiling in hepatocytes of humans and various animal species will assist in deciding which animal species to use during the regulatory toxicology phase, in which the API's toxic potential is assessed. As any metabolite can cause toxicity, the combined metabolites detected in two selected animal species should cover all potential metabolites in humans.
Scaling up of API synthesis according to GMP (good manufacturing practice) standards, acquiring CMC (chemistry and manufacturing controls) information of the API, and determining API stability. Information obtained during these three steps is crucial for defining the formulation of the API and obtain authorization to enter the clinical trial phase. Producing a GMP batch of the API is not mandatory for the non‐clinical toxicology phase but it is strongly advised to use the same GMP batch for both regulatory toxicology studies and the early trial clinical phases. This relates to the fact that it needs to be demonstrated that various GMP batches display an identical (im)purity profile.
Optimization of a good laboratory practice (GLP)‐compliant method of detection of the API in blood or plasma in order to study its pharmaco‐ and toxicokinetic profile during non‐clinical toxicology and clinical trials.
Performing a non‐clinical regulatory toxicology study. This study will be designed based on specific toxicity information obtained during in vitro or in vivo screening and the intended patient populations (e.g., reproductive toxicology or juvenile toxicology). The duration of this study will depend on the expected duration of the treatment in patients during clinical trials and will help to determine the safe starting dose for administration to humans.
Investigating drug–drug interactions (DDI). The different medicine authorities have issued strict guidelines regarding these studies. Although not mandatory for clinical phase 1 studies (healthy volunteers), DDI studies are of crucial importance prior to executing patient trials (phase 2 clinical trials and beyond). This is to avoid potential API toxicity due to interaction with medications or deficiencies in liver or kidney metabolism.
Compiling the investigational brochure (IB). This contains all preclinical (pharmacology and toxicology) data relevant for use of the API in the clinic. The IB will be continuously updated during clinical development.
Compiling the investigational medical product dossier (IMPD). This dossier contains all physicochemical and drug formulation information of the API.
Completing the clinical trial application (CTA) form. The CTA contains information about the drug product, trial design, and investigator details. Together with the IMPD, the CTA is required for approval of the clinical trials.
Designing clinical trials
Before an API obtains market approval and can therefore be prescribed by clinicians, in‐human studies have to be performed. Classically, these studies consist of four phases. In phase 1, API pharmacokinetics and dynamics (including toxicity) are determined in healthy volunteers. During phase 2, the intention is to determine the efficacy of the API in a small and homogeneous patient group. Phase 3 seeks to confirm the effectiveness and safety of the API on clinically relevant outcome measures in a large and more heterogeneous group of patients. The results of phase 3 are mostly used to apply for market authorization. Following market authorization, the (long‐term) safety and efficacy of the drug remain to be monitored (phase 4). One of the most challenging aspects of clinical trials involves the selection, validation, and application of clinically relevant outcome measures.
Outcome measures
Outcome measures provide information about certain disease aspects, such as severity, response to therapy or remission (Brunner & Ravelli, 2009). In case of frequently occurring disease (e.g., cardiovascular disease, cancer), the time‐to‐event (relapse, stroke, myocardial infarction, death), histology, or radiological results are often used as outcome measures (Preiss et al, 2011; Fernandez‐Martos et al, 2012; Etzioni et al, 2015). Obviously, the credibility of a clinical trial greatly depends on the chosen outcome measures and the strategy that is used to quantify them (Tugwell & Boers, 1993). In general, the number of patients required for a clinical trial (sample size) depends on the magnitude of the expected effect relative to the precision of the used measurement. This means that either the treatment should produce a large effect or the used outcome measures should be very precise when only a small patient cohort is available, as is the case for children with OXPHOS disease. Moreover, if the therapy under development slows down disease progression rather than reducing morbidity (Friedman et al, 2010; Kang, 2013), it becomes challenging to identify outcome measures suited for quantitative analysis. In addition, children are continuously growing and developing (meaning that age‐specific tests and age‐ and size‐matched reference values are required) and their understanding and enthusiasm, as well as their cooperation, can affect the test results. Since virtually all the functional tests require the child to perform at his/her maximal level, children with mild mental disability might not give consistent results because of attention deficits and a lack of understanding the importance of the tests. Measuring disease progression in children that are severely mentally disabled and sometimes not able to communicate is virtually impossible. In general, the outcome measures not only depend on the disease stage or disability grade but also on age, gender, intellectual abilities, environment, and personal factors. To the best of our knowledge, there are no established outcome measures that correlate with disease progression in mitochondrial dysfunction (Lee et al, 2010; Augustine et al, 2013; Koene et al, 2014, 2015). In case of mitochondrial OXPHOS disorders with neuromuscular dysfunction, the regulatory authorities [i.e., the European Medicines Agency (EMA) and the American Food and Drug Adminstration (FDA)] advise using the 6‐min walking test (6MWT) as a primary outcome measure. This test measures the total distance an individual is able to walk during 6 min on a hard and flat surface (Balke, 1963). In other disease fields (e.g., Dubrovsky et al, 2013; Alfano et al, 2014), as well as in our own experience with OXPHOS disease patients (Dirks et al, 2015), the 6MWT showed a considerable test–retest variability and lack of sensitivity. Therefore, we expect that other approaches like accelerometry, continuously measuring the physical activity of the child in their home environment, or measuring detailed gait characteristics are more appropriate to detect subtle but clinically relevant changes in disease severity (S. Koene et al, unpublished findings). In addition we have selected several clinically relevant and robust outcome measures to be used in clinical trials with children suffering from mitochondrial disease. These outcome measures were chosen based upon the most burdensome complaints/symptoms according to the patients and their parents (Koene et al, 2013a), and on the results of validation studies in Duchenne muscular dystrophy and cerebral palsy, which were used as a model for mitochondrial myopathy and mitochondrial encephalopathy, respectively. The total set of outcome measures can be divided into a “common core set” including the following: accelerometer test, 6MWT test, Gross Motor Function Measure (GMFM) and Medical Research Council (MRC) scale; and a number of “optional tests” including: forced vital capacity, Child Health Questionnaire, and growth charts (Koene et al, 2013b). The validity of these outcome measures was assessed in several studies (Dirks et al, 2015; S. Koene et al, unpublished findings), which suggested that accelerometry and 2‐dimensional (2D) speckle tracking echocardiography (Blessberger & Binder, 2010) are feasible instruments to monitor disease progression, but the assisted 6‐minute cycling test is not. The currently available diagnostic biomarkers for mitochondrial disease (i.e., GDF15, FGF21) do correlate with disease severity. However, these biomarkers appear not to be suited for monitoring disease progression since the change in the concentration of neither biomarker correlated to the change in disease severity (Koene et al, 2014, 2015). Additional validation studies are required to guide the further selection of outcome measure(s) for use in clinical trials involving children. Obviously, this selection is highly dependent on the mode of action of the API being tested and is preferably made by integrating the expertise of pediatricians and other medical specialists, statisticians, regulatory experts, industry partners, and patient/parent representatives.
Study population
In case of mitochondrial OXPHOS diseases, only a low number of patients are available for phase 2 clinical trials. This means that low patient‐to‐patient variation is required to demonstrate the potential efficacy of a small molecule. In the ideal case, the used patient cohort should display a homogeneous genotype, phenotype, disease stage, and organ specificity. Preferentially, the patients should neither be on medication nor display liver or kidney failure (Pfeffer et al, 2013). However, mitochondrial OXPHOS syndromes with a relatively high level of homogeneity for which a reasonable number of patients are available (e.g., Leber's hereditary optic neuropathy; LHON) are uncommon and hardly any of them is pediatric. Given this fact, the difficulty of assessing outcome measurements in children (see above), and ethical and regulatory considerations, phase 2 clinical trials should preferentially be carried out first in adult mitochondrial disease patients. In addition to mitochondrial syndromes caused by OXPHOS mutations, also other rare pediatric syndromes exist. An example of such a disease is Barth syndrome, caused by a mutation in the TAZ gene. The latter encodes the tafazzin protein, which is involved in the synthesis of the mitochondrial lipid cardiolipin. Clinically, Barth syndrome appears to display a reasonably homogeneous phenotype characterized by skeletal muscle weakness, cardiomyopathy, neutropenia, and growth delay (Barth et al, 1983; Spencer et al, 2006; Roberts et al, 2012; Jefferies, 2013; Rigaud et al, 2013). This suggests that a Barth syndrome patient cohort might be suited for phase 2 clinical trials. On the other hand, because OXPHOS dysfunction in Barth syndrome is a consequence of mitochondrial membrane alterations, such a cohort might not be the best choice for drug efficacy analysis since the obtained results might not be generalizable to primary OXPHOS disorders. Alternatively, Kearns Sayre syndrome is another more or less homogeneous mitochondrial disease that is characterized by ophthalmoparesis, pigmentary retinopathy, deafness, muscle weakness, ataxia, and cardiac conduction block (Yamashita et al, 2008). Although not strictly pediatric (i.e., it can manifest itself in children and adolescents), the progression of Kearns Sayre syndrome appears to be reasonably stable (Grady et al, 2014). Since most Kearns Sayre syndrome patients are mentally competent, the feasibility and ethical acceptability of clinical trial is increased. However, separate studies will have to be performed in neonates, infants, and young children. Another strategy is to include all children diagnosed with a mitochondrial disorder, regardless of their genetic or biochemical diagnosis, using very strict clinical inclusion criteria. The latter can include that the child: (i) has a stable disease course in the past year, (ii) can follow instructions such as hopping on one leg or rotating a pen in the hand, (iii) scores a 6MWT value between the age‐ and height‐matched 5th and 10th percentile, and/or (iv) a GMFM score between 15 and 25%. Such an approach requires a separate neonatal study, including for example neonates with infantile mitochondrial lactic acidosis, since the pharmacokinetics, safety, and efficacy in neonates might differ from the other (pediatric) groups. For all phase 2 trials, it is crucial to realize that children are clinically and physiologically not equivalent to “small adults”. This means that designing a proper drug administration regime requires separate studies on drug formulation, safety, and pharmacokinetics.
Study design
In case of pediatric OXPHOS disease, it is difficult, if not impossible, to carry out classical randomized control clinical trials since these require relatively large homogeneous patient cohorts. However, alternative trial designs are applicable (Chow & Chang, 2008; Gupta et al, 2009; Cornu et al, 2013). In the “crossover” and “n‐of‐1” designs, each patient serves as its own control and receives both placebo and active drug treatment, which facilitates recruitment. Whereas a crossover study aims to determine the efficacy of a drug in a group of patients, an n‐of‐1 study aims to determine drug effects within a single subject, although the results of multiple n‐of‐1 studies can be used for meta‐analysis (Gupta et al, 2009). Formally, crossover and n‐of‐1 designs can only be applied if the disease status is similar between the start of the first and the second treatment phase (i.e., similar baseline measurements before the placebo or active drug phase). In this sense, they are less compatible with pediatric mitochondrial OXPHOS disorders. However, if the duration of the drug treatment and the washout period (being the time between the treatment phases in which the drug and the long‐term drug effects are eliminated from the body before the start of the second phase) is short, and no long‐term effects of treatment are expected (i.e., minimal carryover effect), these designs may be of great value when evaluating drug efficacy in mitochondrial disease. If feasible, larger and phenotypically homogeneous cohorts can be analyzed in crossover studies. In ultra‐rare mitochondrial syndromes or those with a heterogeneous phenotype, n‐of‐1 trials are most suitable. In both cases, patients with an obviously oscillating disease course should be excluded. When choosing a crossover or n‐of‐1 design, several other methodological issues should be covered. First of all, the sequence of the placebo and the active treatment phases should be randomized to correct for confounders such as seasonal influences and (unexpected) learning effects. In case of group comparisons, such as in a crossover study, randomization should be stratified based on predefined criteria, in order to create comparable groups. Secondly, since all of the above‐mentioned study designs are subject to report bias, blinding of both investigators and patients is of great importance, especially in case of subjective end points. Finally, since the outcome measure is applied multiple times within the same subject, the outcome measures should be tested for learning processes, which is expected to be the case for, for example, the 6‐min walking test (Casey et al, 2012). Another design is the “Bayesian trial design” (Berry, 2006), which gives information about the probability that a predefined change occurs (e.g., the chance that the GMFM score is 10% higher after 4 weeks of active treatment compared to the placebo condition). Although the Bayesian design might still be unfamiliar to regulatory authorities and clinical investigators, its flexible design and smaller sample sizes make it a promising design for phase 3 trials.
A case study: the development of KH176
Obviously, results obtained with (patient‐derived) cell lines (as any other model system including mice) should be treated with caution when translating to human pathology. As mentioned above, we hypothesized that treatment with the vitamin E‐derived antioxidant Trolox might be a viable starting point for the rational development of small molecules that correct or mitigate the cellular consequences of nDNA‐encoded complex I mutations. To allow its effective use in animal models and human subjects, we developed a first library of 20 novel Trolox variants, several of which displayed increased potency and efficacy in scavenging cellular ROS. However, small chemical molecules generally induce multiple subtle effects. This means that techniques like single‐parameter high‐throughput cell screening and in vitro potency analysis are not generally predictive to identify the most optimal molecules. By combining automated image quantification and machine learning techniques, we were able to discriminate between the mitochondrial morpho‐functional phenotype of fibroblasts of a healthy individual and a Leigh syndrome patient (Blanchet et al, 2015). Application of this strategy highlighted Trolox ornithylamide hydrochloride (KH003) as a hit compound that effectively scavenged ROS and increased the maximal activity of mitochondrial complex I, complex IV, and citrate synthase. Next, a second generation of compounds was developed to further improve the drug‐like properties and potency of KH003 by modifying its chemical structure through classical medicinal chemistry. The lead optimization was targeted and prioritized toward the improvement of solubility, cell permeability, oral bioavailability, blood–brain barrier permeability, and chemical stability. The potency and efficacy of this 2nd‐generation library in scavenging cellular ROS and protecting cells against oxidative stress was again analyzed in Leigh syndrome patient fibroblasts. This strategy finally yielded KH176 as a lead compound since it displayed the best compromise between potency, drug‐like properties and the in vitro therapeutic window (i.e., the space between potency and toxicity). Following successful regulatory toxicology analysis, KH176 has entered the clinical trial phase ( www.clinicaltrials.gov; NCT02544217) and recently successfully completed the clinical trial phase 1.
Small molecules for treatment of mitochondrial disease
In addition to KH176, various small molecules for treatment of mitochondrial diseases are currently being evaluated in clinical studies (Kerr, 2013; Rai et al, 2015). These include idebenone, EPI‐743, MTP‐131, RP103, coenzyme Q10, bezafibrate, RG2133, DCA, ARG/CIT, lipoic acid, RTA408, and curcumin (Table 2).
Table 2.
Small molecules used in clinical studies for treatment of mitochondrial diseases
| Molecule name | Targeted disease | Clinical phase | Clinical trial identifiera | Primary results | Sponsor |
|---|---|---|---|---|---|
| KH176 | MELAS | Phase 1 double‐blind, randomized, placebo‐controlled study of the safety, tolerability, pharmacokinetics, and pharmacodynamics in healthy volunteers | NCT02544217 | KH176 is well tolerated and displays a promising PK profile | Khondrion, Nijmegen, the Netherlands |
| Idebenone (a.k.a. Catena®, Raxone®, Sovrima®) | LHON | Phase 2 double‐blind, randomized, placebo‐controlled study of the efficacy, safety, and tolerability | NCT00747487 | The primary end point did not reach statistical significance. In a subgroup of patients with discordant visual acuities at baseline, all secondary end points were significantly different between the idebenone and placebo groups | Santhera Pharmaceuticals, Liestal, Switzerland |
| FRDA | Phase 3 double‐blind, randomized, placebo‐controlled study of the efficacy, safety, and tolerability | NCT00537680 | Idebenone did not significantly alter neurological function in FRDA during the 6‐month study | ||
| MELAS | Phase 2a double‐blind, randomized, placebo‐controlled, dose‐finding study | NCT00887562 | No results reported | ||
| EPI‐743 (a.k.a. Vatiquinone®, Vincerinone®) | LS | Phase 2B randomized, placebo‐controlled, double‐blind clinical trial | NCT01721733 | Study status unknown | Edison Pharmaceuticals, Mountain View, USA |
| FRDA | Safety and efficacy study on visual function | NCT01728064 | Ongoing, not recruiting | ||
| MRCD | Emergency use protocol for EPI‐743 in acutely ill patients with inherited MRCD (90 days of end‐of‐life care) | NCT01370447 | Ongoing, not recruiting | ||
| PS | Open‐label phase 2 safety and efficacy study | NCT02104336 | Ongoing, not recruiting | ||
| FRDA | Phase 2A clinical trial on visual function in patients with point mutations | NCT01962363 | Ongoing, not recruiting | University of South Florida, Tampa, USA | |
| MTP‐131 (a.k.a. Bendavia®) | MM | Phase 1/2 multicenter, randomized, double‐blind, placebo‐controlled, multiple ascending‐dose clinical study investigating the safety, tolerability, and efficacy of intravenous MTP‐131 for the treatment of MM in subjects with genetically confirmed MD | NCT02367014 | Recruiting | Stealth BioTherapeutics Inc., Newton, USA |
| SMMDE | Phase 2 randomized, double‐blind, placebo‐controlled study to evaluate the impact of a single intravenous dose | NCT02245620 | Recruiting | ||
| RP103 (Cysteamine bitartrate) | Inherited MD including LS | A phase 2/3 open‐label, dose‐escalating study to assess safety, tolerability, efficacy, PK, and PD of RP103 delayed‐release capsules in children | NCT02023866 RP103‐MITO‐001 | Recruiting | Raptor pharmaceuticals, Novato, USA |
| A phase 2 long‐term open‐label extension study of RP103‐MITO‐001 to assess the safety, tolerability and efficacy of RP103 delayed‐release capsules for treatment of children | NCT02473445 | Recruiting | |||
| Coenzyme Q10 | Children inherited MD due to defects in specific ETC complexes or mtDNA mutations | Phase 3 trial | NCT00432744 | Completed. No results reported yet | University of Florida; FDA office of orphan products development, USA |
| Bezafibrate | MD (Confirmed mt.3243A>G mutation) | Open‐label phase 2 feasibility study | NCT02398201 | Recruiting not started | Newcastle‐upon‐Tyne Hospitals NHS foundation Trust, Newcastle, UK |
| RG2133 (2′,3′,5′‐tri‐O‐acetyluridine) | MD | Open‐label dose‐escalation phase I study to assess the safety, tolerability, PK, and PD of RG2133 in treatment of inherited MD | NCT00060515 | Terminated. No results reported | Repligen Corp, Waltham, USA |
| DCA (dichloroacetate) | MELAS | Phase 2: investigation of clinical syndromes associated with mtDNA point mutations | NCT00068913 | Terminated prematurely because of peripheral nerve toxicity | Eunice Kennedy Shriver NICHD, Bethesda, USA |
| ARG and CIT | MELAS | Open‐label phase 2: ARG flux and NO production in patients and the effect of dietary ARG and CIT supplementation | NCT01339494 | Unknown | Baylor College of Medicine, Houston, USA |
| ARG | MELAS | Open‐label phase 2 on 3 siblings: Efficacy of L‐arginine therapy on endothelium‐dependent vasodilation and mitochondrial metabolism | NCT01603446 | Completed. A significant increase of the maximum work performed at anaerobic threshold was observed | The Hospital for Sick Children, Ontario, Canada |
| Lipoic acid (a.k.a. Thioctic acid) | MM | Pilot compassionate use study | NCT00004770 | No results reported | National Center for Research Resources (NCRR), Bethesda, USA |
| RTA408 | MM | Phase 2 study of the safety, efficacy, and PD of the Nrf2‐activator RTA408 | NCT02255422 | Recruiting | Reata Pharmaceuticals Inc, Irving, USA |
| Curcumin | LHON | Phase 3 randomized, double‐blind, placebo‐controlled trial | NCT00528151 | No results reported | Mahidol University, Salaya, Thailand |
Source: www.clinicaltrials.gov .
ARG, arginine; CIT, citrulline; ETC, electron transport chain; FDA, Food and Drug Administration; FRDA, Friedreich's Ataxia; GLC, glucose; LHON, Leber hereditary optic neuropathy; LS, Leigh syndrome; MD, mitochondrial disease; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes; MM, mitochondrial myopathy; MRCD, mitochondrial respiratory chain diseases; mtDNA, mitochondrial DNA: NICHD, National Institute of Child Health and Human Development; NO, nitric oxide; Nrf2 (a.k.a. NFE2L2), nuclear factor (erythroid‐derived 2)‐like 2; PD, pharmacodynamics; PK, pharmacokinetics; PS, Pearson syndrome; SMMDE, skeletal muscle mitochondrial dysfunction in the elderly.
Idebenone can act as an antioxidant and delivers electrons directly to complex III, thereby bypassing a deficient complex I (Giorgio et al, 2011; Haefeli et al, 2011; Jaber & Polster, 2015). This compound has recently received market approval in Europe for the treatment of LHON. It is also under clinical investigation for the treatment of MELAS (phase 2a studies) and Friedreich ataxia (phase 3 studies).
EPI‐743 functions as a cofactor of the enzyme NAD(P)H dehydrogenase Quinone 1 (NQO1) and enhances the biosynthesis of glutathione (GSH), which is an important cellular antioxidant (Pastore et al, 2013). EPI‐743 is currently evaluated in clinical phase 2 studies in various indications such as Leigh, LHON, and Pearson syndrome. Promising results though from initial single open‐label trials in patients suffering from Leigh syndrome (Enns et al, 2012; Martinelli et al, 2012) or LHON syndrome (Sadun et al, 2012) have been reported.
MTP‐131 is a member of the Szeto‐Schiller (SS) peptide family and binds to the mitochondria‐specific MIM lipid cardiolipin (Alam et al, 2015). It increases OXPHOS efficiency and potentially improves the ability of mitochondria to properly respond to metabolic changes. Using different formulations, MTP‐131 has entered clinical trials for the treatment of mitochondrial myopathies (phase 1/2) and LHON (phase 1/2). In pre‐clinical studies, MTP‐131 corrected excessive ROS generation and increased ATP production in mitochondrial dysfunction (Szeto, 2014).
RP103 is intended to supply the cell with additional cysteine, an important amino acid for GSH neosynthesis (Besouw et al, 2012). RP103 is currently being evaluated in the form of delayed‐release capsules in the treatment of children with inherited mitochondrial disease, including Leigh syndrome (phase 2/3).
Coenzyme Q10 is an ETC component involved in electron transport (Fig 1B), which also displays antioxidant capacity. It is available without prescription and has been evaluated in a phase 2 study on adult patients with mitochondrial cytopathies (MELAS, LHON, and chronic progressive external ophthalmoplegia; CPEO), showing minor improvement (Glover et al, 2010). A phase 3 study (Stacpoole et al, 2012) has been completed in children with primary mitochondrial diseases but, as far as we know, no results have been reported yet.
Bezafibrate is a known activator of the transcription factor peroxisomal proliferator‐activated receptors (PPARs; Wenz et al, 2008). When activated, PPARs induce transcription of genes involved in mitochondrial function including fatty acid metabolism. Effects of bezafibrate are conflicting, since it both displayed adverse effects and positive effects in various studies (Wenz et al, 2008; Viscomi et al, 2011; Yatsuga & Suomalainen, 2012). The therapeutic potential of bezafibrate is currently evaluated in an open‐labeled phase 2 study on patients carrying the mtDNA 3243A>G mutation. The latter mutation is one of the most common mtDNA mutations in mitochondrial disease and is causatively linked to MELAS, CPEO, and maternally inherited deafness and diabetes (MIDD; Nesbitt & McFarland, 2011).
RG2133 or 2′,3′,5′‐tri‐O‐acetyluridine is a precursor of uridine, a pyrimidine nucleotide involved in RNA and DNA synthesis. Endogenous uridine synthesis depends on coenzyme Q10 recycling and normal OXPHOS function. This means that OXPHOS dysfunction can impair uridine synthesis leading to cellular dysfunction. The assessment of RG2133 safety and efficacy in a phase 1 study on patients with mitochondrial disease was terminated. As far as we know, no results have been reported.
DCA (dichloroacetate) is a structural analog of pyruvate. This molecule locks PDHC (which converts pyruvate into acetyl‐coA), in an active state by inhibiting pyruvate dehydrogenase kinase. This might improve OXPHOS function despite the presence of PDHC or OXPHOS defects. A randomized controlled phase 2 trial in patient suffering from MELAS was terminated prematurely because of peripheral nerve toxicity (Kaufmann et al, 2006).
ARG/CIT (L‐arginine/L‐citrulline) are precursors of nitric oxide (NO), an important cellular‐signaling molecule involved in vasodilation. Therefore, increased NO production following supplementation with ARG/CIT is hypothesized to protect MELAS patients against recurrent strokes possibly due to vasoconstriction. Open‐label studies in MELAS patients have shown promising results (Rodan et al, 2015), but still need be performed under well‐controlled conditions to draw final conclusions.
Lipoic acid displays antioxidant activity in vivo (Koufaki, 2014). This compound was evaluated in a combination treatment with creatine and coenzyme Q10 during a phase 2 randomized and controlled trial on patients with mitochondrial cytopathies (Rodriguez et al, 2007). During this trial, positive effects on surrogate markers of oxidative stress were observed. It was also studied alone in a compassionate use program using a single patient with mitochondrial myopathy, but no results are currently available.
RTA408 activates the cytoprotective transcription factor nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2, a.k.a. NFE2L2), a master regulator of cellular antioxidant responses (Reisman et al, 2014). This compound has entered clinical trials for the treatment of mitochondrial myopathy (phase 2) and is reported to stimulate mitochondrial biogenesis and function (Neymotin et al, 2011), and increase the expression of cellular antioxidant defense systems (Saha et al, 2010).
Curcumin, a biomolecule from the golden spice turmeric (Curcuma longa), can modulate multiple cell‐signaling pathways and displays antioxidant properties (Gupta et al, 2011; Trujilo et al, 2014). A randomized and controlled phase 3 study in LHON patients carrying the G11778A mtDNA, mutation has been completed but no results have been reported yet.
Various other small‐molecule treatments are currently under investigation but still at the early preclinical stage. For example, a novel cell‐permeable variant of the complex II substrate succinate (NVP015; Neurovive Pharmaceuticals, Lund, Sweden) has been generated to allow bypassing of dysfunctional complex I. In addition, using a mouse model of isolated complex I deficiency (NDUFS4 −/− mice; Kruse et al, 2008), positive effects of the mTOR inhibitor rapamycin (Johnson et al, 2013, 2015), nicotinamide riboside (a natural precursor of NAD+) (Karamanlidis et al, 2013), and inhibition of poly ADP ribose polymerase (PARP) have been reported (Felici et al, 2014). Both nicotinamide riboside and PARP inhibition are thought to improve mitochondrial function by enhancing intracellular NAD+ levels (Cerutti et al, 2014). Further potential strategies, including mitochondria‐targeted small molecules, have recently been presented elsewhere (e.g., Horobin et al, 2007; Karamanlidis et al, 2013; Apostolova & Victor, 2014; Khan et al, 2014; Malti et al, 2014; Peralta et al, 2015; Rai et al, 2015; Viscomi et al, 2015).
Future perspectives
There is an unmet need for development of clinically relevant treatment strategies for children (and adults) suffering from mitochondrial disease. Developing such a treatment faces many challenges including the small number of established diagnostic and prognostic biomarkers, the lack of truly objective and validated outcome measures that reflect daily life activities/well‐being, and the small number of homogenous patients. To systematically overcome these hurdles, (more) international collaboration between mitochondrial expert centers, patient organizations, industrial partners, and other stakeholders, as well as sufficient funding options, is essential. The tasks ahead include the establishment of guidelines for high‐quality studies, and to improve trial design, for instance, by harmonization of outcome measures.
Conflict of interest
JAMS is the founding CEO of Khondrion ( www.khondrion.com), a Radboud University Medical Center spin‐off biotech company. JB is the COO, and PHGMW and WJHK are scientific consultants of this company.
Pending issues.
Outcome measures: The validation and harmonization of objective clinically relevant outcome measures in children with mitochondrial disease is a crucial step in the interpretation of drug effectiveness. This requires an active stimulation of patient participation and warrants further international collaboration. The same holds true for diagnostic biomarkers suited for use during intervention studies and prognosis.
Natural history studies: Due to the relatively limited number of pediatric patients in which a genetic defect is firmly established, international initiatives are required for proper intervention testing.
Clinical trials: Although many different designs of clinical trials exist, their results should be carefully interpreted and include clinical relevant primary endpoints. The latter are dictated by the available population of affected patients.
Genotype–phenotype relationship: It is unclear why mutations in different genes are associated with similar clinical phenotypes and why similar mutations are associated with different phenotypes. Moreover, it is unknown why certain mutations display tissue‐specific effects. Environmental and/or patient‐specific factors might explain such differences and therefore require further exploration.
Acknowledgements
This research was supported by a PM‐Rare (Priority Medicines Rare disorders and orphan diseases) grant from the Netherlands Organization for Health Research and Development‐Medical Sciences (No: 40‐41900‐98‐033), the Stichting Energy4All ( www.energy4all.eu), and the CSBR (Centres for Systems Biology Research) initiative from the Netherlands Organisation for Scientific Research (NWO; No: CSBR09/013V). The authors express their gratitude to all RCMM and Khondrion team members. We apologize to those authors whose articles we were unable to cite because of space limitations.
EMBO Mol Med (2016) 8: 311–327
See the Glossary for abbreviations used in this article.
See also: A Suomalainen (October 2015)
References
- Alam NM, Mills WC 4th, Wong AA, Douglas RM, Szeto HH, Prusky GT (2015) A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech 8: 701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano LN, Lowes LP, Berry KM, Yin H, Dvorchik I, Flanigan KM, Cripe L, Mendell JR (2014) P.1: Pilot study evaluating motivation on the performance of timed walking in boys with Duchenne muscular dystrophy. Neuromuscul Disord 24: 860 [Google Scholar]
- Apostolova N, Victor VM (2014) Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxid Redox Signal 22: 686–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine EF, Adams HR, Mink JW (2013) Clinical trials in rare disease: challenges and opportunities. J Child Neurol 28: 1142–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balke B (1963) A simple field test for the assessment of physical fitness. Rep Civ Aeromed Res Inst US 1–8 [PubMed] [Google Scholar]
- Barth PG, Scholte HR, Berden JA, Van der Klei‐Van Moorsel JM, Luyt‐Houwen IE, Van‘t Veer‐Korthof ET, Van der Harten JJ, Sobotka‐Plojhar MA (1983) An X‐linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 62: 327–355 [DOI] [PubMed] [Google Scholar]
- Bernier FP, Boheh A, Dennet X, Chow CW, Cleary MA, Thorburn DR (2002) Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 59: 1406–1411 [DOI] [PubMed] [Google Scholar]
- Berry DA (2006) Bayesian clinical trials. Nat Rev Drug Discov 5: 27–36 [DOI] [PubMed] [Google Scholar]
- Besouw M, Tangerman A, Cornelissen E, Rioux P, Levtchenko E (2012) Halitosis in cystinosis patients after administration of immediate‐release cysteamine bitartrate compared to delayed‐release cysteamine bitartrate. Mol Genet Metab 107: 234–236 [DOI] [PubMed] [Google Scholar]
- Blanchet L, Buydens MC, Smeitink JAM, Willems PHGM, Koopman WJH (2011) Isolated mitochondrial complex I deficiency: explorative data analysis of patient cell parameters. Curr Pharm Des 17: 4023–4033 [DOI] [PubMed] [Google Scholar]
- Blanchet L, Smeitink JAM, van Emst‐de Vries SE, Vogels C, Pellegrini M, Jonckheere AI, Rodenburg RJ, Buydens LM, Beyrath J, Willems PHGM et al (2015) Quantifying small molecule phenotypic effects using mitochondrial morpho‐functional fingerprinting and machine learning. Sci Rep 5: 8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessberger H, Binder T (2010) Two dimensional speckle tracking echocardiography: basic principles. Heart 96: 716–722 [DOI] [PubMed] [Google Scholar]
- van Bon BW, Oortveld MA, Nijtmans LG, Fenckova M, Nijhof B, Besseling J, Vos M, Kramer JM, de Leeuw N, Castells‐Nobau A et al (2013) CEP89 is required for mitochondrial metabolism and neuronal function in man and fly. Hum Mol Gen 22: 3138–3151 [DOI] [PubMed] [Google Scholar]
- Bouabe H, Fässler R, Heesemann J (2008) Improvement of reporter activity by IRES‐mediated polycistronic reporter system. Nucl Acids Res 36: e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner HI, Ravelli A (2009) loping outcome measures for paediatric rheumatic diseases. Best Pract Res Clin Rheumatol 23: 609–624 [DOI] [PubMed] [Google Scholar]
- Casey AF, Wang X, Osterling K (2012) Test‐retest reliability of the 6‐minute walk test in individuals with Down syndrome. Arch Phys Med Rehabil 93: 2068–2074 [DOI] [PubMed] [Google Scholar]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J et al (2014) NAD‐dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab 19: 1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF (2015) Mitochondrial disease in adults: what's old and what's new. EMBO Mol Med 7: 1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow SC, Chang M (2008) Adaptive design methods in clinical trials ‐ a review. Orphanet J Rare Dis 3: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu C, Kassai B, Fisch R, Chiron C, Alberti C, Guerrini R, Rosati A, Pons G, Tiddens H, Chabaud S, Aarons L, Bajard A, Ballot C, Bertrand Y, Bretz F, Caudri D, Castellan C, Chabaud S, Cornu C, Dufour F, Dunger‐Baldauf C, Dupont JM, Fisch R, Guerrini R, Jullien V, Kassaï B, Nony P, Ogungbenro K, Pérol D, Pons G, Tiddens H, Rosati A, Alberti C, Chiron C, Kurbatova P, Nabbout R (2013) Experimental designs for small randomised clinical trials: an algorithm for choice. Orphanet J Rare Dis 8: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRe JT, Vasta V, Penn J, Tran NT, Hahn SH (2013) Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med Genet 14: 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud D, Griffin H, Douroudis K, Kleinle S, Eglon G, Pyle A, Chinnery PF, Horvath R (2015) Whole exome sequencing and the clinician: we need clinical skills and functional validation in variant filtering. J Neurol 262: 1673–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks I, Koene S, Verbruggen R, Smeitink JAM, Jansen M, de Groot IJM (2015) The assisted 6‐minute cycling test: An exploratory study in children. Muscle Nerve doi: 10.1002/mus.25021 [DOI] [PubMed] [Google Scholar]
- Distelmaier F, Visch HJ, Smeitink JA, Mayatepek E, Koopman WJH, Willems PHGM (2009) The antioxidant Trolox restores mitochondrial membrane potential and Ca2+‐stimulated ATP production in human complex I deficiency. J Mol Med 87: 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distelmaier F, Valsecchi F, Forkink M, van Emst‐de Vries S, Swarts HG, Rodenburg RJ, Verwiel ET, Smeitink JA, Willems PHGM, Koopman WJH (2012) Trolox‐sensitive reactive oxygen species regulate mitochondrial morphology, oxidative phosphorylation and cytosolic calcium handling in healthy cells. Antioxid Redox Signal 17: 1657–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distelmaier F, Valsecchi F, Liemburg‐Apers DC, Lebiedzinska M, Rodenburg RJ, Heil S, Keijer J, Fransen J, Imamura H, Danhauser K et al (2015) Mitochondrial dysfunction in primary human fibroblasts triggers an adaptive cell survival program that requires AMPK‐α. Biochim Biophys Acta 1852: 529–540 [DOI] [PubMed] [Google Scholar]
- Dubrovsky A, Chloca F, Corderi JR, Jauregui A (2013) P.17.3 Changes in the six minute walk test (6MWT) in Pompe disease. Neuromuscul Disord 23: 827 [Google Scholar]
- Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD et al (2012) Initial experience in the treatment of inherited mitochondrial disease with EPI‐743. Mol Genet Metab 105: 91–102 [DOI] [PubMed] [Google Scholar]
- Etzioni R, Gulati R, Lin DW (2015) Measures of survival benefit in cancer drug development and their limitations. Urol Oncol 33: 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felici R, Cavone L, Lapucci A, Guasti D, Bani D, Chiarugi A (2014) PARP inhibition delays progression of mitochondrial encephalopathy in mice. Neurotherapeutics 11: 651–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Martos C, Guerrero A, Minsky B (2012) Clinically relevant study end points in rectal cancer. Recent Results Cancer Res 196: 3–19 [DOI] [PubMed] [Google Scholar]
- Forkink M, Manjeri GR, Liemburg‐Apers DC, Nibbeling E, Blanchard M, Wojtala A, Smeitink JAM, Wieckowski MR, Willems PHGM, Koopman WJH (2014) Mitochondrial hyperpolarization during chronic complex I inhibition is sustained by low activity of complex II, III, IV and V. Biochim Biophys Acta 1837: 1247–1256 [DOI] [PubMed] [Google Scholar]
- Forkink M, Basit F, Teixeira J, Swarts HG, Koopman WJH, Willems PHGM (2015) Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol 6: 607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LS, Farmer JM, Perlman S, Wilmot G, Gomez CM, Bushara KO, Mathews KD, Subramony SH, Ashizawa T, Balcer LJ et al (2010) Measuring the rate of progression in Friedreich ataxia: implications for clinical trial design. Mov Disord 25: 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V, Petronilli V, Ghelli A, Carelli V, Rugolo M, Lenaz G, Bernardi P (2011) The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta 1817: 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EI, Martin J, Maher A, Thornhill RE, Moran GR, Tarnopolsky MA (2010) A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve 42: 739 [DOI] [PubMed] [Google Scholar]
- Golubitzky A, Phyllis D, Weismann S, Link G, Wikstrom JD, Saada A (2011) Screening for active small molecules in mitochondrial complex I deficient patient's fibroblasts, reveals AICAR as the most beneficial compound. PLoS One 6: e26883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady JP, Campbell G, Ratnaike T, Blakely EL, Falkous G, Nesbitt V, Schaefer AM, McNally RJ, Gorman GS, Taylor RW et al (2014) Disease progression in patients with single, large‐scale mitochondrial DNA deletions. Brain 137: 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta D, Lammersfeld CA, Vashi PG, King J, Dahlk SL, Grutsch JF, Lis CG (2009) Bioelectrical impedance phase angle in clinical practice: implications for prognosis in stage IIIB and IV non‐small cell lung cancer. BMC Cancer 9: 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SC, Patchva S, Aggarwal BB (2011) Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J 15: 195–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Haberberger B, Frisch EM, Wieland T, Iuso A, Gorza M, Strecker V, Graf E, Mayr JA, Herberg U et al (2012) Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet 49: 277–283 [DOI] [PubMed] [Google Scholar]
- Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, Dallmann R, Gueven N (2011) NQO1‐dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS One 6: e17963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V, Debyser Z, Callewaert G, Koopman WJH, Willems PHGM et al (2011) Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci 124: 1115–1125 [DOI] [PubMed] [Google Scholar]
- Hoefs SJ, Dieteren CE, Distelmaier F, Janssen RJ, Epplen A, Swarts HG, Forkink M, Rodenburg RJ, Nijtmans LG, Willems PH et al (2008) NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet 82: 1306–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Honnen S, Mayatepek E, Wätjen W, Koopman WJH, Bossinger O, Distelmaier F (2012) MICS‐1 interacts with mitochondrial ATAD‐3 and modulates lifespan in C elegans . Exp Gerontol 47: 270–275 [DOI] [PubMed] [Google Scholar]
- Horobin RW, Trapp S, Weissig V (2007) Mitochondriotropics: a review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria. J Contr Rel 121: 125–136 [DOI] [PubMed] [Google Scholar]
- Jaber S, Polster BM (2015) Idebenone and neuroprotection: antioxidant, pro‐oxidant, or electron carrier? J Bioenerg Biomembr 47: 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies JL (2013) Barth syndrome. Am J Med Genet C Semin Med Genet 163C: 198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Li J, Xiao X, Li S, Jia X, Sun W, Guo X, Zhang Q (2014) Detection of mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 families with early‐onset high myopia by exome sequencing. Invest Ophthalmol Vis Sci 56: 339–345 [DOI] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A et al (2013) mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342: 1524–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Bitto A, Castanza A, Gagnidze A, Gonzalez B, Gupta K, Hui J, Jarvie C, Johnson BM et al (2015) Dose‐dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet 22: 247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Huigsloot M, Lammens M, Jansen J, van den Heuvel LP, Spiekerkoetter U, von Kleist‐Retzow JC, Forkink M, Koopman WJ, Szklarczyk R et al (2011) Restoration of complex V deficiency caused by a novel deletion in the human TMEM70 gene normalizes mitochondrial morphology. Mitochondrion 11: 954–963 [DOI] [PubMed] [Google Scholar]
- Jonckheere AI, Renkema GH, Bras M, van den Heuvel LP, Hoischen A, Gilissen C, Nabuurs SB, Huynen MA, de Vries MC, Smeitink JA et al (2013) A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 136: 1544–1554 [DOI] [PubMed] [Google Scholar]
- Kang PB (2013) Beyond the gowers sign: measuring outcomes in Duchenne muscular dystrophy. Muscle Nerve 48: 315–317 [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia‐Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, Sendensky MM, Morgan PG, Wang W, Tian R (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X et al (2006) Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66: 324–330 [DOI] [PubMed] [Google Scholar]
- Kerr DS (2013) Review of clinical trials for mitochondrial disorders: 1997‐2012. Neurotherapeutics 10: 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J et al (2014) Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med 6: 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Salemi R, Sugiana C, Othake A, Parry L, Bell KM, Kirk EP, Boneh A, Taylor RW, Dahl HH et al (2004) NDUFS6 mutations are a novel cause of neonatal mitochondrial complex I deficiency. J Clin Invest 114: 83–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisler JE, Whittaker RG, McFarland R (2010) Mitochondrial diseases in childhood: a clinical approach to investigation and management. Dev Med Child Neurol 52: 422–433 [DOI] [PubMed] [Google Scholar]
- Koene S, de Laat P, van Tienoven DH, Vriens D, Brandt AM, Sweep FC, Rodenburg RJ, Donders AR, Janssen MC, Smeitink JAM (2014) Serum FGF21 levels in adult m.3243A>G carriers: clinical implications. Neurology 83: 125–133 [DOI] [PubMed] [Google Scholar]
- Koene S, de Laat P, van Tienoven DH, Weijers G, Vriens D, Sweep FC, Timmermans J, Kapusta L, Janssen MC, Smeitink JAM (2015) Serum GDF15 levels correlate to mitochondrial disease severity and myocardial strain, but not to disease progression in adult m.3243A>G carriers. JIMD Rep 24: 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene S, Wortmann SB, de Vries MC, Jonckheere AI, Morava E, de Groot IJ, Smeitink JA (2013a) Developing outcome measures for pediatric mitochondrial disorders: which complaints and limitations are most burdensome to patients and their parents? Mitochondrion 13: 15–24 [DOI] [PubMed] [Google Scholar]
- Koene S, Jansen M, Verhaak CM, De Vrueh RL, De Groot IJ, Smeitink JAM (2013b) Towards the harmonization of outcome measures in children with mitochondrial disorders. Dev Med Child Neurol 55: 698–706 [DOI] [PubMed] [Google Scholar]
- Koene S, Smeitink JAM (2011) Mitochondrial Medicine, a clinical guideline, ISBN 978‐90‐817737‐0‐6
- Koopman WJH, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JAM, Willems PHGM (2005) Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 289: C881–C890 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Verkaart S, Visch HJ, van Emst‐de Vries S, Nijtmans LG, Smeitink JAM, Willems PHGM (2007) Human NADH: ubiquinone oxidoreductase deficiency: radical changes in mitochondrial morphology? Am J Physiol Cell Physiol 293: C22–C29 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Verkaart S, van Emst‐de Vries SE, Grefte S, Smeitink JAM, Nijtmans LG, Willems PHGM (2008) Mitigation of NADH: ubiquinone oxidoreductase deficiency by chronic Trolox treatment. Biochim Biophys Acta 1777: 853–859 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, Willems PHGM (2010) Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal 12: 1431–1470 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Willem PHGM, Smeitink JAM (2012) Monogenic mitochondrial disorders. N Engl J Med 366: 1132–1141 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Distelmaier F, Smeitink JAM, Willems PHGM (2013) OXPHOS mutations and neurodegeneration. EMBO J 32: 9–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koufaki M (2014) Therapeutic applications of lipoic acid: a patent review (2011‐2014). Expert Opin Ther Pat 24: 993–1005 [DOI] [PubMed] [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD (2008) Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7: 312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake NJ, Compton AG, Rahman S, Thorburn DR (2016) Leigh Syndrome: one disorder, more than 75 monogenic causes. Ann Neurol 79: 190–203 [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim J, Kim HD, Lee JS, Lee YM (2010) Initial experiences with proton MR spectroscopy in treatment monitoring of mitochondrial encephalopathy. Yonsei Med J 51: 672–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman G, Gueguen N, Desquiret‐Dumas V, Kane MS, Wettervald C, Chupin S, Chevrollier A, Lebre AS, Bonnefont JP, Barth M et al (2015) Assembly defects induce oxidative stress in inherited mitochondrial complex I deficiency. Int J Biochem Cell Biol 65: 91–103 [DOI] [PubMed] [Google Scholar]
- Lieber DS, Calvo SE, Shanahan K, Slate NG, Liu S, Hershman SG, Gold NB, Chapman BA, Thorburn DR, Berry GT et al (2013) Targeted exome sequencing of suspected mitochondrial disorders. Neurology 80: 1762–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liemburg‐Apers DC, Imamura H, Forkink M, Nooteboom M, Swarts HG, Brock R, Smeitink JAM, Willems PHGM, Koopman WJH (2011) Quantitative glucose and ATP sensing in mammalian cells. Pharm Res 28: 2745–2757 [DOI] [PubMed] [Google Scholar]
- Liemburg‐Apers DC, Schirris TJ, Russel FG, Willems PHGM, Koopman WJH (2015) Mitoenergetic dysfunction triggers a rapid compensatory increase in steady‐State glucose flux. Biophys J 109: 1372–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightowlers RN, Taylor RW, Turnbull DM (2015) Mutations causing mitochondrial disease: what is new and what challenges remain? Science 349: 1494–1499 [DOI] [PubMed] [Google Scholar]
- Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B (1962) A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest 41: 1776–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malti RH, Jessulat M, Jin K, Musso G, Vlasblom J, Phanse S, Zhang Z, Babu M (2014) Mitochondrial targets for pharmacological intervention in human disease. J Prot Res 14: 5–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi‐Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V et al (2012) EPI‐743 reverses the progression of the pediatric mitochondrial disease‐genetically defined Leigh Syndrome. Mol Genet Metab 107: 383–388 [DOI] [PubMed] [Google Scholar]
- Mayr JA, Haack TB, Freisinger P, Karall D, Makowski C, Koch J, Feichtinger RG, Zimmermann FA, Rolinski B, Ahting U et al (2015) Spectrum of combined respiratory chain defects. J Inherit Metab Dis 38: 629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi‐osmotic type of mechanism. Nature 191: 144–148 [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Thomas KJ, Koopman WJH, Klaffke S, Abou‐Sleiman P, Olpin S, Wood NW, Willems PHGM, Smeitink JAM, Cookson MR et al (2008) Mitochondrial function and morphology are impaired in parkin‐mutant fibroblasts. Ann Neurol 64: 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt V, McFarland R (2011) Phenotypic spectrum of m.3243A>G mitochondrial DNA mutation in children. Arch Dis Child 96: A28 [Google Scholar]
- Neymotin A, Calingasan N, Wile E (2011) Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Rad Biol Med 51: 88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngu LH, Nijtmans LG, Distelmaier F, Venselaar H, van Emst‐de Vries SE, van den Brand MA, Stoltenborg BJ, Wintjes LT, Willems PH, van den Heuvel LP et al (2012) A catalytic defect in mitochondrial respiratory chain complex I due to a mutation in NDUFS2 in a patient with Leigh syndrome. Biochim Biophys Acta 1822: 168–175 [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JAM, Vogel RO (2012) Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain 135: 12–22 [DOI] [PubMed] [Google Scholar]
- Ohtake A, Murayama K, Mori M, Harashima H, Yamazaki T, Tamaru S, Yamashita Y, Kishita Y, Nakachi Y, Kohda M et al (2014) Diagnosis and molecular basis of mitochondrial respiratory chain disorders: exome sequencing for disease gene identification. Biochim Biophys Acta 1840: 1355–1359 [DOI] [PubMed] [Google Scholar]
- Ostergaard E, Weraarpachai W, Ravn K, Born AP, Jonson L, Duno M, Wibrand F, Shoubridge EA, Vissing J (2015) Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J Med Genet 52: 203–207 [DOI] [PubMed] [Google Scholar]
- Palmieri F (2008) Diseases caused by defects of mitochondrial carriers: a review. Biochim Biophys Acta 1777: 564–578 [DOI] [PubMed] [Google Scholar]
- Pastore A, Petrillo S, Tozzi G, Carrozzo R, Martinelli D, Dionisi‐Vici C, Di Giovamberardino G, Ceravolo F, Klein MB, Miller G et al (2013) Glutathione: a redox signature in monitoring EPI‐743 therapy in children with mitochondrial encephalomyopathies. Mol Genet Metab 109: 208–214 [DOI] [PubMed] [Google Scholar]
- Peralta S, Torraco A, Iommarini L, Diaz F (2015) Mitochondrial diseases part III: therapeutic interventions in mouse models of oxphos deficiencies. Mitochondrion 23: 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu‐Wai‐Man P et al (2013) New treatments for mitochondrial disease‐no time to drop our standards. Nat Rev Neurol 9: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MM, Romestaing C, Hepple RT (2011) Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One 6: e18317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss D, Sattar N, McMurray JJ (2011) A systematic review of event rates in clinical trials in diabetes mellitus: the importance of quantifying baseline cardiovascular disease history and proteinuria and implications for clinical trial design. Am Heart J 161: 210–219 [DOI] [PubMed] [Google Scholar]
- Pritchard JF, Jurima‐Romet M, Reimer MLJ, Mortimer E, Rolfe B, Cayen MN (2003) making better drugs: decision gates in non‐clinical drug development. Nat Rev Drug Discov 2: 542–553 [DOI] [PubMed] [Google Scholar]
- van Rahden VA, Fernandez‐Vizarra E, Alawi M, Brand K, Fellmann F, Horn D, Zeviani M, Kutsche K (2015) Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome. Am J Hum Genet 96: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai PK, Russell OM, Lightowlers RN, Turnbull DM (2015) Potential compounds for the treatment of mitochondrial disease. Br Med Bull 116: 5–18 [DOI] [PubMed] [Google Scholar]
- Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW (2014) Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin. Arch Dermatol Res 306: 447–454 [DOI] [PubMed] [Google Scholar]
- Rigaud C, Rigaud C, Lebre AS, Touraine R, Beaupain B, Ottolenghi C, Chabli A, Ansquer H, Ozsahin H, Di FS et al (2013) Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis 8: 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AE, Nixon C, Steward CG, Gauvreau K, Maisenbacher M, Fletcher M, Geva J, Byrne BJ, Spencer CT (2012) The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A 158A: 2726–2732 [DOI] [PubMed] [Google Scholar]
- Rodan LH, Wells GD, Banks L, Thompson S, Schneiderman JE, Tein I (2015) L‐Arginine Affects Aerobic Capacity and Muscle Metabolism in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke‐Like Episodes) Syndrome. PLoS One 10: e0127066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tarnopolsky MA (2007) Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 35: 235–242 [DOI] [PubMed] [Google Scholar]
- Roestenberg P, Manjeri GR, Valsecchi F, Smeitink JAM, Willems PHGM, Koopman WJH (2012) Pharmacological targeting of mitochondrial complex I deficiency: the cellular level and beyond. Mitochondrion 12: 57–65 [DOI] [PubMed] [Google Scholar]
- Sadun AA, Chicani CF, Ross‐Cisneros FN, Barboni P, Thoolen M, Shrader WD, Kubis K, Carelli V, Miller G (2012) Effect of EPI‐743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol 69: 331–338 [DOI] [PubMed] [Google Scholar]
- Saha P, Reddy V, Knopleva M (2010) The triterpenoid 2‐cyano‐3,12‐dioxooleana‐1,9‐dien‐28‐oic‐acid methyl ester has potent anti‐diabetic effects in diet‐induced diabetic mice and Lepr(db/db) mice. J Biol Chem 285: 40581–40592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirris TJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BG, Brandt U, Koopman WJH, Beyrath JD, Rodenburg RJ et al (2015) Statin‐induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab 22: 399–407 [DOI] [PubMed] [Google Scholar]
- Schöckel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G, Algire C, Hägebarth A, Willems PHGM, Kopitz C et al (2015) Targeting mitochondrial complex I using BAY 87‐2243 reduces melanoma tumor growth. Cancer Metab 3: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Dröse S, Brandt U, Hoffmann GF, Ter Laak H et al (2006) Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J 398: 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeitink JAM (2003) Mitochondrial disorders: clinical presentation and diagnostic dilemmas. J Inherit Metab Dis 26: 199–207 [DOI] [PubMed] [Google Scholar]
- Smeitink JAM, van den Heuvel LWPJ, Koopman WJH, Nijtmans LGJ, Ugalde C, Willems PHGM (2004) Cell biological consequences of mitochondrial NADH: ubiquinone oxidoreductase deficiency. Curr Neurovasc Res 1: 29–40 [DOI] [PubMed] [Google Scholar]
- Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, Berthy J, Redfearn SP, Byrne BJ (2006) Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118: e337–e346 [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, deGrauw TJ, Feigenbaum AS, Hoppel C, Kerr DS, McCandless SE, Miles MV, Robinson BH, Tang PH (2012) Design and implementation of the first randomized controlled trial of coenzyme Q10 in children with primary mitochondrial diseases. Mitochondrion 12: 623–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A, Elo JM, Pietiläinen KH, Hakonen AH, Sevastianova K, Korpela M, Isohanni P, Marjavaara SK, Tyni T, Kiuru‐Enari S et al (2011) FGF‐21 as a biomarker for muscle‐manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol 10: 806–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Zoratti M (2014) Mitochondrial channels: ion fluxes and more. Physiol Rev 94: 519–608 [DOI] [PubMed] [Google Scholar]
- Szeto HH (2014) First‐in‐class cardiolipin‐protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171: 2029–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, van den Brand MA, Hendriks‐Franssen MG, Gilissen C, Veltman JA et al (2013) A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet 22: 656–667 [DOI] [PubMed] [Google Scholar]
- Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A et al (2014) Use of whole‐exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312: 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triepels RH, van den Heuvel LP, Loeffen JL, Buskens CA, Smeets RJ, Rubio Gozalbo ME, Budde SM, Mariman EC, Wijburg FA, Barth PG et al (1999) Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol 45: 787–790 [DOI] [PubMed] [Google Scholar]
- Trujilo J, Granados‐Castro LF, Zazueta C, Andérica‐Romero AC, Chirino YI, Pedraza‐Chaverrí J (2014) Mitochondria as a target in the therapeutic properties of curcumin. Arch Pharm (Weinheim) 347: 873–884 [DOI] [PubMed] [Google Scholar]
- Tugwell P, Boers M (1993) OMERACT conference on outcome measures in rheumatoid arthritis clinical trials: introduction. J Rheumatol 20: 528–530 [PubMed] [Google Scholar]
- Vafai SB, Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491: 374–383 [DOI] [PubMed] [Google Scholar]
- Verkaart S, Koopman WJH, van Emst‐de Vries SE, Nijtmans LG, van den Heuvel LW, Smeitink JAM, Willems PHGM (2007a) Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim Biophys Acta 1772: 373–381 [DOI] [PubMed] [Google Scholar]
- Verkaart S, Koopman WJH, Cheek J, van Emst‐de Vries SE, van den Heuvel LW, Smeitink JAM, Willems PHGM (2007b) Mitochondrial and cytosolic thiol redox state are not detectably altered in isolated human NADH: ubiquinone oxidoreductase deficiency. Biochim Biophys Acta 1772: 1041–1051 [DOI] [PubMed] [Google Scholar]
- Valsecchi F, Koopman WJH, Manjeri GR, Rodenburg RJ, Smeitink JAM, Willems PHGM (2010) Complex I disorders: causes, mechanisms, and development of treatment strategies at the cellular level. Dev Disabil Res Rev 16: 175–182 [DOI] [PubMed] [Google Scholar]
- Valsecchi F, Monge C, Forkink M, de Groof AJ, Benard G, Rossignol R, Swarts HG, van Emst‐de Vries SE, Rodenburg RJ, Calvaruso MA et al (2012) Metabolic consequences of NDUFS4 gene deletion in immortalized mouse embryonic fibroblasts. Biochim Biophys Acta 1817: 1925–1936 [DOI] [PubMed] [Google Scholar]
- Valsecchi F, Grefte S, Roestenberg P, Joosten‐Wagenaars J, Smeitink JAM, Willems PHGM, Koopman WJH (2013) Primary fibroblasts of NDUFS4(‐/‐) mice display increased ROS levels and aberrant mitochondrial morphology. Mitochondrion 13: 436–443 [DOI] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M (2011) In vivo correction of COX deficiency by activation of the AMPK/PGC‐1α axis. Cell Metab 14: 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Zeviani M (2015) Emerging concepts in the therapy of mitochondrial disease. Biochim Biophys Acta 1847: 544–557 [DOI] [PubMed] [Google Scholar]
- Wenz T, Diaz F, Spiegelman BM, Moraes CT (2008) Activation of the PPAR/PGC‐1a pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab 8: 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Willems PHGM, Wanschers BF, Esseling J, Szklarczyk R, Kudla U, Duarte I, Forkink M, Nooteboom M, Swarts H, Gloerich J et al (2013) BOLA1 is an aerobic protein that prevents mitochondrial morphology changes induced by glutathione depletion. Antioxid Redox Signal 18: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems PHGM, Rossignol R, Dieteren CE, Murphy MP, Koopman WJH (2015) Redox homeostasis and mitochondrial dynamics. Cell Metab 22: 207–288 [DOI] [PubMed] [Google Scholar]
- Wolf NI, Smeitink JAM (2002) Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology 59: 1402–1405 [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ (2015) Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis 38: 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Nishino I, Nonaka I, Goto Y (2008) Genotype and phenotype analyses in 136 patients with single large‐scale mitochondrial DNA deletions. J Hum Genet 53: 598–606 [DOI] [PubMed] [Google Scholar]
- Yatsuga S, Suomalainen A (2012) Effect of bezafibrate treatment on late‐onset mitochondrial myopathy in mice. Hum Mol Genet 21: 526–535 [DOI] [PubMed] [Google Scholar]
- Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, Kakuma T, Kojima T, Ito M, Tanaka M, Saiki R et al (2015) Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol 78: 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]