

Abstract
Background
Hepatocellular carcinoma (HCC) commonly presents at a late stage when surgery is no longer a curative option. As such, novel therapies for advanced HCC are needed. Oncolytic viruses are a viable option for cancer therapy due to their ability to specifically infect, replicate within, and kill cancer cells. In this study we investigate the ability of GLV-2b372, a novel light-emitting recombinant vaccinia virus derived from a wild-type Lister strain, to kill HCC.
Methods
Four human HCC cell lines were assayed in vitro for infectivity and cytotoxicity. Viral replication was quantified via standard viral plaque assays. Flank HCC xenografts generated in athymic nude mice were treated with intratumoral GLV-2b372 to assess for tumor growth inhibition and viral biodistribution.
Results
Infectivity occurred in a time and concentration dependent manner with 70% cell death in all cell lines by day 5. All cell lines supported efficient viral replication. At 25 days post infection, flank tumor volumes decreased by 50% while controls increased by 400%. Tumor tissue demonstrated substantial GLV-2b372 infection at 24 hours, 48 hours and at 2 weeks.
Conclusions
We demonstrate that GLV-2b372 efficiently kills human HCC in vitro and in vivo and is a viable treatment option for patients with HCC.
Keywords: Oncolytic Virus, Hepatocellular Carcinoma, Vaccinia Virus, Immunotherapy, GLV-2b372, Lister Strain 1.1.1
Introduction
Hepatocellular carcinoma (HCC) is devastating disease with poor prognosis. It is the most common primary malignancy of the liver with approximately 700,000 new cases diagnosed annually worldwide (1). Currently, the only potentially curative treatment for advanced HCC is surgery or ablative therapies for limited disease. Unfortunately, 70–85% of patients are diagnosed at an advanced stage, and are no longer surgical candidates (2). Based on the heterogeneity of HCC and difficulty identifying full extent of tumor burden, traditional systemic chemotherapy and other adjuvant therapies provide poor outcomes (3). The result is that the majority of patients with HCC will die within 6 months of diagnosis (4).
Oncolytic viruses are a promising therapeutic option in cancer treatment due to their ability to selectively infect, replicate within and kill cancer cells. Additionally, they can be used to deliver targeted gene therapies and provide disease progression tracking as well as accurate visualization of tumor burden. For this reason, oncolytic virotherapy is rapidly emerging as a promising frontier in cancer therapy, whether in first-line capacity, or as a secondary alternative when conventional therapeutic options have failed (5).
Among the many viruses that can be used as oncolytic vectors, vaccinia virus (VACV) has emerged as a highly promising candidate. VACV is a double-stranded enveloped lytic DNA virus with a linear genome and a short replication cycle that can infect cells from a variety of animal models and across many cell types. In addition, VACV replicates exclusively in the cytoplasm, thus eliminating any risk of integration. It also bears a large 192-kb genome, making it capable of accepting up to 25-kb extrinsic genetic insertions without compromising viral replication competency. Additionally, VACV immunogenicity concerns were alleviated by the finding that VACV treatment was as effective in vaccinated as non-vaccinated animals, giving it potential to be used in multiple doses, if required (6). Perhaps most importantly, VACV has a well-recognized safety profile, having been administered to millions as a live vaccine in the smallpox eradication campaign of the World Health Organization. Currently, the therapeutic value of VACV is being evaluated in several stage I/II clinical trials.
GLV-2b372 is a novel recombinant VACV derived from the non-engineered Lister LIVP 1.1.1 strain by inserting the TurboFP635 gene expression cassette into the TK locus, which produces a far-red fluorescing protein in infected cells for real-time monitoring of viral infection. Currently in submission for publication from our lab are studies describing the construction of GLV-2b372, and the ability of this virus to treat malignant pleural mesothelioma and ovarian carcinoma. In this study, we evaluate the effects of GLV-2b372 on a panel of human HCC cell lines in vitro and in a xenograft animal model, and explore the potential application of this virus as a clinically relevant therapeutic agent for HCC.
Materials and Methods
Reagents
Carboxymethyl cellulose (CMC) was purchased from MP biomedical (Cat. No. 150560, Solon, OH). CMC medium was prepared prior to each viral titer, and consists of 7.5 g CMC and 500 mL Dulbecco modified Eagle medium supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. Albumin from bovine serum (BSA, Cat. No. A2153), crystal violet powder (Cat. No. C3886) and formaldehyde 37% (Cat. No. 252549) were purchased from Sigma-Aldrich (St. Louis, MO). Ethanol (Cat. No. BP2813) was purchased from Fisher Scientific (Pittsburgh, PA). Crystal violet staining solution is made up of 1.3 g crystal violet powder, 300 mL formaldehyde 37%, 50 mL ethanol and 1 L of water.
Virus
GLV-2b372 is derived from the LIVP 1.1.1 strain, a less virulent wild-type isolate of the LIVP strain (7). The TurboFP635 (Far-red fluorescent protein “katushka”) cDNA was PCR amplified using the plasmid FUKW (kindly provided by Dr. Marco J. Herold, University of Wurzburg) as the template with primers FUKW-5 (5′-GTCGAC(Sal I) CACCATGGTGGGTGAGGATAGCGTGC-3′) and FUKW-3 (5′-TTAATTAA(Pac I) TCAGCTGTGCCCCAGTTTGC-3′). The PCR product was gel-purified, and cloned into the pCR-Blunt II-TOPO vector using Zero Blunt TOPO PCR Cloning Kit (Invitrogen). The resulting construct pCRII-FUKW was sequence confirmed. The TurboFP635 cDNA was then released from pCRII-FUKW with Sal I and Pac I, and subcloned into TK-SEL-hNISa8 with the same cuts, replacing the hNIS cDNA. The resulting sequence confirmed construct TK-SEL-FUKW1 was used to make recombinant virus GLV-2b372 using LIVP 1.1.1 as the parental virus as described previously (8).
Cell culture and cell lines
HCC cell lines SNU-449, Huh-7 and Hep G2 were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA). SNU-739 cell line was obtained via Korean Cell Line Bank (KCLB; Chongno-gu, Seoul, Korea). The Huh-7 cell line was cultured in Roswell Park Memorial Institute medium, and Hep G2, SNU-449 and SNU-739 lines were cultured in Dulbecco modified Eagle medium. African green monkey kidney fibroblast cells (CV-1) were obtained from ATCC and grown in minimum essential medium. All mediums were supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin and all cell lines were incubated at 37 °C in a humidified 5 % CO2 atmosphere.
Turbo fluorescent protein 635 (TurboFP635) expression
SNU-739, SNU-449 and Huh-7 cells were seeded at a density of 2.5×103 cells/well. Hep G2 cells were seeded at 4×103 cells/well. After 24 hours of incubation, cells were infected with oncolytic VACV GLV-2b372 at multiplicities of infection (MOIs) of 0.001, 0.01, 0.1 and 1. 24 hours subsequent to infection, cells were examined for TurboFP635 expression using an inverted fluorescence microscope (Nikon Eclipse TE300; Nikon, Tokyo, Japan). Imaging was performed daily for 5 days.
Viral plaque assay
CV-1 cells were plated at 2×105 cells/well in 24-well plates and incubated overnight. Viral supernatants collected and frozen post cytotoxicity assay were thawed. A serial dilution of each supernatant sample was created to perform standard viral plaque assays on confluent CV-1 cultured wells. Plates were incubated for 4 hours, viral supernatants were aspirated and 1mL of CMC medium was added to each well. The plates were incubated at 37°C in a humidified 5 % CO2 chamber for 2 days. 350 μL of crystal violet staining solution was added to each well and plates were left at room temperature overnight. The following day, the plates were washed with water and viral plaques were counted. All titers were performed in triplicate.
Viral cytotoxicity assay
Cells were plated and infected as mentioned above. Viral supernatants from each infected well were collected daily for 5 days and immediately frozen at −80°C and stored for subsequent viral plaque assays. Viral cytotoxicity was measured at days 0 through 5 for all cell lines using the Dojindo cell counting kit (CCK-8; Rockville, MD). CCK-8 dye was added to phenol red free medium at a ratio of 10:100 and the medium in each well was replaced with 110 μL of the dye solution and incubated for 4 hours. The plate was then read on a spectrophotometer (EL321e; Bio-Tek Instruments, Winooski, VT) at a wavelength of 450 nm. Survival curves are expressed as the percentage of viable cells as compared to uninfected control. All samples were analyzed in triplicate.
Biodistribution
Animals were euthanized at 24 hours, 48 hours and 2 weeks post infection with 1×108 PFUs of GLV-2b372. Animals were harvested for their brain, heart, kidney, lymph nodes, liver, lung, ovary, spleen and tumor tissues. Harvested tissues were frozen and stored at −80°C for subsequent viral plaque assays. Tissues were thawed and underwent mechanical homogenization. Viral plaque assays were performed on homogenized tissues to assess viral presence. All samples were performed in triplicate as described above.
Treatment of HCC in a flank xenograft model
Seven female athymic nude mice (Harlan) aged 6 to 8 weeks were injected with 1×106 Huh-7 cells into the right flank and randomized into groups at 2 weeks subsequent to tumor inoculation. Flank xenografts were treated with 1 intratumoral injection of 1×105 PFUs of GLV-2b372. Tumor size (length and width) and mouse weight were assessed and recorded. Tumor volumes were calculated by using the equation: V (mm3) = (4/3) × (3.14159265) × (a/2)2 × (b/2), where a is the smallest diameter and b the largest diameter.
Statistics
Significance differences among the tumor volume between the control and GLV-2b372 groups were determined via 2-way ANOVA test. Values of p < 0.05 were considered significant. All analyses were performed using Prism version 6.0 statistical software.
Results
GLV-2b372 infects HCC in a time and dose dependent manner
At 24 hours, TurboFP635 expression confirmed viral infection in all cell lines. Infectivity increased with time in all cell lines. Higher initial concentrations of virus resulted in earlier infectivity, demonstrating a dose dependent manner of infectivity. All cell lines demonstrated similar patterns of viral-mediated TurboFP635 expression as demonstrated by representative cell line SNU-739 (Figure 1).
Figure 1. GLV-2b372 infects HCC in a time and dose dependent manner.
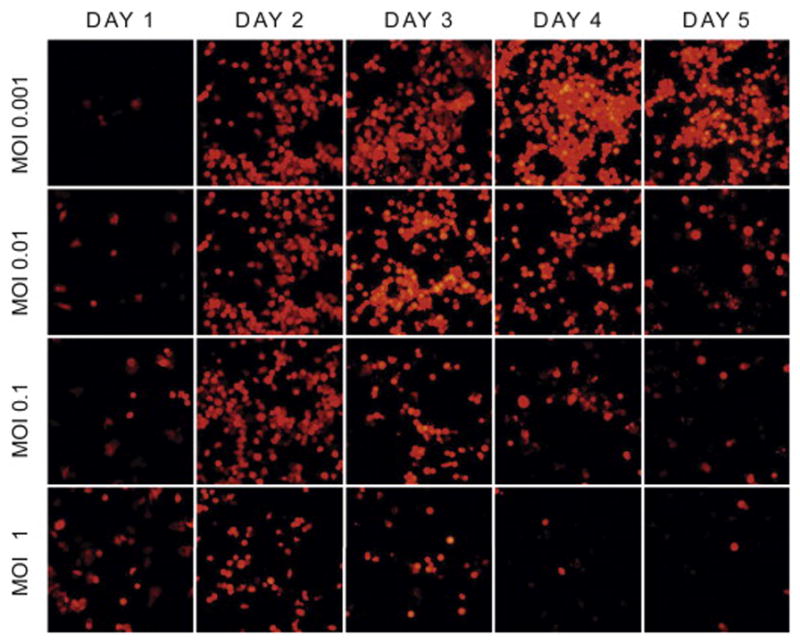
TurboFP635 expression in representative cell line SNU-739 infected at an MOI of 0.001, 0.01, 0.1 and 1 every 24 hours for 5 days. Infectivity occurs in a time and concentration dependent manner. While not shown above, data was collected in all cell lines and was consistent with the above figure.
GLV-2b372 demonstrates efficient viral replication in HCC cells in vitro
Viral proliferation of GLV-2b372 in SNU-739, SNU-449, Hep G2 and Huh-7 cell lines was assessed by standard viral plaque assay of supernatants collected over a 5-day period post infection (Figure 2). All cell lines supported significant GLV-2b372 replication.
Figure 2. GLV-2b372 demonstrates efficient viral replication in HCC cells in vitro.

At an MOI of 1, standard viral plaque assay of the supernatant from infected HCC cells demonstrates an increase in viral titers over 5 days.
GLV-2b372 efficiently kills HCC cell lines
HCC cell lines infected with GLV-2b372 showed efficient cytotoxicity in a dose and concentration dependent manner. By day 5, all cell lines achieved approximately 80% cytotoxicity at an MOI of 1 (Figure 3).
Figure 3. GLV-2b372 efficiently kills HCC cell lines.
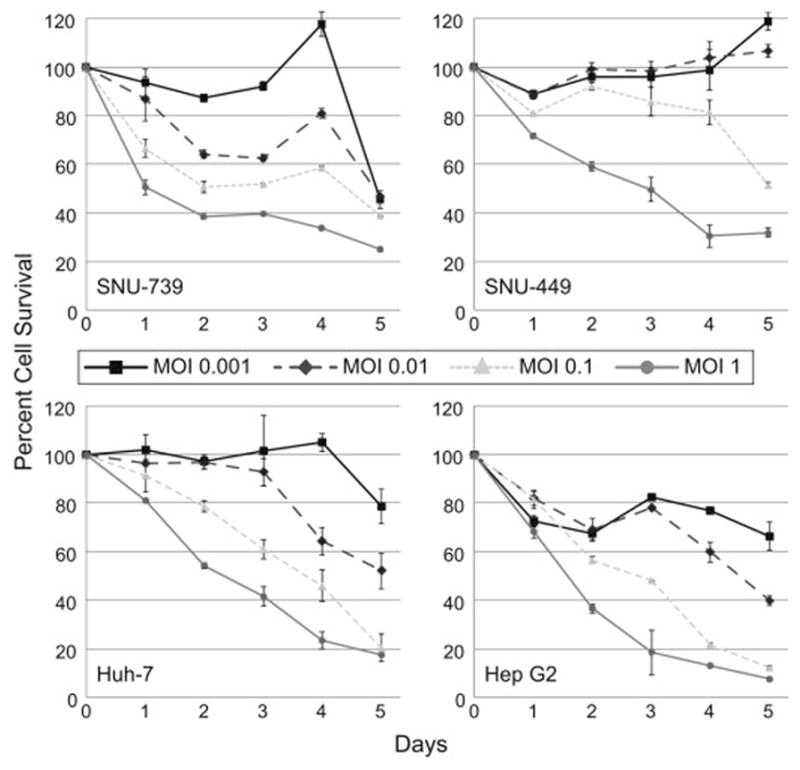
Cytotoxicity assays of 4 HCC cell lines infected with GLV-2b372 at multiplicities of infection (MOIs) of 0.001, 0.01, 0.1 and 1 demonstrate efficient cytotoxicity over 5 days. Higher MOIs of infection resulted in earlier killing of HCC cell lines, with almost all cell lines demonstrating nearly 80% cytotoxicity by day 5.
Biodistribution of GLV-2b372
At sacrifice, organ systems from animals treated with virus were harvested and viral titers were run to quantify viral presence at 24 hours, 48 hours, and at 2 weeks. Minimal virus was found in brain tissue at 24 and 48 hours, but was completely cleared by the 2-week time point. No other organ systems harbored virus at any time point. Significant viral presence was discovered in tumor tissue, particularly at the 48-hour time point, with continued viral intratumoral presence at 2 weeks (Figure 4).
Figure 4. Biodistribution of GLV-2b372 in vivo.
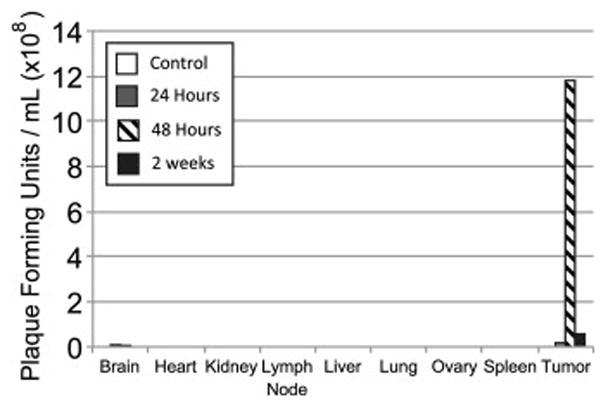
At sacrifice, tumor tissue demonstrated sustained presence of virus for 2 weeks. Minimal virus was found in brain tissue at 24 and 48 hours, but was completely cleared by the 2-week time point. No other organ systems harbored virus at any time point.
GLV-2b372 efficiently kills HCC in vivo
Huh-7 murine flank xenografts infected with GLV-2b372 exhibited a significant reduction in tumor volume when compared to controls (p < 0.05). Those animals treated with GLV-2b372 demonstrated approximately a 70 % reduction in tumor volume over 25 days. While control tumors increased in size by over 300% during that same time period. Three out of four animals treated with GLV-2b372 had complete resolution of tumor. These results indicate that GLV-2b372 can significantly reduce and erradicate HCC tumor volume in vivo (Figure 5).
Figure 5. GLV-2b372 significantly reduces tumor burden in an Huh-7 xenograft mouse model.
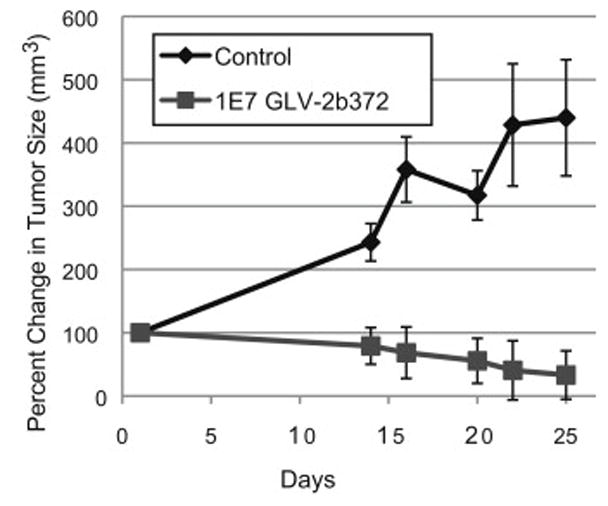
Groups of Huh-7 tumor-bearing nude mice were separated into two groups: control (n=3) and virus (1×105 PFUs GLV-2b372 via intratumoral injection) (n = 4). Tumor burden was significantly decreased in the viral therapy group when compared to control (p < 0.05). At sacrifice, 75 % of mice treated with intratumoral GLV-2b372 were found to have no residual tumor.
Discussion
HCC represents the third most common cause of cancer-related deaths worldwide, claiming an estimated 692,000 lives annually and accounting for approximately 90% of primary liver cancers (1, 9). Due to its silent clinical character, most HCC patients are diagnosed at an advanced stage of disease, when the only curative therapies of surgery and ablation are no longer feasible. The difficulty of identifying the full extent of tumor burden and a lack of consensus in standardizing treatment has resulted in poor prognosis and a paucity of viable therapy options for patients with non-resectable HCC (10).
While several chemotherapeutic agents have exhibited varying degrees of antitumor effects (11), no effective systemic therapy exists for patients with advanced HCC (12). Sorafenib, the only approved drug for treatment of advanced HCC, is a multi-kinase inhibitor prescribed to an estimated 40% of newly diagnosed HCC patients. However, the success of sorafenib is modest at best, only increasing overall survival by approximately 3 months in recent clinical trials (13). The heterogeneous nature of HCC tumors is likely responsible for the high levels of resistance against treatment with sorafenib and chemotherapy. Thus the key to changing the dismal prognosis of advanced HCC is the development of novel first-line and adjuvant treatment options that employ different mechanisms of action. (14).
Oncolytic viruses have demonstrated a marked ability to selectively infect, replicate within and lyse cancer cells. Among the many candidates currently under consideration as ideal oncolytic platforms, we selected vaccinia virus due to several favorable attributes. VACV rapidly reproduces, completing its life cycle within 24 hours from infection, with the release of as many as 10,000 new virions. The extracellular envelope of these newly produced virions is composed directly from the cell membrane. Therefore the VACV extracellular envelope contains several host complement control proteins and only a few exposed viral antigens. This makes VACV efficient at spreading throughout the system by evading both the complement and immune systems, allowing for improved infectivity and replication competency. Due to its large genome of approximately 192-kb, VACV has the capacity to hold up to 25 kb of foreign DNA and simultaneously express multiple therapeutic and imaging transgenes, which is not feasible in other viral vector platforms such as adenoviruses. In contrast to herpes virus the VACV never enters the host nucleus and replication occurs entirely in the cytoplasm. Thus there is no possibility of chromosomal integration. Perhaps most importantly, VACV was safely administered to millions in the World Health Organization’s smallpox eradication campaign, giving it a well-known safety profile in humans. The result being that there is a well-defined list of contraindications to vaccinia use and viable antiviral therapy options such as vaccinia immunoglobulin and cidofovir.
Even after extensive investigation of the use of oncolytic viruses in the treatment of HCC, a complete cure has eluded clinicians. To develop a more efficient oncolytic virus, researchers need to address three issues. First, the virus must be able to reach and infect intended tissue. Second, transgenes inserted into the virus need to be rationally chosen to provide the greatest benefit to treatment of the specific cancer type in question. Finally, the virus must be able to efficiently replicate and spread to surrounding tissue, thus propagating the intended therapeutic effect.
The majority of current studies have emphasized on the development of transgenes that can encode for therapeutic molecules to enhance the antitumor efficacy of oncolytic viruses. Fortunately, these investigations have garnered a great deal of success, and have significantly improved the efficacy of oncolytic vectors to treat cancer. An excellent example is the insertion of the hNIS transporter into both measles and VACV. It facilitates the radiotherapy and imaging of infected tissues, thus greatly increasing the efficacy of the viral therapy (15, 16). In addition to hNIS, a variety of different transgenes have been suggested that both inhibit intracellular pathways and activate prodrugs with varying degrees of success. Unfortunately, without adequate delivery, replication and spread within tumor tissue, the selection and insertion of optimal transgenes tailored to a specific cancer type will not sufficiently augment viral function to completely cure patients with HCC.
For this reason, we believe that focusing on the rational selection of the most optimal viral backbone may have an even more significant impact on tumor eradication than transgene selection. Within the VACV family, the Lister strain alone has thousands of possible variants that could serve as potential oncolytic vectors. One of the most prominent oncolytic VACVs, GLV-1h68 (GLV-ONC), currently involved in Phase I/II clinical trials, required 3 major gene deletions to assure safety of the virus while effectively targeting the majority of solid tumor types in humans. Specifically, Zhang et al found that the triple insertional deletions greatly reduced the replication of GLV-1h68 in normal mouse cells, while the replication of GLV-1h68 in tumor cells was not detrimentally affected (17). Unfortunately, while the insertional deletions increase the selectivity for cancer cells, they also attenuate the effects of the virus.
In our lab, we recently submitted data comparing GLV-1h68 with nonattenuated Lister strain variant GLV-2b372 for the treatment of malignant pleural mesothelioma. GLV-2b372 was derived from a plaque purified isolate of the LIVP 1.1.1 strain, chosen due to its natural tumor selectivity. It was found to have a natural deletion of the VACV thymidine-kinase gene at the J2R locus. Importantly, we found that when compared to GLV-1h68, GLV-2b372 was able to achieve equivalent in vitro killing of malignant pleural mesothelioma cells with 10–100 times fewer plaque forming units (PFUs) of virus. This result confirms that fewer gene deletions results in a more potent virus.
In this study, we examined the ability of GLV-2b372 to destroy HCC, both in vitro and in vivo. We demonstrate that GLV-2b372 effectively kills HCC cells in vitro and exhibits robust viral replication. More importantly though, our in vivo experiment shows that GLV-2b372 and GLV-1h68, its more attenuated cousin have similar biodistribution (17). Importantly, this result shows that even though GLV-2b372 is a more virulent vector for cancer treatment, it is selective for tumor tissue even with fewer gene deletions.
Previous studies using GLV-1h68 for the treatment of HCC were promising. Ghentschev et al were able to demonstrate significant suppression of tumor growth as compared to controls. However, by 30 days post infection, tumors treated with virus had approximately doubled in size (18). In our study, we demonstrate that treatment with GLV-2b372 not only inhibited tumor growth, but actually reduced tumor volume by 75 % over the same period of time. In addition, we were able to show complete eradication of tumor in 75% of mice treated with GLV-2b372. This evidence further supports the idea that GLV-2b372 is a more potent viral vector for oncolytic therapy.
In conclusion, GLV-2b372, a derivative of the less attenuated LIVP 1.1.1 isolate, demonstrates significant oncolysis in an HCC model in vitro. More importantly though, we demonstrate a similar biodistribution in vivo as compared to the more attenuated GLV-1h68. We further demonstrate that GLV-2b372, in contrast to other viral vectors, not only significantly reduces tumor burden, but can completely eradicate tumor. We believe that further modification of GLV-2b372 with carefully selected therapeutic genes could result in a superior oncolytic viral platform for the treatment of HCC.
Acknowledgments
Supported in part by training grants T 32 CA09501 (J.C.), a grant for rare diseases research from “Cycle for Survival”, a grant from Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center. The research grade virus production was supported by the Research and Development Division of Genelux Corporation.
Footnotes
This was presented at the Academic Surgical Congress in Las Vegas, NV on Feb. 3, 2015.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greten TF, Duffy AG, Korangy F. Hepatocellular carcinoma from an immunologic perspective. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(24):6678–85. doi: 10.1158/1078-0432.CCR-13-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Worns MA, Weinmann A, Schuchmann M, Galle PR. Systemic therapies in hepatocellular carcinoma. Dig Dis. 2009;27(2):175–88. doi: 10.1159/000218351. [DOI] [PubMed] [Google Scholar]
- 4.Liu M, Liu X, Ren P, Li J, Chai Y, Zheng SJ, et al. A cancer-related protein 14-3- 3zeta is a potential tumor-associated antigen in immunodiagnosis of hepatocellular carcinoma. Tumour Biol. 2014 doi: 10.1007/s13277-013-1555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert opinion on biological therapy. 2011;11(5):595–608. doi: 10.1517/14712598.2011.558838. [DOI] [PubMed] [Google Scholar]
- 6.Lee SS, Eisenlohr LC, McCue PA, Mastrangelo MJ, Lattime EC. Intravesical gene therapy: in vivo gene transfer using recombinant vaccinia virus vectors. Cancer research. 1994;54(13):3325–8. [PubMed] [Google Scholar]
- 7.Advani SJ, Buckel L, Chen NG, Scanderbeg DJ, Geissinger U, Zhang Q, et al. Preferential replication of systemically delivered oncolytic vaccinia virus in focally irradiated glioma xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(9):2579–90. doi: 10.1158/1078-0432.CCR-11-2394. [DOI] [PubMed] [Google Scholar]
- 8.Chen N, Zhang Q, Yu YA, Stritzker J, Brader P, Schirbel A, et al. A novel recombinant vaccinia virus expressing the human norepinephrine transporter retains oncolytic potential and facilitates deep-tissue imaging. Molecular medicine (Cambridge, Mass) 2009;15(5–6):144–51. doi: 10.2119/molmed.2009.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 10.Gish RG, Finn RS, Marrero JA. Extending survival with the use of targeted therapy in the treatment of hepatocellular carcinoma. Gastroenterology & hepatology. 2013;9(4 Suppl 2):1–24. [PMC free article] [PubMed] [Google Scholar]
- 11.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nature reviews Drug discovery. 2012;11(3):215–33. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 12.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 13.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine. 2008;359(4):378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 14.Bertino G, Di Carlo I, Ardiri A, Calvagno GS, Demma S, Malaguarnera G, et al. Systemic therapies in hepatocellular carcinoma: present and future. Future Oncol. 2013;9(10):1533–48. doi: 10.2217/fon.13.171. [DOI] [PubMed] [Google Scholar]
- 15.Blechacz B, Splinter PL, Greiner S, Myers R, Peng KW, Federspiel MJ, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology. 2006;44(6):1465–77. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 16.Haddad D, Chen NG, Zhang Q, Chen CH, Yu YA, Gonzalez L, et al. Insertion of the human sodium iodide symporter to facilitate deep tissue imaging does not alter 16 oncolytic or replication capability of a novel vaccinia virus. Journal of translational medicine. 2011;9:36. doi: 10.1186/1479-5876-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Liang C, Yu YA, Chen N, Dandekar T, Szalay AA. The highly attenuated oncolytic recombinant vaccinia virus GLV-1h68: comparative genomic features and the contribution of F14.5L inactivation. Molecular genetics and genomics : MGG. 2009;282(4):417–35. doi: 10.1007/s00438-009-0475-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentschev I, Muller M, Adelfinger M, Weibel S, Grummt F, Zimmermann M, et al. Efficient colonization and therapy of human hepatocellular carcinoma (HCC) using the oncolytic vaccinia virus strain GLV-1h68. PloS one. 2011;6(7):e22069. doi: 10.1371/journal.pone.0022069. [DOI] [PMC free article] [PubMed] [Google Scholar]