

Abstract
Mitochondrial dysfunction has been linked to both cellular senescence and ageing. Despite the relationship, it is still unclear whether mitochondria have a causal role in senescence. In this issue of The EMBO Journal, Correia‐Melo et al (2016) combine targeted depletion of mitochondria with impairment of their biogenesis to demonstrate that decreased numbers of mitochondria impair the senescence response. Their results suggest that targeting mitochondria could reduce the detrimental effects of senescence during ageing.
Subject Categories: Ageing, Cell Cycle, Metabolism
Cellular senescence is a stress response in which damaged or ageing cells stop proliferating and undergo distinct changes in their transcription, chromatin organization and meta‐bolism. Senescent cells also produce a complex mix of secreted factors including matrix metalloproteinases, growth factors and pro‐inflammatory cytokines, collectively termed the senescence‐associated secretory phenotype (SASP) (Salama et al, 2014). The importance of mitochondria in cellular senescence has been linked to their ability to generate reactive oxygen species (ROS). ROS can affect cellular senescence by inducing or stabilizing the DNA damage response (DDR), leading to a permanent growth arrest. However, little is known about how mitochondrial ROS affect other features of senescence like the SASP. Moreover, the role of mitochondria in senescence extends beyond ROS production and other sources of ROS can also be important for the development of senescence. Therefore, it needs to be clarified whether mitochondria can be effectors of senescence, and if so, by which mechanisms.
Passos and colleagues (Correia‐Melo et al, 2016) illuminated this matter by studying senescence in fibroblasts lacking mitochondria. This was achieved by taking advantage of an elegant approach: Parkin‐mediated mitophagy allows the generation of cells that are viable but depleted of mitochondria (Narendra et al, 2008). Combining this with complementary approaches, the authors found that the absence of mitochondria affects multiple features of senescence induced by ionizing radiation, such as the production of ROS and the SASP. Moreover, many transcriptional changes observed during senescence were reversed upon depletion of mitochondria. Paradoxically, the reduction in the expression of the cell cycle inhibitors p16INK4a and p21CIP1 was not sufficient to rescue cell proliferation.
This impairment of senescence was not due to insufficient energy production. Indeed, senescent cells lacking mitochondria displayed higher ATP levels due to increased glycolysis. Therefore, the execution of the senescence programme does not depend on ATP levels per se, but rather on the status of mitochondrial oxidative metabolism. This is in agreement with previous results from the Peeper laboratory, which reported that an increase of mitochondrial respiration through activation of pyruvate dehydrogenase (PDH) was required for the execution of oncogene‐induced senescence (OIS) (Kaplon et al, 2013). Importantly, Kaplon et al also showed that senescence could be reversed in primary fibroblasts by blocking this OIS‐specific metabolic rewiring towards mitochondrial oxidative metabolism.
Mitochondrial mass and mitochondrial oxidation control the senescence phenotype, but the mechanisms have remained elusive. Passos and colleagues (Correia‐Melo et al, 2016) identify a novel pathway which links the DDR with increased mitochondrial mass through transcriptional activation of the mitochondrial biogenesis regulator PGC‐1β. This increase in mitochondrial mass is dependent on ATM, AKT and mTORC1. Therefore, the authors propose a model in which the engagement of the DDR by different senescent triggers initiates a positive feedback loop that drives mTOR activation, PGC‐1β‐dependent mitochondrial biogenesis and ROS production. The production of ROS maintains the persistent activation of the DDR, stabilizing cellular senescence and its pro‐oxidant and pro‐inflammatory phenotypes (Fig 1).
Figure 1. Mitochondria are important effectors of the senescence response.
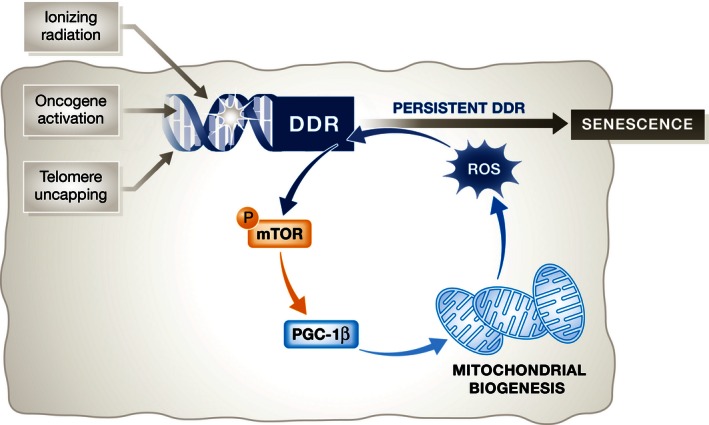
Passos and colleagues (Correia‐Melo et al, 2016) describe that mitochondria form part of an autoregulatory loop involving the DNA damage response (DDR), mTOR and the production of ROS that results in persistent DDR and has a stabilizing effect on senescence.
Mitochondria and cellular senescence are important contributors to ageing and age‐related disease (Lopez‐Otin et al, 2013). Therefore, Passos and colleagues also analysed whether there is a link between mitochondrial content and senescence during ageing (Correia‐Melo et al, 2016). In agreement with their in vitro results, they observed that increased DNA damage in aged hepatocytes correlates with higher mitochondrial content in an mTOR‐dependent fashion. Interestingly, Correia‐Melo et al (2016) suggest that the inhibition of mTOR, which is known to increase lifespan in mice (Harrison et al, 2009), alleviates the age‐promoting features of senescence by affecting mitochondrial mass rather than mitochondrial function.
Since mTOR controls many processes, ranging from protein translation to lipid biosynthesis, its effects on lifespan are probably multifactorial. Among those, the ability of mTOR to control the senescent secretome could explain some of its effects in ageing. The authors propose here that mTOR could regulate the SASP via a complex pathway that involves mitochondrial biogenesis, ROS production and increased DNA damage (Correia‐Melo et al, 2016). Eventually, this activates NF‐κB to induce the SASP. On the other hand, recent work suggests that mTOR controls the translation of known SASP regulators such as MAPKAPK2 (a kinase downstream of p38) (Herranz et al, 2015) or IL1A (Laberge et al, 2015) to directly regulate the SASP. The relative contribution of these factors, as well as yet undiscovered mechanisms by which mTOR can control the SASP, will have to be established.
Previously, the role of mitochondria in ageing has been associated with the accumulation of dysfunctional mitochondria. In this regard, it was recently shown that mitochondrial defects, often associated with ageing, can themselves cause a distinct type of senescence termed MiDAS (mitochondrial dysfunction‐associated senescence; Wiley et al, 2015). Adding to this, the present study suggests that increased mitochondrial mass may also drive age‐related diseases and senescence phenotypes.
It is now apparent that in addition to being a central part of the metabolic reprogramming occurring during senescence (Kaplon et al, 2013), mitochondria can both induce (Wiley et al, 2015) and mediate (Correia‐Melo et al, 2016 ) senescence, so it will be necessary to integrate all these different observations. Moreover, a recent study has shown that autophagy contributes to maintain muscle function during ageing by preventing senescence (Garcia‐Prat et al, 2016). Interestingly, the main consequence of autophagy loss is the accumulation of dysfunctional mitochondria and ROS due to defective mitophagy (Garcia‐Prat et al, 2016). Therefore, identifying ways of targeting mitochondria could be useful to design novel therapies to treat the many age‐related pathologies in which senescence plays a role.
See also: C Correia‐Melo et al (April 2016)
References
- Correia‐Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly‐Y M, Rodriguez‐Cuenca S, Vidal‐Puig A et al (2016) Mitochondria are required for pro‐ageing features of the senescent phenotype. EMBO J 35: 724–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Prat L, Martinez‐Vicente M, Perdiguero E, Ortet L, Rodriguez‐Ubreva J, Rebollo E, Ruiz‐Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Munoz‐Canoves P (2016) Autophagy maintains stemness by preventing senescence. Nature 529: 37–42 [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, Dharmalingam G, Faull P, Carroll T, Martinez‐Barbera JP, Cutillas P, Reisinger F, Heikenwalder M et al (2015) mTOR regulates MAPKAPK2 translation to control the senescence‐associated secretory phenotype. Nat Cell Biol 17: 1205–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene‐induced senescence. Nature 498: 109–112 [DOI] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson‐Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS et al (2015) MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153: 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama R, Sadaie M, Hoare M, Narita M (2014) Cellular senescence and its effector programs. Genes Dev 28: 99–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J (2015) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23: 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]