

Abstract
Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is a central regulator of cellular and mitochondrial metabolism. Cellular bioenergetics are critically important in “energy-guzzling” neurons, but the components and wiring of the transcriptional circuit through which PGC-1α regulates the neuronal electron transport chain have not been established. This information may be vital for restoring neuronal bioenergetics gene expression that is compromised during incipient Parkinson's neuropathology and in aging-dependent brain diseases. Here we delineate a neuronal transcriptional circuit controlled by endogenous PGC-1α. We show that a feed-forward circuit of endogenous neuronal PGC-1α and the orphan nuclear estrogen-related receptor α (ERRα) activates the nuclear-encoded mitochondrial electron transport chain. PGC-1α not only trans-activated expression of ERRα, but also coactivated ERRα target genes in complexes I, II, IV, and V of the neuronal electron transport chain via association with evolutionary conserved ERRα promoter binding motifs. Chemical activation of this transcriptional program induced transcription of the neuronal electron transport chain. These data highlight a neuronal transcriptional circuit regulated by PGC-1α that can be therapeutically targeted for Parkinson's and other neurodegenerative diseases.
1. Introduction
PGC-1α is a central regulator of cellular and mitochondrial metabolism in metabolically highly active nonneuronal cell types—brown fat cells, cardiomyocytes, and muscle cells [1]. PGC-1α dysfunction is linked to diseased states of these cell types such as diabetes [2], cardiomyopathy [3], and sarcopenia [4]. PGC-1α orchestrates a remodeling of cells to increase “clean energy” production [5]. It quantitatively and qualitatively increases energy production as well as the detoxifying enzymes necessary to remove the reactive oxygen species that are the byproduct of increased ATP production [1]. PGC-1α induces mitochondrial biogenesis in response to a number of physiological clues such as exercise, cold, and fasting [1]. It remodels individual organelles by increasing levels of electron transport chain (ETC) complexes as well as ATP synthase within isolated mitochondria [4, 6].
The brain is the most energy-demanding organ [7], but the components and wiring of the transcriptional circuits through which PGC-1α regulates energy production in brain have not been dissected. This is in contrast to other cell types and organs for which considerable progress has been made in elucidating PGC-1α function [1, 4, 6, 8–13]. This information may be vital for restoring the neuronal bioenergetics that are compromised in several brain diseases, including Parkinson's (PD) [14], Huntington's (HD) [15, 16], and amyotrophic lateral sclerosis (ALS) [17].
We previously meta-analyzed laser-captured human dopamine neuron and substantia nigra transcriptomes of hundreds of individuals with Parkinson's and controls, followed by two-stage replication [14]. We found ten gene sets (i.e., groups of transcripts that encode the same biological pathway) with previously unknown associations with PD [14]. These gene sets pinpointed defects in mitochondrial electron transport, glucose utilization, and glucose sensing and indicated that these systems changes may occur already at earliest, subclinical stages of Lewy body neuropathology. Genes controlling cellular bioenergetics that are expressed in response to PGC-1α were underexpressed in dopaminergic neurons laser-captured from substantia nigra of motor PD patients [14]. Mechanistically, transduction with PGC-1α blocked mutant α-synuclein and rotenone toxicity in rat primary mesencephalic cultures [14]. Other laboratories showed that PGC-1α potently modulates dopaminergic neurodegeneration in two mouse models of PD [18–20]. The findings in sporadic PD are supported in a PARK2-linked, autosomal recessive variant of PD [19], where repression of PGC-1α by the parkin substrate PARIS contributes to neurodegeneration [19].
Here we set out to clarify a specific, open question: the transcriptional circuit through which endogenous PGC-1α regulates the neuronal electron transport chain in neuronal cells and brain. Our data indicate that endogenous PGC-1α and estrogen-related receptor α (ERRα) coactivate the nuclear-encoded electron transport chain in neuronal cells through a feed-forward loop. This transcriptional network can now be further defined and therapeutically exploited as chemical activation induced a pervasive increase in endogenous neuronal electron transport chain gene expression.
2. Materials and Methods
2.1. Mouse Brains
Snap-frozen whole brain tissue from PGC-1α KO mice, originally characterized by Dr. Bruce Spiegelman (Dana-Farber Cancer Institute, Harvard Medical School), were obtained from Jackson Laboratory (stock number 008597). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local animal care committee.
2.2. Cell Culture
SK-N-MC neuroblastoma cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) FCS. All cells were cultured in the presence of 100 U/mL penicillin and 100 μg/mL of streptomycin sulfate in 5% CO2 at 37°C.
2.3. Transfections and Adenoviral Transductions
Low passage SK-N-MC cells were plated at 8 × 105 cells/well in a 6-well plate the day before transfection in media lacking antibiotics. Routinely, cells were transfected with a total of 1 to 5 μg of plasmid DNA using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. For adenoviral transductions, SK-N-MC cultures were transduced with adenovirus encoding PGC-1α or LacZ (50 MOI) for 24 hours as described elsewhere [12]. Cells were harvested after 48 hours of treatment.
2.4. RNA Isolation and Quantitative Real-Time PCR
RNA was extracted from SK-N-MC cells or snap-frozen brain tissue samples by TRIzol (GIBCO/BRL) extraction similar to what we describe in [12]. RNA quality was determined by spectrophotometry and by visual inspection of electropherograms using the RNA 6000 NanoChip Kit on the Agilent 2100 Bioanalyzer (Agilent Technologies). For quantitative gene expression analysis in human biospecimens, TaqMan Assay-on-demand primers and probes (Applied Biosystems) were used. Amplification products were analyzed for specificity by agarose gel electrophoresis. To detect PGC-1α mRNA, we have used TaqMan probe Hs01016719_m1, which does not differentiate between various PGC-1α isoforms. The comparative threshold cycle method was used for analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and RPL13 ribosomal RNA were used as RNA loading controls. Equal amplification efficiencies were confirmed for target and reference genes.
2.5. siRNA Transfection
Low passage SK-N-MC cells were seeded into 6-well dishes at 40% confluency. The required amount of target siRNA (Invitrogen) and 9 μL of Lipofectamine RNAi MAX (Invitrogen) were each diluted into a final volume of 250 μL in Opti-MEM (GIBCO), then combined, gently mixed, and incubated at room temperature for 25 min. 500 μL of this transfection solution was overlaid onto cells at a final concentration of 80 nM siRNA. Transfection of SK-N-MC cells with RNAi Negative Control (Dharmacon, with no significant homology to any known gene sequences from mouse, rat, or human) served as a negative control. After 48 hr incubation at 37°C in the presence of 5% CO2, cells were lysed by TRIzol reagent, and total RNA was isolated by chloroform/isopropanol precipitation. To detect PGC-1α protein levels by Western blot analysis we used a rabbit polyclonal antibody (H300, Santa Cruz, CA, USA).
2.6. Quantitative Chromatin Immunoprecipitation Analysis
Chromatin immunoprecipitation assays (ChIPs) were performed in asynchronously growing SK-N-MC cells transfected with the myc-PGC-1α construct or the empty vector. Cross-linking was carried out with 1% formaldehyde for 10 min at room temperature. Cross-linking was subsequently quenched by adding glycine to a final concentration of 250 mM for 10 min. Cells were collected and washed twice with PBS and then resuspended in 2.5 mL of lysis buffer (150 mM NaCl, 50 mMTris-HCl pH 8.0, 1% NP-40, 25 μM MG-132, and 1x Complete® Protease inhibitor cocktail). After 10 min on ice, cells were sonicated to obtain DNA fragments of ~500 bp as determined by agarose gel electrophoresis with ethidium bromide staining. Protein-DNA complexes were isolated by centrifugation at 15,000 rpm for 20 min. Supernatants with protein-DNA complexes were incubated for 16 hrs with rabbit polyclonal antibody directed against PGC-1α. Normal rabbit IgG was used as a control. Antibody-protein-DNA complexes were further incubated with 100 μL of magnetic DYNA beads (Invitrogen) to isolate antibody bound fractions of chromatin. Immunocomplexes were washed with the following buffers: low salt (20 mM Tris-Cl, pH 8.1, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, and 1x complete protease inhibitor), high salt (20 mM Tris-Cl, pH 8.1, 500 mM NaCl, 1% Triton X-100, and 2 mM EDTA), LiCl (10 mM Tris-Cl, pH 8.1, 250 mM LiCl, 1% deoxycholate, 1% NP-40, and 1 mM EDTA), and twice in TE (10 mM Tris-Cl, pH 8.1, and 1 mM EDTA). Protein-DNA complexes were eluted in 1% SDS and 100 mM NaHCO3. Cross-links of pulldown fractions and inputs (2% of total IP fraction) were reversed by overnight incubation in elution buffer and 0.2 M NaCl. DNA was then extracted, purified, precipitated, and resuspended in TE for qPCR. Immunoprecipitated DNA was analyzed by real-time PCR as previously described. The primer sequences are available in supplement. The dissociation curves showed that PCRs yielded single products. Samples from three or more independent immunoprecipitation assays were analyzed.
2.7. Statistical Analysis
Values were expressed as mean ± standard error of the mean (SEM). Differences between groups were examined for statistical significance using one-way ANOVA or two-tailed Student's t-tests, using GraphPad Prism 5 software. A P value less than 0.05 denoted the presence of a statistically significant difference.
3. Results
3.1. Endogenous PGC-1α Regulates Nuclear-Encoded Electron Transport Chain Genes in Neuronal Cells and in Brain
To determine whether endogenous PGC-1α systematically regulates the expression of the endogenous, neuronal electron transport chain, we silenced native PGC-1α using small interfering RNA (siRNA) in dopaminergic SK-N-MC neuroblastoma cells. Transfection with 100 nM PGC-1α siRNA reliably knocked down PGC-1α mRNA abundance by 80% compared to cells transfected with negative control siRNA (Supplementary Figure S1, in Supplementary Material available online at http://dx.doi.org/10.1155/2016/2405176). Similar results were obtained when UBC instead of the ribosomal gene RPL13 was used to control for RNA loading. Silencing of endogenous PGC-1α repressed the relative abundance of 14 of 18 nuclear-encoded electron transport chain genes (ETC) analyzed chosen to representing complexes I, II, III, IV, and V of the electron transport chain with P values below 0.05 (Figure 1(a)). Importantly, similar results were observed in brain of PGC-1α null mice [21]. Expression of 5 out of 6 ETC subunits probed was significantly decreased in PGC-1α knockout mice (Figure 1(b)) compared to age- and sex-matched wild-type littermates (N = 3) with P values below 0.05.
Figure 1.

Endogenous PGC-1α regulates the nuclear-encoded electron transport chain genes in neuronal cells and in mice. (a) Silencing of endogenous PGC-1α repressed the expression of nuclear-encoded electron transport chain genes representing complexes I, II, III, IV, and V of the electron transport chain in SK-N-MC cells by quantitative PCR analysis (note log2 scale). Model circuit and observed effects in circuit components are shown (top). Subunits of complexes I to V of the electron transport chain are color-coded in panels (a)–(c) in accordance with the color legend shown in (b). Means ± SEM are shown (N = 3 for each treatment). The ribosomal gene RPL13 was used to control for input RNA. (b) Expression of representative electron transport chain genes was similarly reduced in brain of PGC-1α null mice [21] compared to age- and sex-matched wild-type littermates (N = 3). The ribosomal gene RPL13 was used to control for input RNA. (c) Transduction with adenovirus carrying PGC-1α trans-activated the expression of endogenous genes encoding nuclear subunits of complexes I, II, IV, and V of the mitochondrial respiratory chain in SK-N-MC cells compared controls transduced with the LacZ gene. Model circuit and observed effects in circuit components are shown (top).
Conversely, we previously showed that transduction with adenovirus carrying PGC-1α (but not transduction with the control LacZ gene) trans-activated the expression of endogenous genes encoding nuclear subunits of complexes I, II, IV, and V of the mitochondrial respiratory chain in primary rat midbrain cultures [14]. We independently confirm this here in an additional cell line, SK-N-MC cells (Figure 1(c)). In the catecholaminergic SK-N-MC cells, 15 of 18 ETC genes analyzed were overexpressed in response to transduction with PGC-1α (Figure 1(c)). Collectively, these data show that endogenous PGC-1α regulates electron transport chain gene expression in neuronal cells and in brain.
3.2. The Orphan Nuclear Estrogen-Related Receptor α (ERRα) Is an Early Target of Endogenous, Neuronal PGC-1α
ERRα was identified on the basis of its sequence similarity to classical, hormone-regulated steroid receptors [23]. It recognizes similar DNA motifs as the estrogen receptors but does not bind naturally secreted estrogens in animals [24]. However, PGC-1α is a peptide ligand for ERRα in nonneuronal cells [13]. There, PGC-1α induces the expression of ERRα and potently converts ERRα from a factor with little or no transcriptional activity to a potent regulator of gene expression via interaction with leucine-rich motifs in the PGC-1α peptide [13]. To determine whether PGC-1α similarly exerts its regulatory control on the neuronal electron transport chain genes in coordination with endogenous ERRα, we silenced endogenous PGC-1α in SK-N-MC cells. Knockdown of PGC-1α dramatically repressed endogenous ERRα expression (Figure 2(a)) by more than 90% and also repressed the late target gene nuclear respiratory factor-1 (NRF1) by more than 50% (Figure 2(c)) compared to controls transfected with scrambled siRNAs. To further delineate the underlying transcriptional program, we then silenced endogenous ERRα (Supplementary Figure S2). Silencing ERRα not only repressed the NRF1 gene expression (Figure 2(d)) but also recapitulated the reduction in electron transport chain gene expression observed in response to PGC-1α-silencing (with the exception of COX7A2 expression) (Figure 2(b)). This is consistent with previous studies in nonneuronal cells, that is, murine myoblasts [8] and human osteosarcoma cells [13].
Figure 2.

The orphan nuclear estrogen-related receptor α (ERRα) is an early target of endogenous, neuronal PGC-1α. (a) Silencing of neuronal PGC-1α repressed the expression of endogenous ERRα by more than 90%, respectively, compared to controls transfected with scrambled siRNAs (NS). Model circuit and observed effects in circuit components are shown. (b) Silencing of ERRα largely recapitulated the reduction in electron transport chain gene expression observed in response to PGC-1α-silencing (note log2 scale). NRF1 gene expression was downregulated by more than 50% by silencing PGC-1α (c) or ERRα (d). The ribosomal gene RPL13 was used as control for input RNA. Mean ± SEM shown (N = 3 for each set). ∗ denotes P value ≤ 0.05.
Collectively, these data suggest that in neuronal cells, endogenous PGC-1α is a potent transcriptional coactivator of the early target gene ERRα and that both endogenous PGC-1α and ERRα activity modulate the late target gene NRF1 and the expression of most components of the human neuronal electron transport chain.
3.3. PGC-1α Physically Associates with Evolutionary Conserved ERRα Binding Motifs in the Promoters of Neuronal Electron Transport Chain Genes That Are Dysregulated in Parkinson's Disease
Transcriptional coregulators like PGC-1α exert their function through transcriptional complexes that occupy the promoters of distinct target genes. Transcription factors direct these complexes (including the transcriptional coregulator) to specific target sequences. The transcription factor ERRα occupies a nine-nucleotide extended half-site sequence with the consensus TNAAGGTCA, referred to as ERRα response element (ERRE) [25, 26]. These ERRα binding motifs are evolutionary conserved and enriched in electron transport chain genes (Figure 3(a) and Supplementary Figure S3) [8, 27]. In order to evaluate whether PGC-1α regulation of ETC genes is the result of an interaction of its transcriptional complex with these evolutionarily conserved ERRα binding motifs, we performed quantitative chromatin immunoprecipitation (ChIP) analyses in SK-N-MC neuroblastoma cells overexpressing PGC-1α protein.
Figure 3.

PGC-1α physically associates with evolutionary conserved ERRα binding motifs in the promoters of neuronal electron transport chain genes that are dysregulated in Parkinson's disease. (a) The VISTA plot of a 2-kb promoter region of the SDHB gene is shown with percentage identity of the human and mouse sequences. Small vertical bars indicate the location of conserved predicted ERRE binding motifs and the asterisk indicates the binding motif assayed by quantitative chromatin immunoprecipitation. (b) Quantitative chromatin immunoprecipitation (qChIP) analyses in SK-N-MC neuroblastoma cells transfected with a myc-tagged PGC-1α plasmid construct were performed. Promoter fragments for ATP5A1 (complex V), COX5B (complex IV), NDUFB5 (complex I), and SDHB (complex II) were specifically enriched in the IP fraction of PGC-1α compared to IgG control indicating PGC-1α occupancy of the conserved ERRE motifs. UCP-2, a known transcriptional target of PGC-1α [22], was used as positive control. No PGC-1α occupancy was seen in intergenic regions lacking a predicted ERRα binding site that was included as a negative control. Quantitative PCR data were normalized to genomic DNA and visualized as percent input. ∗ denotes P value ≤ 0.05.
We evaluated PGC-1α cooccupancy of conserved ERRα in the promoters of electron transport chain genes that are underexpressed in laser-captured nigral dopamine neurons of patients with symptomatic PD neuropathology as well as in individuals with incipient, subclinical PD neuropathology [14]. One gene representative for each of complexes I, II, IV, and V of the electron transport chain was investigated. ATP5A1 (complex V), COX5B (complex IV), NDUFB5 (complex I), and SDHB (complex II) were evaluated. Promoter fragments were specifically enriched in the IP fraction of PGC-1α compared to IgG control indicating PGC-1α occupancy of the conserved ERRE motifs (Figure 3(b)). UCP-2, a known transcriptional target of PGC-1α [22], was used as positive control. No PGC-1α occupancy was seen in intergenic regions lacking a predicted ERRα binding site that were included as negative controls (Figure 3(b)).
These results indicate that PGC-1α not only trans-activates expression of the transcription factor ERRα but also coactivates its target genes in the neuronal electron transport chain via occupancy of conserved ERRα binding motifs in their promoters.
3.4. The Endogenous PGC-1α and ERRα-Regulated Feed-Forward Circuit Can Be Targeted through Systems Pharmacology
Pioglitazone, a thiazolidinedione approved for the treatment of diabetes, is a synthetic ligand for Peroxisome proliferator-activated receptor γ (PPARγ) and to a lesser extent PPARα [28]. PPARγ trans-activates PGC-1α thereby activating mitochondrial biogenesis in human subcutaneous tissue [29]. Importantly, for Parkinson's disease [30], treatment with pioglitazone or with related thiazolidinediones is protective in multiple animal models of PD [31–33]. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone are linked to parkinsonism in humans and rodents. Thiazolidinediones strongly suppress MPTP-induced-loss of tyrosine hydroxylase-positive cells in the substantia nigra pars compacta [31, 32] as well as motor and olfactory dysfunctions in animal models [32]. Pioglitazone also suppressed rotenone-induced reduction in striatal dopamine levels and locomotor activity in rats [34].
To test the idea that the PGC-1α and ERRα-regulated feed-forward circuit can be exploited as a target system for therapeutics, we evaluated the endogenous transcriptional response to pioglitazone treatment in neuronal cells. A dose response curve with increasing concentrations of pioglitazone was performed (Supplementary Figure S4). At a concentration of 10 μM, 48-hour treatment with pioglitazone pervasively activated the PGC-1α-ERRα circuit (Figure 4). It induced a statistically significant 5-fold increase in expression of endogenous PGC-1α, a significant 2-3 fold increase in endogenous ERRα, and a correlated, significant 2–5-fold-trans-activation of their electron transport chain target genes (Figure 4). These data confirm that the PGC-1α and ERRα-regulated feed-forward circuit is druggable for early intervention in PD and other brain diseases.
Figure 4.
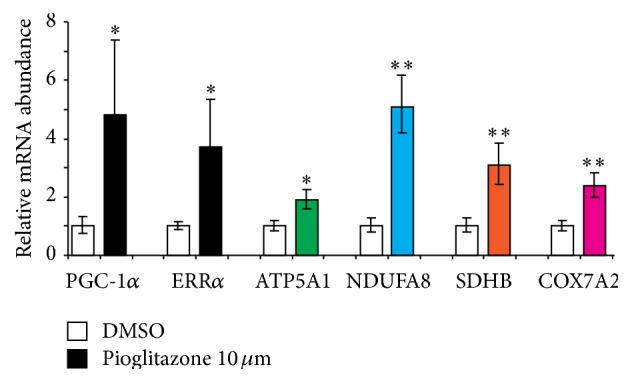
Endogenous PGC-1α is a target for drugs designed to restore electron transport chain expression. Treatment with pioglitazone (concentration of 10 μM, 48 hours of treatment) pervasively activated the PGC-1α-ERRα circuit. It induced a statistically significant 5-fold increase in expression of endogenous PGC-1α, a significant 2-3-fold increase in endogenous ERRα, and a resulting significant 2–5-fold-trans-activation of their endogenous electron transport chain target genes compared to cells treated with vehicle alone. Mean ± SEM shown (N = 10 for each treatment). ∗ denotes P value ≤ 0.05. ∗∗ denotes P value ≤ 0.005.
4. Discussion
Cellular bioenergetics are particularly important in “energy-guzzling” neurons, but the role of PGC-1α in regulating the neuronal electron transport chain has not previously been clarified. In this study we delineate a previously unconfirmed neuronal transcriptional circuit controlled by endogenous PGC-1α. By combining gene silencing and gene expression with quantitative chromatin immunoprecipitation analysis in neuronal cells and mouse brain, and taken together with our previous studies in primary mesencephalic cultures [14], we show evidence for a feed-forward circuit of endogenous neuronal PGC-1α and ERRα that activates the nuclear-encoded mitochondrial electron transport chain via occupancy of evolutionary conserved ERRα motifs. PGC-1α-induced ETC gene expression has been previously linked to mitochondrial respiration [35]. In muscle cells, for example, PGC-1α-induced ETC gene expression results in increased mitochondrial respiration [35].
Mitochondrial dysfunction is impaired in common and rare neuronal diseases. Recent studies have shown that genes involved in the nuclear-encoded electron transport chain exhibit reduced expression in dopamine neurons and substantia nigra of humans with symptomatic and subclinical Parkinson's neuropathology. Systems biology analysis of human brains revealed a pervasive expression defect of PGC-1α-linked bioenergetics genes in laser-captured dopamine neurons of Parkinson's patients and substantia nigra of individuals with subclinical, brainstem-predominant Lewy body neuropathology [14] that likely represent preclinical PD [36]. These findings were replicated in an independent population [37]. The gene sets identified pinpointed defects in mitochondrial electron transport, glucose utilization, and glucose sensing early in the disease course [14]. Conversely, activating the PGC-1α-regulated program ameliorated mutant α-synuclein- and rotenone-induced loss of dopamine neurons in primary midbrain cultures [14]. In mouse models of PD, the PGC-1α transgene suppressed MPTP-induced dopaminergic neurodegeneration [18]. Conversely, deletion of PGC-1α dramatically enhanced MPTP-induced degeneration of nigral dopamine neurons in a mouse model of PD [20]. In mice carrying mutant PARK2-linked familial PD repression of PGC-1α by the parkin substrate PARIS contributes to neurodegeneration, while increased PGC-1α expression suppressed mutant parkin-induced neurodegeneration [19]. In an isogenic human induced Pluripotent Stem Cell model of Parkinson's PGC-1α suppressed cell loss in response to environmental toxins and mutant α-synuclein [38]. In short, evidence in human brain and in multiple cellular, human stem cell, genetic and toxic animal models of PD link PGC-1α-regulated programs to an onset mechanism of Parkinson's. Beyond Parkinson's there are clues to suggest that a PGC-1α-regulated transcriptional program is more generally involved in aging-related diseases such as ALS and HD [16, 39]. Mildly increased PGC-1α expression in skeletal muscle protects from sarcopenia during aging [4].
Is this pathway a tractable target for gene therapy? In mice, both too little and too much PGC-1α are detrimental. PGC-1α knockout leads to cardiomyopathy [12], but forced overexpression of PGC-1α at supraphysiologic levels induces uncontrolled mitochondrial proliferation and cardiomyopathy [12]. Analogously, adenoassociated virus- (AAV-) mediated overexpression of PGC-1α in the substantia nigra induces a loss of dopaminergic markers and enhances nigral vulnerability [40, 41].
Chemically restoring the activity of the endogenous PGC-1α-regulated circuit (i.e., reduced in Parkinson's neuropathology) back to normal may be a more advantageous strategy for early intervention in incipient PD than forced overexpression of exogenous PGC-1α. This could be accomplished through small molecule drugs that modulate any of the switches in the neuronal circuit we here delineated. PGC-1α expression can be activated through molecules acting upstream of the PGC-1α gene such a glitazones. For example, pioglitazone confers neuroprotection in mouse models of PD [32] and activates the entire neuronal PGC-1α-ERRα-regulated feed-forward circuit in neuronal cells through activation of the nuclear receptor PPARγ, the transcription factor of PGC-1α. Because PPARγ regulates numerous transcriptional cascades in addition to the PGC-1α-regulated circuit, this approach carries the risk of side effects through broad activation of unwanted programs. Moreover, initiation of treatment during earliest, preclinical disease stages might be necessary to achieve meaningful effects. In patients with clinically manifest PD (indicating advanced underlying Lewy body neuropathology and substantial loss of dopamine neurons), no efficacy was found for pioglitazone in slowing disease progression in a clinical trial [42]. However, a large, recent epidemiologic study suggested a beneficial effect for glitazones such as pioglitazone in reducing risk of PD in neurologically normal individuals with diabetes [43]. This study found an incidence rate of PD in the glitazone-exposed group of 6.4 per 10,000 patient years compared with 8.8 per 10,000 patient years in those prescribed other antidiabetic treatments [43]. ERRα is another switch in the circuit that could be targeted. We show that endogenous PGC-1α regulates neuronal ERRα transcription (Figure 2) and that silencing neuronal ERRα recapitulates the effect of PGC-1α knockdown on endogenous electron transport chain expression (Figure 2). ERRα may be both sufficient and necessary for mediating the action of PGC-1α on mitochondrial biogenesis as in muscle cells induction of mitochondrial biogenesis by PGC-1α was largely suppressed when ERRα was inhibited [8]. Targeting ERRα directly with small molecules is an attractive strategy for drug development, although the ligand-binding pocket is small [44]. Phytoestrogens activate ERRα [45] and a synthetic compound that inhibits ERRα has been reported [8, 45]. There is also precedent for a promising third strategy, targeting the ERRα-PGC-1α interaction with small molecules [8, 46].
These data clarify a transcriptional network regulated by neuronal PGC-1α that now can be therapeutically targeted for common neurodegenerative diseases. Novel chemical modulators tailored to this circuit together with a transformed clinical trial paradigm directed at individuals with earliest, preclinical stages of neuropathology will be positioned to modify neuronal bioenergetics defects and potentially achieve substantial clinical benefits for patients with neurodegenerative disease.
Supplementary Material
The Supplementary Material includes Supplementary Figures and a Supplementary Table.
Acknowledgments
This work was supported by NIH Grant U01 NS082157, the US Department of Defense, and the M.E.M.O. Hoffman Foundation.
Conflict of Interests
The authors have no conflict of interests to report.
References
- 1.Austin S., St-Pierre J. PGC1α and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. Journal of Cell Science. 2012;125(21):4963–4971. doi: 10.1242/jcs.106625. [DOI] [PubMed] [Google Scholar]
- 2.Mootha V. K., Lindgren C. M., Eriksson K.-F., et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 3.Handschin C., Spiegelman B. M. Peroxisome proliferator-activated receptor γ coactivator 1 coactivators, energy homeostasis, and metabolism. Endocrine Reviews. 2006;27(7):728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 4.Wenz T., Rossi S. G., Rotundo R. L., Spiegelman B. M., Moraes C. T. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(48):20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Finkel T. Cell biology: a clean energy programme. Nature. 2006;444(7116):151–152. doi: 10.1038/444151a. [DOI] [PubMed] [Google Scholar]
- 6.Austin S., Klimcakova E., St-Pierre J. Impact of PGC-1α on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radical Biology and Medicine. 2011;51(12):2243–2248. doi: 10.1016/j.freeradbiomed.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 7.Raichle M. E., Gusnard D. A. Appraising the brain's energy budget. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10237–10239. doi: 10.1073/pnas.172399499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mootha V. K., Handschin C., Arlow D., et al. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(17):6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arany Z., He H., Lin J., et al. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metabolism. 2005;1(4):259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Rohas L. M., St-Pierre J., Uldry M., Jäger S., Handschin C., Spiegelman B. M. A fundamental system of cellular energy homeostasis regulated by PGC-1α . Proceedings of the National Academy of Sciences of the United States of America. 2007;104(19):7933–7938. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin J., Handschin C., Spiegelman B. M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metabolism. 2005;1(6):361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Lehman J. J., Barger P. M., Kovacs A., Saffitz J. E., Medeiros D. M., Kelly D. P. Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. Journal of Clinical Investigation. 2000;106(7):847–856. doi: 10.1172/jci10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schreiber S. N., Knutti D., Brogli K., Uhlmann T., Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor α (ERRα) The Journal of Biological Chemistry. 2003;278(11):9013–9018. doi: 10.1074/jbc.m212923200. [DOI] [PubMed] [Google Scholar]
- 14.Zheng B., Liao Z., Locascio J. J., et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Science Translational Medicine. 2010;2(52) doi: 10.1126/scitranslmed.3001059.52ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui L., Jeong H., Borovecki F., Parkhurst C. N., Tanese N., Krainc D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Weydt P., Pineda V. V., Torrence A. E., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1α in Huntington's disease neurodegeneration. Cell Metabolism. 2006;4(5):349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Thau N., Knippenberg S., Körner S., Rath K. J., Dengler R., Petri S. Decreased mRNA expression of PGC-1α and PGC-1α-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. Journal of Neuropathology and Experimental Neurology. 2012;71(12):1064–1074. doi: 10.1097/nen.0b013e318275df4b. [DOI] [PubMed] [Google Scholar]
- 18.Mudò G., Mäkelä J., Di Liberto V., et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cellular and Molecular Life Sciences. 2012;69(7):1153–1165. doi: 10.1007/s00018-011-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin J.-H., Ko H. S., Kang H., et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144(5):689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.St-Pierre J., Drori S., Uldry M., et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Lin J., Wu P.-H., Tarr P. T., et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell. 2004;119(1):121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Valle I., Álvarez-Barrientos A., Arza E., Lamas S., Monsalve M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovascular Research. 2005;66(3):562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 23.Giguere V., Yang N., Segui P., Evans R. M. Identification of a new class of steroid hormone receptors. Nature. 1988;331(6151):91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 24.Ranhotra H. S. Estrogen-related receptor alpha and mitochondria: tale of the titans. Journal of Receptor and Signal Transduction Research. 2015;35(5):386–390. doi: 10.3109/10799893.2014.959592. [DOI] [PubMed] [Google Scholar]
- 25.Johnston S. D., Liu X., Zuo F., et al. Estrogen-related receptor alpha 1 functionally binds as a monomer to extended half-site sequences including ones contained within estrogen-response elements. Molecular Endocrinology. 1997;11(3):342–352. doi: 10.1210/me.11.3.342. [DOI] [PubMed] [Google Scholar]
- 26.Vanacker J.-M., Bonnelye E., Chopin-Delannoy S., Delmarre C., Cavaillès V., Laudet V. Transcriptional activities of the orphan nuclear receptor ERR α (estrogen receptor-related receptor-α) Molecular Endocrinology. 1999;13(5):764–773. doi: 10.1210/mend.13.5.0281. [DOI] [PubMed] [Google Scholar]
- 27.Deblois G., Hall J. A., Perry M.-C., et al. Genome-wide identification of direct target genes implicates estrogen-related receptor α as a determinant of breast cancer heterogeneity. Cancer Research. 2009;69(15):6149–6157. doi: 10.1158/0008-5472.can-09-1251. [DOI] [PubMed] [Google Scholar]
- 28.Smith U. Pioglitazone: mechanism of action. International Journal of Clinical Practice. 2001;(121):13–18. [PubMed] [Google Scholar]
- 29.Bogacka I., Xie H., Bray G. A., Smith S. R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54(5):1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 30.Carta A. R., Simuni T. Thiazolidinediones under preclinical and early clinical development for the treatment of Parkinson's disease. Expert Opinion on Investigational Drugs. 2015;24(2):219–227. doi: 10.1517/13543784.2015.963195. [DOI] [PubMed] [Google Scholar]
- 31.Dehmer T., Heneka M. T., Sastre M., Dichgans J., Schulz J. B. Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with IκBα induction and block of NFκB and iNOS activation. Journal of Neurochemistry. 2004;88(2):494–501. doi: 10.1046/j.1471-4159.2003.02210.x. [DOI] [PubMed] [Google Scholar]
- 32.Schintu N., Frau L., Ibba M., et al. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson's disease. European Journal of Neuroscience. 2009;29(5):954–963. doi: 10.1111/j.1460-9568.2009.06657.x. [DOI] [PubMed] [Google Scholar]
- 33.Hunter R. L., Dragicevic N., Seifert K., et al. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. Journal of Neurochemistry. 2007;100(5):1375–1386. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- 34.Ulusoy G. K., Celik T., Kayir H., Gürsoy M., Isik A. T., Uzbay T. I. Effects of pioglitazone and retinoic acid in a rotenone model of Parkinson's disease. Brain Research Bulletin. 2011;85(6):380–384. doi: 10.1016/j.brainresbull.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Wu Z., Puigserver P., Andersson U., et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98(1):115–124. doi: 10.1016/s0092-8674(00)80611-x. [DOI] [PubMed] [Google Scholar]
- 36.Dickson D. W., Fujishiro H., DelleDonne A., et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson's disease. Acta Neuropathologica. 2008;115(4):437–444. doi: 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
- 37.Elstner M., Morris C. M., Heim K., et al. Expression analysis of dopaminergic neurons in Parkinson's disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathologica. 2011;122(1):75–86. doi: 10.1007/s00401-011-0828-9. [DOI] [PubMed] [Google Scholar]
- 38.Ryan S. D., Dolatabadi N., Chan S. F., et al. Erratum: isogenic human iPSC parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell. 2013;155(7):1652–1653. doi: 10.1016/j.cell.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Da Cruz S., Parone P. A., Lopes V. S., et al. Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metabolism. 2012;15(5):778–786. doi: 10.1016/j.cmet.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciron C., Lengacher S., Dusonchet J., Aebischer P., Schneider B. L. Sustained expression of PGC-1α in the rat nigrostriatal system selectively impairs dopaminergic function. Human Molecular Genetics. 2012;21(8):1861–1876. doi: 10.1093/hmg/ddr618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark J., Silvaggi J. M., Kiselak T., et al. Pgc-1α overexpression downregulates Pitx3 and increases susceptibility to MPTP toxicity associated with decreased Bdnf. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0048925.e48925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson's disease: a phase 2, multicentre, double-blind, randomised trial. The Lancet Neurology. 2015;14(4):795–803. doi: 10.1016/S1474-4422(15)00144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brauer R., Bhaskaran K., Chaturvedi N., Dexter D. T., Smeeth L., Douglas I. Glitazone treatment and incidence of Parkinson’s disease among people with diabetes: a retrospective Cohort study. PLoS Medicine. 2015;12(7) doi: 10.1371/journal.pmed.1001854.e1001854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kallen J., Schlaeppi J.-M., Bitsch F., et al. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor α (ERRα): crystal structure of ERRα ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1α . The Journal of Biological Chemistry. 2004;279(47):49330–49337. doi: 10.1074/jbc.m407999200. [DOI] [PubMed] [Google Scholar]
- 45.Suetsugi M., Su L., Karlsberg K., Yuan Y.-C., Chen S. Flavone and isoflavone phytoestrogens are agonists of estrogen-related receptors. Molecular Cancer Research. 2003;1(13):981–991. [PubMed] [Google Scholar]
- 46.Handschin C., Mootha V. K. Estrogen-related receptor α (ERRα): a novel target in type 2 diabetes. Drug Discovery Today: Therapeutic Strategies. 2005;2(2):151–156. doi: 10.1016/j.ddstr.2005.05.001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplementary Material includes Supplementary Figures and a Supplementary Table.