

Abstract
Animal skin, which is the tissue that directly contacts the external surroundings, has evolved diverse functions to adapt to various environments. Amphibians represent the transitional taxon from aquatic to terrestrial life. Exploring the molecular basis of their skin function and adaptation is important to understand the survival and evolutionary mechanisms of vertebrates. However, comprehensive studies on the molecular mechanisms of skin functions in amphibians are scarce. In this study, we sequenced the skin transcriptomes of seven anurans belonging to three families and compared the similarities and differences in expressed genes and proteins. Unigenes and pathways related to basic biological processes and special functions, such as defense, immunity, and respiration, were enriched in functional annotations. A total of 108 antimicrobial peptides were identified. The highly expressed genes were similar in species of the same family but were different among families. Additionally, the positively selected orthologous groups were involved in biosynthesis, metabolism, immunity, and defense processes. This study is the first to generate extensive transcriptome data for the skin of seven anurans and provides unigenes and pathway candidates for further studies on amphibian skin function and adaptation.
The adaptation of organisms to their environments is the basis of evolutionary biology. This adaptation occurs at many levels of biological organization, from molecules to organs1. Animal skin, acting as the direct medium interacting with external surroundings, has experienced distinctly adaptive evolution. Numerous shifts have occurred in animal skin functions since the habitats transformed from an aquatic to a terrestrial environment. For aquatic species, the skin mainly possesses basic functions, including protection, sensing, and communication with other organisms, although several specific functions have evolved (e.g., locomotion)2. Conversely, the skin of species living on land has developed complex functions. Faced with arid environments, terrestrial species have had to adapt their skin to retain moisture. Thus, stratum corneum has developed in their skin, with special structures and compositions3,4. Moreover, the skin of homeothermic terrestrial animals has developed thermal receptors to deliver signals toward the thermoregulation system5. Therefore, the skin of animals has undergone marked changes due to shifts in environmental backgrounds; investigating the differences in skin function among species may shed light on the adaptive evolution of organisms.
Amphibian represents a valuable taxon that links the evolutionary gap between aquatic and terrestrial animals. Their skin performs versatile functions that are important for survival6 and possesses not only basic functions, such as acting as the boundary tissue, but also complex structures and adaptive functions. For example, the defensive barriers of amphibian skin are more complex than those of aquatic organisms2. The corneous cell envelope, which is an important composition of the amniote stratum corneum, initially occurred in the adult amphibian epidermis4. Homologous keratohyalin granules have also increased in the epidermis of amphibians7. Furthermore, amphibian skin has developed abundant glands (primarily including mucous and granular glands) that secrete diverse biologically active compounds for protection against predators or microorganisms8. The mucous glands usually secrete mucus containing a variety of mucins, which help defend against potential predators9. Different from the mucous glands, the granular glands are reservoirs of antimicrobial peptides (AMPs), essential molecules in innate immunity10, which provide an effective and fast-acting defense against microorganisms6. Additionally, amphibian skin also participates in respiration. In amphibians, at least 30 percent of the oxygen exchange and most or all of the carbon dioxide elimination occurs through the skin11. The skin of lungless salamanders (e.g., all species in Plethodontidae and caecilians) even serves as their only respiratory organ12.
Although adaptive functions of amphibian skin have been reported, the underlying molecular basis and functional mechanisms are still unclear. To date, several genes and AMPs have been identified in amphibian skin. For instance, genes of AMP families, such as the temporin13, esculetin14 and brevinin superfamilies15, have been discovered in different taxa. However, most of these researches have used traditional low-throughput methods, such as screening of presumed genes from cDNA libraries, on the basis of conserved regions of precursors. Recently, a high-throughput proteomic analysis on the skin of the Chinese giant salamander has identified several proteins involved in skin functions, including keratins, beta-actins, annexins, and respiration-related proteins16; however, this study was restricted to one species and was unable to comprehensively elucidate adaptive molecular evolution. Thus, it is urgent to utilize high-throughput techniques with ample species to explore the molecular basis of amphibian skin functions and adaptation from a holistic perspective.
Emerging transcriptome sequencing technology and bioinformatics tools are undoubtedly the most appropriate methods to address this problem. These tools can obtain the total RNA (including mRNA and noncoding RNA) transcripts from particular cells or tissues17. Although many studies have adopted this method to elucidate scientific questions, the existing research on non-model amphibians is insufficient. Most previous studies have focused only on particular development stages or specific tissues18,19. Species living in different environments evolve diverse skin phenotypes; therefore, comparing the expressed genes in the skin of different amphibians may help us understand the genetic basis of skin functions and adaptation. In this study, we sought to sequence and compare the skin transcripts of different species, to investigate the functions of amphibian skin, and to further reveal the evolutionary significance of this organ. Specifically, we collected seven species widely distributed in the Badagongshan Nature Reserve (Zhangjiajie, Hunan Province). These species belong to three families, Ranidae, Megophryidae and Rhacophoridae, which are semi-aquatic, terrestrial and arboreal, respectively. In the Ranidae family, Pelophylax nigromaculatus and Odorrana margaretae were selected. Leptobrachium boringii and Megophrys sangzhiensis from the Megophryidae family, and Rhacophorus omeimontis, R. dennysi and Polypedates megacephalus from the Rhacophoridae family were also chosen. By sequencing and analyzing the transcripts of skin samples from these species, we sought to acquire ample information about the genetic basis of skin functions and adaptation in amphibians.
Results
Illumina sequencing and de novo assembly
The seven species (four individuals per species) yielded a number of raw reads, ranging from 23,940,046 in R. omeimontis to 34,471,587 in R. dennysi (Table 1). After removing adapters, primers and low-quality reads, we obtained 19,792,130–33,156,716 clean reads (Table 1). These clean reads were subsequently assembled into transcripts. Finally, the transcripts were assembled into putative unigenes. Detailed information about the putative unigenes in seven species is shown in Table 1. To verify the assembly quality, we mapped the clean reads to the assembled unigenes and BLASTed the available protein sequences of a closely related species (Xenopus tropicalis) with assembled unigenes. The ranges of mapping rates and BLAST rates in seven species were 72.99–84.01% and 50.02–56.94%, respectively. The raw reads were available in the NCBI SRA browser (Bioproject accession number: SRR2768939), and the sequences of assemblies were deposited in the Transcriptome Shotgun Assembly (TSA) Database under accession numbers: GEGF00000000 for R. omeimontis, GEGG00000000 for R. dennysi, GEGH00000000 for P. megacephalus, GEGI00000000 for P. nigromaculatus, GEGJ00000000 for O. margaretae, GEGK00000000 for L. boringii and GEGL00000000 for M. sangzhiensis.
Table 1. Statistical summary of the assembled unigenes in the skin of seven frogs.
| Species | Raw reads | Clean reads | Total numbers | Min Length | Mean Length | Max Length | N50 | BLAST rates | Mapping rates |
|---|---|---|---|---|---|---|---|---|---|
| M. sangzhiensis | 33,897,689 | 32,703,903 | 48,794 | 201 | 868 | 15,756 | 1,652 | 55.32% | 84.01% |
| L. boringii | 30,103,264 | 29,086,874 | 49,267 | 201 | 840 | 18,108 | 1,578 | 56.94% | 80.23% |
| P. megacephalus | 31,428,218 | 30,529,842 | 69,425 | 201 | 699 | 17,281 | 1,142 | 51.29% | 80.42% |
| R. dennysi | 34,471,587 | 33,156,716 | 63,614 | 201 | 770 | 17,309 | 1,387 | 54.78% | 82.26% |
| R. omeimontis | 23,940,046 | 19,792,130 | 55,841 | 201 | 626 | 15,364 | 893 | 50.02% | 72.99% |
| O. margaretae | 24,211,333 | 19,888,107 | 54,093 | 201 | 687 | 15,166 | 1,046 | 51.82% | 81.41% |
| P. nigromaculatus | 32,146,280 | 31,173,113 | 61,691 | 201 | 690 | 15,507 | 1,127 | 54.80% | 78.71% |
Note: BLAST rates were generated by BLASTing the closely related X. tropicalis protein databases with the assembled unigenes. Mapping rates were generated by mapping clean reads to the assembled unigenes using the Bowtie mode of RSEM 1.2.0.
Functional annotation of the unigenes
The entire set of unigenes was annotated in seven frequently used databases, including the NCBI non-redundant (Nr) protein and nucleotide (Nt) databases, the Protein family (Pfam) database, the Cluster of Orthologous Groups of proteins (KOG/COG) database, the SwissProt protein database, the Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog (KO) database, and the Gene Ontology (GO) database. Among the total unigenes, approximately 42.99% to 50.38% had been annotated in at least one database according to BLAST searches (Table 2).
Table 2. Numbers of unigenes annotated in the skin of the seven anuran species against seven databases.
| Megophryidae |
Rhacophoridae |
Ranidae |
|||||
|---|---|---|---|---|---|---|---|
| M. sangzhiensis | L. boringii | P. megacephalus | R. dennysi | R. omeimontis | O. margaretae | P. nigromaculatus | |
| NR | 21,500(44.06%) | 21,728(44.1%) | 27,028(38.93%) | 23,269(36.57%) | 24,514(43.89%) | 22,532(41.65%) | 23,069(37.39%) |
| NT | 10,875(22.28%) | 10,935(22.19%) | 11,609(16.72%) | 11,270(17.71%) | 11,226(20.1%) | 11,055(20.43%) | 11,528(18.68%) |
| KO | 11,318(23.19%) | 11,237(22.8%) | 13,742(19.79%) | 11,847(18.62%) | 12,912(23.12%) | 11,865(21.93%) | 11,829(19.17%) |
| SP | 19,873(40.72%) | 19,924(40.44%) | 23,936(34.47%) | 21,008(33.02%) | 22,424(40.15%) | 20,670(38.21%) | 20,989(34.02%) |
| Pfam | 16,476(33.76%) | 16,590(33.67%) | 21,712(31.27%) | 18,438(28.98%) | 17,171(30.74%) | 16,747(30.95%) | 17,432(28.25%) |
| GO | 18,776(38.48%) | 18,874(38.3%) | 24,555(35.36%) | 20,818(32.72%) | 20,510(36.72%) | 19,540(36.12%) | 20,209(32.75%) |
| KOG | 11,083(22.71%) | 11,008(22.34%) | 13,810(19.89%) | 11,356(17.85%) | 11,536(20.65%) | 10,970(20.27%) | 11,000(17.83%) |
| one | 24,583(50.38%) | 24,749(50.23%) | 32,176(46.34%) | 27,352(42.99%) | 27,657(49.52%) | 25,908(47.89%) | 27,182(44.06%) |
| Total | 48,794(100%) | 49,267(100%) | 69,425(100%) | 63,614(100%) | 55,841(100%) | 54,093(100%) | 61,691(100%) |
The percentages of unigenes annotated against each database among all unigenes are shown in parentheses.
Note: SP, SwissProt; one indicates unigenes annotated in at least one database.
To identify the functional distribution of unigenes, we collected GO annotations for seven species. The annotated unigenes ranged from 18,776 in M. sangzhiensis to 24,555 in P. megacephalus (Table 2). Intriguingly, the distribution of annotated unigenes at level two GO terms in the seven species showed highly similar patterns (Fig. 1). All of the unigenes annotated in the GO database were classified into three main categories (Fig. 1). Among them, “biological processes” contained the largest numbers of annotated unigenes, followed by “cellular components” and “molecular functions”. Within the biological processes, “cellular process” and “metabolic process” were the two most common GO terms. For the cellular components category, most of the unigenes were related to “cell part” and “cell”. In the category of molecular functions, unigenes were mostly involved in “binding”, followed by “catalytic activity” (Fig. 1). These highly enriched GO terms mainly referred to the maintenance of the basic regulation and metabolic functions of amphibian skin. Additionally, GO terms related to special skin functions, such as “response to stimulus” in the biological process category and “oxidoreductase activity” in the molecular function category, were also enriched. To further annotate the gene functions, we analyzed the GO annotations at level three. “Oxidoreductase activity” accounted for 16.9–18.6% of unigenes in the seven anurans.
Figure 1. Histogram of the GO annotations of unigenes in seven species.

The x-axis represents GO terms belonging to three categories, and the y-axis represents gene percentages of each term. Bars with different colors represent different species.
In addition to the GO annotation, we mapped the unigenes in the KOG categories and found similar distribution patterns across the seven anurans. The percentage of annotated unigenes in the “general function prediction only” (“R” term) and “signal transduction mechanisms” (“T” term) categories were far higher than those in the other categories, containing more than 15% of the unigenes (Fig. 2). Moreover, several terms relating to skin-specific functions, such as intracellular trafficking, secretion, and vesicular transport (U) and defense mechanisms (V), were also enriched (Fig. 2).
Figure 2. KOG annotation results of the skin of seven anuran species.

The capital letters on the x-axis symbolize 26 biological processes, as shown on the right side, and the y-axis represents the gene percentage of each process. Bars with different colors represent different species.
Additionally, we performed KEGG pathway analysis to understand the biological functions and interactions among gene products. Unigenes annotated in the Nr and Pfam databases were searched in the KEGG database. Five types of hierarchy one pathways were involved in the KEGG annotation in the seven species (Fig. 3). Among them, the “organismal systems” pathway, including the categories “circulatory system”, “development”, “digestive system”, “endocrine system”, “environmental adaptation”, “excretory system”, “immune system”, “nervous system”, and “sensory system”, contained the largest number of annotated sequences. In hierarchy two pathways, most of the unigenes were annotated in signal transduction pathways across seven species. Notably, the PI3K-Akt signaling pathway (ko: 04151, Supplementary Fig. S1) was the top enriched pathway in all seven species.
Figure 3. KEGG pathway annotation for the skin of seven anuran species.
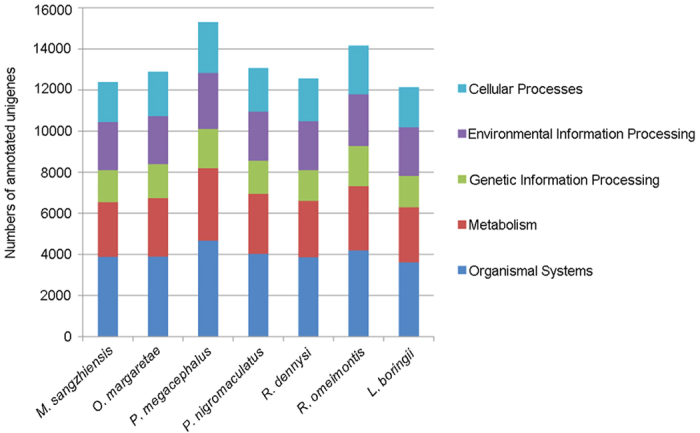
Bars with different colors represent five pathways in KEGG annotations. The x-axis represents the seven species, and the y-axis represents the total annotated transcript numbers.
The immunome and antimicrobial peptides (AMPs) of seven anurans
For amphibian skin, immune functions, especially AMP secretion, are among the most important functions. KEGG pathway analysis also revealed the highly enriched immune-related pathways in seven species. Therefore, we sought to explore the molecular basis of skin immune function in amphibians. First, the unigenes in immune system processes (GO annotation) were counted in the seven species (Table 3). Then, the differences in numbers of unigenes annotated in each process were evaluated among three families by using the analysis of variance (ANOVA). Among the 16 processes of the immune system (level two GO categories), “immune system development”, “regulation of immune system process” and “immune response” contained the three largest amounts of unigenes (Table 3), indicating their important roles in the immune system. For the differences among the three families, the numbers of unigenes annotated in “activation of immune response” (level two category) and “immune response-activating signal transduction” (level three category) differed significantly among the three families, with ANOVA F = 8.399, P = 0.037 and ANOVA F = 10.156, P = 0.027, respectively. Second, all unigenes were mapped in the database of anuran defense peptides (DADP) to identify AMPs across seven species. In total, 108 putative AMPs were identified in these skin samples. The numbers of AMP unigenes identified in single species varied from 4 in L. boringii to 28 in O. margaretae (Table 4). Notably, the midkine family was present in the skin of all anurans, thus indicating its potential role in defense against microorganisms. The overall types of AMPs showed differences among the three families, but these differences were marginally non-significant (ANOVA F = 6.094, P = 0.061), and species in the Ranidae family produced more AMPs than did species in the other families.
Table 3. Numbers of unigenes annotated for immune functions and their percentages in all processes in the seven anuran species.
| Immune system process | Megophryidae |
Rhacophoridae |
Ranidae |
||||
|---|---|---|---|---|---|---|---|
| M. sangzhiensis | L. boringii | P. megacephalus | R. dennysi | R. omeimontis | O. margaretae | P. nigromaculatus | |
| Immune system development | 90(52.02%) | 81(56.64%) | 95(52.49%) | 90(56.60%) | 101(56.42%) | 89(57.42%) | 88(62.86%) |
| Regulation of immune system process | 84(48.55%) | 79(55.24%) | 91(50.28%) | 80(50.31%) | 94(52.51%) | 77(49.68%) | 73(52.14%) |
| Immune response | 69(39.88%) | 55(38.46%) | 81(44.75%) | 63(39.62%) | 67(37.43%) | 55(35.48%) | 52(37.12%) |
| Positive regulation of immune system process | 55(31.79%) | 43(30.07%) | 54(29.83%) | 48(30.19%) | 46(25.70%) | 39(25.16%) | 43(30.71%) |
| Leukocyte activation | 53(30.64%) | 33(23.08%) | 59(32.60%) | 55(34.59%) | 53(29.61%) | 38(24.52%) | 43(30.71%) |
| Immune effector process | 36(20.81%) | 23(16.08%) | 40(22.10%) | 30(18.87%) | 35(19.55%) | 28(18.06%) | 26(18.57%) |
| Activation of immune response | 31(17.92%) | 24(16.78%) | 28(15.47%) | 22(13.84%) | 24(13.41%) | 20(12.90%) | 20(14.29%) |
| Myeloid cell homeostasis | 19(10.98%) | 19(13.29%) | 20(11.05%) | 20(12.58%) | 25(13.97%) | 21(13.55%) | 24(17.14%) |
| Negative regulation of immune system process | 13(7.51%) | 18(12.59%) | 12(6.63%) | 12(7.55%) | 12(6.70%) | 12(7.75%) | 18(12.86%) |
| Leukocyte migration | 14(8.09%) | 11(7.69%) | 11(6.08%) | 11(6.92%) | 13(7.27%) | 11(7.10%) | 14(10.00%) |
| Production of molecular mediator of immune response | 11(6.36%) | 8(5.59%) | 10(5.52%) | 8(5.03%) | 10(5.59%) | 9(5.81%) | 10(7.14%) |
| Antigen processing and presentation | 10(5.78%) | 3(2.10%) | 7(3.87%) | 6(3.77%) | 5(2.79%) | 6(3.87%) | 5(3.57%) |
| Somatic diversification of immune receptors | 7(4.05%) | 3(2.10%) | 7(3.87%) | 5(3.14%) | 9(5.03%) | 6(3.87%) | 8(5.71%) |
| Leukocyte homeostasis | 6(3.47%) | 3(2.10%) | 3(1.66%) | 3(1.89%) | 4(2.23%) | 6(3.87%) | 3(2.14%) |
| T cell selection | 4(2.31%) | 2(1.40%) | 3(1.65%) | 3(1.89%) | 0 | 1(0.65%) | 1(0.71%) |
| Tolerance induction | 1(0.58%) | 0 | 1(0.55%) | 1(0.63%) | 3(1.68%) | 2(1.29%) | 0 |
| All | 173 | 143 | 181 | 159 | 179 | 155 | 140 |
Table 4. Numbers of AMP transcripts in the seven anuran species.
| AMP families | Megophryidae |
Rhacophoridae |
Ranidae |
||||
|---|---|---|---|---|---|---|---|
| M. sangzhiensis | L. boringii | P. megacephalus | R. dennysi | R. omeimontis | O. margaretae | P. nigromaculatus | |
| Antimicrobial peptide precursor | 1(1) | ||||||
| Andersonin | 4(1) | 8(1) | 2(1) | 4(1) | |||
| Bombesin | 1(1) | 1(1) | 1(1) | ||||
| Brevinin | 1(1) | 1(1) | 1(1) | ||||
| Bradykinin | 2(2) | ||||||
| Cathelicidin | 2(1) | 1(1) | 3(2) | 4(3) | 3(3) | 4(4) | |
| Esculentin | 1(1) | 1(1) | 2(2) | 1(1) | 2(2) | 1(1) | |
| Immuno regulatory | 1(1) | ||||||
| Lividin | 1(1) | 1(1) | |||||
| Midkine | 1(1) | 1(1) | 1(1) | 1(1) | 1(1) | 1(1) | 1(1) |
| Nigrocin | 1(1) | 1(1) | 1(1) | 1(1) | 1(1) | ||
| Odorranain | 1(1) | 8(6) | 1(1) | ||||
| Palustrin | 1(1) | 1(1) | 2(2) | 1(1) | |||
| Polypedatein | 2(1) | ||||||
| Pelophylaxin | 1(1) | 1(1) | 1(1) | 1(1) | 1(1) | 1(1) | |
| Pancreatic | 2(2) | 1(1) | 1(1) | 1(1) | |||
| Pleurain | 1(1) | ||||||
| Peptide YY | 1(1) | 1(1) | 2(1) | 1(1) | 1(1) | ||
| Serine protease inhibitor | 1(1) | 1(1) | |||||
| Uncharacterized | 1(1) | 1(1) | 1(1) | 1(1) | |||
| All | 11(10) | 4(4) | 15(12) | 24(14) | 8(6) | 28(23) | 18(18) |
The category of each AMP is shown in parentheses.
Quantification of gene expression in seven anurans
To quantify the expressed levels of unigenes, we separately calculated the FPKM (fragments per kb per million reads) values and ranked the values from high to low in seven species (Supplementary Dataset 1). For O. margaretae and P. nigromaculatus, which belong to the Ranidae family, AMP unigenes (such as esculentin-2-MG1, esculentin-1-MG1, and pelophylaxin-2 protein precursor) were expressed with extremely high FPKM values (Supplementary Dataset 1). However, similar results were not found for species in the other two families (Supplementary Dataset 1). Furthermore, unigenes of keratin-associated proteins (type I and II) were extremely highly expressed in the skin of the Rhacophoridae and Megophryidae families, but the expression levels of the same unigenes were relatively low in species of the Ranidae family. Additionally, the oxidative cytochrome c oxidase 1 (COX1) unigenes were most highly expressed in the skin of only the Megophryidae family, and ferritin genes were most highly expressed in the skin of species from both the Rhacophoridae and Ranidae families. Next, we searched these special unigenes in the GO annotation results and found that they mainly involved “metabolic process”, “binding”, “response to stimulus”, “catalytic activity”, and “enzyme regulator activity”. The AMPs were involved in “defense response” (level four), “response to stress” (level three), and “response to bacterium” (level three), whereas keratins were involved in the level four GO term “response to wounding” and the level three GO terms “response to stress” and “cytoskeletal protein binding”. Both COX1 and ferritin were involved in “oxidoreductase activity”. The most highly expressed genes were related to the respiration (“catalytic activity”), protection (“response to wounding”) and immunity (“response to stimulus”) functions of amphibian skin.
Selection detection
A total of 3,280 orthologous groups (OGs) were identified across seven species. Then, a phylogenetic tree of these OGs was constructed to detect selective genes (Supplementary Fig. S2). Seven OGs were detected under strong positive selection, with false discovery rate (FDR) corrected q values lower than 0.05 (Table 5). Subsequently, we annotated the protein functions of these OGs against the Nr database and found that these OGs were predicted to be TCEA [transcription elongation factor A (SII)], ATPIF1 (ATPase inhibitory factor 1), EIF3C (eukaryotic translation initiation factor 3 subunit C), ANXA1 (annexin A1), CAPZB (F-actin capping protein subunit beta 2), CD9 (CD9 antigen) and putative RPL26L1 (ribosomal protein l26-like 1). Furthermore, we searched these seven OGs against the KEGG database to annotate the pathways in which they are involved. Only three pathways were predicted: ribosomes (ko: 03010), hematopoietic cell lineage (ko: 04640), and RNA transport (ko: 03013). Among them, the hematopoietic cell lineage pathway, in which the CD9 antigen gene is predicted to function (Supplementary Fig. S3), is involved in the KEGG (hierarchy two) immune system pathway.
Table 5. Seven OGs under strong positive selection in the seven anuran species identified in Site Models of PAML 4.7.
| OGs ID | M. sangzhiensis | O. margaretae | P. megacephalus | P. nigromaculatus | R. dennysi | R. omeimontis | L. boringii | Corrected q value | NR annotation |
|---|---|---|---|---|---|---|---|---|---|
| OG02954 | MsS|comp35721_c0 | OmS|comp91984_c0 | PmS|comp62038_c0 | PnS|comp51049_c0 | RdS|comp53254_c0 | RoS|comp205006_c0 | VbS|comp39811_c0 | 0 | eIF3C |
| OG04000 | MsS|comp24234_c0 | OmS|comp60913_c0 | PmS|comp57210_c0 | PnS|comp40616_c0 | RdS|comp32925_c0 | RoS|comp199691_c0 | VbS|comp32105_c0 | 0.003 | CAPZB |
| OG04340 | MsS|comp13846_c0 | OmS|comp76568_c0 | PmS|comp14060_c0 | PnS|comp46670_c2 | RdS|comp57643_c0 | RoS|comp153345_c0 | VbS|comp35227_c0 | 0.007 | CD9 |
| OG05658 | MsS|comp29431_c0 | OmS|comp41010_c0 | PmS|comp47485_c0 | PnS|comp45618_c0 | RdS|comp38617_c0 | RoS|comp153557_c1 | VbS|comp33211_c0 | 0.042 | ANXA1 |
| OG05703 | MsS|comp36086_c0 | OmS|comp91549_c0 | PmS|comp54574_c0 | PnS|comp40281_c0 | RdS|comp45282_c0 | RoS|comp202769_c0 | VbS|comp35512_c0 | 0.005 | TCEA |
| OG06215 | MsS|comp13717_c0 | OmS|comp41023_c0 | PmS|comp35291_c0 | PnS|comp39934_c0 | RdS|comp33168_c0 | RoS|comp190201_c0 | VbS|comp20632_c0 | 0.008 | ATPIF 1 |
| OG06474 | MsS|comp17232_c0 | OmS|comp105337_c0 | PmS|comp60614_c5 | PnS|comp21297_c0 | RdS|comp57304_c0 | RoS|comp153994_c0 | VbS|comp41648_c0 | 0.005 | Putative RPL26L1 |
Note: TCEA, transcription elongation factor A (SII); ATPIF1, ATPase inhibitory factor 1; EIF3C, eukaryotic translation initiation factor 3 subunit C; ANXA1, annexin A1; CAPZB, F-actin capping protein subunit beta 2; CD9, CD9 antigen; and putative RPL26 L1, putative ribosomal protein l26-like 1.
Discussion
Exploring the molecular basis of skin function and adaptation in amphibians is essential to understand the survival and evolutionary mechanisms of amphibians and amniotes. In this study, we generated extensive transcriptome datasets of the skin in seven anurans, compared the unigene populations and expression patterns, and finally inferred the candidate genes involved in skin adaptive evolution. By sampling the skin of four individuals per species and sequencing their transcriptomes, we acquired more than 20 million reads for each species and over 210 million reads for the skin of seven species. This large dataset allowed us to examine the skin function of amphibians in detail and to identify novel genes and proteins involved in skin adaptation.
According to the functional annotation results, unigenes that participated in skin’s basic biological and metabolic processes were largely enriched. In the level two GO categories of cellular component and biological process, most of the unigenes were involved in maintaining basic biological functions, such as “cell part” and “metabolic process”, indicating the basic functions of amphibian skin. In molecular function terms, many of the identified unigenes participate in “binding” processes, including ion binding, heterocyclic compound binding, and organic cyclic compound binding, thus implying that amphibian skin is active in synthesizing the proteins involved in these processes. Similar findings have been reported in Chinese brown frogs20. Furthermore, unigenes of the ribosomal proteins were highly expressed in the skin of all seven frogs. Ribosome proteins (40S and 60S) are similarly abundantly expressed in the skin of channel catfish21 and sheep22. The abundant expression of these ribosomal protein genes is presumably due to increased peptide synthesis.
In addition to its basic survival functions, amphibian skin has evolved complex structures and functions that allow amphibians to live between water and land2. The skin acts as the first-line defensive barrier against harmful external factors. In this study, two types of keratins (type I and II) were identified on the basis of their high expression levels in species of the Megophryidae and Rhacophoridae families, thus indicating their important biological functions. In GO annotation, keratin unigenes were annotated as “response to stress” and “response to wounding processes”. In addition, previous studies have recorded that keratins are the most versatile, plentiful constituents involved in cell binding, adhesion, and signal transport functions23. These proteins may also play general roles in skin integrity and disease defense24. Therefore, we presumed that the keratins in amphibian skin play an essential role in protection, and further studies on these types of genes are needed.
Furthermore, amphibian skin has developed functions in immunity to defend against various external microbes. As the annotation results showed, the abundant processes related to “response to stimulus” of the biological process term in the GO classification, “defense mechanisms (V)” in the KOG annotation, and “immune system” of the organismal systems term in the KEGG pathways demonstrated the functions of immunity and defense in anuran skin, although the expression levels of these genes were not the highest among the studied genes. This phenomenon is plausible because the immune system involves many types of tissues, such as the thymus, spleen and skin, and the number of contigs annotated for the immune system function in the skin occupies a relatively small proportion (28.48%) of the total contigs in immune tissues19. In addition, the immune-related PI3K-Akt signaling pathway, which plays critical roles in immune cell development25, was the top enriched pathway in the seven anurans. Therefore, we presumed that immune cell development is a common process in amphibian skin. The skin’s immune functions are executed mostly by AMPs, which are secreted and released by skin granular glands and protect organisms against harmful pathogens6. Because of their important role in immunity, AMPs have been characterized across diverse amphibians through traditional low-throughput methods6,13,14,15. However, these methods are limited and are not comprehensive, owing to the presumably targeted AMP genes. In this study, a high-throughput transcriptome sequencing method was used to identify as many AMPs as possible at one time. Overall, we identified 108 AMP unigenes belonging to 19 families. Among these unigenes, only the MDK (midkine) family was identified in all the seven species. MDK was first characterized in murine carcinoma cells, and it plays important roles in the release of growth factors during inflammation26. Its ortholog was also present in X. laevis27 but was not present in other amphibians. Thus, this study provided evidence that MDK exists in the skin of these seven non-model species; further research on its potential functions is needed.
Respiration is another important function of amphibian skin11,12. As shown in this study, the process of “catalytic activity” was detected in the seven frog skin samples according to the GO annotation at level two. In this activity, oxidoreductases, such as mitochondrial complex I: NADH-ubiquinone oxidoreductase, have been represented as an important enzyme type in the respiratory chain28. Moreover, three types of oxidoreductase activity genes [COX1, COX3 (cytochrome c oxidase 3) and GAPDH (glyceraldehydes-3-phosphate dehydrogenase)] possess relatively high FPKM values in Ranidae and Megophryidae species. It has been reported that the mitochondrial COX proteins are important electron-driven proton pumps and participate in cell respiration and aerobic energy metabolism29. In addition, recent studies on the proteome of the Chinese giant salamander have identified several proteins involved in respiration, such as NADH-ubiquinone oxidoreductase chain 5, cytochrome b and ATPase subunit 616. Therefore, we suggest that future research on the relationships among these genes is necessary to elucidate the molecular mechanism of skin respiration in amphibians.
To deduce the adaptability of skin, we compared the similarities and differences in the unigene expression levels among species from three families. The types of highly expressed genes were different but showed similar functions among the three families. Cystatins (CSTs) and AMPs, which were, highly expressed in species of Rhacophoridae and Ranidae, respectively, are all related to immune functions. An earlier report has noted that CSTs, including cystatin and stefin, have important physiological functions in the immune system, including stabilizing host-parasite interactions and defending against pathogens30. Similarly, both COX genes and ferritin subunits, which were highly expressed in species of Megophryidae and the other two families, respectively, are involved in the GO term “oxidoreductase activity”, indicating their similar roles in respiration. Therefore, all species exhibited highly expressed genes in defense, immune functions, and respiration, but each exhibited different types of genes and proteins. It has been noted that frogs’ phenotypes are correlated with their ecological habitats31; thus, we hypothesized that these discrepancies among families may be linked to their diverse habitats. Megophryidae, Rhacophoridae and Ranidae species live in terrestrial, arboreal, and semi-aquatic habitats, respectively. Semi-aquatic species are exposed to more pathogens than terrestrial and arboreal species because water often serves as a transmission medium for pathogens such as the zoospore of Batrachochytrium dendrobatidis32. Hence, AMPs were highly expressed in the skin of only the Ranidae family species (O. margaretae and P. nigromaculatus), and the AMP categories also suggested that the Ranidae family expressed and produced more antimicrobial proteins than the other two families did. Nevertheless, this finding does not mean that terrestrial and arboreal species lack immune defense capacity; instead, they have evolved with different protection mechanisms to adapt to complex environments. As shown in the FPKM ranking of seven species, keratins, which are associated with maintaining cell binding and adhesion23, were all highly expressed in Megophryidae and Rhacophoridae species. Tight cytoskeletal structures protect organisms against microorganisms and help heal wounds. The high expression of keratin genes in Megophryidae and Rhacophoridae species indicates that these species possess strong skin defense functions that differ from those of Ranidae species. Furthermore, for semi-aquatic species of Ranidae, two types of genes related to respiration were highly expressed, thus suggesting that these species require these highly expressed genes to ensure skin respiration, especially when they reside in water with low oxygen concentrations. Thus, these different patterns of highly expressed genes among families may tightly correlate with their habitats.
During evolution, positive selection, which suggests an excess of high-frequency variants leading to changes in encoded proteins, is a frequent event for the formation of new genes and functions33. Recently, high-throughput transcriptome sequencing has offered a new technique for discovering positively selected genes; many genes have been identified through this method33,34. In this study, seven OGs exhibited strong positive selection, with functions involving biosynthesis, metabolism, immunity and defense processes. Specifically, TCEA, EIF3C and RPL26 all play important roles in translation and protein synthesis35,36,37, whereas ATPIF1 is an essential element regulating ATP synthesis, hydrolysis, and energy metabolism38. In immunity and defense processes, three proteins were identified to be positively selected: CAPZB, ANXA1 and CD9. CAP (F-actin capping protein) is an actin-binding protein that regulates the function of the actin system and maintains cytoskeletal organization39. This protein affects the length and number of actin filaments via binding to the barbed end of actin filaments39. Therefore, CAP is involved in skin structure maintenance and stable wound healing. ANXA1 might effectively mediate the anti-inflammatory activity of glucocorticoids, thereby affecting the innate and adaptive immune systems40. These two proteins are both involved in maintaining the defense barrier functions of skin. CD9 was originally described as a surface marker of leukemia and lymphohematopoietic cells; it participates in T cell activation, which serves as the initiation process of the immune response41. A recent study has indicated that this marker may be involved in the anti-bacterial immune response in turtles42. KEGG pathway annotation also indicated that CD9 participates in the hematopoietic cell pathway of the immune system (as shown in Supplementary Fig. S3). Therefore, it is likely that these positively selected genes, which are involved in biosynthesis, metabolism, immunity, and defense processes, are targets of adaptive molecular evolution. New traits resulting from changes in these proteins may allow species to adapt to their habitats34.
In summary, we sequenced the skin transcriptomes of seven species belonging to three families to explore the molecular mechanisms and adaptive evolution of amphibian skin. Many genes and proteins involved in basic and specific skin functions were identified. The highly expressed genes in different species were related to their corresponding habitats. Species living in different environments may evolve diverse adaptive mechanisms with different molecular bases. Seven OGs involved in biosynthesis, metabolism, immunity, and defense processes were presumed to be candidates of adaptive molecular evolution. This study provides molecular data for exploring the mechanisms of skin function in amphibians and may serve as a guide for researchers studying the adaptation of skin at the molecular level.
Methods
Ethics statement
All samples used in this study were collected with the permission of the management bureau of the Badagongshan National Nature Reserve. The animal experiments were performed under an animal ethics approval granted by the Central China Normal University (CCNU).
Sample collection and RNA extraction
A total of 28 frogs belonging to the three families were collected from Badagongshan National Nature Reserve (with permission) in Hunan province during the 2013 breeding season. They were separately placed in single plastic boxes (173 × 115 × 68 mm) containing the same plant leaves and water as their natural environment, and they were brought back to the laboratory. The living frogs were then sacrificed and dissected with scissors soaked in a 0.1% diethyl pyrocarbonate (DEPC) solution in the laboratory. For each species, the skin tissue was carefully removed from the underlying tissues and flash-frozen in liquid nitrogen until needed. These experiments were performed in accordance with relevant animal ethics guidelines and regulations granted by CCNU. To eliminate the individual differences and to identify as many expressed genes as possible, we mixed skin tissues from four individuals (two female and two male) per species and extracted the total RNA using an RNA extraction kit (Omega Bio-Tek, USA) together with TRIzol (Invitrogen, USA). RNA degradation and contamination was assessed with 1% agarose gels. The purity, on the basis of optical densities of the samples, was examined using a NanoPhotometer® spectrophotometer (Implen, USA). RNA concentration was measured with a Qubit® RNA Assay Kit with a Qubit® 2.0 Fluorometer (Life Technologies, USA). The RNA integrity was assessed using an RNA Nano 6000 Assay Kit with a Bioanalyzer 2100 system (Agilent Technologies, USA). Additionally, to measure the RNA quality of each sample, the RNA Integrity Number (RIN) was calculated43. All seven samples had RIN values larger than 7.8 (7.8–9.4), indicating the high quality of the input RNA samples.
cDNA library construction and sequencing
A total amount of 3 μg of RNA per species was used to construct sequencing libraries. The mRNAs were enriched with oligo - (dT) beads and were then broken into short fragments with NEBNext First Strand Synthesis Reaction Buffer (5×). These fragments were subsequently used as templates to synthesize the first-strand and second-strand cDNAs. After adenylating the 3′ ends of the cDNA fragments, NEBNext Adaptors were ligated to the cDNA fragments. The cDNA fragments of 150–200 bp were preferentially purified with an AMPure XP system (Beckman Coulter, USA). Subsequently, the purified adaptor-ligated cDNAs were successively treated with USER Enzyme (NEB, USA) at 37 °C for 15 min and 95 °C for 5 min. Then, PCR was conducted using Phusion High-Fidelity DNA polymerase, universal PCR primers and an Index (X) primer. PCR products were purified using an AMPure XP system, and the library quality was evaluated using an Agilent Bioanalyzer 2100 system. Subsequently, the cDNA samples were clustered on a cBot Cluster Generation System using a TruSeq PE Cluster Kit (Illumina, USA). After cluster generation, the library preparations were sequenced on a HiSeqTM 2000 platform (Illumina, USA), and the stranded paired-end reads were generated.
Sequence assembly and annotation
All of the raw data were initially processed using in-house Perl scripts (Supplementary Methods). Prior to assembly, clean reads were obtained by removing the sequencing adapters, primers, and low-quality reads (reads including bases greater than 50% with a Q-value ≤ 5 and ambiguous bases (N) more than 10%). The clean reads were assembled de novo in Trinity (r2012-10-05)44 with 2 for the min_kmer_cov setting and default settings for all other parameters. The unigenes were identified as the longest transcript for each gene with in-house Perl scripts to avoid redundant transcripts (Supplementary Methods). Unigenes generated by assembly were subsequently used for downstream analyses.
All assembled unigenes were annotated using NCBI-BLAST-2.2.28 + with E-values ≤ 1E-5 in seven databases, including the Nr and Nt databases in NCBI (http://www.ncbi.nlm.nih.gov), the Pfam database (http://en.wikipedia.org/wiki/Protein_family), the KOG (http://www.ncbi.nlm.nih.gov/COG), the SwissProt protein database (http://www.expasy.ch/sprot), the KO database (http://www.genome.jp/kegg/pathway.html), and the GO database (http://www.geneontology.org/). To assess the quality of assembled reads, we mapped the total clean reads of seven species to the assembled unigenes using the Bowtie mode of RSEM 1.2.045,46. Furthermore, we BLASTed the available protein sequences of X. tropicalis (ftp://ftp.ensembl.org/pub/release-75/fasta/xenopus_tropicalis/) with our assembled unigenes using NCBI-BLAST-2.2.28 + with E-values ≤ 1E-5. Additionally, we performed GO annotation47 to classify the functions of our unigenes utilizing the Blast2GO pipelines48 with E-values ≤ 1E-3, retaining the top 20 hits. Then, we searched against the eukaryotic KOG database49 to annotate the unigenes with E-values ≤ 1E-5. Additionally, we searched sequences against the KEGG database to understand the functions and pathway annotations using KOBAS 2.050.
AMP identification
The AMPs were identified by scanning the database of anuran defense peptides (DADP)51 using NCBI-BLAST-2.2.28 + with E-values ≤ 1E-5. Additionally, AMPs that were not found in the DADP database but that have been described in the literature for amphibians, such as the cathelicidin family52, were set as keywords when searching against the Nr database. Furthermore, the keyword “andersonin-9” (GenBank: GU134093.1), which is a type of AMP found in Odorrana andersonii and our species but was absent in the DADP database, was also included. The types of AMPs were determined by the Nr annotation and DADP scanning results. Then, we assessed the significant differences in the categories of AMPs of frogs in the three families using one-way ANOVA analysis.
Quantification of gene expression levels
We used the software package RSEM 1.2.0 to estimate the gene expression levels for each species. First, read_count values of all genes were obtained by mapping clean data back onto the transcripts. Then, we calculated the FPKM values, the most common method to estimate gene expression levels53. After estimating the FPKM values, we searched the Nr database to predict proteins and domains and then annotated the biological functions using GO classifications. The FPKM values were ranked from high to low for each species.
Selection analyses
To recognize the molecular basis of adaptive selection, we conducted selection detection across all orthologous genes. OrthoMCL 2.0.354, which is based on protein similarity graph methods, was used to identify putative OGs from assembled sequences among the seven species. To identify positively selected genes, we reconstructed the orthologous genes tree using maximum likelihood (ML) methods in PhyML 3.055. The outgroups were X. tropicalis and Latimeria chalumnae. Site models in the “Codeml” application of PAML 4.756 were then used to detect the positively selected genes among putative OGs of the seven species. Generally, q values that were corrected by FDRs lower than 0.05 indicated significant evidence of positive selection.
Additional Information
How to cite this article: Huang, L. et al. Comparative transcriptome analyses of seven anurans reveal functions and adaptations of amphibian skin. Sci. Rep. 6, 24069; doi: 10.1038/srep24069 (2016).
Supplementary Material
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 31201713, No. 31270425 and No. 31470442) and research funds from CCNU (No. CCNU14B01001).
Footnotes
Author Contributions H.W. designed the research and provided financial support. L.H. and J.L. performed the experiments, analyzed and interpreted the data. L.H., J.L. and H.W. wrote the manuscript. H.A. collected samples and conducted a portion of the laboratory work. Z.L. and M.Z. supervised the study and revised the manuscript. All authors read and approved the final manuscript.
References
- Laughlin S. B. The role of sensory adaptation in the retina. J Exp Biol. 146, 39–62 (1989). [DOI] [PubMed] [Google Scholar]
- Chuong C. M. et al. What is the ‘true’ function of skin? Exp Dermatol. 11, 159–187 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts R. O. & Francoeur M. L. Lipid biophysics of water loss through the skin. Proc Natl Acad Sci USA 87, 3871–3873 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alibardi L. Adaptation to the land: the skin of reptiles in comparison to that of amphibians and endotherm amniotes. J. Exp Zool. 298, 12–41 (2003). [DOI] [PubMed] [Google Scholar]
- Romanovsky A. A. Skin temperature: its role in thermoregulation. Acta Physiol. 210, 498–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. & Lai R. The chemistry and biological activities of peptides from amphibian skin secretions. Chem Rev. 115, 1760–1846 (2015). [DOI] [PubMed] [Google Scholar]
- Alibardi L. Keratinization in the epidermis of amphibians and the lungfish: comparison with amniote keratinization. Tissue Cell. 33, 439–449 (2001). [DOI] [PubMed] [Google Scholar]
- Simmaco M., Mignogna G. & Barra D. Antimocrobial peptides from amphibian skin: what do they tell us? Peptide Science. 47, 435–450 (1998). [DOI] [PubMed] [Google Scholar]
- Toledo R. D. & Jared C. Cutaneous granular glands and amphibian venoms. Comp Biochem Physiol. 111, 1–29 (1995). [Google Scholar]
- Rinaldi A. C. Antimicrobial peptides from amphibian skin: an expanding scenario: Commentary. Curr Opin Chem Biol. 6, 799–804 (2002). [DOI] [PubMed] [Google Scholar]
- Jørgensen C. B. Amphibian respiration and olfaction and their relationships: from Robert Townson (1794) to the present. Biol Rev. 75, 297–345 (2000). [DOI] [PubMed] [Google Scholar]
- Nussbaum R. A. & Wilkinson M. A new genus of lungless tetrapod: a radically divergent caecilian (Amphibia: Gymnophiona). Proc Roy Soc Lond B Biol. 261, 331–335 (1995). [Google Scholar]
- Ohnuma A. et al. Antimicrobial peptides from the skin of the Japanese mountain brown frog Rana ornativentris: evidence for polymorphism among preprotemporin mRNAs. Peptides. 28, 524–532 (2007). [DOI] [PubMed] [Google Scholar]
- Reshmy V., Preeji V., Parvin A., Santhoshkumar K. & George S. Three novel antimicrobial peptides from the skin of the Indian bronzed frog Hylarana temporalis (Anura: Ranidae). J. Pept Sci. 17, 342–347 (2011). [DOI] [PubMed] [Google Scholar]
- Wang H. et al. Novel antimicrobial peptides isolated from the skin secretions of Hainan odorous frog. Odorrana hainanensis. Peptides. 35, 285–290 (2012). [DOI] [PubMed] [Google Scholar]
- Geng X. et al. Proteomic analysis of the skin of Chinese giant salamander (Andrias davidianus). J. Proteomics. 119, 196–208 (2015). [DOI] [PubMed] [Google Scholar]
- Morozova O., Hirst M. & Marra M. A. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genom Hum G. 10, 135–151 (2009). [DOI] [PubMed] [Google Scholar]
- Eo S. H. et al. Comparative transcriptomics and gene expression in larval tiger salamander (Ambystoma tigrinum) gill and lung tissues as revealed by pyrosequencing. Gene. 492, 329–338 (2012). [DOI] [PubMed] [Google Scholar]
- Savage A. E., Kiemnec-Tyburczy K. M., Ellison A. R., Fleischer R. C. & Zamudio K. R. Conservation and divergence in the frog immunome: pyrosequencing and de novo assembly of immune tissue transcriptomes. Gene. 542, 98–108 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang Z. et al. Transcriptome analysis and identification of genes related to immune function in skin of the Chinese brown frog. Zool Sci. 26, 80–86 (2009). [DOI] [PubMed] [Google Scholar]
- Karsi A. et al. Transcriptome analysis of channel catfish (Ictalurus punctatus): initial analysis of gene expression and microsatellite-containing cDNAs in the skin. Gene. 285, 157–168 (2002). [DOI] [PubMed] [Google Scholar]
- Fan R. et al. Skin transcriptome profiles associated with coat color in sheep. BMC genomics. 14, 389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragulla H. H. & Homberger D. G. Structure and functions of keratin proteins in simple, stratified, keratinized and cornified epithelia. J. Anat. 214, 516–559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum E. B., Poorten T. J., Settles M. & Murdoch G. K. Only skin deep: shared genetic response to the deadly chytrid fungus in susceptible frog species. Mol Ecol. 21, 3110–3120 (2012). [DOI] [PubMed] [Google Scholar]
- Juntilla M. M. & Koretzky G. A. Critical roles of the PI3K/Akt signaling pathway in T cell development. Immunol Lett. 116, 104–110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson S. L. et al. Midkine and pleiotrophin have bactericidal properties. J. Biol Chem. 285, 16105–16115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi K. et al. Restricted expression of Xenopus midkine gene during early development. J. Biochem. 118, 94–100 (1995). [DOI] [PubMed] [Google Scholar]
- Efremov R. G. & Sazanov L. A. Respiratory complex I: ‘steam engine’ of the cell? Curr Opin Struc Biol. 21, 532–540 (2011). [DOI] [PubMed] [Google Scholar]
- Soto I. C., Fontanesi F., Liu J. & Barrientos A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. BBA-Bioenergetics. 1817, 883–897 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magister Š. & Kos J. Cystatins in immune system. J. Cancer. 4, 45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy C. R., Tixier T., Le Nöene C. & Christian K. A. Field hydration state varies among tropical frog species with different habitat use. Physiol Biochem Zool. 87, 197–202 (2014). [DOI] [PubMed] [Google Scholar]
- Morgan J. A. et al. Population genetics of the frog-killing fungus Batrachochytrium dendrobatidis. Proc Natl Acad Sci USA 104, 13845–13850 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J. et al. Transcriptome analysis revealed positive selection of immune-related genes in tilapia. Fish Shellfish Immun. 44, 60–65 (2015). [DOI] [PubMed] [Google Scholar]
- Heras J., Koop B. F. & Aguilar A. A transcriptomic scan for positively selected genes in two closely related marine fishes: Sebastes caurinus and S. rastrelliger. Mar Genom. 4, 93–98 (2011). [DOI] [PubMed] [Google Scholar]
- Trachsel H. & Staehelin T. Initiation of mammalian protein synthesis. The multiple functions of the initiation factor eIF-3. BBA-Nucleic Acids and Protein Synthesis. 565, 305–314 (1979). [DOI] [PubMed] [Google Scholar]
- Reinberg D. & Roeder R. G. Factors involved in specific transcription by mammalian RNA polymerase II. Transcription factor IIS stimulates elongation of RNA chains. J. Biol Chem. 262, 3331–3337 (1987). [PubMed] [Google Scholar]
- Ofir-Rosenfeld Y., Boggs K., Michael D., Kastan M. B. & Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 32, 180–189 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezón E., Butler P. J. G., Runswick M. J., Carbajo R. J. & Walker J. E. Homologous and heterologous inhibitory effects of ATPase inhibitor proteins on F-ATPases. J. Biol Chem. 277, 41334–41341 (2002). [DOI] [PubMed] [Google Scholar]
- Hartmann H., Noegel A. A., Eckerskorn C., Rapp S. & Schleicher M. Ca2+ -independent F-actin capping proteins. Cap 32/34, a capping protein from Dictyostelium discoideum, does not share sequence homologies with known actin-binding proteins. J. Biol Chem. 264, 12639–12647 (1989). [PubMed] [Google Scholar]
- Perretti M. & D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 9, 62–70 (2009). [DOI] [PubMed] [Google Scholar]
- Boucheix C. et al. Molecular cloning of the CD9 antigen. A new family of cell surface proteins. J. Biol Chem. 266, 117–122 (1991). [PubMed] [Google Scholar]
- Zhou X., Feng H., Guo Q. & Dai H. Identification and characterization of the first reptilian CD9, and its expression analysis in response to bacterial infection. Dev Comp Immunol. 34, 150–157 (2010). [DOI] [PubMed] [Google Scholar]
- Mueller O., Lightfoot S. & Schroeder A. RNA Integrity Number (RIN)–Standardization of RNA Quality Control. Agilent Application Note, Publication 1–8 (2004). [Google Scholar]
- Grabherr M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M. & Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. & Dewey C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC bioinformatics. 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. Gene Ontology: tool for the unification of biology. Nat Genet. 25, 25–99 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 21, 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- Tatusov R. L. et al. The COG database: an updated version includes eukaryotes. BMC bioinformatics. 4, 41 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., Cai T., Olyarchuk J. G. & Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 21, 3787–3793 (2005). [DOI] [PubMed] [Google Scholar]
- Novković M., Simunić J., Bojović V., Tossi A. & Juretić D. DADP: the database of anuran defense peptides. Bioinformatics. 28, 1406–1407 (2012). [DOI] [PubMed] [Google Scholar]
- Hao X. et al. Amphibian cathelicidin fills the evolutionary gap of cathelicidin in vertebrate. Amino Acids. 43, 677–685 (2012). [DOI] [PubMed] [Google Scholar]
- Trapnell C. et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28, 511–555 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Mackey A. J., Stoeckert C. J. & Roos D. S. OrthoMCLDB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, D363–D368 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. & Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52, 696–704 (2003). [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.