

Abstract
Synthetic full agonists of PPARγ have been prescribed for the treatment of diabetes due to their ability to regulate glucose homeostasis and insulin sensitization. While the use of full agonists of PPARγ has been hampered due to severe side effects, partial agonists have shown promise due to their decreased incidence of such side effects in preclinical models. No kinetic information has been forthcoming in regard to the mechanism of full versus partial agonism of PPARγ to date. Here, we describe the discovery of a partial agonist, SR2067. A co-crystal structure obtained at 2.2 Å resolution demonstrates that interactions with the β-sheet are driven exclusively via hydrophobic interactions mediated through a naphthalene group, an observation that is unique from other partial agonists. Surface plasmon resonance revealed that SR2067 binds to the receptor with higher affinity (KD = 513 nM) as compared to that of full agonist rosiglitazone, yet it has a much slower off rate compared to that of rosiglitazone.
Graphical abstract
PPARs (peroxisome proliferator-activated receptors) are ligand-activated transcription factors that belong to the nuclear receptor superfamily.1 Three isoforms of PPAR exist (PPARα, PPARδ, and PPARγ), and these receptors have been shown to control the transcription of gene programs involved in fatty acid metabolism, glucose homeostasis, and adipogenesis. 2 Functioning as obligate heterodimers with the retinoid X receptor (RXRα, β, and γ), these transcriptional complexes can be stimulated by PPAR or RXR agonists such as retinoic acid, fatty acids, prostaglandins, or eicosanoids.3 Synthetic ligands for PPARα have been used clinically for the treatment of hypertriglyceridemia, and synthetic agonists of PPARγ (a.k.a. insulin sensitizers) have been used in the treatment of type II diabetes mellitus due to their ability to sensitize peripheral tissues to the action of insulin.4
Many synthetic full agonists of PPARγ5 are thiazolidinediones (TZDs). Rosiglitazone (Avandia, GlaxoSmithKline) and pioglitazone (Actos, Takeda Pharmaceuticals) are TZDs that have been prescribed for treating hyperglycemia in diabetics.6 While TZDs have been useful in lowering glucose levels and improving lipid profiles in diabetic patients, their use has been limited due to adverse side effects. Side effects of TZD treatment include weight gain, increased adiposity, and increased risk of bone fractures.7 More troublesome, however, is the association of TZD use with renal fluid retention, edema, and plasma volume expansion (hemodilution), which is a driver of undesirable cardiovascular events including cardiomegaly and congestive heart failure.8 The TZDs hyperactivate the receptor, leading to supraphysiological expression of target genes. While side effects have been postulated to result from PPARγ transcriptional targets, the antidiabetic effects of full agonists have been shown to correlate with inhibition of phosphorylation of PPARγ residue Ser273 by CDK5/ERK rather than transcriptional activation of target genes.9 This observation supports the usefulness of antagonists and partial agonists of PPARγ in the treatment of diabetes.10
PPARγ has a canonical nuclear receptor domain architecture consisting of a disordered activation function 1 domain (AF1) for interaction with coregulators, a zinc-finger DNA-binding domain (DBD), and a ligand binding domain (LBD), also known as AF2.11 Binding of ligands to the LBD causes dissociation of co-repressors from AF1 and AF2 and recruitment of co-activators to AF1 and AF2. Co-activator recruitment, in turn, allows for other transcriptional machinery such as chromatin remodelers and RNA polymerase to be assembled on the transcriptional start site.12 Of particular interest is the LBD of PPARγ, where agonists and antagonists bind in a largely hydrophobic pocket of approximately 1200 Å3.13 The PPARγ LBD is an ~30 kDa domain consisting of 13 α-helices (H1–H12, H2′) and one 3-stranded parallel β-sheet region. The AF2 surface is formed by a hydrophobic patch from helix 12 and helix 11. Full agonists such as rosiglitazone form a hydrogen-bond network involving the TZD headgroup with side chains of residues His323 (helix 5), His449 (helix 11), and Tyr473 (helix 12) to stabilize the AF2 surface, allowing co-activator docking.
In contrast to full agonists such as rosiglitazone, partial agonists of PPARγ have shown promise as therapeutic agents for type II diabetes mellitus due to their reduced side effects and improved therapeutic index in humans.14,15 These compounds show decreased transcriptional activity when compared to that of TZD full agonists in reporter assays, yet they still maintain potent insulin-sensitizing effects in animal models. Additionally, these compounds are not adipogenic and do not increase plasma volume when administered chronically in rodents. Several partial agonist compounds, such as nTZDpa, GW0072, INT131, FK614, and SR202, have shown insulin-sensitizing properties with fewer adverse side effects.16–20 However, the mechanism of action of these compounds has not been fully delineated.
While the underlying mechanism of PPARγ partial agonism is not fully characterized, phosphorylation of the receptor is involved in obesity and insulin sensitization. Phosphorylation of residue Ser273 of PPARγ by ERK has been shown to be increased in the obese state.9,21 Furthermore, the insulin-sensitizing effects of PPARγ modulators correlates with the reduction of Ser273 phosphorylation and not with the magnitude of agonism. It was demonstrated that Ser273 phosphorylation resulted in the repression of a subset of PPARγ target genes, genes most associated with insulin sensitivity, and blockage of this PTM resulted in normalization of these dysregulated genes. It is important to note that the blockage of Ser273 phosphorylation was not via kinase inhibition but was the result of alteration in the LBD surface involved in kinase interaction. Inhibition of phosphorylation at Ser273 is independent of stabilization of helix 12 (Tyr473, His449, and His323), suggesting that partial agonists and antagonists could also be insulin sensitizers. Many PPARγ agonists contain an acidic group key to their function. Full agonists often use an acid group to stabilize AF2 (Tyr473, His449, and His323), whereas most partial agonists use an acidic group to stabilize the β-sheet region. However, PPARγ modulators with acidic groups have often displayed unfavorable pharmacokinetic properties.22
Here, we present a novel potent partial agonist of PPARγ, SR2067. SR2067 differs from other partial agonists in that it is a non-acid agonist of PPARγ. Two X-ray crystal structures reveal that the compound interacts largely through hydrophobic interactions mediated near and around the β-sheet region and helices 3 and 7, with few interactions near helix 12. Additionally, we present surface plasmon resonance kinetic data showing that the compound exhibits high affinity for the receptor and, unlike rosiglitazone, exhibits a slow off rate from the receptor, which could prove to be beneficial from a pharmacokinetic perspective.
RESULTS AND DISCUSSION
SR2067 Is a Non-acid PPARγ Partial Agonist
Favorable pharmacokinetic properties have been observed with non-acid agonists of PPARγ, particularly in regard to a lack of partitioning to the liver. To further explore novel chemical space of this class of agonists and better understand modes of structural interaction with the receptor, we synthesized a novel non-acid partial agonist of PPARγ, SR2067 (Figure 1A). The SR2067 agonist bears a central indole moiety similar to GSK compound 7b and the benzimidazole moiety of telmisartan, a sulfonamide linker related to partial agonist INT131, and additional hydrophobic groups unique to SR2067.23–25
Figure 1.

Binding Mode of SR2067. (A) Chemical structure of SR2067 and its molecular contacts with PPARγ (generated using Poseview). (B) Ribbon diagram of the PPARγ ligand binding domain (red) subunit A in complex with SR2067 (depicted as sticks; carbon atoms are colored blue).
To confirm the ability of SR2067 to agonize the PPARγ receptor and measure the degree of transcriptional activation, we performed a cell-based co-transfection luciferase reporter transactivation assay using HEK293 cells. In addition to activation of the receptor by SR2067, we performed the transactivation assay with full agonist rosiglitazone and with antagonist SR1664.10,26 Conducting the experiment with rosiglitazone allowed us to compare transactivation of the receptor by SR2067 with full agonism of the receptor by model full-agonist rosiglitazone and to antagonism of the receptor by model antagonist SR1664. This data is presented in Figure 2A. On the basis of the results in Figure 2A, SR2067 is defined as a partial agonist because it demonstrates a transcriptional potency of ~60% as compared to that of rosiglitazone. To probe the ability of SR2067 to partially agonize the receptor in other cellular environments, we measured the ability of SR2067 to induce lipid accumulation in differentiated 3T3-L1 preadipocyctes as well as performed qPCR on an adipogenic PPARγ target gene, aP2 (FABP4), to measure transcript levels in the presence and absence of SR2067 in murine SVF cells (stromal vascular fraction, preadipocytes). As shown in Figure 2B and consistent with partial agonism, SR2067 induced levels of oil red staining for lipid accumulation less than that of the full agonist rosiglitazone but higher than that of the antagonist SR1664. Additionally, expression levels of the PPARγ target gene aP2 in SVF cells induced by treartment with SR2067 were lower than those observed in cells treated with the full agonist rosiglitazone but higher than those in cells exposed to the antagonist SR1664. The relative mRNA levels of aP2 from treated SVF cells are shown in Figure 2C.
Figure 2.

Transcriptional activation data. (A) EC50 values were derived from dose–response curves of full agonist rosiglitazone, partial agonist SR2067, and antagonist 1664. (B) Oil red staining of differenrtiated 3T3-L1 cells after treatement with either the full agonist rosiglitzaone, partial agonist SR2067, or antagonist SR1664. (C) qPCR analysis of the PPARγ target gene aP2 (FABP4) in SVF cells treated with either the full agonist rosiglitazone, partial agonist SR2067, or antagonist SR1664.
Crystallization and Structure Determination
To gain an understanding of the structural mechanism of SR2067, partial agonism of PPARγ co-crystallization trials were performed on SR2067 with the PPARγ LBD. Residues 205–477 of the PPARγ LBD were overexpressed in Escherichia coli and extensively purified, and a ligand–protein complex was formed and purified by means of size-exclusion chromatography. Crystallization trials yielded two crystal forms. Data sets were collected with synchrotron radiation, which revealed one crystal to be of space group P21 (diffraction to 2.8 Å) and one crystal to be of space group C2 (diffraction to 2.2 Å). The phase problem was overcome by means of molecular replacement. Initial difference Fourier maps revealed clear electron density for the SR2067 ligand. Given the higher quality of the C2 crystal diffraction in comparison to that of the P21 crystal diffraction, only the C2 crystal form was refined to completion, and all further analysis in this article refers to the C2 crystal form. Data processing and refinement parameters are found in Table 1. The SR2067–PPARγ complex structure was refined to a final R/Rfree of 18.6/24.0. Reduced model bias electron density maps of the SR2067 ligand can be found in Figure S1. The ribbon diagram of the receptor bound to SR2067 can be found in Figure 1B.
Table 1.
Crystallographic Data Tablea
| PPARγ–SR2067 | |
|---|---|
| wavelength (Å) | 0.9795 |
| resolution range (Å) | 40–2.2 |
| space group | C2 |
| unit cell a, b, c (Å) | 92.8, 62.6, 118.6 |
| unique angle β (deg) | 101.9 |
| unique reflections | 30423 (2796) |
| multiplicity | 3.6 (3.6) |
| completeness (%) | 92.3 (95.5) |
| mean I/sigma(I) | 9.9 (1.5) |
| Wilson B-factor | 47.5 |
| Rmerge | 6.8 (98.0) |
| Rpim | 4.3 (63.1) |
| Rfactor | 18.5 |
| Rfree | 24.0 |
| protein subunits in ASU | 2 |
| total number of atoms | 4292 |
| macromolecule atoms | 4075 |
| ligands atoms | 39 |
| water atoms | 178 |
| protein residues | 505 |
| RMS bonds (Å) | 0.008 |
| RMS angles (deg) | 1.127 |
| Ramachandran favored (%)b | 97.8 |
| average B-factor total | |
| macromolecule B-factor | 67.5 |
| solvent B-factor | 61.3 |
Values in parentheses correspond to the last shell.
Ramachandran values calculated using MolProbity.
Consistent with previous structures of PPARγ, the asymmetric unit contained two PPARγ subunits (homodimer), with chain A forming a canonical LBD and chain B demonstrating an alternate conformation of the AF2 surface due to crystal contacts.27 Only chain A had electron density of sufficient quality to model the SR2067 ligand. Chain A could be modeled from residues 206–238, 241–259, and 276–475, whereas chain B could be modeled from residues 207–240, 245–268, 276–461, and 467–475. Superimposition of chain A to chain B revealed an RMSD of 0.73 Å over 232 Cα atoms. The ligand binding domain forms a globular structure consisting of 13 α-helices and a 3-stranded β-sheet. The ω-loop joining helices 2′ and 3 was disordered and could not be modeled. A ribbon diagram of chain A bound to SR2067 is shown in Figure 1B. Chain A has a total surface area of 13 430 Å2, and chain B has a total surface area of 13 052 Å2, creating 1013 Å2 of buried surface area at the dimer interface (PISA).
Superimposition of the SR2067 ligand bound structure with other PPARγ agonist bound structures revealed few differences in global conformation (Figure 3). Superimposition of the SR2067 bound structure (chain A) to the full agonist rosiglitazone structure (chain A) resulted in an RMSD of 0.59 Å over 246 Cα atoms, whereas superimposition to the partial agonist bound structure (chain A) INT131 resulted in an RMSD of 0.89 Å over 246 Cα atoms and that to the partial agonists bound structure (chain A) MRL24 resulted in an RMSD of 0.45 Å over 243 Cα atoms. Consistent with previous findings, agonist and partial agonist mechanisms are mediated through distinct stabilization of different regions of the ligand binding site rather than global structural alterations.
Figure 3.

SR2067 induces a global fold similar to that by other PPARγ partial agonists. Several crystal structures of the PPARγ LBD were superimposed (Cα shown as wires). The rosiglitazone structure is shown in yellow (PDB: 4EMA), the INT131 bound structure is shown in green (PDB: 3FUR), the SR2067 structure is shown in red (PDB: 4R06), and the MRL24 bound structure is shown in cyan (PDB: 2Q5P).
Structural Details of the SR2067 Binding Mode
The ligand binding pocket of PPARγ is an approximately 1200 Å3 cavity, largely hydrophobic in nature, that is Y-shaped and formed by contributions from helices 3, 5, 6, 7, 11, and 12 and the β-sheet, as shown in Figure 4. SR2067 has an accessible surface area of 698 Å2, and binding results in the burying of 381 Å2 of chain A from the PPARγ LBD. Several full agonists such as rosiglitazone extend from the β-sheet to the AF2 surface (Figure 4A), whereas several partial agonists such as nTZDpa and intermediate agonist BVT.13 occupy only the cavity proximal to the β-sheet and helix 3 (Figure 4B). In contrast, SR2067 has a binding mode more similar to rosiglitazone in that it occupies not only the cavity proximal to the β-sheet region and helix 3 but also proximal to the AF2 surface (Figure 4B).
Figure 4.

Comparison of SR2067 to the PPARγ full agonist and partial agonist binding modes. The PPARγ LBD is shown as ribbons (red), and SR2067 (blue), rosiglitazone (yellow), and nTZDpa (white) are shown as sticks. (A) Comparison by superimposition of the rosiglitazone and SR2067 binding modes (rosiglitazone PDB: 2PRG). (B) Comparison by superimposition of the nTZDpa and SR2067 binding modes (nTZDpa PDB: 2Q5S).
The interactions of SR2067 with PPARγ can roughly be divided into three categories: interactions with the AF2 residues, interactions of the central indole moiety with residues including or near helix 3, and interactions with the β-sheet mediated through contacts with the naphthalene group. SR2067 makes several contacts with AF2 residues and adjoining residues of the proximal portion of the ligand binding cavity (Figure 5). AF2 residues stabilized by hydrogen bonding by full agonists such as rosiglitazone can be viewed in Figure 5A. Two hydrogen bonds are formed between SR2067 and PPARγ in this region (Figure 5B). The Tyr327 side chain is a hydrogen-bond donor to the carbonyl oxygen of SR2067 (2.6 Å). The SR2067 amine is a hydrogen-bond donor to the side chain oxygen acceptor of Ser289 (2.6 Å). The phenyl group and terminal ethyl moiety of SR2067 make hydrophobic contacts with His449 (helix 11) and Leu453 (helix 11). π–π contacts are formed between the phenyl group of SR2067 and Phe282 of helix 3 in the cavity proximal to AF2. The conserved residues of the AF2 form a hydrogen-bond network between side chains: Tyr473 to His449 (3.0 Å), Tyr473 to His323 (2.9 Å), and Tyr 327 to H449 (3.4 Å); however, no hydrogen bonds were found between SR2067 and Tyr473, His323, or His449.
Figure 5.

AF2 binding pocket. The PPARγ LBD is shown as ribbons (yellow for the rosiglitazone bound structure, and red for the SR2067 bound structure) and rosiglitazone (carbon atoms are colored yellow) and SR2067 (carbon atoms are colored blue) are depicted as sticks. (A) Rosiglitazone hydrogen-bond network in the AF2 pocket. (B) SR2067 hydrogen-bond network in the AF2 pocket. (C) Superimposition of the rosiglitazone and SR2067 structures in the AF2 region.
The SR2067 naphthalene group lies nearly equidistant between the β-sheet and helix 3 (Figures 1B and 6). The naphthalene group makes hydrophobic contacts and van der Waals contacts with the side chain of Ile341 of the β-sheet, Met348 of the β-sheet, Cys285 of helix 3, and Ile281 of helix 3 (Figure 6A,B). This position also allows for hydrophobic contacts to the Met364 side chain located on helix 7 as well as the Arg288 side chain of helix 3. SR2067 is positioned such that the two oxygen atoms and sulfur atom of the sulfonamide are in a trans conformation with respect to the side chain of residue Val339 on the β-sheet, so as to avoid a steric clash. The sulfonamide group of SR2067 provides no stabilizing interactions with the β-sheet; instead, interactions with the β-sheet are mediated solely through the naphthalene group.
Figure 6.

SR2067 stabilization of the β-sheet region. The PPARγ LBD is shown as ribbons (red) and SR2067 is represented as sticks with carbon atoms colored blue. (A) Surface representation of the β-sheet pocket with green color used to signify hydrophobicity. (B) Hydrophobic contacts of SR2067 with the PPARγ β-sheet region. SR2067 is shown in sticks and with transparent van der Waals radii. PPARγ residues engaged in hydrophobic contacts have side chains displayed as sticks.
The indole ring of SR2067 lies between helices 3 and 5, allowing for the stabilization of both through hydrophobic contacts. Specifically, the indole moiety makes hydrophobic contacts with the side chains of Arg288 and Cys285 of helix 3 and Ile326 and Leu330 of helix 5.
Kinetic Analysis of SR2067 and Rosiglitazone
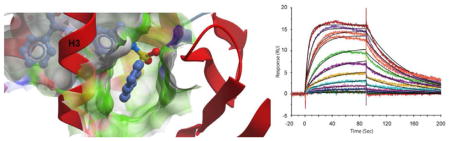
The mechanism of PPARγ partial agonism has not been previously attributed to differences in affinity among partial and full agonists; partial agonists with similar affinity to full agonists still demonstrate significantly lower transcriptional activation in comparison to that of full agonists. However, kinetic constants (on and off rates) have not been determined for partial agonists. To date, association and dissociation rates have been derived only for the full agonist rosiglitazone. To probe whether significant differences in on and off rates can be attributed to partial agonists versus full agonists, we performed surface plasmon resonance experiments using the PPARγ ligand binding domain and SR2067. The rate constants for the PPARγ–SR2067 interaction could be calculated by fitting a simple 1:1 interaction model. Since the interaction reached equilibrium rapidly, a steady-state affinity model was also fitted to the data, and KD values from kinetic and affinity fits are in good agreement (Table 2 and Figure 7). The equilibrium constant for the SR2067 interaction is approximately 500 nM and has rate constants of ka = 6.91 × 104 1/M·s and kd = 0.0158 1/s (Figure 7C–E).
Table 2.
Affinity and Kinetic Data for PPARγ Interactionsa
| compound | ka (1/M·s) | kd (1/s) | KD (M) | χ2 (RU2) |
|---|---|---|---|---|
| Rosi: Affinity | 1.39 × 10−5 (±1.1 × 10−6) | 0.911 (±0.46) | ||
| SR2067: Kinetics | 3.17 × 104 (±3.9 × 102) | 1.63 × 10−2 (±1.6 × 10−2) | 5.13 × 10−7 (±3.2 × 10−8) | 0.368 (±0.17) |
| SR2067: Affinity | 5.65 × 10−7 (±1.8 × 10−8) | 0.327 (±0.07) |
Due to the fast interaction between Rosi and PPARγ, no rate constants could be calculated. For the SR2067 interaction, both kinetic and steady-state affinity analyses were performed. Numbers in parentheses indicate the standard errors calculated from triplicate experiments.
Figure 7.

SPR analysis of PPARγ with rosiglitazone and SR2067. (A) Interaction between PPARγ and rosiglitazone. Samples were serially diluted from 30 to 0.5 μM. (B) Steady-state affinity of PPARγ and rosiglitazone fitted with a constant Rmax obtained from the SR2067 steady-state affinity fit. (C) Overlay plot for the kinetic interaction between PPARγ and SR2067. Sensorgrams (colored) show the experimental data overlaid with the fitted data (black lines). (D) Residuals plot for the data show in panel C shows the accuracy of the fit. (E) Steady-state affinity fit for the PPARγ–SR2067 interaction shown in panel C.
To compare the partial agonist SR2067 to full agonist rosiglitazone, we performed surface plasmon resonance with rosiglitazone (Figure 7A,B). Rosiglitazone demonstrated off rates that were too fast to be measured (in contrast to Li et al.14). However, an equilibrium affinity constant could be measured; the KD for rosiglitazone was found to be 14 μM. In addition to being more potent in cellular assays, SR2067 is also a much higher affinity binder to PPARγ, demonstrating a nearly 25-fold higher affinity for PPARγ than rosiglitazone. These results not only confirm that partial agonism is not simply an artifact of lower affinity compounds but also that hydrogenbond networks such as with the AF2 residues are not needed for high-affinity binding to PPARγ.
While kinetics is often neglected in the drug discovery process, several groups have noted a need for kinetics early in the drug discovery process.28–30 This is especially true with respect to off rates, which can often be correlated with potency. Novel lead compounds and scaffolds targeting PPARγ are typically screened for overall affinity and EC50 values early in the drug discovery process, which overlooks the respective on and off rates that contribute to the overall affinity. In fact, multiple examples exist outside of PPAR that show that compounds with similar affinities but large differences in off rates can have different biological outcomes and potencies. Specifically, longer residence times of a drug occupying a target (slower off rates) often yield better duration of action times for that drug, which has been found to be true of several drug such as the M3 antagonists for use in COPD.31 Given that PPARγ agonists/antagonists have been shown to exert their insulinsensitizing effects by means of blocking CDK5/ERK phosphorylation rather than by means of transactivation, we hypothesize that ligand occupancy time on the receptor (slower off rates) would improve the potency of PPARγ modulators by means of creating extended times of protection from kinase activity. To this end, we compared the partial agonist SR2067 off rates to those of other PPARγ modulators of the full agonist and antagonist classes. While the full agonist rosiglitazone had no measurable off rate, the antagonist CAB was reported to have a kd of 3.24 × 10−3 (1/s).32 Another antagonist, G3335, was reported to have a kd of 2.8 × 10−2 (1/s).33 While all compounds for which off rates have been measured lie in a fairly tight range, patterns can be found among them. Both our partial agonist SR2067 and antagonists G3335 and CAB have much slower off rates than full agonist rosiglitazone. SR2067 has a 1.8-fold slower off rate compared to that of antagonist G3335 and a 5-fold faster off rate in comparison to that of CAB. Consistent with our hypothesis, there is a trend toward slower off rates with our partial agonist SR2067 and antagonists as compared to that with a model full agonist. Given that transcriptional activation of the receptor is not necessary for insulin sensitization, antagonizing the receptor by means of longer residency times may be a key to improved therapeutics of the receptor. More elaborate SAR and future drug discovery efforts will need to be undertaken to verify the role of off rates in the mechanism of partial agonism and antagonism and to understand the pharmacologic profiles produced in conjunction with slower off rates.
An interesting discovery is the divergence in EC50 values in our transactivation assays from the KD values; generally, these values would be expected to be in a more similar range. Rosiglitazone demonstrated an EC50 value of 495 nM in our transactivation assay but a KD value of 14 μM. Similarly, SR2067 demonstrated an EC50 value of 16 nM in our transactivation assay but a KD of 513 nM. Both of these compounds display a nearly 30-fold divergence between the EC50 and KD values. Due to variables such as cell penetration, divergence in affinity and cellular potency is usually explained by higher affinity and lower cellular activation, the opposite of what is observed here. This implies that there may be a mechanism in which PPAR ligands can be made more potent in cells. While the mechanism of this needs future investigation, a recent study by Hughes et al. showed that fatty acid binding proteins 1 and 2 not only bind PPAR ligands at high affinity but also stimulate ligand-dependent transcriptional activity by 10- fold.34
Implications for the Development of Non-acid PPARγ Partial Agonists
Several groups have investigated the mechanism of PPARγ partial agonism using small numbers of selected test compounds.22,27,35–38 Initial studies have revealed that partial agonists can operate through at least two different structural mechanisms defined by the portions of the binding pockets that they occupy. The first class binds only in the portion of the pocket proximal to the β-sheet (Figure 4B); the compounds are positioned between the β-sheet and helix 3. We have termed this portion of the ligand binding pocket the β-sheet pocket. Compounds of this first class make no contact with the residues involved with or near the AF2 surface. The second class of compounds not only occupies the β-sheet pocket but also extends to the portion of the pocket where the TZD headgroup of full agonists stabilizes the AF2 residues (Figure 4A). This region of the pocket we have coined the AF2 pocket. No full agonists have been found to occupy only the AF2 pocket; they also extend to other regions of the LBD pocket. Partial agonists that occupy the AF2 pocket do so without stabilizing the residues critical to stabilization of helix 12 and the AF2 surface (His449, Tyr473, and His323). However, one common aspect between both classes of partial agonists has been contact with and stabilization of the β-sheet region. This is not surprising as this is proximal to the phosphorylation site of CDK5/ERK, the blocking of which has been correlated with insulin sensitization.
Comparison of SR2067 to the full agonist rosiglitazone reveals differences in the AF2 pocket, highlighting the mechanism of partial agonism of SR2067. Rosiglitazone forms a hydrogen-bond network from the TZD head to stabilize the AF2 surface (Figure 5A). This network extends among residues Tyr327, His323, His449, and Tyr327. Superimposition of the rosiglitazone bound structure and the SR2067 bound structure show that the His449 and Tyr327 side chains are located in similar positions between both structures, whereas the His323 side chain is forced to shift nearly 2 Å, and the Tyr473 side chain is located nearly 1 Å shifted in comparison to the SR2067 structure (Figure 5C). The alteration in His323 in the SR2067 structure (in comparison to the rosiglitazone bound structure) is a result of the ethyl group of SR2067 imposing a steric clash with the His323 side chain. Interestingly, the hydrogen-bond network among the side chains of the AF2 pocket is preserved among the rosiglitazone bound structure and the SR2067 bound structure, and it is even tighter in the SR2067 structure in some cases: His323 hydrogen bonds to Tyr473 (3.1 Å in the rosiglitazone bound structure and 2.9 Å in the SR2067 structure), His323 hydrogen bonds to His449 (3.7 Å in the rosiglitazone bound structure and 3.4 Å in the SR2067 bound structure), and Tyr473 hydrogen bonds to His449 (3.4 Å in the rosiglitazone bound structure and 3.0 Å in the SR2067 bound structure). Additionally, SR2067 forms a hydrogen bond with Tyr327 (2.6 Å) that is not possible with rosiglitazone, perhaps explaining the tighter hydrogen bond in the SR2067 bound structure between Tyr327 and His449. From this, it can be deduced that the hydrogen-bond pattern among the residues of the AF2 pocket are not stabilizing enough on their own to allow for full agonism. Instead, the full agonism of rosiglitazone is perpetuated by the hydrogen-bond network of the side chains directly with the ligand itself: 2.9 Å to His323, 2.6 Å to His449, and 2.7 Å to Tyr473. Although the hydrogen-bond network among the side chains is intact in comparison to rosiglitazone, the partial agonist SR2067 does not form any of these direct hydrogen bonds to His323, His449, and Tyr473.
Previous studies have shown that partial agonists of PPARγ preferentially stabilize the β-sheet region of the LBD. Most PPARγ partial agonists stabilize the β-sheet region by means of hydrogen bonding through an acidic group. These acidic groups have been found to stabilize the β-sheet through hydrogen-bonding networks to the backbone atoms of the β- sheet, especially the backbone nitrogen of Ser342. Only a handful of non-acid examples of partial agonists have recently been synthesized that have corresponding structural information: GSK1997132B, PA-082, Cerco-A, and INT131.14,22,39,40 However, the moieties of these compounds that occupy the β-sheet pocket generally have polar atoms of some sort to achieve stabilization of the β-sheet. For GSK1997132B, this includes a chlorine atom to make a halogen bond to the sulfur atom of β-sheet residue Met348. For PA-082, this includes two ether bonds that limit interaction with the hydrophobic side chains of the β-sheet. For Cerco-A, this includes three oxygen atoms that allow for the formation of water-mediated hydrogen-bonding networks to the β-sheet region (backbone nitrogen atoms of Ser342, Ile 341, and side chain oxygen atom Ser342). In the case of INT131, a quinolone group attached to an ether connecting group occupies the β-sheet cavity with the lone pair of the quinolone nitrogen atom poised to be a hydrogen-bond acceptor to the backbone of residue Ser342 (however, this interaction is weak, with a distance of 3.8 Å) and the oxygen atom of the ether forms a water-mediated hydrogen bond to the backbone carbonyl of Leu340.
A naphthalene group, as evidenced by SR2067, presents a unique interaction mechanism for stabilization of the β-sheet region of the PPARγ LBD, which could be used in future drug discovery efforts. Not only does SR2067 lack an acidic group, in contrast to other non-acid partial agonists for which structural data is available, but also the naphthalene group allows for a completely hydrophobic interaction with the β-sheet region by means of interactions with the hydrophobic side chains such as Leu340, Ile341, and Met348 (Figure 6). In addition to the favorable interactions with the β-sheet, the hydrophobicity of the naphthalene group also aids in advantageous interactions with helix 3 via the side chains of Cys285 and Arg288, which allows for the naphthalene group to be in a hydrophobic sandwich between helix 3 and the β-sheet region. The use of an entirely hydrophobic moiety such as naphthalene to mediate the interactions of the β-sheet pocket (in contrast to mixed hydrophobic/polar groups in other partial agonists) is further highlighted by the presence of multiple water molecules in the β-sheet pocket of the apo crystal structure of the PAPRγ LBD; the β-sheet pocket is hypothesized to be better accommodated by completely hydrophobic groups such as naphthalene due to the favorable energy of displacement of these water molecules in this largely hydrophobic environment.
Conclusions
In this work, we report the first co-crystal structure of a non-acid PPARγ partial agonist that modulates the β-sheet pocket of the LBD with completely nonpolar atoms (a naphthalene group). The structural implications for future drug design efforts not only lie in the use of non-acid interactions but we additionally present evidence that hydrophobic stabilization of the β-sheet is not only possible but is perhaps preferable. We also present the first full kinetic characterization of a PPARγ partial agonist; the data presented show that partial agonist SR2067 has decreased off rates in comparison to that of full agonist rosiglitazone. Further work will need to be undertaken to determine if decreased off rates are a defining property of partial agonists. Finally, while the promise of partial agonists offers decreased side effects as compared to those of TZD-based full agonists, the decrease in side effects of these modulators may not be extensive enough for clinical purposes. In such cases, SR2067 offers a novel scaffold for the design of non-agonist/antagonist modulators with even fewer side effects than partial agonists.
METHODS
Chemical Synthesis
The synthesis of SR2067 was conducted as described in ref 41. Briefly, 5-indolecarboxylic acid 1 was coupled to 1- phenylpropan-1-amine in the presence of triethylamine and 1- [bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) to provide amide 2. Sulfonylation of the indole nitrogen was carried out chemoselectively using phase-transfer conditions as describes in Scheme 1 to provide SR2067 (3) as a colorless solid after purification.
Scheme 1. Synthesis of SR2067a.

aReaction conditions: (a) 1-phenylpropan-1-amine; HATU, CH2Cl2, triethylamine; (b) 1-naphthalenesulfonyl chloride, BnNEt3+Cl−, CH2Cl2, KOH.
N-(1-Phenylpropyl)-1H-indole-5-carboxamide (2)
To a mixture of 1H-indole-5-carboxylic acid (1, 1 equiv) in DMF were added DIEA (10 equiv) and HATU (1 equiv). The mixture was stirred for 5 min, and then 1-phenylpropan-1-amine (1 equiv) was added. The reaction mixture was stirred at rt for 30 min. The completion of the reaction was monitored by reverse-phase analytical HPLC. The solvent was removed in vacuo to obtain the crude product, which was purified by flash chromatography (EtOAc/hex) to provide the title compound as a pale yellow powder.
1-(Naphthalen-1-ylsulfonyl)-N-(1-phenylpropyl)-1H-indole- 5-carboxamide (3, SR2067)
A mixture of N-(1-phenylpropyl)-1H-indole- 5-carboxamide (2, 1 equiv), benzyltriethylammonium chloride (0.1 equiv), potassium hydroxide (2 equiv), 1-naphthalenesulfonyl chloride (1.2 equiv) was stirred in DCM at RT. When the reaction was judged to be complete by TLC analysis, water was added to the mixture, which was extracted with DCM (3×). The combined organics were washed with brine (1×), dried (MgSO4), and concentrated in vacuo. Purification by silica gel chromatography (EtOAc/hex) provided the title compound. 1H NMR (DMSO-d6, 400 MHz) δ: 8.70 (d, J = 8.5 Hz, 1H), 8.60 (dd, J = 0.8, 8.5, 1H), 8.47 (dd, J = 1.3, 7.7 Hz, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.22 (d, J = 3.7 Hz, 1H), 8.11 (m, 1H), 8.08 (d, J = 0.8, 8.5 Hz, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.76–7.71 (m, 2H), 7.66 (dt, J = 1.1, 7.0, 1H), 7.34 (dd, J = 1.7, 7.7 Hz, 2H), 7.30–7.26 (m, 2H), 7.19 (dt, J = 1.3, 7.2, 1H), 6.93 (dd, J = 0.6, 3.7, 1H), 4.86 (quint, J = 8.55 Hz, 1H), 1.78 (hept, J = 7.0 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H). MS: m/z 469 [M + H]+.
Cell-Based Transactivation Assay
HEK 293T cells (ATCC; cat. no. CRL-3216) were cotransfected in batch by adding 4.5 μg of human GAL4-PPARγ-Hinge-LBD with 4.5 μg of 5× multimerized UASluciferase reporter and 27 μL of X-treme Gene 9 transfection reagent in serum-free Opti-MEM reduced serum media (Gibco). Cells cotransfected with Gal4-pBind vector and UAS-LUC were used as a nonreceptor control. After 18 h of incubation at 37 °C in a 5% CO2 incubator, transfected cells were plated in quadruplicate in white 384- well plates (PerkinElmer) at a density of 10 000 cells per well. After replating, cells were treated with either DMSO only or the indicated compounds in increasing doses from 2 pM to 10 μM. After an 18 h incubation, treated cells were developed with Brite Lite Plus (PerkinElmer) and read in 384-well Luminescence PerkinElmer EnVision Multilabel plate reader. Graphs were plotted as fold change of treated cells over DMSO-treated control cells. Experiments were performed in triplicate, and statistical analysis including the incorporation of error bars was carried out in GraphPad Prism.
In Vitro Adipocyte Differentiation and Oil Red Staining
3T3- L1 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) (Gibco BRL/Life Technologies, Grand Island, NY). Twenty-four hours postconfluence, the 3T3-L1 cells were differentiated with isobutylmethylxanthine (0.5 mM), dexamethasone (1 μM), insulin (10 μg/mL), and 10% FBS for 48 h. After induction of adipogenesis, the cells were maintained with insulin (10 μg/mL) and were treated with PPARγ modulators or DMSO only for 2 or 3 days. Lipid accumulation in the cells was detected by Oil Red O staining.
Quantitative Real-Time Polymerase Chain Reaction
Immortalized stromal vascular fraction (SVF) cells were exposed to DMEM/F-12 GlutaMAX (Invitrogen) containing dexamethasone (5 μM, Sigma), insulin (0.5 μg mL−1, Sigma), isobutylmethylxanthine (0.5 mM, Sigma), rosiglitazone (1 μM, Cayman), T3 (1 nM, Sigma), and 10% FBS for 2 days. After induction, cells were maintained in media containing insulin (0.5 μg mL−1), T3 (1 nM), 10% FBS, and 5 μM of each compounds for 3 days. Total RNA was extracted from 3T3-L1 cells or immortalized SVF cells. The RNA was reverse-transcribed using the ABI reverse transcription kit (Applied Biosystems/Thermo Fisher Scientific, Waltham, MA). Quantitative polymerase chain reactions were performed with SYBR green fluorescent dye using an ABI9300 polymerase chain reaction machine. Relative mRNA expression was determined by the ΔΔ−Ct method normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels. Primer sequences are as follows. aP2 primers: 5′-AAG GTG AAGAGC ATC ATA ACC CT-3′ (forward), 5′-TCA CGC CTT TCA TAA CAC ATT CC-3′ (reverse); GAPDH primers: 5′-ACA CAT TGG GGG TAG GAA CA-3′ (forward), 5′-ACC CAG AAG ACT GTG GAT GG-3′ (reverse).
Protein Expression and Purification
An expression vector (pET11) encoding the ligand binding domain of PPARγ (residues 205–477) with an N-terminal hexa-histidine tag was transformed into E. coli strain BL21 (DE3). Cells were grown at 37 °C in LB medium with 50 μg/mL ampicillin. PPARγ protein expression was induced by the addition of 0.5 mM isopropyl β-D-thiogalactoside (IPTG) when the cell density reached an optical density of 0.5 OD. Protein was expressed at 16 °C for 16 h. Cells were harvested by centrifugation, resuspended in storage buffer (20 mM Tris 8.0, 0.5 M NaCl, 10 mM imidazole, 10% glycerol, and 2 mM β-mercaptoethanol), and stored at −80 °C.
Cells were lysed by two passages through a French press and clarified by centrifugation at 12 000g for 1 h. The supernatant was applied to a 5 mL His-Trap FF crude column (GE Healthcare), washed with 75 mL of storage buffer, and eluted with a 75 mL linear elution gradient (0.01–0.5 M imidazole). Fractions containing PPARγ were pooled and dialyzed to 20 mM Tris, pH 8.0, 20 mM NaCl, and 1 mM DTT. For co-crystallography, 10 mg of PPARγ at a concentration of 10 mg mL−1 was mixed with a 10:1 molar ratio of SR2067 (PPARγ/SR2067) and incubated at 4 °C for 30 min. The PPARγ–SR2067 complex was then applied to a Superdex 75 size-exclusion column (GE Healthcare) and purified using a buffer composed of 20 mM Tris, pH 8.0, 10 mM NaCl, and 1 mM TCEP. Fractions containing dimeric PPARγ–SR2067 complex were then concentrated to 10 mg mL−1 using a 10 000 molecular weight cutoff spin concentrator (Millipore). The purified PPARγ–SR2067 complex was not flash cooled prior to use and was used immediately after purification for crystallization trials.
Crystallization, Data Collection, and Model Refinement
Two crystal forms of PPARγ in complex with SR2067 were formed: one in space group C2 and one in space group P21. Both crystal forms were produced from a protein solution of 10 mg mL−1. All crystals were grown using the hanging drop vapor diffusion crystallization method with 0.5 mL in the well; 1 μL of the complex was mixed with 1 μL of well solution to form the hanging drop. Co-crystals of space group C2 were produced using a well solution composed of 2 M ammonium sulfate, and co-crystals of space group P21 were produced using a well solution of 1 M sodium citrate. Crystallization trials were conducted at 289 K. Crystals appeared and grew to full size within 1 week. The approximate dimensions of the cubic-shaped crystals were 250 μm in each dimension. Crystals were transferred using to the well solution containing 15% ethylene glycol for cryoprotection. Crystals were harvested with a cryo-loop (Hampton Research) and were flash cooled to 100 K. All data collection was carried out at 100 K. Data for both crystals was collected at SSRL beamline 11.1 with use of a MAR Mosaic 325 mm CCD detector. A total of 220° of data was collected for each crystal using 0.2° oscillations and 1.2 s exposures. Indexing, integration, and scaling were carried out using HKL2000.42 Molecular replacement was carried out using Phaser and PDB 2Q59 as a search model with water molecules and ligands removed.27,43 After molecular replacement, unambiguous electron density in an Fo – Fc electron density map (contoured at 3.5σ) was visible for ligands in one subunit of both crystal forms. Data quality was significantly higher for the C2 crystal data set largely determined by resolution; the C2 data set was 2.2 Å, whereas the P21 data set was 2.8 Å. Given this, only the C2 data set was refined to completion. Consistent with previous PPARγ LBD crystal structures and Matthew’s coefficient calculations, two subunits (one homodimer) were found per asymmetric unit. Refinement was carried out using iterative cycles of Phenix.refine and manual model building in Coot.44,45 Structure validation was carried out using MolProbity.46 Kicked 2Fo – Fc electron density maps were calculated using Phenix to help reduce model bias;47 an electron density quality is shown in Supporting Information Figure S1. Coordinates and structure factors were deposited in the Protein Data Bank under accession code 4R06. Data processing and refinement statistics can be found in Table 1.
Surface Plasmon Resonance: PPARγ Immobilization
Interaction studies between PPARγ, SR2067 (MW = 469 g/mol), and rosiglitazone (MW = 357 g/mol) were performed at 25 °C using a Biacore T100 V2.0.4 instrument (GE Healthcare, Uppsala, Sweden). Purified PPARγ protein was covalently attached to a CM5 chip (GE Healthcare) via amine coupling chemistry. The carboxymethyl groups present on the dextran layer of the CM5 chip were activated by a 7 min injection of 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) (GE Healthcare, Uppsala, Sweden). PPARγ was injected onto the activated surface for 900 s at 10 μL/min in 10 mM sodium acetate (pH 5.2) at a final concentration of 50 μg/mL, resulting in a surface density increase of approximately 5700 RU of PPARγ. Any remaining unoccupied reactive sites were blocked by a 7 min injection of 1 M ethanolamine hydrochloride-NaOH, pH 8.5.
Surface Plasmon Resonance: PPARγ–SR2067 Interactions
For each PPARγ–SR2067 interaction, a serial dilution of analyte covering a concentration range of ~10 × KD to 0.1 × KD was prepared in phosphate buffered saline (PBS; 20 mM phosphate buffer, pH 7.4, 2.7 mM KCl, 137 mM NaCl, and 0.05% Surfactant P20) with 5% DMSO. Samples were 2-fold serially diluted from 6240 to 6 nM. Samples were injected onto the surface at 30 μL/min for 90 s, and formed complexes were allowed to dissociate for 120 s. Since SR2067 completely dissociated from the surface, no regeneration steps were required. All sample responses were double referenced using a blank immobilized reference surface (NHS + EDC-activated/deactivated), and a series of blank injections was included to account for any system and injection artifacts. At least one concentration was injected in duplicate. Triplicate experiments on two surface densities were performed for each analyte, and the double referenced data was globally fitted using a simple 1:1 Langmuir model using the Biacore T200 V3.0 evaluation software (GE Healthcare, Uppsala, Sweden). Both 1:1 kinetic and steady-state affinity analysis were performed on the SR2067 data sets.
Surface Plasmon Resonance: PPARγ–Rosiglitazone Interactions
Due to the fast rate constants of the PPARγ–Rosi interaction, it was not possible to obtain the individual rate constants, ka and kd; hence, equilibrium analyses were performed. Due to the observed concentration-dependent aggregation of rosiglitazone occurring above 30 μM in PBS + 5%DMSO, no saturating concentrations could be used for the equilibrium analysis. Samples were serially diluted from 30 to 0.5 μM and were injected onto the surface at 30 μL/min for 60 s, and formed complexes were allowed to dissociate for 30 s. Triplicate experiments were performed, and the double referenced data was globally fitted using a simple 1:1 steady-state interaction with a constant Rmax obtained from the SR2067 steady-state affinity fits. The Rmax was MW corrected, resulting in an Rmax of 3.63 RU/100 Da. Steady-state affinity levels were calculated at 20 s after the injection started, and the average response of a 5 s window was used in the fit. Data was fitted using the Biacore T200 V3.0 evaluation software (GE Healthcare, Uppsala, Sweden). All SPR experiments were carried out at 25 °C, and the binding data was solvent-corrected using an eight-point solvent correction curve ranging from 4.5 to 5.8% DMSO that was prepared according to the Biacore laboratory guideline 29-0918-63 AA.
Supplementary Material
Acknowledgments
We would like to thank R. Stanfield and the I. Wilson group for help with data collection and synchrotron time at SSRL. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Footnotes
Author Contributions
L.M.v.M. carried out protein purification, SPR experiments, manuscript preparation, and crystal structure interpretation. J.B.B. directed research, participated in manuscript preparation, and crystal structure solution. S.W.P. and M.H. conducted SPR experiments, data fitting, and manuscript preparation. P.R.G., T.M.K., and Y.S. synthesized compounds and tested compounds in transactivation assays.
Notes
The authors declare no competing financial interest.
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio. 5b00580.
Reduced model bias electron density map (PDF)
References
- 1.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 2.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 3.Miyata KS, McCaw SE, Marcus SL, Rachubinski RA, Capone JP. The peroxisome proliferator-activated receptor interacts with the retinoid X receptor in vivo. Gene. 1994;148:327–330. doi: 10.1016/0378-1119(94)90707-2. [DOI] [PubMed] [Google Scholar]
- 4.Akiyama TE, Meinke PT, Berger JP. PPAR ligands: potential therapies for metabolic syndrome. Curr Diabetes Rep. 2005;5:45–52. doi: 10.1007/s11892-005-0067-3. [DOI] [PubMed] [Google Scholar]
- 5.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARgamma agonists: time for a reassessment. Trends Endocrinol Metab. 2012;23:205–215. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Doggrell SA. Clinical trials with thiazolidinediones in subjects with Type 2 diabetes–is pioglitazone any different from rosiglitazone? Expert Opin Pharmacother. 2008;9:405–420. doi: 10.1517/14656566.9.3.405. [DOI] [PubMed] [Google Scholar]
- 7.Rizos CV, Elisaf MS, Mikhailidis DP, Liberopoulos EN. How safe is the use of thiazolidinediones in clinical practice? Expert Opin Drug Saf. 2009;8:15–32. doi: 10.1517/14740330802597821. [DOI] [PubMed] [Google Scholar]
- 8.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 9.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidovic D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPARgamma- RXR- nuclear receptor complex on DNA. Nature. 2008;456:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta, Mol Cell Biol Lipids. 2007;1771:936–951. doi: 10.1016/j.bbalip.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 14.Motani A, Wang Z, Weiszmann J, McGee LR, Lee G, Liu Q, Staunton J, Fang Z, Fuentes H, Lindstrom M, Liu J, Biermann DH, Jaen J, Walker NP, Learned RM, Chen JL, Li Y. INT131: a selective modulator of PPAR gamma. J Mol Biol. 2009;386:1301–1311. doi: 10.1016/j.jmb.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 15.Higgins LS, Depaoli AM. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am J Clin Nutr. 2010;91:267S–272S. doi: 10.3945/ajcn.2009.28449E. [DOI] [PubMed] [Google Scholar]
- 16.Berger JP, Petro AE, Macnaul KL, Kelly LJ, Zhang BB, Richards K, Elbrecht A, Johnson BA, Zhou G, Doebber TW, Biswas C, Parikh M, Sharma N, Tanen MR, Thompson GM, Ventre J, Adams AD, Mosley R, Surwit RS, Moller DE. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol Endocrinol. 2003;17:662–676. doi: 10.1210/me.2002-0217. [DOI] [PubMed] [Google Scholar]
- 17.Oberfield JL, Collins JL, Holmes CP, Goreham DM, Cooper JP, Cobb JE, Lenhard JM, Hull-Ryde EA, Mohr CP, Blanchard SG, Parks DJ, Moore LB, Lehmann JM, Plunket K, Miller AB, Milburn MV, Kliewer SA, Willson TM. A peroxisome proliferator-activated receptor gamma ligand inhibits adipocyte differentiation. Proc Natl Acad Sci U S A. 1999;96:6102–6106. doi: 10.1073/pnas.96.11.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee DH, Huang H, Choi K, Mantzoros C, Kim YB. Selective PPARgamma modulator INT131 normalizes insulin signaling defects and improves bone mass in diet-induced obese mice. American journal of physiology Endocrinology and metabolism. 2012;302:E552–560. doi: 10.1152/ajpendo.00569.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minoura H, Takeshita S, Ita M, Hirosumi J, Mabuchi M, Kawamura I, Nakajima S, Nakayama O, Kayakiri H, Oku T, Ohkubo-Suzuki A, Fukagawa M, Kojo H, Hanioka K, Yamasaki N, Imoto T, Kobayashi Y, Mutoh S. Pharmacological characteristics of a novel nonthiazolidinedione insulin sensitizer, FK614. Eur J Pharmacol. 2004;494:273–281. doi: 10.1016/j.ejphar.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 20.Santini E, Fallahi P, Ferrari SM, Masoni A, Antonelli A, Ferrannini E. Effect of PPAR-gamma activation and inhibition on glucose-stimulated insulin release in INS-1e cells. Diabetes. 2004;53:S79–S83. doi: 10.2337/diabetes.53.suppl_3.s79. [DOI] [PubMed] [Google Scholar]
- 21.Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2014;517:391–395. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sime M, Allan AC, Chapman P, Fieldhouse C, Giblin GM, Healy MP, Lambert MH, Leesnitzer LM, Lewis A, Merrihew RV, Rutter RA, Sasse R, Shearer BG, Wilson TM, Xu RX, Virley DJ. Discovery of GSK1997132B a novel centrally penetrant benzimidazole PPARgamma partial agonist. Bioorg Med Chem Lett. 2011;21:5568–5572. doi: 10.1016/j.bmcl.2011.06.088. [DOI] [PubMed] [Google Scholar]
- 23.Lamotte Y, Martres P, Faucher N, Laroze A, Grillot D, Ancellin N, Saintillan Y, Beneton V, Gampe RT., Jr Synthesis and biological activities of novel indole derivatives as potent and selective PPARgamma modulators. Bioorg Med Chem Lett. 2010;20:1399–1404. doi: 10.1016/j.bmcl.2009.12.107. [DOI] [PubMed] [Google Scholar]
- 24.Amano Y, Yamaguchi T, Ohno K, Niimi T, Orita M, Sakashita H, Takeuchi M. Structural basis for telmisartan-mediated partial activation of PPAR gamma. Hypertens Res. 2012;35:715–719. doi: 10.1038/hr.2012.17. [DOI] [PubMed] [Google Scholar]
- 25.Taygerly JP, McGee LR, Rubenstein SM, Houze JB, Cushing TD, Li Y, Motani A, Chen JL, Frankmoelle W, Ye G, Learned MR, Jaen J, Miao S, Timmermans PB, Thoolen M, Kearney P, Flygare J, Beckmann H, Weiszmann J, Lindstrom M, Walker N, Liu J, Biermann D, Wang Z, Hagiwara A, Iida T, Aramaki H, Kitao Y, Shinkai H, Furukawa N, Nishiu J, Nakamura M. Discovery of INT131: a selective PPARgamma modulator that enhances insulin sensitivity. Bioorg Med Chem. 2013;21:979–992. doi: 10.1016/j.bmc.2012.11.058. [DOI] [PubMed] [Google Scholar]
- 26.Marciano DP, Kuruvilla DS, Boregowda SV, Asteian A, Hughes TS, Garcia-Ordonez R, Corzo CA, Khan TM, Novick SJ, Park H, Kojetin DJ, Phinney DG, Bruning JB, Kamenecka TM, Griffin PR. Pharmacological repression of PPARgamma promotes osteogenesis. Nat Commun. 2015;6:7443. doi: 10.1038/ncomms8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, Griffin PR. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 28.Andersson K, Karlsson R, Lofas S, Franklin G, Hamalainen MD. Label-free kinetic binding data as a decisive element in drug discovery. Expert Opin Drug Discovery. 2006;1:439–446. doi: 10.1517/17460441.1.5.439. [DOI] [PubMed] [Google Scholar]
- 29.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discovery. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 30.Zhang R, Monsma F. The importance of drugtarget residence time. Curr Opin Drug Discov Devel. 2009;12:488–496. [PubMed] [Google Scholar]
- 31.Disse B, Speck GA, Rominger KL, Witek TJ, Jr, Hammer R. Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci. 1999;64:457–464. doi: 10.1016/s0024-3205(98)00588-8. [DOI] [PubMed] [Google Scholar]
- 32.Yen HR, Harris TJ, Wada S, Grosso JF, Getnet D, Goldberg MV, Liang KL, Bruno TC, Pyle KJ, Chan SL, Anders RA, Trimble CL, Adler AJ, Lin TY, Pardoll DM, Huang CT, Drake CG. Tc17 CD8 T cells: functional plasticity and subset diversity. J Immunol. 2009;183:7161–7168. doi: 10.4049/jimmunol.0900368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye F, Zhang ZS, Luo HB, Shen JH, Chen KX, Shen X, Jiang HL. The dipeptide H-Trp-Glu-OH shows highly antagonistic activity against PPARgamma: bioassay with molecular modeling simulation. ChemBioChem. 2006;7:74–82. doi: 10.1002/cbic.200500186. [DOI] [PubMed] [Google Scholar]
- 34.Hughes ML, Liu B, Halls ML, Wagstaff KM, Patil R, Velkov T, Jans DA, Bunnett NW, Scanlon MJ, Porter CJ. Fatty Acid-binding Proteins 1 and 2 Differentially Modulate the Activation of Peroxisome Proliferator-activated Receptor alpha in a Ligand-selective Manner. J Biol Chem. 2015;290:13895–13906. doi: 10.1074/jbc.M114.605998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu IL, Huang CF, Peng YH, Lin YT, Hsieh HP, Chen CT, Lien TW, Lee HJ, Mahindroo N, Prakash E, Yueh A, Chen HY, Goparaju CM, Chen X, Liao CC, Chao YS, Hsu JT, Wu SY. Structure-based drug design of a novel family of PPARgamma partial agonists: virtual screening, X-ray crystallography, and in vitro/in vivo biological activities. J Med Chem. 2006;49:2703–2712. doi: 10.1021/jm051129s. [DOI] [PubMed] [Google Scholar]
- 36.Furukawa A, Arita T, Fukuzaki T, Satoh S, Mori M, Honda T, Matsui Y, Wakabayashi K, Hayashi S, Araki K, Ohsumi J. Substituents at the naphthalene C3 position of (−)-Cercosporamide derivatives significantly affect the maximal efficacy as PPARgamma partial agonists. Bioorg Med Chem Lett. 2012;22:1348–1351. doi: 10.1016/j.bmcl.2011.12.066. [DOI] [PubMed] [Google Scholar]
- 37.Montanari R, Saccoccia F, Scotti E, Crestani M, Godio C, Gilardi F, Loiodice F, Fracchiolla G, Laghezza A, Tortorella P, Lavecchia A, Novellino E, Mazza F, Aschi M, Pochetti G. Crystal structure of the peroxisome proliferator-activated receptor gamma (PPARgamma) ligand binding domain complexed with a novel partial agonist: a new region of the hydrophobic pocket could be exploited for drug design. J Med Chem. 2008;51:7768–7776. doi: 10.1021/jm800733h. [DOI] [PubMed] [Google Scholar]
- 38.Einstein M, Akiyama TE, Castriota GA, Wang CF, McKeever B, Mosley RT, Becker JW, Moller DE, Meinke PT, Wood HB, Berger JP. The differential interactions of peroxisome proliferator-activated receptor gamma ligands with Tyr473 is a physical basis for their unique biological activities. Mol Pharmacol. 2007;73:62–74. doi: 10.1124/mol.107.041202. [DOI] [PubMed] [Google Scholar]
- 39.Burgermeister E, Schnoebelen A, Flament A, Benz J, Stihle M, Gsell B, Rufer A, Ruf A, Kuhn B, Marki HP, Mizrahi J, Sebokova E, Niesor E, Meyer M. A novel partial agonist of peroxisome proliferator-activated receptor-gamma (PPARgamma) recruits PPARgamma-coactivator-1alpha, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol Endocrinol. 2006;20:809–830. doi: 10.1210/me.2005-0171. [DOI] [PubMed] [Google Scholar]
- 40.Wakabayashi K, Hayashi S, Matsui Y, Matsumoto T, Furukawa A, Kuroha M, Tanaka N, Inaba T, Kanda S, Tanaka J, Okuyama R, Wakimoto S, Ogata T, Araki K, Ohsumi J. Pharmacology and in vitro profiling of a novel peroxisome proliferator-activated receptor gamma ligand, Cerco-A. Biol Pharm Bull. 2011;34:1094–1104. doi: 10.1248/bpb.34.1094. [DOI] [PubMed] [Google Scholar]
- 41.Kamenecka TM, Griffin PR, et al. Patent US2012/0309769 A1. N-Benzylindole modulators of PPARG. 2012
- 42.Otwinowski Za, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Praznikar J, Afonine PV, Guncar G, Adams PD, Turk D. Averaged kick maps: less noise, more signal··· and probably less bias. Acta Crystallogr, Sect D: Biol Crystallogr. 2009;65:921–931. doi: 10.1107/S0907444909021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.