

Abstract
Ku heterodimer is a DNA binding protein with a prominent role in DNA repair. Here, we investigate whether and how Ku impacts the DNA damage response by acting as a post‐transcriptional regulator of gene expression. We show that Ku represses p53 protein synthesis and p53‐mediated apoptosis by binding to a bulged stem‐loop structure within the p53 5′ UTR. However, Ku‐mediated translational repression of the p53 mRNA is relieved after genotoxic stress. The underlying mechanism involves Ku acetylation which disrupts Ku–p53 mRNA interactions. These results suggest that Ku‐mediated repression of p53 mRNA translation constitutes a novel mechanism linking DNA repair and mRNA translation.
Keywords: mRNA translation, Ku, p53, DNA damage
Subject Categories: DNA Replication, Repair & Recombination; Protein Biosynthesis & Quality Control
Introduction
Genomic instability leads to genetic alterations contributing to cancer development and progression and is therefore considered as a crucial hallmark of cancer. The DNA damage response (DDR) is central for the maintenance of genome stability and includes transcriptional regulation, cell cycle arrest, DNA repair, and apoptosis when DNA breaks are not repaired. The DDR also triggers a broad post‐transcriptional reprogramming modulating the expression of genes involved in DNA repair, cell cycle control, and/or apoptosis. Among the post‐transcriptional processes, mRNA translation emerges as a critical regulator of DDR since it offers several benefits, including response rapidity, reversibility, and control at both global and mRNA‐specific levels. Emerging evidence strongly supports the view that, besides effects on DNA and signaling, DNA repair proteins are involved in the DDR by acting as post‐transcriptional regulators of gene expression (e.g., 1, 2).
The main sensor of double‐stand breaks (DSBs), the most toxic form of DNA damage, in mammalian cells is the protein Ku, a highly abundant protein made up of two subunits, Ku70 and Ku80. Ku and the DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) together form the DNA‐PK complex that plays key roles in the repair of DSBs through the non‐homologous end‐joining (NHEJ) pathway. Accumulating evidences suggest that Ku is an RNA binding protein (RBP) and plays a role in RNA metabolism. Indeed, Ku associates with a specific stem‐loop of the RNA component of the telomerase ribonucleoprotein complex, named hTR/TLC1 3, 4. Importantly, the Ku heterodimer has been found to bind mature polyadenylated transcripts in three recent studies uncovering the in vivo mRNA interactomes of human/mouse cells 5, 6, 7. However, the post‐transcriptional role of Ku and its specific mRNA targets have been poorly investigated. One study found that Ku binds internal ribosomal entry sites that are complex RNA structures mediating translation of specific proteins during cell stress situations 8. In addition, DNA‐PKcs is required for translational reprogramming following DNA damage 9, leading to speculate that Ku might be involved in translational regulation in stress conditions. Intriguingly, both Ku subunits were identified in a screen for proteins binding to the 5′ UTR of the mRNA encoding the tumor suppressor protein p53, a master gene regulator of the DDR 10. While it is now widely accepted that p53 mRNA translational regulation by RBPs contributes to keep p53 protein low in normal conditions (e.g., 10, 11, 12) and to significantly increase p53 levels upon stress (e.g., 13, 14), it has never been investigated whether p53 mRNA translation is coordinated to other steps of the DDR.
Here, we investigated whether, when, and how the DNA repair protein Ku impacts DDR by acting as a post‐transcriptional regulator. We show that Ku represses p53 mRNA translation and function by binding to a stem‐loop structure within the p53 5′ UTR under normal conditions. Post‐translational modifications of Ku after DNA damage relieve this inhibitory mechanism by dissociating Ku from the p53 mRNA.
Results and Discussion
Knockdown of Ku increases p53 expression levels
Since Ku is an essential 15, abundant protein in which the two subunits Ku70 and Ku80 stabilize each other, we set out to define whether endogenous Ku regulates p53 expression by performing siRNA‐mediated depletion of Ku70 and measuring p53 expression. As expected from the well‐known reciprocal stabilization of both Ku subunits 15, Ku80 degradation paralleled siRNA‐mediated silencing of Ku70 (Fig 1A). We found that the level of p53 protein was markedly and significantly increased (up to 3‐fold) upon transient knockdown of Ku70 in HCT116 cells (Fig 1A). Concomitantly, the protein levels of p21 and GADD45α, two main p53‐target genes, were increased (Fig EV1A). A similar effect of Ku on p53 accumulation was observed using the same siRNA in A549 cells (Fig EV1B), a different Ku70 siRNA in HCT116 cells (Fig EV1C) and an siRNA targeting Ku80 (Fig EV1D), ruling out potential cell type‐specific or siRNA off‐target effects. As Ku is involved in the repair of DSBs through NHEJ, Ku depletion could result in unrepaired DNA damage and the activation of DDR pathway evidenced by the phosphorylation of phosphatidylinositol‐3 kinase‐related kinases (PIKKs, including ATM, ATR, and DNA‐PK), γ‐H2AX, and p53. However, siRNA‐mediated depletion of Ku caused neither the phosphorylation of ATM (S1981) (Figs 1B and EV1E) nor the one of p53 (S15) (Figs 1B and EV1E) or of γ‐H2AX (Figs 1C and EV1F). In addition, p53 accumulation after Ku silencing was not suppressed by wortmannin (an inhibitor of PIKKs) (Fig 1D) or by NU‐7441 (a highly potent and selective DNA‐PKcs inhibitor) (Fig 1E), indicating that DNA damage signaling was not the cause of p53 stabilization and that DNA‐PKcs is not involved in this effect. The observation that inhibition of factors involved in DNA repair (specifically, XRCC4 or DNA‐PKcs) did not result in the accumulation of p53 (Fig 1F) further supports our conclusion that partial loss of Ku directly modifies the expression of p53 and that the latter is not the consequence of unrepaired DNA or of siRNA off‐target effects. Previous studies established a functional link between Ku and p53 impacting cancer development. Indeed, Ku deletion exacerbates oncogenesis in p53‐defective mice but reduces oncogenesis in p53‐intact mice 16, 17. Low cancer levels in Ku−/− mice have been attributed to persistent unrepaired DSBs that elevated a p53‐dependent DNA damage response 16. Our results show that transient and partial reduction in Ku levels modifies p53 accumulation, but this is not associated to constitutive cellular damage responses induced by deficiency in repairing spontaneous DNA damage.
Figure 1. Ku downregulation increases p53 expression.

-
A–ITreatment of HCT116 cells with siRNA targeting Ku70 (A–I), ASF (C) (positive control for γ‐H2AX activation 29), XRCC4 (F) or control (A–I) combined or not with a treatment with doxorubicin (Doxo) (positive control for phosphorylation of ATM and p53) (B), wortmannin (WTN) (D), NU‐7441 (E, F), camptothecin (CPT), MG132 (H), or cycloheximide (CHX) (I) followed by Western blot analysis (A–F, H–I) or RT–qPCR analysis (G). (A) The basal levels of p53 normalized to GAPDH were arbitrarily set at 1.0 and the fold change is shown in (ii) (n = 4). In RT–qPCR analysis (G), mRNA levels were standardized against HPRT mRNA (n = 3). The two panels of (C) are portion of the same gel. Statistical analysis by unpaired t‐test (*P < 0.05; **P < 0.01). All error bars reflect SEM.
Figure EV1. Ku knockdown induces p53 accumulation.
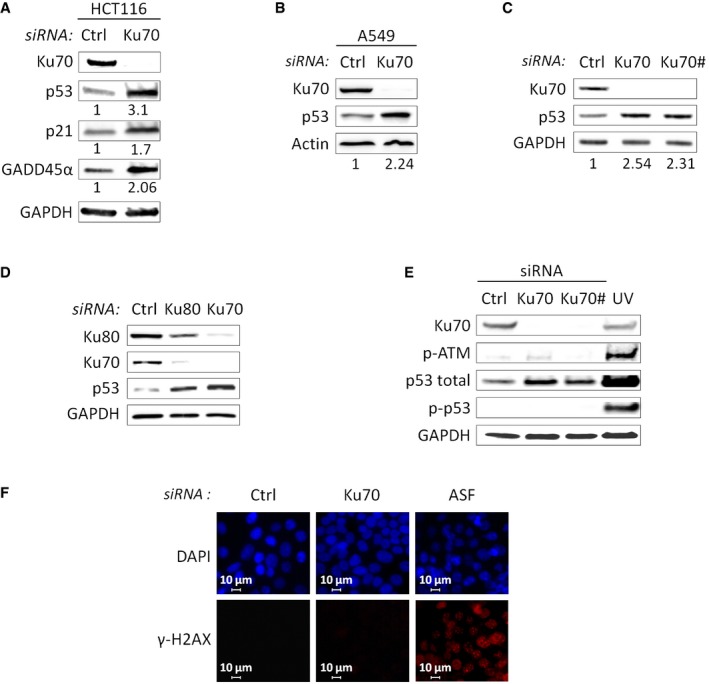
- Western blot analysis of HCT116 cells after Ku70 depletion. The basal levels of p53, p21, and GADD45α normalized to GAPDH were arbitrarily set at 1.0, and the fold change is shown below each lane.
- Western blot analysis of A549 cells after siRNA‐mediated Ku70 or control depletion. The basal levels of p53 normalized to actin were arbitrarily set at 1.0, and the fold change is shown below each lane.
- Western blot analysis of HCT116 cells after Ku70 depletion using two different siRNAs (Ku70 and Ku70#) or after control depletion. The basal levels of p53 normalized to GAPDH were arbitrarily set at 1.0, and the fold change is shown below each lane.
- Western blot analysis of HCT116 cells after Ku70 or Ku80 depletion or after control depletion.
- Western blot analysis of HCT116 cells after siRNA‐mediated Ku70 depletion using two different siRNAs (Ku70 and Ku70#) or control depletion, or after UV irradiation (positive control for phosphorylation of ATM and p53).
- Immunofluorescence experiments were performed using γ‐H2AX antibody and DAPI staining on HCT116 cells treated with siRNA targeting Ku70, ASF (positive control for γ‐H2AX activation 29) or control. Images of a single representative field were shown.
To provide further evidence that Ku directly impacts p53, we defined whether Ku depletion upregulated p53 by transcriptional or post‐transcriptional mechanisms. Knockdown of Ku in HCT116 cells resulted in a 1.3‐fold increase in p53 mRNA levels (Fig 1G) that alone cannot account for more than threefold increase in protein levels (Fig 1A), but did not modify p53 protein stability (Fig 1H). However, when HCT116 cells were treated with the translational inhibitor cycloheximide, p53 upregulation after Ku silencing was lost (Fig 1I), suggesting that Ku likely modifies p53 expression by regulating mRNA translation.
Ku interacts with a stem‐loop structure within the p53 5′ UTR
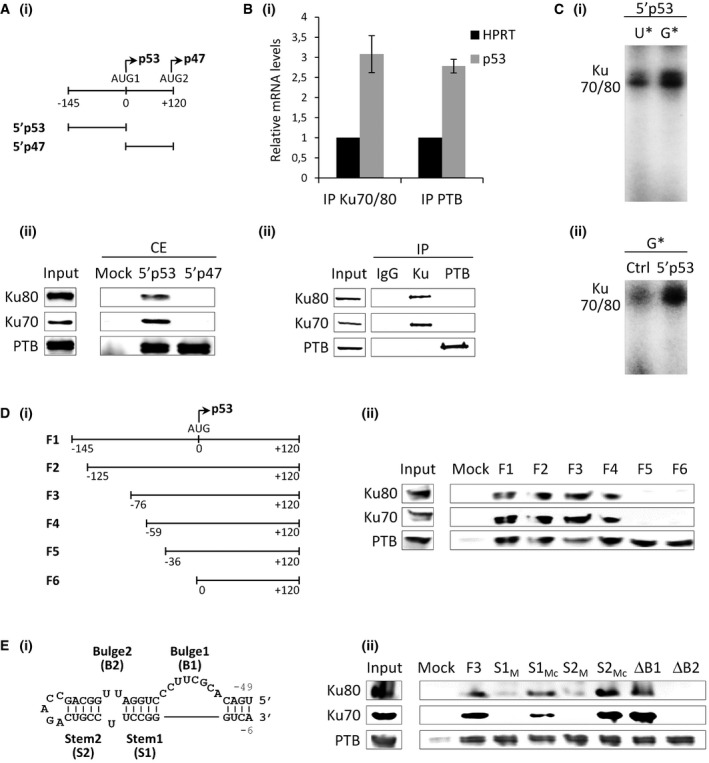
To explore the potential role of Ku in p53 mRNA translation, we first investigated the ability of Ku to bind the p53 transcript. Both subunits of Ku heterodimer were previously shown to interact with the p53 mRNA in a screen for proteins that specifically bind to the p53 5′ UTR 10. The p53 mRNA includes two start codons: AUG1 for synthesis of the full‐length p53 and AUG2 for translation of the N‐truncated p53 isoform (p47) (Fig 2A). To verify that Ku interacts with p53 mRNA, we performed RNA affinity chromatography using in vitro‐transcribed RNAs corresponding to the p53 5′ UTR (5′p53) or to the sequence in between the two AUG codons (5′p47). As shown in Fig 2A, Ku was pulled down only by the p53 5′ UTR RNA either with nuclear (Fig EV2A) or cytoplasmic extracts of HeLa cells. Conversely, the RBP PTB that has been previously shown to regulate p53 translation by binding both regions of the p53 5′‐sequence 18 was retained by both RNA fragments (Figs 2A and EV2A). In addition, Ku did not bind the RNA fragments corresponding to the 5′ UTR of c‐myc and EMCV (Fig EV2B), suggesting that this interaction is specific to the p53 mRNA. To determine whether Ku binds to the p53 mRNA in cellulo, we performed an RNP immunoprecipitation (RIP) assay using cell extracts from HCT116 cells and monitored the endogenous p53 transcript co‐immunoprecipitated with an antibody recognizing only the conformational epitope of the Ku heterodimer or, as a positive control, with the PTB antibody. As shown in Fig 2B, endogenous p53 mRNA was immunoprecipitated to a similar extent by the Ku and PTB antibody compared to HPRT mRNA. Then, UV cross‐linking was used to provide further evidence that Ku interacts with the p53 5′ UTR without requirement of additional protein factors (Figs 2C and EV3A). UV cross‐linking using 32P‐labeled RNA substrates corresponding to the 5′ UTR of p53 in the presence of purified recombinant human Ku gave two bands of ≈70 and 80 kDa (Fig 2C). The UV cross‐linking signal obtained with the p53 5′ UTR was reduced when incubating Ku with a control RNA corresponding to the p53 5′ UTR antisense sequence. These findings together with data showing that the UV cross‐linked complexes were immunoprecipitated with an antibody recognizing the Ku heterodimer (Fig EV3B) suggest that the binding of Ku is direct and specific (Fig 2C). Next, to further delineate the binding sites of Ku in the p53 5′ UTR, we created a series of RNA constructs in which the 5′ terminal region of the p53 transcript was progressively deleted in a 5′‐to‐3′ direction and analyzed Ku binding by RNA affinity chromatography (Fig 2D). The region encompassing nucleotides −145/120 and 0/120 were used as positive and negative controls, respectively. We showed that the association of Ku with the p53 transcript was not affected by deleting the sequence encompassing nucleotides −145 to −59 (Fig 2D). However, the sequence downstream position −59 appeared to be essential for Ku binding to the RNA. This sequence folds in a bulged stem‐loop structure whose folding is preserved when the p53 5′ UTR is extended in the 3′ direction 19. Since Ku was found to bind stem‐loop structures 3, 4, 20, we asked whether it could bind the hairpin‐loop encompassing nucleotides −49 to −6 within the 5′‐terminal region of p53 (Figs 2E and EV2C). To this end, we created RNA constructs in which the two stems (S1 and S2) were mutated by replacing 3 nucleotides in one strand of each stem (S1M UGG to ACC; S2M GCA to CGU). RNA constructs with a compensatory double mutation that should restore the base‐pairing in each stem (S1Mc and S2Mc) were also generated to define whether the formation of the stem was important for Ku–RNA interaction. Since the bulge in the stem‐loop of telomerase RNA is essential for proper Ku binding 4 and the stem‐loop structure at the p53 contains two bulges, we created two additional constructs in which the two bulge sequences were deleted (ΔB1 and ΔB2). As shown in Fig 2E, Ku–RNA interaction was lost with the two stem mutations S1M and S2M but the compensatory mutants S1Mc and S2Mc restored Ku binding to the RNA. Deletion of the loop proximal bulge (ΔB2), but not of the distal one (ΔB1), was detrimental to the Ku–RNA association, as revealed by both RNA affinity chromatography (Fig 2E) and EMSA analysis (Fig EV4). Overall, these results show that Ku interacts specifically and directly with p53 mRNA both in vitro and in cellulo. The identification of the Ku binding site at p53 5′ UTR adds to the growing evidence that RNA binding is a conserved function of the Ku heterodimer. Similar to the interaction of Ku with hTR/TLC1 3, 4 and with the HIV‐1 TAR RNA 20, the Ku binding site at the p53 5′ UTR forms a stem‐loop structure with a bulge motif. These three RNA hairpins show little or no sequence similarity but share similar secondary structures, suggesting that Ku recognizes RNAs with a conserved fold. In agreement with previous findings 21, the RNA stem‐loop conformation might have the ability to mimic a B‐form DNA helix and thereby associate with Ku's preformed ring.
Figure 2. Ku interacts with the p53 mRNA .
-
ARNA affinity chromatography using the p53 5′ UTR (5′p53) or a portion of the p47 5′ UTR (5′p47) (depicted in (i)) and HeLa cytoplasmic extracts (CE), followed by Western blot analysis (ii).
-
BIP of in cellulo RNA‐protein complexes (RIP) in HCT116 cells, followed either by RT–qPCR analysis (i) or by Western blot analysis (ii). The height in bar graphs (i) represents the mean and the bars indicate single data points of two independent experiments. The relative p53 mRNA levels for each IP sample were normalized to the corresponding IP IgG and to the corresponding input sample and were plotted relatively to the HPRT mRNA.
-
CUV cross‐linking of recombinant Ku70/Ku80 with the in vitro‐transcribed 5′p53 with alpha 32P‐GTP or alpha 32P‐UTP (i). UV cross‐linking of recombinant Ku70/Ku80 with the in vitro‐transcribed 5′p53 and the Ctrl (5′p53 antisense) with alpha 32P‐GTP (ii).
-
D, ERNA affinity chromatography using the different RNA constructs depicted in (i), as described in (A).
Figure EV2. Ku specifically interacts with the p53 mRNA.
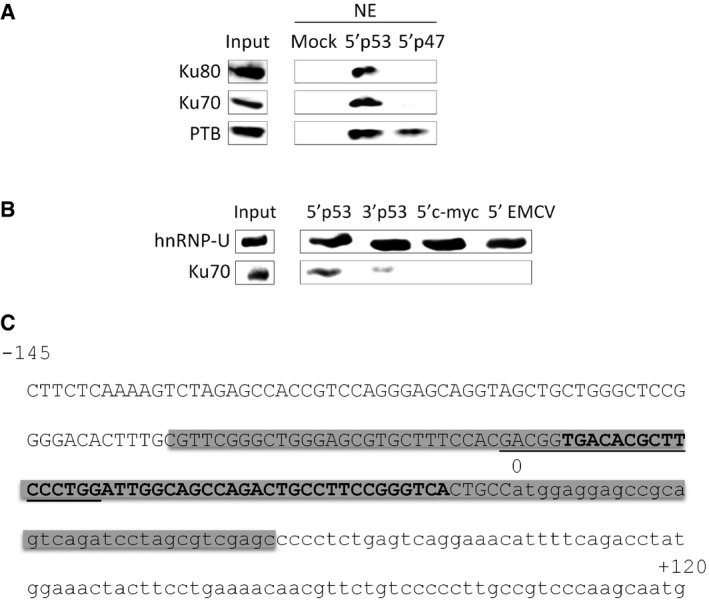
- RNA affinity chromatography using the p53 5′ UTR WT (5′p53) or a portion of the p47 5′ UTR (5′p47) (depicted in Fig 2Ai) and HeLa nuclear extracts (NE), followed by Western blot analysis with the Ku70, Ku80, or PTB antibodies.
- RNA affinity chromatography using the p53 5′ UTR WT (5′p53), p53 3′ UTR WT (3′p53), c‐myc 5′ UTR (5′c‐myc) or the encephalomyocarditis virus (EMCV) 5′ UTR (5′EMCV), and HeLa cytoplasmic extracts, followed by Western blot analysis with the hnRNP‐U (loading control) and Ku70 antibodies.
- 5′‐terminal sequence of p53 mRNA. Gray box: large hairpin domain reported in 19 (−80/+34). Capital letter: 5′ UTR of p53 (5′p53; −145/0). Small letter: sequence in between AUG1 and AUG2 (5′p47; 0/+120). Bold: stem‐loop sequence in the large hairpin domain bound by Ku and represented in Fig 2E (−49/−6). Underlined: sequence involved in 5′‐3′ base‐pairing 13.
Figure EV3. Ku directly interacts with the p53 mRNA.
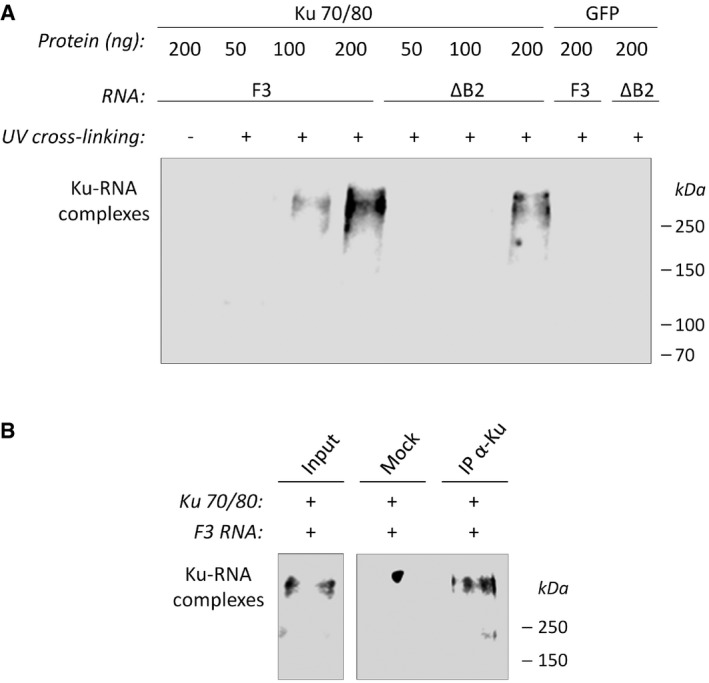
- Increasing amounts of purified recombinant Ku 32 (or GFP, negative control) were incubated with the p53 RNA fragments containing the stem‐loop WT (F3) or mutated in the loop proximal bulge (ΔB2), followed or not by UV cross‐linking. After SDS–PAGE analysis, transfer on membrane, and UV cross‐linking, the position of Ku‐RNA complexes was revealed by chemiluminescence.
- UV cross‐linked complexes processed as in (A) were immunoprecipitated with an antibody recognizing the conformational epitope of the Ku heterodimer (clone 162) and processed as above.
Figure EV4. Quantitative binding of Ku to the p53 mRNA.
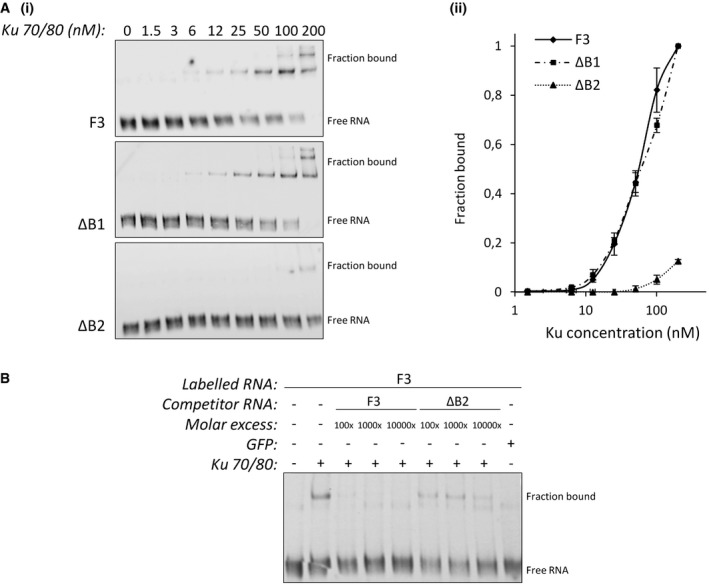
- Increasing concentrations of purified recombinant Ku were incubated with trace amounts of p53 RNA fragments containing the stem‐loop WT (F3) or mutated in the loop proximal bulge (ΔB1 or ΔB2). After acrylamide gel electrophoresis, transfer on membrane, and UV cross‐linking, the position of Ku‐RNA complexes was revealed by chemiluminescence (i). The fraction of RNA (F3, ΔB1 or ΔB2) bound to Ku [bound/(bound + free)] was quantitated and plotted as a function of the Ku concentration (n = 3) (ii). All error bars reflect SEM.
- EMSA analysis as described in (A) with a constant amount of Ku (100 nM) and trace amounts of labeled F3 RNA in the presence of increasing concentrations of unlabeled F3 or ΔB2 competitors. The specificity of binding was demonstrated further by adding purified recombinant GFP (100 nM).
Ku regulates p53 translation and function
To explore the hypothesis that p53 mRNA is a translational target of Ku, we transfected HCT116 cells with Ku‐specific or control siRNAs and then fractionated cytoplasmic extracts of these cells by sucrose density gradient centrifugation. RNA was isolated from pooled non‐polysomal and polysomal fractions, and the distribution of p53 mRNAs between these fractions was determined by RT–qPCR. This allowed us to determine whether the amount of p53 mRNA engaged in translation (i.e., associated with polysomes) was affected by the Ku knockdown. We showed that the profile of polysomes was only slightly altered by Ku depletion, indicating that Ku does not impact global mRNA translation (Fig EV5A). However, the relative amount of p53 mRNA (but not of HPRT mRNA) shifted from non‐polysomal toward polysomal fractions upon knockdown of Ku (Fig 3A). A second Ku‐targeting siRNA also showed an increased accumulation of the p53 mRNA in the polysomal fraction (Fig EV5B). This clearly demonstrated that knockdown of Ku results in increased translation of p53 mRNA.
Figure EV5. Ku decreases p53 mRNA translation.
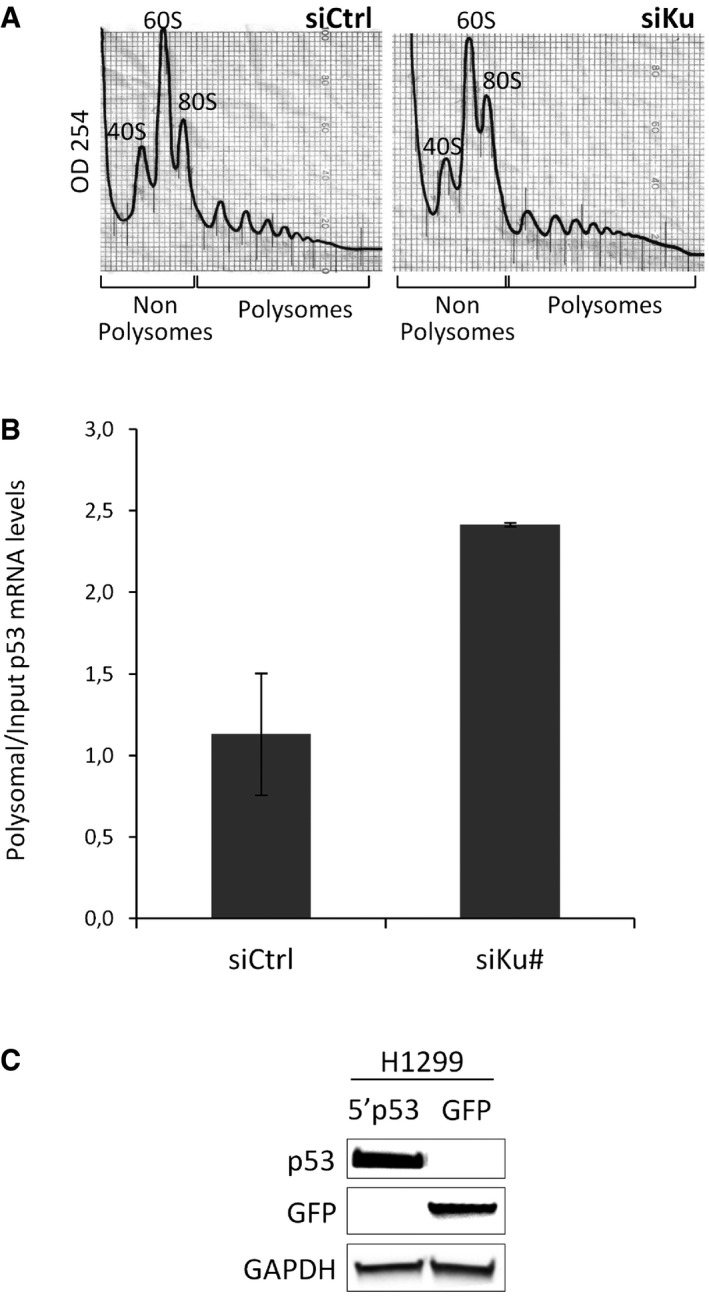
- Polysome distribution profile of cytoplasmic lysates after siRNA‐mediated Ku or control depletion. The positions of the 40S, 60S, and 80S ribosomal subunits and polysomal peaks are indicated, as well as fractions and polysomal fractions.
- Non‐polysomal and polysomal fractions were extracted from HCT116 cells treated with the siRNA Ku70# and quantitative RT–qPCR was performed using specific primers for p53 mRNAs. The p53 mRNA levels in P and NP were normalized to the input and HPRT mRNA. The height in bar graphs represents the mean and the bars indicate single data points of two independent experiments.
- Western blot from H1299 (p53‐null) cells transfected with p53 or GFP reporters, showing that p53 can be detected in H1299 cells only when ectopically expressed. GAPDH: loading control.
Figure 3. Ku regulates p53 mRNA translation and function.
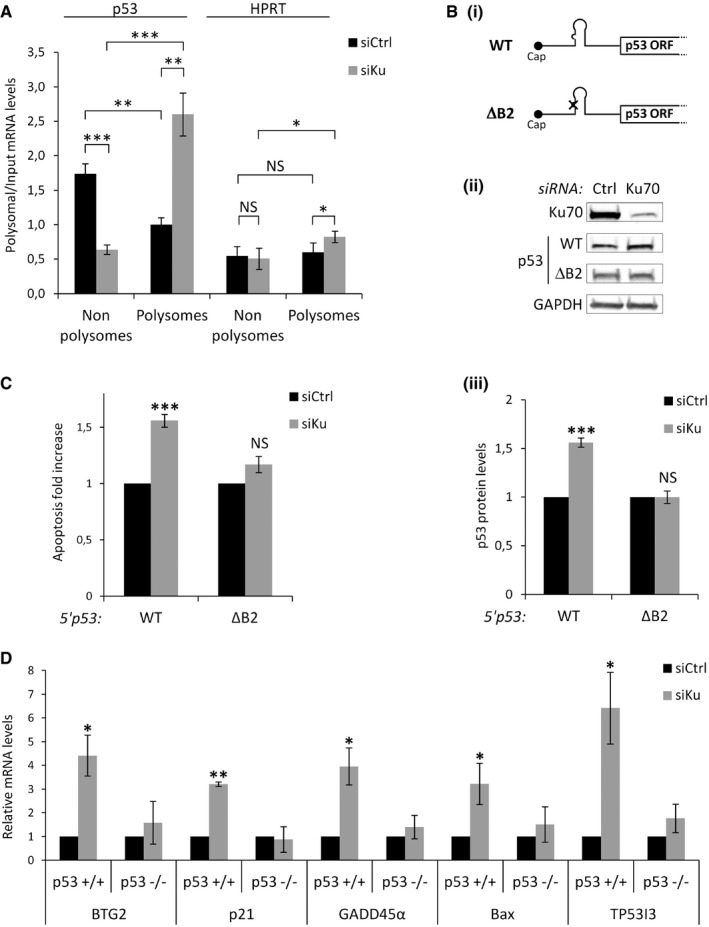
- Non‐polysomal (NP) and polysomal (P) fractions were extracted from HCT116 cells transfected with the siRNA Ku70 and quantitative RT–qPCR was performed using specific primers for p53 and HPRT mRNAs. The p53 mRNA levels in P and NP fractions were normalized to the input (n = 3).
- Western blot from H1299 (p53‐null) cells treated with siRNA Ku70 or control (ii), followed by transfections of RNA constructs depicted in (i). p53 protein levels from the WT and the ΔB2 reporters depicted in (i) normalized to firefly luciferase (transfection control reporter) (iii, n = 3).
- Annexin V/PI staining and flow cytometry experiments performed as in (B) to assess cell death (n = 3).
- RT–qPCR analysis of Bax, TP53I3, BTG2, GADD45α, and p21 after siRNA‐mediated Ku or control depletion in HCT116 p53+/+ or HCT116 p53−/− (n = 3). Relative mRNA levels were standardized against GAPDH mRNA.
To further investigate whether p53 mRNA is a translational target of Ku, we transfected p53‐null (Fig EV5C) H1299 cells with in vitro‐transcribed reporter RNAs containing the p53 5′ UTR, either wild‐type (5′p53 WT) or mutated in the bulge motif (5′p53 ΔB2) (Fig 3B). We showed that Ku depletion led to a statistically significantly increase in p53 accumulation of the wild‐type compared to the mutated construct (Fig 3B). As Ku affected p53 protein expression and p53 has well‐documented proapoptotic activities, we asked whether this effect had consequences on p53 function in apoptosis. To this end, we performed siRNA‐mediated depletion of Ku in H1299 cells followed by transfection with 5′p53 WT or mutated 5′p53 ΔB2 reporters and we measured apoptosis by flow cytometry and annexin V/propidium iodide staining analysis. We found that Ku depletion induces a statistically significant increase in p53‐dependent apoptosis only with the 5′p53 WT construct (Fig 3C). Finally, we showed that Ku depletion increases the mRNA expression of p53 target genes (p21, GADD45α, Bax, TP5313, BTG2) in HCT116 p53+/+ but not in HCT116 p53−/− cells (Fig 3D), indicating that the effect of Ku on the expression of these genes is p53‐dependent. Taken together, these observations identify p53 mRNA as a physiological target of Ku and implicate Ku as a novel factor involved in translational regulation of p53 mRNA. Ku represses p53 protein synthesis and p53‐mediated apoptosis by binding to a bulged stem‐loop structure within the p53 5′ UTR. This stem‐loop is included in a large hairpin domain that is relatively stable (ΔG = −52.9 kcal/mol) and is preserved in the context of the full‐length p53 transcript (19; Fig EV2C). Since Ku did not regulate translation of reporters in which the 5′ UTR is attached to a luciferase ORF (data not shown), we concluded that the formation of this large hairpin domain including sequences within the p53 ORF is required for proper binding of Ku. However, Chen and Kastan proposed that the sequence encompassing nucleotides −54 and −34 (Fig EV2C) forms a double‐stranded structure with the 3′ UTR that is critical for both translational repression and stress induction of p53 by Nucleolin and RPL26, respectively 13. Moreover, RPL26 binding to the p53 mRNA was not affected by mutations of the stem‐loop and did not interact with Ku (data not shown), suggesting that the Ku‐dependent and Nucleolin/RPL26‐dependent mechanisms are physically distinct. Switching from one mechanism to another might depend on whether the 5′ UTR folds with the coding sequence to form the hairpin bound by Ku or with the 3′ UTR bound by Nucleolin and RPL26.
Ku repression of p53 expression is relieved after DNA damage
Our data suggest a novel role of Ku in reducing the level of p53 expression in cells grown under normal growth conditions. We therefore wondered whether and how the suppressive effect exerted by Ku is relieved upon DNA damage, when higher levels of p53 are required. As shown in Fig 4A, p53 accumulation after Ku depletion was lost after treatment with etoposide or bleomycin, suggesting that p53 translational repression by Ku is reverted by DNA damage. However, this effect was not related to a modification of Ku sub‐cellular localization or to Ku degradation after DDR (Fig EV6A). Since previous studies showed that Ku cytoprotective functions are affected by Ku acetylation 22, 23, we tested the role of this post‐translational modification in the Ku‐mediated regulation of p53 mRNA translation. To this end, we first determined the effect of the histone deacetylase inhibitor, trichostatin A (TSA), on the expression of p53 after Ku silencing. Similar to what we observed with etoposide and bleomycin, Ku depletion did not result in the accumulation of p53 after treatment with TSA (Fig 4B). In agreement with previous studies 22, 23, we found that Ku was acetylated after treatment with TSA or upon DNA damage (Fig 4C), indicating that acetylation of Ku might be involved in the derepression of p53 mRNA translation. In support of this possibility, RNA pull‐down experiments revealed that the association of Ku with the p53 5′ UTR was significantly reduced after etoposide, bleomycin, or TSA treatments (Fig 4D). Overexpression of PCAF, an acetyltransferase that binds Ku and increases its acetylation in vivo and in vitro 23, also reduced Ku binding to the p53 5′ UTR (Fig 4E), further suggesting that Ku acetylation plays a role in derepressing p53 mRNA translation by abrogating Ku–p53 mRNA interactions. Since Ku binds DNA and RNA using the same binding site 24 and acetylation of lysine residues located within the region of Ku DNA binding cradle interferes with the ability to bind DNA 22, we made the hypothesis that Ku acetylation in the DNA binding domain interfered with the association of Ku to the RNA. To test this possibility, we performed RNA pull‐down assays using total extracts from HCT116 cells transfected with Ku80 and either wild‐type Ku70 (Ku70 WT) or a mutated Ku70 predicted to be defective in DNA binding but not altered in the overall architecture of the Ku70–Ku80 complex (Ku70 Mut6E) 25. As shown in Fig 4F, unlike Ku70 WT, Ku70 Mut6E was not retained either by the p53 5′ UTR or a dsDNA fragment, suggesting that Ku binding to the p53 5′ UTR is mediated by its DNA binding domain. Then, we carried out site‐directed mutagenesis to replace lysines located within the Ku70 DNA binding cradle to glutamines (K to Q), to mimic constitutive acetylation. We focused on K282 and K338 of Ku70 since constitutive acetylation of these residues was previously shown to suppress the activity of Ku70 to bind DNA 22. We showed that, similarly to Ku70 Mut6E, Ku70 K282Q, and Ku70 K282/338Q, but not an acetylation‐deficient mutant Ku70 K282/338R, reduced the ability of Ku to bind the p53 5′ UTR in RNA pull‐down assays (Figs 4G and EV6B). Overall, these results suggest that Ku acetylation within the DNA binding domain interferes with the ability of Ku to bind and regulate p53 mRNA expression in DNA damage conditions.
Figure 4. Ku acetylation relieves repression of p53 mRNA translation.
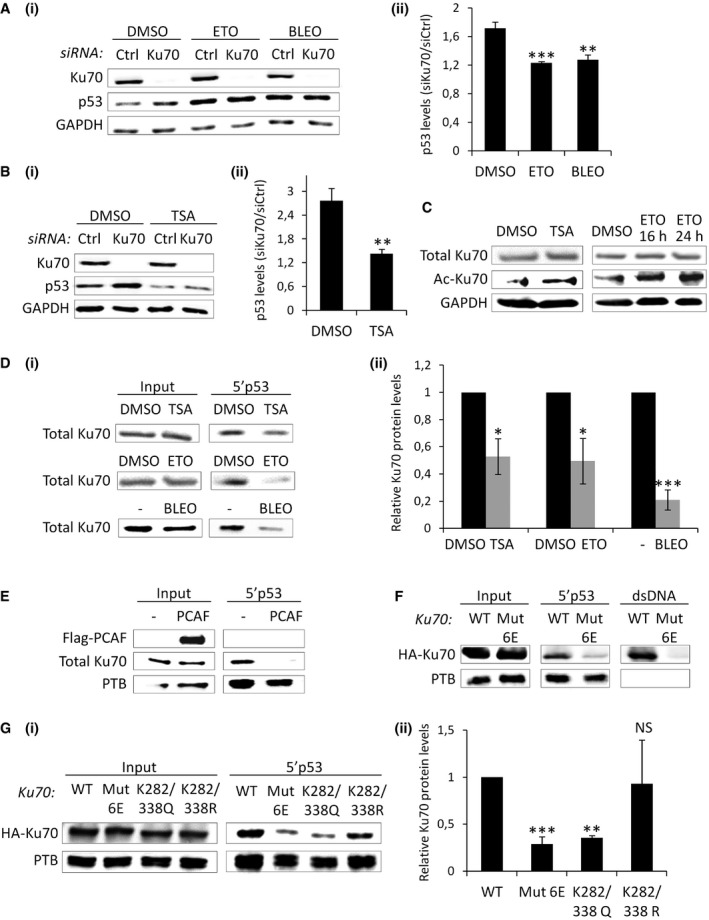
- Western blot analysis after treatment with Ku70 or control siRNA, followed by incubation with DMSO, etoposide (ETO), or bleomycin (BLEO) for 16 h (i). p53 levels after Ku70 depletion normalized to siRNA control for each condition (ii, n = 3).
- Western blot analysis as in (A), except that trichostatin A (TSA), was used after Ku/control knockdown (n = 4).
- Western blot in cells treated either with DMSO, TSA, or ETO for 16 or 24 h.
- RNA affinity chromatography using the 5′p53 RNA and total extracts from cells treated with TSA, ETO, or BLEO for 24 h, followed by Western blot analysis of Ku70 quantified and normalized to the input (ii, n = 3).
- RNA affinity chromatography using the 5′p53 RNA and total extracts from cells transfected or not with PCAF encoding plasmid for 48 h, followed by Western blot analysis.
- RNA affinity chromatography using the 5′p53 RNA or a dsDNA and total extracts from cells transfected with HA‐tagged Ku70 wild‐type (WT) or mutated (Mut6E) Ku70 25 for 48 h, followed by Western blot analysis.
- RNA affinity chromatography as in (F), but with two additional Ku mutants (K282/338Q and K282/338R), followed by Western blot of Ku70 quantified and normalized to the input and to the loading control, PTB (ii, n = 3).
Figure EV6. Ku acetylation relieves repression of p53 mRNA translation.
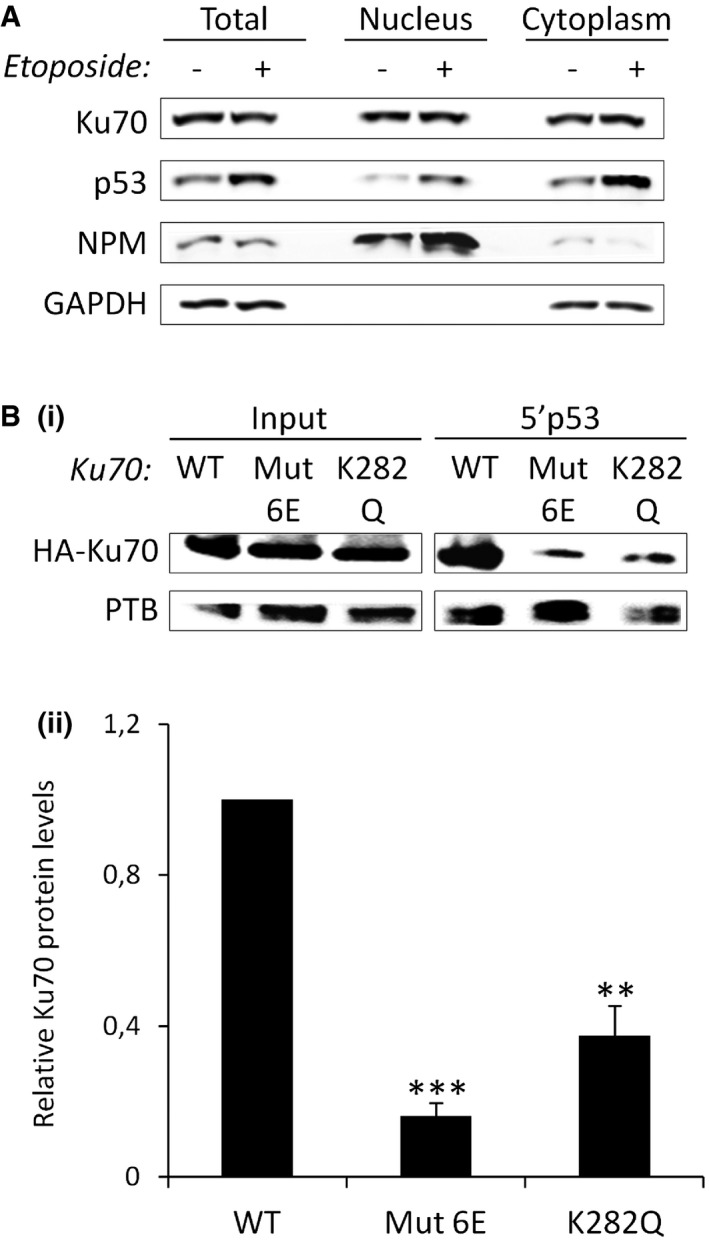
- Western blot analysis after treatment with ETO for 16 h, followed or not by nuclear/cytosolic fractionation. NPM (nucleophosmin) and GAPDH are used as markers of the nuclear and cytosolic fractions, respectively.
- RNA affinity chromatography using the 5′p53 RNA and total extracts from cells transfected with HA‐tagged wild‐type (WT) or mutated (Mut6E or K282Q) Ku70 for 48 h, followed by Western blot analysis of Ku70 (i) quantified and normalized to the input and to the loading control, PTB (ii, n = 3).
Our results support a model in which Ku suppresses p53 mRNA translation in cells grown under normal growth conditions, thereby contributing to the low steady‐state level of p53. In cells stressed with DNA‐damaging agents, Ku acetylation abrogates the Ku‐dependent suppression of translation and permits increased translation of p53 mRNA (Fig 5). Transiently blunting the potential detrimental effects of p53 induction could be beneficial in reducing p53‐mediated cell death in settings of physiological DNA damage, as recently proposed for the action of the ribosomal protein RPL22 on p53 expression in cells undergoing V(D)J recombination 26. Besides repairing DNA, Ku protects cells from death by regulating the synthesis (e.g., APAF 27) or the activity of factors involved in apoptosis (e.g., Bax 23). Although the specific physiological or pathological settings involving Ku repression of p53 function remains to be investigated, our data extend the notion of a link between Ku and apoptosis to its role in post‐transcriptional regulation and unraveled a new level of complexity in the p53 regulation of expression in normal and stress conditions.
Figure 5. Ku impacts mRNA translation and function during DDR.

Schematic model illustrating that Ku contributes to the low steady‐state level of p53 under normal growth conditions by suppressing p53 mRNA translation and that this inhibitory mechanism is abrogated during DNA damage due to Ku acetylation, thereby allowing p53 upregulation.
In conclusion, we provide evidence of a new role of Ku in regulating p53 mRNA translation and function that is distinct from its known function in DNA repair. The proposed mechanism provides fertile ground for further investigation on the interplay between Ku and p53 in cancer development and treatment, and more generally, on the post‐transcriptional role of Ku in cancer cells. It also uncovers a novel mechanism by which acetyltransferases and histone deacetylase inhibitors might impact tumorigenesis and suggest that acetylation may be an important player in the regulation of mRNA translation. Finally, our findings provide interesting insights into the link between mRNA translation and DNA repair, and support the concept that DNA binding proteins involved in repair also bind RNA and regulate gene expression at the post‐transcriptional level, reminiscent to the REM (RNA, enzyme, and metabolite) model 28. The network based on proteins with a dual function in DNA repair and RNA metabolism may offer the advantage to promptly activate the gene expression response required to overcome the DNA insults soon after detection.
Materials and Methods
Cell culture and transfections
Colon carcinoma (HCT116; Sigma) and lung cancer cells (A549 or H1299; ATCC) were, respectively, grown in DMEM media (4.5 g/l glucose) or in DMEM media (1 g/l glucose) supplemented with 10% FBS, 2 mM L‐glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were tested for mycoplasma contamination using the Mycoalert mycoplasma detection kit (Lonza LT07‐218) according to the manufacturer's instructions. siRNAs were transfected using the Interferin reagent (Polyplus) according to the manufacturer's instructions. In brief, cells at 60% confluency were transfected twice with 50 nM siRNA in a 72 h time interval. Post‐transfection time for optimal depletion was 120 h after the second transfection. For transient RNA transfection experiments, 100 ng of in vitro‐transcribed capped and polyadenylated mRNAs (mMESSAGE mMACHINE® T7 Transcription Kit; Thermo Fisher Scientific) were transfected in 6‐well plates using Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. For DNA plasmid transfections, 7.5 μg of plasmids (including Ku70 mutants generated with Quick Change II XL site‐directed mutagenesis (Agilent technologies)) was transfected in 10‐cm2 dishes using jet‐PEI reagent (Polyplus) according to the manufacturer's instructions. Cells were subsequently incubated at 37°C for 16 h following RNA transfection and 48 h following DNA plasmid transfections before harvesting and analysis.
RNA chromatography
Two hundred micrograms of nuclear or cytoplasmic extract from HeLa cells or 200 μg of extracts from HCT116 treated with TSA, bleomycin, or etoposide and lysed in a buffer containing 20 mM Tris pH 8, 1% NP‐40, 150 mM NaCl, 2 mM EDTA, and 10% glycerol were precleared with 20 μl of streptavidin acrylamide beads (Thermo Fisher Scientific) in the binding buffer containing 20 mM Tris pH 8, 1 mM DTT, 100 mM KCl, 0.2 mM EDTA for 1 h at 4°C. In vitro‐transcribed biotinylated RNA (T7‐flash Biotin RNA transcription Kit; Ampliscribe) was fixed on 20 μl of streptavidin acrylamide beads by incubation in the binding buffer for 1 h at 4°C. The RNA fixed on beads was then combined to the precleared extracts for 3 h at 4°C. The beads were collected by centrifugation, washed five times with 1 ml of the binding buffer, resuspended in 5 μl of loading buffer, and boiled for 5 min. After centrifugation, the supernatant was loaded onto an SDS–PAGE gel and analyzed by Western blot.
RNA‐protein cross‐linking
One hundred nanograms of recombinant Ku70/Ku80 proteins was incubated with in vitro‐transcribed 32P‐labeled RNAs (150,000 cpm; MEGAshortscript™ T7 Kit, Thermo Fisher Scientific) for 20 min at room temperature. Following UV‐C irradiation (4,000 J/m2), 10 U RNase One and 1 μg RNase A/T1 were added and the mix incubated for 45 min at 37°C. The reaction was boiled for 5 min and loaded onto an SDS–PAGE gel.
siRNA
Sense sequences are the following for: control (5′‐GGUCCGGCUCCCCCAAAUGtt‐3′), Ku70 (Ku70: iGenome SMART pool or Ku‐70#: 5′‐GAGUGAAGAUGAGUUGACAtt‐3′ 25), ASF (5′‐AGUUAUGGAAGAUCUCGAUtt‐3′ 29), XRCC4 (5′‐AUAUGUUGGUGAACUGAGAtt‐3′), and Ku80 (5′‐CAGAGAAGAUUCUUCAUGGGtt‐3′30).
Drug treatment
Before recovering for analysis, the cells were incubated/exposed with: 10 μM etoposide (Sigma‐Aldrich E1383) for 16 h or 24 h; 1 μM trichostatin A (TSA, Sigma‐Aldrich T1952) for 24 h; 0.1 μM doxorubicin (Sigma‐Aldrich D1515) for 16 h; 30 μM wortmannin (Millipore #12‐338) for 1 h; 2 μM NU‐7441 (Tocris Bioscience #3712) for 2 h; 20 μM MG132 (Peptanova 3175‐v) for 2 h; 300 μM cycloheximide (CHX, Sigma‐Aldrich C4859) for 1 h; 30 μg/ml bleomycin (Calbiochem #203401) for 24 h; 50 J/m2 UV‐C (UV Stratalinker 1800, Stratagene) followed by a 16 h post‐incubation.
Preparation of cell extracts and immunoblotting
Total cell extracts were prepared and analyzed by Western blot with antibodies against Ku70 (NB100‐1915, clone S10B1; Novus Biologicals), Ku80 (ab3107, Clone 111; Abcam), p53 (sc‐126, clone DO‐1; Santa Cruz), p47 (VP‐P955, clone CM1; Vector laboratories), GAPDH (sc‐32233, clone 6C5; Santa Cruz), p21 (sc‐397, clone C‐19; Santa Cruz), GADD45α (#4632, clone D17E8; Cell Signaling), P‐Ser1981 ATM (ab81292, clone EP1890Y; Abcam), P‐Ser15 p53 (#9284; Cell Signaling), ASF (32‐9500, Zymed), γ‐H2AX (16‐193, clone JBW301; Millipore), P‐Ser473 Akt (#9271; Cell Signaling Technology), DNA‐PK (sc‐5282, clone G‐4; Santa Cruz), P‐Ser2056 DNA‐PK (Santa Cruz Biotech sc‐5282), XRCC4 (rabbit polyclonal anti‐XRCC4 31), PTB (HB‐94, cloneBB7.7; ATCC), NPM (sc‐56622, FC82291; Santa Cruz), ac‐Ku70 (ATB‐K0033, clone K542; Ameritech Biomedicines), HA (H9658, clone HA‐7; Sigma‐Aldrich), FLAG (F3165, clone M2; Sigma‐Aldrich), Actin (A3853; Sigma‐Aldrich).
In cellulo RNA immunoprecipitation (RIP)
The RIP assay kit (RN 1001, MBL international) was used according to the manufacturer's instructions. The antibodies used for the IP are directed against Ku70/80 (GTX23108, clone 162; GeneTex) and PTB (HB‐94, clone BB7.7; ATCC).
EMSA
Indicated amounts of purified human Ku70/80 protein (or GFP, negative control) were incubated for 20 min on ice with 0.8 fmol biotinylated RNA probe in 10 μl reaction containing 20 mM Tris–HCl, pH 7.5, 2 mM MgCl2, 90 mM potassium acetate, 0.1 mM EDTA, 0.3 mM DTT, 5% glycerol, 25 μg/ml tRNA, and 0.1 mg/ml BSA. The mixture was loaded on a 5% polyacrylamide gel, electrophoresed at 4°C for 1 h at 200 V, and then transferred to a Biodyne B nylon membrane (Thermo Scientific). After cross‐link under UV light (UV Stratalinker 1800), signals of probe were detected with the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific) according to the manufacturer's instructions.
UV cross‐linking using biotinylated RNA
Indicated amounts of purified recombinant Ku70/80 were incubated with biotinylated RNAs for 20 min at 4°C in 10 μl reaction containing 20 mM Tris–HCl, pH 7.5, 2 mM MgCl2, 90 mM KOAc, 0.1 mM EDTA, 0.3 mM DTT, and 5% glycerol. Following UV irradiation (4,000 J/m2), the cross‐linking reaction was processed as indicated for EMSA except that cross‐linked complexes were loaded onto a 7.5% SDS–PAGE gel. For immunoprecipitation of UV cross‐linked complexes, cross‐linking reactions were incubated for 4 h at 4°C with 20 μl protein G Sepharose beads, preincubated overnight at 4°C with protease inhibitor cocktail and 50 μl of Ku70/80 mAb (sc‐162, Genetex) in 500 μl of PBS, NP‐40 0.1%. The beads were collected by centrifugation, washed four times with 1 ml of the binding buffer, resuspended in 10 μl of loading buffer, and heated at 90°C for 5 min. After centrifugation, the supernatant was loaded onto a 7.5% SDS–PAGE gel and analyzed as indicated for EMSA.
FACS
APC annexin V/Dead Cell apoptosis Kit (V35113, Thermo Fisher Scientific) was used according to the manufacturer's instructions. Briefly, cells transfected or not with siRNA Ctrl or Ku70 and the different constructs (5′p53 WT or ΔB2) were collected, washed in PBS, and incubated in 100 μl of Annexin‐binding buffer 5× (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), containing 5 μl of Annexin V‐FITC and 1 μl of propidium iodide (PI) solution at 100 μg ml (AnnexinV‐FITC/Dead Cell Apoptosis Kit, Thermo Fisher Scientific) during 15 min at room temperature (RT) in the dark. Four hundred microliters of Annexin‐binding buffer 5× were then added after washes with PBS‐BSA 1%. Labeled cells were preserved on ice and run on a flow cytometer (FACS Calibur, Becton‐Dickinson, Franklin Lakes, NJ, USA).
Cell fractionation
PARIS kit Protein and RNA isolation system (AM‐1921; Thermo Fisher Scientific) was used according to the manufacturer's instructions.
RT–qPCR
Total RNA was purified from HeLa cells using TRI Reagent solution (Applied Biosystems) according to the manufacturer's instructions. After DNase treatment (DNA free, Thermo Fisher Scientific) and quantification, reverse transcription was done with 1 μg of total RNA using RevertAid First‐strand cDNA Synthesis Kit (Thermo Fisher Scientific) and random hexamers according to the manufacturer's instructions. Primers to amplify specific genes are listed in Table 1.
Table 1.
Real‐time PCR oligonucleotides
| Gene | Primer | Sequence (5′–3′) |
|---|---|---|
| p53 | Forward | TGGGCTTCTTGCATTCTGG |
| Reverse | GCTGTGACTGCTTGTAGATGGC | |
| Ku70 | Forward | ATGGCAACTCCAGAGAGCAGGTG |
| Reverse | AGTGCTTGGTGAGGGCTTCCA | |
| Ku80 | Forward | TGACTTCCTGGATGCACTAATCGT |
| Reverse | TTGGAGCCAATGGTCAGTCG | |
| BTG2 | Forward | TTCCCAGACCTGCTTCCAGTCTTT |
| Reverse | ACAAGATGCAAGAACACAGCCTGC | |
| TP53I3 | Forward | AAGCGAGGAAGTCTGATCACCAGT |
| Reverse | AGGCAGAATTTGCTCCGTGAAAGC | |
| Bax | Forward | TCTACTTTGCCAGCAAACTGGTGC |
| Reverse | TGTCCAGCCCATATGGTTCTGAT | |
| GADD45α | Forward | GATGCCCTGGAGGAAGTGCT |
| Reverse | AGCAGGCACAACACCACGTT | |
| p21 | Forward | GGAAGACCATGTGGACCTGT |
| Reverse | ACTGCAGGCTTCCTGTGG | |
| HPRT | Forward | TGCTTTCCTTGGTCAGGCAGT |
| Reverse | CTTCGTGGGGTCCTTTTCACC |
Polysomes fractionation
HCT116 cells (40 millions) were treated with 0.1 mg/ml CHX for 15 min at 37°C. The cells were lysed in hypotonic buffer (5 mM Tris pH 7.5, 2.5 mM MgCl2, 1.5 mM KCl, and 100 U/ml RNAsine). Cells were incubated on ice for 5 min and vortexed. Triton X‐100 and sodium deoxycholate were added each at 0.5% final concentration. Cells were incubated on ice for 5 min and centrifuged for 5 min at 16,000 g. The supernatants were layered on a continuous sucrose gradient (15–50% sucrose) and ultracentrifugated at 250,000 g in an SW41‐Ti rotor at 4°C for 2 h. Fractions were collected with an ISCO density gradient fractionation system (Foxy Jr fraction collector coupled to UA‐6UV detector, Lincoln, NE) to follow the absorbance at 254 nm. The fractions recovered from the gradient were then divided into two groups, the fractions containing actively translated mRNAs, polysomes, and the fractions containing untranslated mRNAs, the non‐polysomes. RNAs from each fraction were extracted using the TRIzol LS reagent (Thermo Fisher Scientific) according to the manufacturer's instructions and RT–qPCR was performed.
Author contributions
AL performed the majority of the experiments and analyzed data; MLB, NS, AC, and SM helped with some experiments; HP and PC contributed with helpful discussion; PC, SB, and PF provided material and advices; AC helped with the design of several experiments; SM conceived and supervised all experiments, and wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Olivier Sordet, Stéphane Pyronnet, Yvan Martineau, Didier Trouche, Robin Fåhraeus for materials and discussions. We are grateful to Anne Willis, Jean‐Christophe Bourdon for providing the PTB and the p47 antibodies, respectively. This work was supported by INSERM, Ligue Nationale Contre le Cancer (to A.C.), ARC (Association pour la Recherche contre le Cancer) (to S.M) and Emergence GSO (to S.M). A.C was supported by FRM (Fondation pour la Recherche Medicale) and A.L. by Ligue Nationale Contre le Cancer. M.LB was supported by the Midi‐Pyrénées Region/INSERM. P Calsou's team is supported by Ligue Nationale Contre le Cancer (Equipe labellisée 2013).
EMBO Reports (2016) 17: 508–518
References
- 1. Savage KI, Gorski JJ, Barros EM, Irwin GW, Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ, McCluggage WG et al (2014) Identification of a BRCA1‐mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol Cell 54: 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kleiman FE, Manley JL (2001) The BARD1‐CstF‐50 interaction links mRNA 3' end formation to DNA damage and tumor suppression. Cell 104: 743–753 [DOI] [PubMed] [Google Scholar]
- 3. Ting NS, Yu Y, Pohorelic B, Lees‐Miller SP, Beattie TL (2005) Human Ku70/80 interacts directly with hTR, the RNA component of human telomerase. Nucleic Acids Res 33: 2090–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dalby AB, Goodrich KJ, Pfingsten JS, Cech TR (2013) RNA recognition by the DNA end‐binding Ku heterodimer. RNA 19: 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwon SC, Yi H, Eichelbaum K, Fohr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN (2013) The protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20: 1122–1130 [DOI] [PubMed] [Google Scholar]
- 6. Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM et al (2012) Insights into RNA biology from an atlas of mammalian mRNA‐binding proteins. Cell 149: 1393–1406 [DOI] [PubMed] [Google Scholar]
- 7. Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold‐Brown D, Drew K, Milek M et al (2012) The mRNA‐bound proteome and its global occupancy profile on protein‐coding transcripts. Mol Cell 46: 674–690 [DOI] [PubMed] [Google Scholar]
- 8. Silvera D, Koloteva‐Levine N, Burma S, Elroy‐Stein O (2006) Effect of Ku proteins on IRES‐mediated translation. Biol Cell 98: 353–361 [DOI] [PubMed] [Google Scholar]
- 9. Powley IR, Kondrashov A, Young LA, Dobbyn HC, Hill K, Cannell IG, Stoneley M, Kong YW, Cotes JA, Smith GC et al (2009) Translational reprogramming following UVB irradiation is mediated by DNA‐PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev 23: 1207–1220 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Takagi M, Absalon MJ, McLure KG, Kastan MB (2005) Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 123: 49–63 [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Cho SJ, Shu L, Yan W, Guerrero T, Kent M, Skorupski K, Chen H, Chen X (2011) Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev 25: 1528–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang A, Zhou N, Huang J, Liu Q, Fukuda K, Ma D, Lu Z, Bai C, Watabe K, Mo YY (2013) The human long non‐coding RNA‐RoR is a p53 repressor in response to DNA damage. Cell Res 23: 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J, Kastan MB (2010) 5′‐3′‐UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev 24: 2146–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gajjar M, Candeias MM, Malbert‐Colas L, Mazars A, Fujita J, Olivares‐Illana V, Fahraeus R (2012) The p53 mRNA‐Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 21: 25–35 [DOI] [PubMed] [Google Scholar]
- 15. Li G, Nelsen C, Hendrickson EA (2002) Ku86 is essential in human somatic cells. Proc Natl Acad Sci U S A 99: 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holcomb VB, Rodier F, Choi Y, Busuttil RA, Vogel H, Vijg J, Campisi J, Hasty P (2008) Ku80 deletion suppresses spontaneous tumors and induces a p53‐mediated DNA damage response. Cancer Res 68: 9497–9502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A (2000) DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404: 510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grover R, Ray PS, Das S (2008) Polypyrimidine tract binding protein regulates IRES‐mediated translation of p53 isoforms. Cell Cycle 7: 2189–2198 [DOI] [PubMed] [Google Scholar]
- 19. Blaszczyk L, Ciesiolka J (2011) Secondary structure and the role in translation initiation of the 5′‐terminal region of p53 mRNA. Biochemistry 50: 7080–7092 [DOI] [PubMed] [Google Scholar]
- 20. Kaczmarski W, Khan SA (1993) Lupus autoantigen Ku protein binds HIV‐1 TAR RNA in vitro. Biochem Biophys Res Commun 196: 935–942 [DOI] [PubMed] [Google Scholar]
- 21. Bullock SL, Ringel I, Ish‐Horowicz D, Lukavsky PJ (2010) A'‐form RNA helices are required for cytoplasmic mRNA transport in Drosophila. Nat Struct Mol Biol 17: 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, Lu YS, Matsuyama S, Chen CY (2007) Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double‐strand breaks by targeting Ku70 acetylation. Cancer Res 67: 5318–5327 [DOI] [PubMed] [Google Scholar]
- 23. Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA (2004) Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax‐mediated apoptosis. Mol Cell 13: 627–638 [DOI] [PubMed] [Google Scholar]
- 24. Pfingsten JS, Goodrich KJ, Taabazuing C, Ouenzar F, Chartrand P, Cech TR (2012) Mutually exclusive binding of telomerase RNA and DNA by Ku alters telomerase recruitment model. Cell 148: 922–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Britton S, Coates J, Jackson SP (2013) A new method for high‐resolution imaging of Ku foci to decipher mechanisms of DNA double‐strand break repair. J Cell Biol 202: 579–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rashkovan M, Vadnais C, Ross J, Gigoux M, Suh WK, Gu W, Kosan C, Moroy T (2014) Miz‐1 regulates translation of Trp53 via ribosomal protein L22 in cells undergoing V(D)J recombination. Proc Natl Acad Sci USA 111: E5411–E5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Zio D, Bordi M, Tino E, Lanzuolo C, Ferraro E, Mora E, Ciccosanti F, Fimia GM, Orlando V, Cecconi F (2011) The DNA repair complex Ku70/86 modulates Apaf1 expression upon DNA damage. Cell Death Differ 18: 516–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hentze MW, Preiss T (2010) The REM phase of gene regulation. Trends Biochem Sci 35: 423–426 [DOI] [PubMed] [Google Scholar]
- 29. Li X, Manley JL (2005) Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122: 365–378 [DOI] [PubMed] [Google Scholar]
- 30. Rampakakis E, Di Paola D, Zannis‐Hadjopoulos M (2008) Ku is involved in cell growth, DNA replication and G1‐S transition. J Cell Sci 121: 590–600 [DOI] [PubMed] [Google Scholar]
- 31. Wu PY, Frit P, Malivert L, Revy P, Biard D, Salles B, Calsou P (2007) Interplay between Cernunnos‐XLF and nonhomologous end‐joining proteins at DNA ends in the cell. J Biol Chem 282: 31937–31943 [DOI] [PubMed] [Google Scholar]
- 32. Bombarde O, Boby C, Gomez D, Frit P, Giraud‐Panis MJ, Gilson E, Salles B, Calsou P (2010) TRF2/RAP1 and DNA‐PK mediate a double protection against joining at telomeric ends. EMBO J 29: 1573–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File