

Abstract
A major objective of nanomedicine is to combine in a controlled manner multiple functional entities into a single nanoscale device to target particles with great spatial precision, thereby increasing the selectivity and potency of therapeutic drugs. A multifunctional nanoparticle is described for controlled conjugation of a cytotoxic drug, a cancer cell targeting ligand, and an imaging moiety. The approach is based on the chemical synthesis of polyethylene glycol that at one end is modified by a thioctic acid for controlled attachment to a gold core. The other end of the PEG polymers is modified by a hydrazine, amine, or dibenzocyclooctynol moiety for conjugation with functional entities having a ketone, activated ester, or azide moiety, respectively. The conjugation approach allowed the controlled attachment of doxorubicin through an acid-labile hydrazone linkage, an Alexa Fluor dye through an amide bond, and a glycan based ligand for the cell surface receptor CD22 of B cells using strain promoted azide-alkyne cycloaddition. The incorporation of the ligand for CD22 led to rapid entry of the nanoparticle by receptor-mediated endocytosis. Covalent attachment of doxorubicin via hydrazone linkage caused pH responsive intracellular release of doxorubicin and significantly enhanced cytotoxicity of nanoparticles. A remarkable 60-fold enhancement in cytotoxicity of CD22 (+) lymphoma cells was observed compared to non-targeted nanoparticles.
Keywords: gold nanoparticles, doxorubicin, B-cell lymphoma, pH responsive drug release
Graphical Abstract
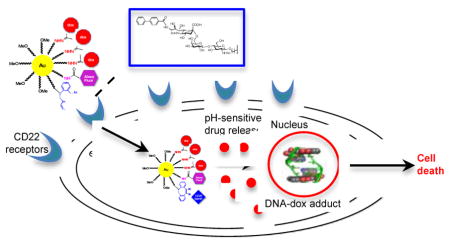
Controlled multi-functionalization of nanoparticles with a cytotoxic drug, a glycan ligand for the cell surface receptor CD22, and an imaging moiety led to a remarkable 60-fold enhancement in cytotoxicity of CD22 (+) lymphoma cells compared to non-targeted nanoparticles.
Introduction
Nanomaterials are emerging as promising devices for drug delivery.[1] These carriers can increase longevity of a drug in the blood stream, solubilize hydrophobic drugs, offer controlled release by environmental sensitive or external stimuli, and accumulate in solid tumors by enhanced permeability and retention effect.[2] The therapeutic efficiency of nanomaterials can further be improved by surface functionalization by for example a tissue targeting ligand,[3] a cell-penetrating molecule,[4] or by a signaling peptide for organelle targeting.[5] Moreover, therapeutic targeting can be combined with imaging by attachment of an appropriate contrast agent.[6]
Polymeric micelles are especially promising for targeted drug delivery because of their chemical versatility, stealth properties, and their ability to carry high payloads.[3] The most commonly used polymeric micelles are composed of polyethylene glycol (PEG) grafted to poly-D,L-lactide-co-glycolide (PLGA), polylactic acid, poly-γ-caprolactone (PCL), and poly-alkyl-cyanoacrylates.[7] These amphiphilic molecules self assemble in water to create micelles that have an apolar core that can be used for drug loading and a polar corona that provides stealth properties.[8]
Polymeric micelles are entering clinical evaluation and for example PEG–poly (glutamic acid) polymeric micelles carrying cisplatin (NC-6004, Nanoplatin®)[9] were examined in a phase 1 clinical trial. Compared to the free drug, the nano-delivery device was associated with less toxicity and nausea, and the disease control rate was encouraging. A number of other synthetic polymer nanocarriers have been evaluated in clinical trials, including doxorubicin-loaded polymeric micelles[10] and mitoxantrone-loaded polybutylcyanoacrylate nanoparticles.[11]
Although polymeric micelles are effective at encapsulation of hydrophobic drugs, the entrapment of hydrophilic drugs leads in general to poor drug loading.[12] Furthermore, these delivery systems suffer from premature drug release,[12–13] resulting in rather modest increase of selectivity over free drug. In addition, these formulations may can cause burst release of a drug leading to a reduced therapeutic efficiency.[14] These issues have been addressed by the covalent attachment of drugs to polymeric nanoparticles. Although promising results have been achieved by using this type of nanoparticle, it has been difficult to combine covalent drug attachment with targeted delivery due to a lack of orthogonal conjugation chemistries. This challenge has been addressed by individual attachment of drug and a targeting module to a polymer followed by self-assembly. For example, doxorubicin and a folate receptor[15] were each attached to PLGA-amino-PEG through amide coupling and the resulting polymers employed for micelle formation. Drawbacks of this approach include possible interference of the functional groups on micelle formation and amide bond chemistry can only be employed for the attachment of a limited number of entities.[16] Moreover, attachment of doxorubicin through acylation is not ideal as it can not readily be released and therefore compromises its activity.[17] Post-nanoparticle functionalization is a more attractive approach, and for example random amide coupling has been used to attach a peptide that binds the urokinase plasminogen activator receptor (Upar)[18] and functionalized gemcitabine to the polymeric surface of iron oxide nanoparticles. This type of conjugation lacks selectivity and the resulting particles do not have a corona that provides stealth properties. A more elegant and controlled coupling approach involved the preparation of amphiphilic copolymers that contain several reactive groups for selective functionalization. For example, a polymer was prepared having pendant enol ethers and a terminal furan for coupling of drugs and a targeting agent by thiol-ene and reverse Diels Alder reactions, respectively.[13b] Although conceptionally elegant, having both functionalities at the same polymer may make it difficult to generalize the approach because of difficulties of properly presenting a targeting ligand at the surface of the particles.
We report here a novel approach for the controlled covalent attachment of a drug and targeting ligand by employing poly (ethylene glycol) (PEG) functionalized at one end with a thioctic acid for covalent attachment to a gold core to form stable nanoparticles and at the other end by a reactive functional group for drug, probe, or targeting ligand attachment. We have found that hydrazine, amine, and dibenzocyclooctynol (DIBO),[19] which can be ligated to ketones, activated esters, and azides, respectively (Figure 1), are attractive for attachment of three different entities in a controlled manner. These conjugation reactions are orthogonal and do not require toxic reagents. The methodology was applied to the preparation of a multifunctional nanoparticle that is modified by a carbohydrate-based ligand for CD22,[20] which is expressed on B-cells, and is attractive for the treatment of B-cell lymphomas. Additionally, the reactive groups were exploited for the attachment of doxorubicin and Alexa Fluor 568. It was found that attachment of the glycan ligand for CD22 and a cytotoxic drug resulted in a remarkable ~60-fold increase in cytotoxicity. An additional advantage of such a delivery devise containing drug molecules covalently bound to a gold core is that it may overcome multidrug resistance.[21]
Figure 1.

Chemical synthesis of multifunctional AuNPs targeting CD22 receptors of B cells. a) One end of PEG is modified with thioctic acid for tethering to AuNPs and another end by a functional group for post-synthesis modification. b) Multifunctional AuNPs were synthesized by a modified Burst method. Three bioorthogonal functional groups are present for post-synthesis modification. Hydrazines can selectively react with the ketone of doxorubicin to give a hydrazone-linked drug. Amines can react with an active ester of Alexa Fluor 568 to give an amide bound fluorophore, and finally, DIBO can react with an azide of the CD22 targeting ligand by strain promoted azide-alkyne cycloaddition (SPAAC) to provide a triazole-linked module.
Results and discussion
Chemical synthesis of heterobifunctional PEG derivatives
Gold nanoparticles (AuNPs) are attractive drug delivery vehicles owing to their ease of synthesis, chemical inertness, and flexibility of covalent surface functionalization that can offer high-affinity binding interactions through multivalent display of therapeutic molecules.[1b] Heterobifunctional polymers 1, 2 and 3 were prepared starting from a α-hydroxy-ω-azido-poly(ethylene glycol) (6, HO-PEG-N3, MW = ~2,000 Da) for attachment to a gold core. Thus, the alcohol of 6 was esterified with thioctic acid in presence of N,N′-dicyclohexylcarbodiimide (DDC), 4-(dimethylamino) pyridine (DMAP), and triethylamine to give PEG derivative 7 having a terminal thioctic acid moiety (Scheme 1). The azide of 7 was reduced to an amine using triphenylphosphine in a mixture of THF and water at 50 °C to give polymer 2. FTIR spectra of 7 confirmed the presence of azido moiety (2098 cm−1) and this signal had disappeared in the reduced product 2 (Supporting Information, Page S5). Polymer 2 was modified with activated carbonate 5 in presence of triethylamine to yield polymer 1. 1H NMR spectrum of 1 showed aromatic proton signals typical for DIBO (7.18–7.38 ppm) that had appropriate integrations compared to the PEG signals (3.58–3.75 and 4.12–4.25 ppm), indicating complete functionalization (Supporting Information, Page S7).
Scheme 1.
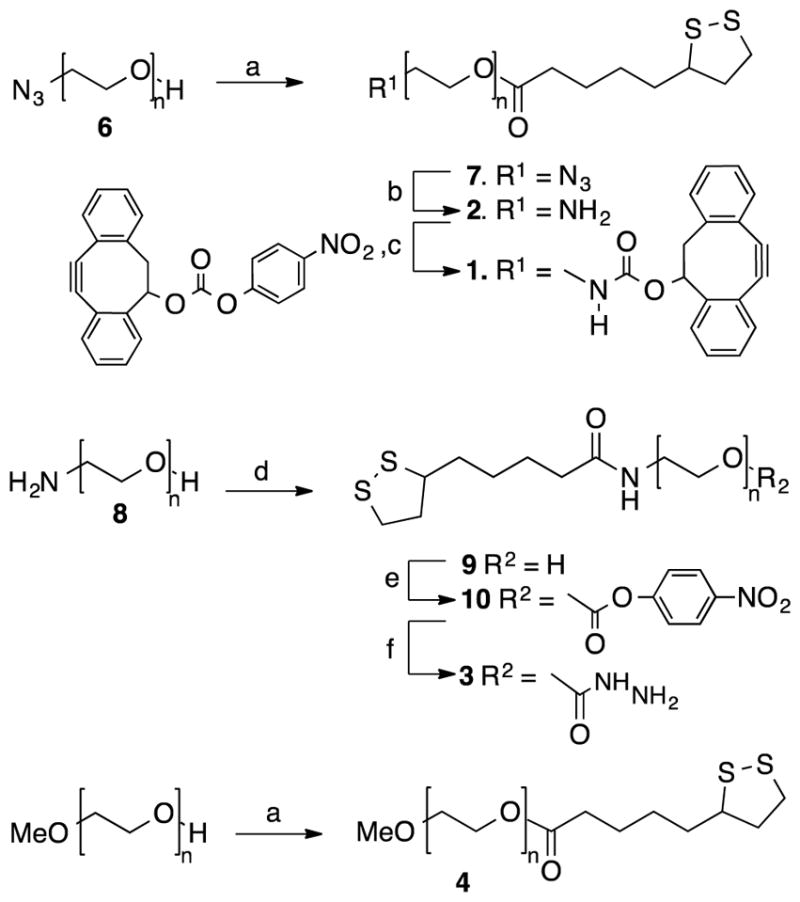
Synthesis of heterobifunctional polymers 1–4. Reagents and conditions: a) thioctic acid, DCC, DMAP, Et3N, CH2Cl2; b) PPh3, THF, 50 °C, H2O; c) 5, Et3N, CH2Cl2; d) thioctic acid, DCC, NHS, Et3N, CH2Cl2; e) p-nitrophenyl chloroformate, Et3N, CH2Cl2; f) hydrazine monohydrate, CH2Cl2.
Polymer 3 was synthesized starting from α-hydroxy-ω-amino-poly (ethylene glycol) (8, HO-PEG-NH2, MW = ~2,000 Da) that was coupled with thioctic acid to give 9 (TA-PEG-OH). The hydroxyl group of compound 9 was activated with p-nitrophenyl chloroformate to generate compound 10, which was reacted with hydrazine monohydrate to provide hydrazine functionalized polymer 3. Finally, polymer 4 containing non-reactive methyl ether was synthesized using a reported approach to control the density of various reactive functionalities at nanoparticle surface.[13d]
Chemoenzymatic synthesis of CD22 targeting ligand for strain promoted azide-alkyne cycloaddition (SPAAC)
Trisaccharide 14, which contains a sialic acid modified at C-9 by a biphenylcarbonyl moiety, is a high affinity ligand for CD22.[20] This compound contains an anomeric azidopentyl linker for conjugation to the DIBO moiety[19] of the nanoparticles. The target glycan was synthesized by a chemoenzymatic approach employing acceptor 12 and a convenient one-pot two-enzyme sialylation system using a modified literature procedure (Scheme 2).[22] This enzyme system does, however, not tolerate bulky substituents at C-9 of sialic acid and therefore a strategy was used by which CMP-sialic acid modified by a C-9 amino function was employed for transfer which was followed by acylation of the amine of sialic acid to give target compound 14.
Scheme 2.

Chemoenzymatic synthesis of the glycan ligand of CD22. Reagents and conditions: a) TMSOTf, 1,2-dichloroethane, 5-azidopentanol, 60 °C, 18 h; b) NaOMe, MeOH, 1 h, (74%, two steps); c) Neu5Ac9NH2, CTP, Pd(2,6)ST, NmCSS, pH ≈ 9.5, 37 °C, overnight, (53%); d) 4-biphenylcarbonyl-NHS ester, Et3N, DMF, 24 h, (81%).
Acceptor 12 was prepared in high overall yield by reaction of per-O-acetylated LacNAc (11) with 5-azidopentanol in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) to give a glycoside that was deacetylated with sodium methoxide in methanol. Next, C9-amino sialic acid was introduced by in-situ formation of CMP-Neu5Ac9NH2 by condensation of Neu5Ac9NH2 with CTP in the presence of N. meningitidis CMP-sialic acid synthetase (NmCSS) followed by the addition of compound 12 and α(2,6)-sialyltransferase (Pd(2,6)ST) derived from P. damselae to give the desired sialoside 13 in a yield of 53%. Treatment of 13 with 4-biphenylcarbonyl-N-hydroxysuccinimide in DMF gave 14 in an excellent yield of 81%.
Synthesis and characterization of multifunctional AuNPs
AuNPs A containing PEG derivatives (Figure 1) having three different surface reactive functional groups, was prepared by a modified literature procedure,[23] whereby LiBH4 was added drop wise to a vigorously stirred mixture of polymers 1–4 (ratio of 1/2/3/4 is 10/10/30/50) and HAuCl4 in anhydrous THF under atmosphere of nitrogen in the dark. After a reaction time of 3 h, methanol was added to quench the excess of reducing agent. The PEG stabilized nanoparticles were soluble in THF, which made it possible to remove uncomplexed polymers and inorganic salts by dialysis applying a 12–14 kDa molecular weight cut-off membrane. An aqueous solution of AuNPs was obtained by concentrating the THF solution under reduced pressure, which was followed by the addition of water and extensive dialysis against water for 3 days. The resulting nanoparticles were characterized by ultraviolet-visible (UV-Vis) absorption spectroscopy, showing a λmax at 518 nm (Supporting Information, Figure S1), which is a characteristic surface plasmon resonance band for AuNPs. Dynamic light scattering of the resulting AuNPs showed a mean diameter of 21 ± 1 nm (Supporting Information, Figure S2 and Table S1). Furthermore, transmission electron microscopy (TEM) images revealed that the gold core had an average diameter of 5–8 nm (Supporting Information, Figure S4). Thermogravimetric analysis (TGA) of nanoparticle A gave a weight ratio of gold to organic matter of approximately 22:78 (Supporting Information, Figure S5). Interestingly, the heterobifunctional PEG derivatives lacking hydrophobic PCL did not show any structural heterogeneity and instability that we observed for PCL-PEO based block polymers.[13d]
Multi-functionalization
Multifunctional nanoparticle D was prepared by subsequent conjugations of doxorubicin (15), Alexa Fluor 568 (16), and glycan ligand (14) (Figure 3a). It was anticipated that the ketone moiety of doxorubicin could be selectively ligated to the hydrazine moiety of particle A to give hydrazone-linked doxorubicin.[24] Such linker was expected to be stable at physiological pH but hydrolyzes and releases free doxorubicin after endocytosis and entering endosomes and lysosomes that have an acidic environment. The amines of the particles can be reacted with activated esters such as Alexa Fluor derivative 16, and finally glycan ligand 14 is modified by an azido moiety that can be exploited for strain-promoted cycloaddition with the DIBO moiety of the particles.
Figure 3.

Compounds for surface modification and chemical composition of AuNPs. a) The chemical structures of compounds 14–16 used for surface modification of the nanoparticles. b) Chemical composition of AuNPs A–D.
Thus, particle A was incubated with doxorubicin in presence of catalytic amount of trifluoroacetic acid (TFA) in the dark for 48 h to form hydrazone linked AuNPs B. The solution was dialyzed against water to remove free doxorubicin. To quantify the conjugated doxorubicin, AuNPs B were suspended in acetate buffer at pH 5 and stirred for 30 h. The resulting nanoparticle solution was centrifuged (Milipore, centrifugation filters) to remove the gold particles and the free doxorubicin was quantified by HPLC analysis, which revealed a conjugation efficiency of 86% (Supporting Information, Figure S6).
To examine drug release in more detail, nanosurface energy transfer (NSET) effect was utilized.[25] As previously reported, the emission spectrum of doxorubicin (λemi at 565 nm) overlaps with the UV-Vis absorption spectrum of AuNPs, resulting in a decrease of fluorescence intensity of doxorubicin due to energy transfer to AuNPs.[21] The fluorescence of doxorubicin will, however, recover once it is released from nanoparticles by hydrolysis of the hydrazone bond. Thus, AuNPs B was incubated in acetate buffer of pH 5 and PBS buffer of pH 7.4 and fluorescence emission was measured over different time intervals. As shown in Figure 2, incubation of AuNPs B at pH 5 resulted in rapid recovery of fluorescence, whereby after 15 h no further increase was observed, indicating doxorubixin had been completely released. However, incubation of AuNPs B at pH 7.4 did not exhibit significant fluorescence recovery even after prolonged periods of time, indicating that doxorubicin will only be released when entering acidic compartments of cells.
Figure 2.

Schematic illustration of fluorescence recovery after the hydrolysis of hydrazone bonds of AuNPs B. a) Quantitative analyses of the cumulative release of doxorubicin at 37 °C from AuNPs B at pH 7.4 or 5.0 (complete release of doxorubixin was assumed when no further increase of fluorescence was observed over time and set at 100%) b) Fluorescence emission spectra of AuNPs B in acetate buffer (pH 5.0) over time. Note: spectra at 15 and 20 h overlap.
Next, attention was focused on further conjugation reactions to obtain AuNPs D. Thus, the solution containing AuNPs B was adjusted to pH 8 by the addition of aqueous NaHCO3, and active ester 16 was added. After a reaction time of 24 h, the solution was dialyzed against water to remove free Alexa Fluor 568 (AuNPs C), and then azide 14 was added for a strain promoted azide-alkyne cycloaddition with the DIBO moieties of nanoparticle C to attach the glycan ligand through a triazole moiety. After 24 h, the solution was dialyzed to give nanoparticle D (Figure 3). The degree of functionalization for Alexa Fluor 568 was determined by measuring the fluorescence intensity showing a 94% efficiency of conjugation. It is important to note that the degree of functionalization may even be higher as some fluorescence quenching by the gold core of the nanoparticles is possible.[26] Quantitative monosaccharide analysis was performed by treatment of the particles with TFA at 100 °C to cleave glycosidic linkages followed by analysis by high pH anion exchange chromatography (HPAEC) (Supporting Information, Figure S7). It was found that the level of glycan functionalization was approximately 85%. Collectively, these results demonstrate that the conjugation approach is highly efficient. Dynamic light scattering (Supporting Information, Figure S2 and Table S1) showed a slight increase in size (29 ± 2 nm) due to the three consecutive chemical transformations on the surface of nanoparticles, while TEM indicated no significant change in the size and morphology of the gold core (Supporting Information, Figure S4).
Previous studies, in which liposomes were modified with a glycan such as 14, had indicated that optimal targeting was achieved when approximately 5% of the surface molecules were modified with the targeting agent.[20c] Furthermore, previously we had found that 10% surface modification of polymeric micelles with a fluorescent tag is appropriate for various visualization purposes.[13d] Thus, the accomplished conjugation efficiencies for 14 and the fluorophore was expected to be appropriate. Furthermore, the drug loading was limited to 30% of the surface molecules to avoid unwanted effects due to the hydrophobicity of the drug.
In vitro cytotoxicity, cellular uptake, and intracellular localization of AuNPs
CD22, which is a validated target for the treatment of B-cell lymphoma, undergoes receptor-mediated endocytosis,[27] and hence is an attractive target for delivery of drug loaded nanoparticles.[20b] The glycan moiety of AuNPs D is a high affinity ligand of CD22, and thus it was anticipated that particles endowed with this functionality should preferentially be endocytosed by cells expressing CD22. Daudi Burkitt’s lymphoma cells that express CD22 were incubated with AuNPs B (non-targeted NPs), AuNPs D (targeted NPs), and free doxorubicin at varying concentrations. After 48 h, cell viability was measured by MTT assay. As can be seen in Figure 4a, targeted AuNP D (IC50 = 0.48 μM) exhibited a 60-fold increase in cytotoxicity compared to AuNP B (IC50 = 27 μM) indicating that the glycan moiety greatly facilitates selective uptake and that the hydrazone-linked doxorubicin can be cleaved intra-cellularly to cause cytotoxicity. In respect to the latter, the basicity of the amine of doxorubicin is important for toxicity,[28] and therefore, the hydrazone linkage needs to be cleaved before the drug can exert its effect. The low toxicity of AuNP B is probably caused by non-specific cellular uptake. Importantly, control AuNPs modified only with targeting glycan 14 did not show any toxicity at corresponding concentrations (Supporting Information, Figure S8).
Figure 4.

Biological examination of AuNPs B, C, and D. a) Cytotoxicity of doxorubicin tethered AuNPs B and D on Daudi Burkitt’s cells. Data shown are mean ± SD (n=3). b) Daudi Burkitt’s cells were exposed to nanoparticles C and D at 5–20 μg mL−1 gold for 2 h. After the cells were washed and lysed, fluorescence intensity (absorbance 578 nm, emission 603 nm) was measured and using corresponding calibration curves uptake was calculated as mean ± SD (n=3). c, d) TEM images of representative sections of Daudi Burkitt’s cells that were incubated with AuNPs C and D at 100 μg mL−1 gold for 10 h. c) Most AuNPs D were freely dispersed in the cytoplasm (shown with red arrow) as single nanoparticles, while some were found to be aggregated. d) AuNPs C did not exhibit any internalization.
Alexa Fluor 568-conjugated AuNPs C and D were employed to study in more detail cellular uptake. CD22-expressing Daudi Burkitt’s lymphoma cells and CD22 non-expressing Jurkat cells were exposed to different concentrations of AuNPs C and D, and after incubation time of 2 h, cell lysates were analyzed for fluorescence intensity. As expected, the glycan ligand (14) of AuNPs D led to a significant increase in cellular uptake compared to the treatment with non-targeted nanoparticles C (Figure 4b and Supporting Information, Figure S9). Importantly, the Jurkat cells did not show significant uptake of AuNPs D under similar experimental conditions, demonstrating excellent targeting properties of the new AuNPs.
The intracellular localization of nanoparticles D and C was examined by TEM, which can visualize the gold core of the nanoparticles. Daudi Burkitt’s cells were exposed to AuNPs C and D for 10 h. The use of targeted AuNPs D showed significant number of internalized nanoparticles (Figure 4c and Supporting Information, Figure S10). The internalized nanoparticles D were predominantly dispersed in the cytosol as individual nanoparticles, whereas few were found in aggregated form. Thus, it appears that the particles can escape vesicular structures after internalization.[29] As expected, cells treated with AuNPs C did not show internalized nanoparticles (Figure 4d and Supporting Information, Figure S11).
Conclusion
Although active targeted delivery of cytotoxic drugs to cancer cells is an attractive concept to overcome poor selectivity’s of cytotoxic drugs, results are often disappointing due to premature release of a drug from a nanocarrier delivery devise.[12–13] Covalent attachment of a drug to a nanoparticle is an attractive approach to overcome this problem.[30] This technology has, however, not matured due to difficulties of preparing nanoparticles that have good stability, a polar corona for stealth properties and allow controlled conjugation of a drug and targeting device. The multifunctional gold nanoparticle platform described here has a unique feature that it is composed of a polar PEG corona modified with hydrazine, amine and azide reactive functional groups for post-synthesis modification by hydrazone bond formation, amide bond chemistry, and SPAAC, respectively. These conjugation methods are highly selective and allow the attachment of three different functional entities with high efficiency. The particles exhibit excellent stability because at one end they are modified by thiotic acid for attachment to a gold core by a modified Brust approach. The three functional groups of the polymers are compatible with the reducing conditions employed in this reaction. The resulting nanoparticles are small (5–8 nm), which is attractive for drug delivery and exhibit excellent water solubility and were stable for a prolonged period of time. In this respect, recent studies have shown that PEG-coated gold nanoparticles with size ranging from 15–60 nm exhibit liver, kidney and spleen toxicity in mice.[31]. Nanoparticles that have diameters of ≤5.5 nm are also not attractive for in vivo use because these are rapidly cleared by the renal route.[32] Other attractive features of the new platform include excellent control over ligand density and the targeting ligand is well assessable by binding to cell surface receptors. As a proof of principle, we have demonstrated that a nanoparticle modified by a glycan ligand for CD22 for targeting B-cells, doxorubicin attached via a pH-sensitive hydrazone linkage for cytotoxicity, and a fluorophore for measuring uptake, exhibits a 60-fold increase in cytotoxicity of CD22 expressing B-cells compared to similar non-targeting particles. The pH responsive nature of drug release was studied in detail and free doxorubicin was only observed when applied to acidic conditions. It is the expectation that new nanoparticle platform can be employed for the combinatorial attachment of various cell targeting devices and drug molecules.
Experimental Section
General reagents and materials
Polyethylene glycol (PEG, number average molecular weight, Mn ~2,000 Da), polyethylene glycol monomethyl ether (Mn ca. 2,000 Da), p-toluenesulfonyl chloride, sodium azide, dimethylformamide (DMF, 99.8%), 4-(dimethyl amino) pyridine (DMAP, 99%), 1,3-dicyclohexyl carbodiimide (DCC, 99%), hydrogen tetrachloroaurate (III) trihydrate (HAuCl4·3H2O, 99.9%), triethylamine (99.5%), thioctic acid (98%), N-hydroxysuccinamide (NHS, 99%), p-nitrophenyl chloroformate (96%), hydrazine monohydrate (98%), 1,2 dichloroethane, sodium methoxide, and 4-biphenyl-carboxylic acid, Neu5Ac were purchased from Carbosynth LLC and lithium aluminium hydride (LiAlH4, 95%) was obtained from Aldrich. Alexa Fluor 568 carboxylic acid succinimidyl ester was obtained from Invitrogen. Doxorubicin hydrochloride was purchased from LC Laboratories. A 0.25 M LiBH4 solution in tetrahydrofuran (THF) was freshly prepared by diluting commercial 2 M LiBH4 (Aldrich) with freshly distilled THF. PEG was dried by azeotropic distillation from toluene followed by storage in vacuo at 60 °C for 24 h. Dichloromethane (CH2Cl2, 99%+, Fisher Scientific) was distilled from CaH2 prior to use. All other chemicals and solvents were of analytical grade and were used as received. Column chromatography was performed on 70–230-mesh silica gel. Thin-layer chromatography was performed using Kieselgel 60 F254 (Merck) and visualized using UV, I2 adsorption, and/or H2SO4/heat. Dialysis membrane Spectra/Pro 2 (molecular weight cut-off 12–14 kDa) and Spectra/pro 7 (molecular weight cut-off 50 kDa) were purchased from Spectrum Laboratories, Inc. All other reagents and solvents were of analytical grade.
General methods for compound characterization
1H and 13C NMR spectra (CDCl3 or D2O) were recorded using a Varian Merc-300 spectrometer equipped with Sun workstations at 300K with the residual 1H solvent peak as reference and the solvent carbon signal as standard, respectively. COSY, HSQC, and HMBC experiments were used to assist assignment of the sugar products. Multiplicities are quoted as singlet (s), doublet (d), doublet of doublets (dd), triplet (t), or multiplet (m). NMR signals were assigned on the basis of 1H NMR, 13C NMR, gCOSY, and gHSQC experiments. Chemical shifts are quoted on the δ-scale in parts per million (ppm). Residual solvent signals were used as an internal reference. FT-IR spectra were recorded on a Shimadzu IR Prestige-21 spectrophotometer. MALDI-TOF MS spectra were recorded on an Applied Biosystems 5800 MALDI-TOF in the positive ion mode by using 2,5-dihydroxy-benzoic acid in acetonitrile (10 mg mL−1) as a matrix. Thermogravimetric analyses (TGA) were conducted on a Perkin-Elmer Pyris 1 Thermogravimetric Analyzer at a heating rate of 10 °C min−1 under nitrogen. UV-Vis spectra were recorded on a Beckman Coulter DU 800 spectrophotometer, between 200 and 800 nm wavelength. Fluorescent spectroscopy was carried on a BMG Labtech POLARstar OPTIMA reader. Reverse phase HPLC studies were carried out using Agilent 1100 series (Eclipse XDB-C18 column, 5 μm, 4.6*250 mm). Sugar analysis was conducted by high-pH anion-exchange chromatography (HPAEC) using an ICS-3000 Ion Chromatography System (Dionex, Sunnyvale, CA, USA) with deionized water and 200 mM NaOH as eluent. TEM observations were made using a Philips/FEI Tecnai 20 instrument operating at an accelerating voltage of 200 kV. DLS measurements were performed on a zeta potential and particle size analyzer (Malvern Zetasizer Nano ZS system).
General procedure for the preparation of AuNPs A
Glassware used for the preparation of Au nanoparticles was washed three times with aqua regia followed by copious amounts of nano-pure water and finally dried in an oven at 150 °C for 24 h. To a solution of HAuCl4•3H2O (16 mg, 0.040 mmol) in anhydrous THF (12 mL) was added polymer 1 or a mixture of 1–4 (0.200 g, 1:2:3:4, 1:1:3:5, w/w/w/w). After stirring the mixture in the dark under a nitrogen atmosphere for 22 h, LiBH4 in THF (0.25 M, 0.5 mL, ~3 eq. to HAuCl4) was added dropwise under vigorous stirring. After stirring the reaction mixture for 3 h, ethanol (3 mL) was added and stirring was continued for 12 h. The solution was dialyzed against tetrahydrofuran (THF) applying a 50 KDa molecular weight cut-off membrane (Spectra/pro 7) until no unassociated polymer was detected by thin-lay chromatography (eluent: CHCl3/CH3OH, 9/1). The solution was concentrated to a small volume (1.5–3.5 wt%) and then slowly added (25 μL min−1) to a 4-fold excess of sterile water with vigorous stirring. The mixture was then dialyzed (12–14 KDa MWCO) against sterile water and then lyophilized. UV-Vis, TEM, dynamic light scattering, TGA, and zeta potential were for particle characterization.
Procedure for the preparation of AuNPs B–D
30 mg of AuNPs A stabilized by polymers 1–4 (1:2:3:4, 1:1:3:5, w/w/w/w), compound 15 (5.31 mg, 3–4 equiv), and TFA (25 μL) were dissolved in anhydrous 6 mL (1:1) THF/MeOH. The reaction mixture was stirred in the dark at RT for 48 h. The solution was then dialyzed against tetrahydrofuran (THF) applying a 50 KDa molecular weight cut-off membrane (Spectra/pro 7) until no fluorescence intensity (from doxorubicin) was detected in the dialysate. The solution was concentrated to a small volume (1.5–2%) and then slowly added to a 4-fold excess of sterile water with vigorous stirring. The mixture was stirred for another hour and then dialyzed (12–14 KDa MWCO) against sterile water to obtained AuNPs B. The pH of the solution was then adjusted to 8 by the addition of NaHCO3. To the solution, N-hydroxysuccinamide-modified Alexa Fluor 568 (compound 16, 1.1 mg, 1.5 equiv) was added to react to the amines. The reaction mixture was stirred in the dark at room temperature at 24 h and was then dialyzed against water for 2 days until no fluorescence was detected. Compound 14 (1.9 mg, 2 equiv) was added and stirred for 24 h in dark at RT and resulting mixture was dialyzed against water for 2 days to give nanoparticle D. Dynamic light scattering, TEM, zeta potential, sugar and HPLC analysis were used to characterize these AuNPs.
Measurement of release of doxorubicin from AuNPs B
To quantitatively determine the release of doxorubicin, AuNPs B were suspended in phosphate buffer (pH 7.4) or acetate buffer (pH 5.0) at 1.5 mg mL−1 and stirred for 30 h. The fluorescence emission spectra were recorded every 2 h. Next the nanoparticle solution was centrifuged (Milipore, centrifugation filters) to remove supernatant that contained released doxorubicin. This was further lyophilized and the amount of doxorubicin in given nanoparticles preparation was quantified using HPLC with solvent gradient from 20 to 50% (0.01M TFA:acetonitrile) for 25 min/injection. The amount of doxorubicin was in agreement with the fluorescence intensity measurements.
Quantitative sugar analysis
Sugar analysis was conducted by high-pH anion-exchange chromatography (HPAEC) using an ICS-3000 Ion Chromatography System (Dionex, Sunnyvale, CA, USA) with deionised water and 200 mM NaOH as eluent. The system consists of a SP gradient pump with an AS autosampler, ICS-3000 thermal compartment, and an ICS-3000 electrochemical detector equipped with an amperometry cell. The cell consists of a gold electrode, a combination reference electrode of glass and Ag/AgCl (3 M KCl) and titanium counter electrode consisting of the cell body. Separation was carried out using the CarboPac PA 20 column set consisting of a guard column (50 mm × 4 mm I.D.) and an analytic column (150 mm × 3 mm I.D.) The column and the electrochemical detection cell were placed inside the ICS-3000 thermal compartment for temperature control. The chromatographic system control, data acquisition, and analysis were carried out using Chromeleon Software (Dionex). Sample preparation: 0.5–1.0 mg of nanoparticle sample and sugar standard, such as compound 12 were treated with 2 M TFA in water (250 μL) at 100 °C for 4 h. Sample and standard were dried by spin-vacuo centrifugation, resuspended in 0.5 mL of water and passed through a C18 Cartridge (Waters), dried by spin-vacuo centrifugation, redissolved in quantitative volume of water. The sugar content in the sample was determined based on the calibration curves of standards.
Cell line and culture conditions
Human B lymphoblasts Daudi Burkitt’s cells (CCL-213, ATCC) and human Jurkat cells (Clone E6-1; ATCC) were cultured in ATCC-formulated RPMI-1640 medium with L-glutamine (2 mM), sodium bicarbonate (1.5 g L−1), glucose (4.5 g L−1), HEPES (10 mM) and sodium pyruvate (1.0 mM). The media was supplemented with penicillin (100 U mL−1) and streptomycin (100 μg mL−1, Mediatech) and fetal bovine serum (FBS, 10%, BenchMark). Cells were maintained in a humid 5% CO2 atmosphere at 37 °C and subcultured every 2–3 days.
Cytotoxicity assay
Cytotoxicity was determined using the MTT assay. On the day of exposure, exponentially growing cells were plated as 1×105 cells/well in 180 μL in 96-well tissue culture plates (Nunc). Cells were then incubated with medium (control), free doxorubicin, AuNPs B, and AuNPs D (20 μL, 10X in cell culture medium) for 2 h at 37 °C. Next the plate was centrifuged and supernatant was replaced with fresh medium (200 μL/well), which allowed for an additional 48 h of incubation. The viability was measured by quantifying the cellular ability to reduce the water-soluble tetrazolium dye 3–4,5-dimethylthiazole-2, 5-diphenyl tetrazolium bromide (MTT) to its insoluble formazan salt as follows. At 44 h, MTT (5 mg mL−1 in PBS, 20 μL/well) was added to the wells and the cells were further incubated for 4 h. At 48 h the supernatant was carefully removed and the water-insoluble formazan salt was dissolved in DMSO (120 μL/well). The absorbance was measured at 545 nm using a microplate reader (BMG Labtech). Data points were collected in triplicate and expressed as normalized values for untreated control cells (100%). Data were fitted using Prism software (GraphPad Software, Inc).
Measurement of cellular uptake
Daudi Burkitt’s cells and Jurkat cells as control were harvested and added to tubes as 1×106 cells in 320 μL medium. AuNPs C or D (80 μL, 5X in cell culture medium) was added to give final volume of 400 μL/tube. After an incubation of 2 h, the supernatant was discarded and the cells were washed three times with Dulbecco’s phosphate buffered saline. Next, the cells were lysed in Passive Lysis Buffer 1X (100 μL; Promega Corporation) and the fluorescence (excitation 578 nm, emission 603 nm) was measured in black 96-well plates using a fluorescent microplate reader (BMG Labtech). Calibration curves of the appropriate Alexa Fluor 568 conjugated AuNPs C or D in Passive Lysis Buffer 1X were used to calculate total cellular uptake. The data are presented as mean ± SD of triplicate treatments, with each experiment being repeated three times.
Intracellular localization of AuNPs by transmission electron microscopy (TEM)
Daudi Burkitt’s cells were plated at 1×106 cells/well in 96-well plates and the cells were incubated with AuNPs C or D in culture medium (100 μg mL−1 of Au). After 10 h, the medium was removed and the cells were washed twice with Dulbecco’s phosphate buffered saline and then fixed with 2% glutaraldehyde in phosphate buffered saline (PBS; 0.1 M, pH 7.4) for 1 h in Eppendorf tube. Samples were then washed three times with PBS to remove excess glutaraldehyde (10 min each wash). Cells were then post fixed with 1% OsO4/0.1 M PBS for 1 h, pelleted, and rinsed with buffer and then washed 3 times for 10 min with distilled water to remove excess of salts before dehydration in ethanol series. Next, the cells were dehydrated through a graded ethanol series of 25, 50, and 75 for 10 min each and 100% for 10 min thrice. This was followed by transition into (1:1) propylene oxide (PO):ethanol twice for 10 min. The cells were infiltrated with Embed 812 through graded (resin:PO) series of 25, 50, and 75 with 1 h between each step. Samples were then kept in fresh 100% Embed 812 resin by polymerization at 60 °C for 48 h. Samples were then sectioned with a Diatome diamond knife on a RMC MT-X ultramicrotome (Boeckeler Instruments Inc.). Ultrathin sections (~100 nm) were collected on 300 mesh Cu grids. Sections were then post-stained with uranyl acetate (30 min) and lead citrate (5 min). The samples were viewed with a JEOL JEM-1200 TEM equipped with an AMT XR41C Bottom-Mount CCD camera using 80 kV accelerating voltage.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. John Shields (Center for Advanced Ultrastructure Research, University of Georgia) for TEM assistance, Prof. Suraj Sharma (Department of Textiles, University of Georgia) for TGA measurements. The National Cancer Institute of the U.S. National Institute of Health supported this research (R01CA088986 to G.-J.B.).
Footnotes
Supporting information for this article is available. Provided are: synthesis procedures and characterization of polymers 1–4 and compound 14; nanoparticle characterization including DLS, TEM, and zeta potential; sugar, doxorubicin, and Alexa Fluor 568 quantification, standard curves for cellular uptake studies; intracellular localization of nanoparticle by TEM.
References
- 1.a) Hawker CJ, Wooley KL. Science. 2005;309:1200–1205. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]; b) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; c) Torchilin VP. Pharm Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]; d) van Dongen SFM, de Hoog HPM, Peters RJRW, Nallani M, Nolte RJM, van Hest JCM. Chem Rev. 2009;109:6212–6274. doi: 10.1021/cr900072y. [DOI] [PubMed] [Google Scholar]; e) Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Angew Chem Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Petros RA, DeSimone JM. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]; g) Elsabahy M, Wooley KL. Chem Soc Rev. 2012;41:2545–2561. doi: 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Hubbell JA, Chilkoti A. Science. 2012;337:303–305. doi: 10.1126/science.1219657. [DOI] [PubMed] [Google Scholar]; i) Wang AZ, Langer R, Farokhzad OC. Annu Rev Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]; j) Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Chem Soc Rev. 2013;42:1147–1235. doi: 10.1039/c2cs35265f. [DOI] [PubMed] [Google Scholar]; k) Zhang G, Zeng X, Li P. J Biomed Nanotechnol. 2013;9:741–750. doi: 10.1166/jbn.2013.1583. [DOI] [PubMed] [Google Scholar]
- 2.a) Maeda H, Bharate GY, Daruwalla J. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]; b) Fang J, Nakamura H, Maeda H. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]; c) Brannon-Peppas L, Blanchette JO. Adv Drug Delivery Rev. 2012;64:206–212. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Phillips MA, Gran ML, Peppas NA. Nano Today. 2010;5:143–159. doi: 10.1016/j.nantod.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J. Angew Chem Int Ed. 2004;43:6323–6327. doi: 10.1002/anie.200460800. [DOI] [PubMed] [Google Scholar]; b) Torchilin VP. Biopolymers. 2008;90:604–610. doi: 10.1002/bip.20989. [DOI] [PubMed] [Google Scholar]
- 5.Rajendran L, Knolker HJ, Simons K. Nat Rev Drug Discov. 2010;9:29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 6.Boisselier E, Astruc D. Chem Soc Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 7.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Chem Soc Rev. 2012;41:2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu FX, Karnik R, Wang AZ, Alexis F, Levy-Nissenbaum E, Hong S, Langer RS, Farokhzad OC. Nano Today. 2007;2:14–21. [Google Scholar]
- 9.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ, Matsumura Y, Kataoka K, Nishiya T. Br J Cancer. 2011;104:593–598. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Watanabe N. Br J Cancer. 2004;91:1775–1781. doi: 10.1038/sj.bjc.6602204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Q, Sun X, Zeng L, Liu J, Zhang Z. Nanomed Nanotechnol Biol Med. 2009;5:419–423. doi: 10.1016/j.nano.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Matsumura Y. Adv Drug Deliv Rev. 2008;60:899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 13.a) Bromberg L. J Control Release. 2008;128:99–112. doi: 10.1016/j.jconrel.2008.01.018. [DOI] [PubMed] [Google Scholar]; b) Shi M, Ho K, Keating A, Shoichet MS. Adv Funct Mater. 2009;19:1689–1696. [Google Scholar]; c) Attia ABE, Ong ZY, Hedrick JL, Lee PP, Ee PLR, Hammond PT, Yang YY. Curr Opin Colloid Interface Sci. 2011;16:182–194. [Google Scholar]; d) Li X, Guo J, Asong J, Wolfert MA, Boons GJ. J Am Chem Soc. 2011;133:11147–11153. doi: 10.1021/ja2012164. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cai K, He X, Song Z, Yin Q, Zhang Y, Uckun FM, Jiang C, Cheng J. J Am Chem Soc. 2015;137:3458–3461. doi: 10.1021/ja513034e. [DOI] [PubMed] [Google Scholar]
- 14.Zeng J, Yang L, Liang Q, Zhang X, Guan H, Xu X, Chen X, Jing X. J Control Release. 2005;105:43–51. doi: 10.1016/j.jconrel.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 15.Yoo HS, Park TG. J Control Release. 2004;96:273–283. doi: 10.1016/j.jconrel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Ulbrich K, Etrych T, Chytil P, Jelinkova M, Rihova B. J Control Release. 2003;87:33–47. doi: 10.1016/s0168-3659(02)00348-6. [DOI] [PubMed] [Google Scholar]
- 17.Wong PT, Choi SK. Chem Rev. 2015;115:3388–3432. doi: 10.1021/cr5004634. [DOI] [PubMed] [Google Scholar]
- 18.Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M, Nie S, Mao H, Yang L. ACS Nano. 2013;7:2078–2089. doi: 10.1021/nn3043463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ning XH, Guo J, Wolfert MA, Boons GJ. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Zaccai NR, Maenaka K, Maenaka T, Crocker PR, Brossmer R, Kelm S, Jones EY. Structure. 2003;11:557–567. doi: 10.1016/s0969-2126(03)00073-x. [DOI] [PubMed] [Google Scholar]; b) Collins BE, Blixt O, Han S, Duong B, Li H, Nathan JK, Bovin N, Paulson JC. J Immunol. 2006;177:2994–3003. doi: 10.4049/jimmunol.177.5.2994. [DOI] [PubMed] [Google Scholar]; c) Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. Blood. 2010;115:4778–4786. doi: 10.1182/blood-2009-12-257386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. ACS Nano. 2011;5:3679–3692. doi: 10.1021/nn200007z. [DOI] [PubMed] [Google Scholar]
- 22.a) Yu H, Chokhawala HA, Huang S, Chen X. Nat Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kaltgrad E, O’Reilly MK, Liao L, Han S, Paulson JC, Finn MG. J Am Chem Soc. 2008;130:4578–4579. doi: 10.1021/ja077801n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) O’Reilly MK, Collins BE, Han S, Liao L, Rillahan C, Kitov PI, Bundle DR, Paulson JC. J Am Chem Soc. 2008;130:7736–7745. doi: 10.1021/ja802008q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Brust M, Fink J, Bethell D, Schiffrin DJ, Kiely C. J Chem Soc-Chem Comm. 1995:1655–1656. [Google Scholar]; b) Azzam T, Eisenberg A. Langmuir. 2007;23:2126–2132. doi: 10.1021/la0627563. [DOI] [PubMed] [Google Scholar]
- 24.Lau A, Berube G, Ford CH, Gallant M. Bioorg Med Chem. 1995;3:1305–1312. doi: 10.1016/0968-0896(95)00126-2. [DOI] [PubMed] [Google Scholar]
- 25.a) Darbha GK, Ray A, Ray PC. ACS Nano. 2007;1:208–214. doi: 10.1021/nn7001954. [DOI] [PubMed] [Google Scholar]; b) Lee H, Lee K, Kim IK, Park TG. Biomaterials. 2008;29:4709–4718. doi: 10.1016/j.biomaterials.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 26.a) Pustovit VN, Shahbazyan TV. J Chem Phys. 2012;136:204701. doi: 10.1063/1.4721388. [DOI] [PubMed] [Google Scholar]; b) Ledin PA, Kolishetti N, Hudlikar MS, Boons GJ. Chem-Eur J. 2014;20:8753–8760. doi: 10.1002/chem.201402225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molina A. Annu Rev Med. 2008;59:237–250. doi: 10.1146/annurev.med.59.060906.220345. [DOI] [PubMed] [Google Scholar]
- 28.Priebe W. Current Pharmaceutical Design. 1995;1:51–68. [Google Scholar]
- 29.Nativo P, Prior IA, Brust M. ACS Nano. 2008;2:1639–1644. doi: 10.1021/nn800330a. [DOI] [PubMed] [Google Scholar]
- 30.a) Zhang XQ, Xu X, Lam R, Giljohann D, Ho D, Mirkin CA. ACS Nano. 2011;5:6962–6970. doi: 10.1021/nn201446c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vigderman L, Zubarev ER. Adv Drug Deliv Rev. 2013;65:663–676. doi: 10.1016/j.addr.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Zhang XD, Wu D, Shen X, Liu PX, Yang N, Zhao B, Zhang H, Sun YM, Zhang LA, Fan FY. Int J Nanomed. 2011;6:2071–2081. doi: 10.2147/IJN.S21657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.