

Abstract
The smooth muscle cell directly drives the contraction of the vascular wall and hence regulates the size of the blood vessel lumen. We review here the current understanding of the molecular mechanisms by which agonists, therapeutics, and diseases regulate contractility of the vascular smooth muscle cell and we place this within the context of whole body function. We also discuss the implications for personalized medicine and highlight specific potential target molecules that may provide opportunities for the future development of new therapeutics to regulate vascular function.
I. Introduction
A. Scope and Limitations
The smooth muscle cells of blood vessels display considerable plasticity in their phenotype. In healthy, young blood vessels, the phenotype is largely contractile and blood pressure is well autoregulated. However, during the life span of an individual, vascular cells can switch to a synthetic, largely noncontractile phenotype in response to proinflammatory stimuli, diet or other factors that result in the development of atherosclerosis or vessel remodeling. We will not focus on these processes here but refer the reader to several recent reviews on this topic (Heusch et al., 2014; Brown and Griendling, 2015; Tabas et al., 2015).
Here we will focus on the contractile phenotype, which also can display plasticity of function through a range of more subtle adaptations to aging, biomechanical stress, and vasoactive physiologic and pathophysiologic molecules. The current review will focus on these responses and especially focus, as a prototype disease of contractile vascular smooth muscle, on the complex role of this cell type in hypertension and where many opportunities exist for the exploration of untapped potential therapeutic targets.
B. Overview of Regulation of Blood Pressure/Vascular Tone
1. Guyton View of Regulation Blood Pressure, Kidney Role, Volume Regulation.
In humans, the diagnosis of hypertension is widespread, but typically asymptomatic; 20–50% of the world’s population has hypertension and in the United States ∼30% of the population is hypertensive (Hajjar et al., 2006). Furthermore, hypertension is a major risk factor for cardiovascular disease, stroke, and end-stage renal disease, and thus, there is significant morbidity and mortality associated with this disease. Because blood pressure (BP) is related to the cardiac output (CO) and systemic vascular resistance (SVR) by the equation BP = CO × SVR, increases in either CO or SVR should produce hypertension. Thus, although the molecular mechanism(s) that produce hypertension would be expected to be relatively straightforward, over 50 years of investigation have not defined the molecular mechanism(s) that underlies this medical condition.
The control of blood pressure is an integrated response that includes regulation by neural receptors, hormones, and renal fluid balance (Guyton, 1991). However, the handling of sodium within the kidney is well accepted to be the major factor that regulates blood pressure (Fig. 1), and hence, in the pathogenesis of hypertension renal Na+ excretion, which regulates intravascular volume, is the primary determinant of cardiac output (CO) and therefore blood pressure (Guyton, 1991). The role of control of intravascular volume by the kidney for the pathogenesis of hypertension is supported by the results of an elegant series of studies by Lifton’s group (reviewed in Lifton et al., 2001). These investigators demonstrated that in humans, rare genetic causes of hypertension all arise from a defect in the handling of Na+ in the kidney; mutations that increase Na+ reabsorption (volume expansion) result in severe hypertension, whereas mutations that decrease Na+ resorption (volume contraction) produce hypotension. We will not discuss the well-accepted role of renal fluid balance in regulation blood pressure, because this topic has been the subject of a number of reviews (Lifton et al., 2001; Oparil et al., 2003; Coffman and Crowley, 2008; Johnson et al., 2008).
Fig. 1.

SVR versus kidney: Blood pressure is the product of systemic vascular resistance and cardiac output (BP = SVR × CO). Changes in Na+ reabsorption will increase or decrease intravascular volume and result in an increase or decrease cardiac output, which will alter blood pressure. Similarly, alterations in vascular tone can either increase or decrease SVR, which leads to an increase or decrease in blood pressure (see text for details).
2. Recent Direct Confirmation of Changes in Vascular Tone/Resistance Related to Changes in Systemic Vascular Resistance and Blood Pressure and the Importance of Vascular Smooth Muscle Contraction in both Normal Physiology and Pathophysiology—Hypertension.
More than 90% of patients are diagnosed with essential hypertension, or hypertension of unknown etiology (Oparil et al., 2003). Fortunately, despite the lack of a clear mechanism, there are a number of classes of antihypertensive agents that effectively lower blood pressure. Intuitively, one would expect that changes in vascular tone would result in changes in systemic vascular resistance (SVR) and result in either hyper- and/or hypotension. And, although a number of the classes of antihypertensive agents target the vascular smooth muscle [α-blockers, angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), calcium channel blockers (CCBs)], until recently, there was little experimental evidence consistent with the regulation of vascular tone being an important factor for the molecular mechanism that produces hypertension (Fig. 1). However, a number of studies have demonstrated the importance of changes in vascular reactivity or the regulation of vascular smooth muscle contraction and/or vascular tone for the control of blood pressure. For these experiments, investigators have genetically modified a mouse to produce abnormalities in the regulation of vascular tone and/or vascular dysfunction; these mouse models include the BKCa2+ channel β1 subunit knockout (KO) (Brenner et al., 2000), estrogen receptor β KO (Zhu et al., 2002), vascular smooth muscle cell Sur2 K(ATP) channel KO (Chutkow et al., 2002), endothelial nitric oxide synthase (eNOS) KO (Huang et al., 1995), RGS2 KO (Tang et al., 2003), PKGI KO (Tang et al., 2003), PKGIα leucine zipper mutant (Michael et al., 2008), and the MYPT1 KO (Qiao et al., 2014). All of these mice have both vascular dysfunction and hypertension, and these data suggest that vascular dysfunction produces hypertension. However, in these transgenic models, vascular dysfunction within the kidney could alter fluid balance and a resulting increase in intravascular volume and the resulting increase in CO could be responsible for producing hypertension. The most compelling argument that isolated vascular dysfunction results in hypertension are the results of Crowley et al. (2005). These investigators demonstrated that mice with a KO of the angiotensin type 1 (AT1) receptor were hypotensive. Furthermore, these investigators produced mice with the KO of the AT1 receptors in the kidney with normal AT1 expression in the peripheral vasculature, as well as the KO of AT1 receptors in the peripheral vascular smooth muscle with normal AT1 expression in the kidney. The blood pressure in these two strains was equal and intermediate between the AT1 KO and wild-type (WT) mice. These results demonstrate that in isolation, an abnormality in the regulation of vascular smooth muscle contraction produces a change in blood pressure, and therefore, an isolated increase in vascular smooth muscle tone will produce hypertension. Thus the regulation of vascular smooth muscle contraction is important in both health and disease.
3. Racial Differences/Personalized Medicine.
Further complicating investigation of the mechanism underlying the pathogenesis of hypertension are racial differences in the effectiveness of the various classes of antihypertensives (Cushman et al., 2000; Johnson, 2008; Gupta, 2010), including the response to β-blockers, ACE inhibitors, and ARBs. White compared with black patients with hypertension are more likely to respond to β-blockers, ACE inhibitors, and ARBs, whereas for black patients, treatment with a diuretic or calcium channel blocker (CCB) is more likely to be effective (Johnson et al., 2008). Additionally, there also appears to be regional differences in the response to antihypertensive agents; there is a 10 state region in the Southeastern U.S., referred to as the Stroke Belt, in which the mortality from cerebral vascular accidents is 10% greater than the rest of the country. In this region, compared with the rest of the U.S., treatment of hypertension with diuretics, β-blockers, ACE inhibitors, and clonidine is less effective, whereas there is no difference in the effectiveness of CCBs and prazosin (Cushman et al., 2000). After controlling for race, the differences in the therapeutic success of diuretics and clonidine is still present. Furthermore, for black patients with hypertension in this region, similar to the rest of the U.S., CCBs are more likely to control blood pressure and the effectiveness of β-blockers and prazosin therapy is poor.
These racial differences in response to therapy are also present for the treatment of heart failure. Analysis of the results of the V-HeFT (Vasodilator-Heart Failure) trial demonstrated that treatment of black patients with heart failure with the combination of hydralazine and isosorbide dinitrate reduced mortality, whereas this regimen did not change mortality compared with placebo for white patients (Carson et al., 1999). In contrast to these results, treatment of heart failure with enalapril reduced mortality in white, but not black, patients, and in white patients, enalapril produced a larger reduction in blood pressure and regression of cardiac size than hydralazine and isosorbide dinitrate (Carson et al., 1999).
These racial and regional differences in the response to antihypertensive regimens could be due to polymorphisms. A number of genome wide-association studies (GWAS) have investigated this question (reviewed in Cushman et al., 2000; Johnson et al., 2008), and these studies as well as their implications will be discussed later in this review. However, changes in the vascular smooth muscle phenotype could be responsible for diversity in the effectiveness of the different classes of antihypertensive agents. Defining the role of the vascular smooth muscle phenotype in the pathogenesis of hypertension could identify novel therapeutic targets, which could be exploited in rational drug design. Furthermore, comparing the vascular smooth muscle phenotype between races and regions could potentially define the mechanism that governs the heterogeneity in the response to antihypertensive therapy and form the basis for an individualized approach for selecting an effective antihypertensive regimen.
II. Regulation of Ca2+
A. Ca2+ Determines Vascular Smooth Muscle Cell Contractility and Phenotype
Vascular smooth muscle cells (VSMC), like all other muscle cells, depend on Ca2+ influx to initiate contraction. However, the VSMC intracellular Ca2+ concentration does not only determine the contractile state, but also affects the activity of several Ca2+ dependent transcription factors and thereby determines VSMC phenotype. To govern the various Ca2+-dependent functions and in reaction to different stimuli, VSMCs use a variety of plasmalemmal and sarcoplasmic reticulum (SR) Ca2+ channels to produce a large repertoire of Ca2+ signals, which differ in their spatial and temporal distribution (reviewed by Amberg and Navedo, 2013). These signals range from cell-wide changes in [Ca2+] to highly localized Ca2+ entry or release events. Ca2+ can enter the cell from the extracellular space or be released from the largest intracellular Ca2+ store, the sarcoplasmic reticulum (SR). Extracellular Ca2+ influx is mainly mediated by the opening of voltage dependent L-type Ca2+ channels (LTCC), but there are a number of other channels that modulate intracellular Ca2+, including transient receptor potential (TRP) cation channels. Because of their high single-channel conductance and expression in VSMCs, LTCCs have the largest influence on global [Ca2+]i, and their activity largely determines the VSMC’s contractile state and ultimately vessel diameter (Knot and Nelson, 1998). In response to agonist activation of SR-bound inositol trisphosphate (IP3) or ryanodine receptors (RyR), Ca2+ is released into the cytoplasm from the SR. Local Ca2+ signals from the plasmalemma or the junctional SR can augment or oppose increases in global Ca2+ through the activation of Ca2+-dependent ion channels and their regulatory signaling molecules that ultimately affect plasma membrane potential and therefore LTCC activity.
B. Compartmentalization of Ca2+ Signaling
The concept of Ca2+ compartmentalization was introduced when it was demonstrated that local increases in Ca2+ could activate the contractile apparatus without influencing other Ca2+-dependent signaling pathways (Karaki, 1989). Ca2+ is slow to diffuse across the cytoplasm (Berridge, 2006) and a large flux of Ca2+ is required to achieve the high Ca2+ concentration necessary for activation of Ca2+-dependent processes. Therefore, to compartmentalize and regulate Ca2+ signals, VSMCs arrange their organelles in a fashion that limits the space for diffusion and thereby increases the effect of local changes in [Ca2+] (Kargacin, 1994; Poburko et al., 2004) (Fig. 2). The effects of Ca2+ entry hence depend on the way that organelles, Ca2+ pumps, channels, and Ca2+-dependent signaling molecules are organized in signaling microdomains around the source of the Ca2+ signal, as well as its duration and amplitude. More on the organization of such microdomains in VSMCs and how they affect VSMC contractility and phenotype can be found in the review on regulation of cellular communication by signaling microdomains by Billaud et al. (2014).
Fig. 2.

Compartmentalization of Ca signaling.
1. Ca2+ Sparklets.
Local increases in cytoplasmic Ca2+ resulting from influx through single or small clusters of LTCCs are called Ca2+ sparklets (reviewed by Navedo and Amberg, 2013). Because of the steep voltage sensitivity of LTCCs, the sparklet frequency and persistence are closely linked to membrane potential. Thus local changes in membrane potential will result in alterations of local sparklet activity, whereas cell-wide depolarization leads to extensive opening of LTCCs and global influx of Ca2+ (Navedo et al., 2005; Amberg et al., 2007). Increases in [Ca2+]i and Ca2+ sensitivity of the contractile apparatus in VSMCs are considered hallmarks of essential hypertension, and it has been widely assumed that the increase in intracellular Ca2+ is mediated by increased influx through LTCCs. Consistent with this are results in the rat where banding was used to produce a sudden high intravascular pressure in the right renal artery. After only 2 days, VSMCs from the right renal artery showed increased expression of α1C subunits of the LTCC and increased Ca2+ currents compared with VSMCs from the left renal artery (Pesic et al., 2004). However, the ratio of right renal artery/left renal artery α1C subunit expression decreased over time, which may indicate a dynamic adjustment to this sudden pressure overload occurring within the VSMCs.
Surprisingly, overall LTCC expression and cell-wide Ca2+ influx was recently found to be decreased in a mouse model of essential hypertension (Tajada et al., 2013). However, although there was a decrease in the number of LTCCs present on the plasma membrane, the LTCCs showed increased local sparklet activity. These investigators demonstrated that fewer, but highly active, LTCCs were able to increase [Ca2+]i locally as well as cell-wide. The activity of Ca2+ sparklets has been shown to depend on whether the LTCC is part of a pentad complex bound to the plasma membrane by the scaffolding protein AKAP150 (Navedo et al., 2008). LTCCs that are not coupled in such complexes have a higher probability of producing stochastic sparklets with low flux and short duration, whereas AKAP-associated channels can produce high-activity persistent sparklets.
The dynamics of these persistent sparklets are regulated by kinases and phosphatases that are targeted to a subpopulation of LTCCs by the plasmalemmal anchor AKAP150. Under physiologic conditions in these signaling microdomains, the formation of persistent sparklets mainly relies on protein kinase C (PKC) activity and is counteracted by the serine phosphatase calcineurin. In pathologic conditions such as diabetes, however, protein kinase A (PKA) becomes a mediator of enhanced sparklet activity (Navedo et al., 2010). In a study by Navedo et al. (2008) it was shown that the inhibition of cytoplasmic calcineurin with cyclosporine A in AKAP−/− mice had no effect on LTCC sparklet activity, whereas the inhibition of AKAP150-anchored calcineurin in wild-type mice yielded an increase in persistent sparklets. This confirmed the hypothesis that there was a negative relationship between calcineurin and LTCC sparklet activity but highlighted the importance of calcineurin being targeted to the plasmalemma by AKAP150. The relevance of PKC interaction with LTCCs in the development of ATII-induced hypertension has been demonstrated in a number of experiments, in which not only the KO of PKC but also the ablation of AKAP150 lead to an inability of ATII infusion to produce hypertension (Navedo et al., 2008). In this model, the level of cellular PKC was unchanged in AKAP150−/− VSMCs. These data suggest that recruitment of PKC to the LTCC by AKAP150 is crucial for the development of this form of hypertension. AKAP150 is also thought to play a role in the functional coupling of LTCCs to each other, which amplifies Ca2+ influx and is, similar to persistent sparklet activity, increased in hypertension (Nieves-Cintron et al., 2008). Although the mechanism of coupled gating is still under investigation, a model has been proposed by which coupled gating is mediated by calmodulin (CaM)-dependent interactions between the carboxy-terminals of AKAP150-coupled LTCCs and is increased with PKC activation and calcineurin inhibition (Navedo et al., 2010; Cheng et al., 2011).
It should be noted, that in some studies, there was significant PKC activation in agonist-mediated vasoconstriction, but not in pressure-mediated vasoconstriction (Jarajapu and Knot, 2005; Ito et al., 2007), suggesting that PKC has a negligible role in myogenic tone. However, other groups reported PKC involvement in the modulation of the arteriolar myogenic response to increased intravascular pressure (Hill et al., 1990). This study demonstrated that inhibition of PKC led to inhibition of the myogenic response, whereas a stimulator of PKC activity increased myogenic responsiveness.
In a recent study (Mercado et al., 2014), investigators demonstrated that AKAP150-recruited PKC also regulates the activity of Ca2+-permeable, nonselective TRPV4 channels. These channels can produce Ca2+ sparklets with 100-fold higher Ca2+ flux compared with LTCCs, yet they have been linked to VSMC relaxation (Earley et al., 2009). This association results from the high Ca2+ flux that enables TRPV4 to stimulate SR-membrane bound RyRs in relative proximity to the plasmalemma as a form of Ca2+-induced Ca2+ release (CICR) found in VSMCs.
In contrast to TRPV4 sparklets, LTCC sparklet flux is much lower and therefore not sufficient to trigger Ca2+ release from the SR, but overall LTCC sparklet activity is higher and hence LTCCs have a much greater effect on global Ca2+. Through the effect on global Ca2+, LTCC sparklet activity determines the rate at which the SR can refill its Ca2+ stores. However, neither the SR Ca2+ content nor the number and amplitude of SR Ca2+ release events appear to be directly linked to LTCC sparklet activity (Collier et al., 2000; Essin et al., 2007).
Other important members of the TRP channel family include TRPC1, TRPC3, TRPC6, and TRPM4. They have been found to have a role in regulating myogenic tone as well as the myogenic response and are known to be involved in the mechanism of action of vasoconstrictors (refer to reviews by Beech, 2005, 2013) and will be discussed in the section II.B.4. However, for details on the role of TRP channels in vascular function and how the dysregulation of vascular as well as endothelial TRP channels is related to vascular-related pathologies, please see the recent review by Earley and Brayden (2015).
2. Ca2+ Sparks.
Highly restricted and large Ca2+ release events through SR RyRs are called Ca2+ sparks, and Ca2+ sparks have an important regulatory role in VSMCs. Similar to sparklets, their spatial reach is small, so they have no direct effect on contractility; however, the proximity of RyRs to the plasma membrane allows them to affect global [Ca2+]i indirectly (reviewed by Amberg and Navedo, 2013). The nature of the VSMC’s response to Ca2+ sparks depends on the Ca2+-activated plasmalemmal ion channels that are spatially coupled to the RyRs. In many VSMC, tissues sparks are targeted to large conductance Ca2+-dependent K+ channels (BK) that oppose vasoconstriction by allowing hyperpolarizing outward K+ currents (Nelson et al., 1995). On the other hand, Ca2+ gated Cl− channels (CaCCs) depolarize the plasmalemma and thereby enhance Ca2+ influx through LTCCs (Kitamura and Yamazaki, 2001; Leblanc et al., 2005).
a. Ca2+-dependent K+ channel-coupled sparks.
A single Ca2+ spark increases the open probability of about 30 BK channels in its proximity by 100-fold (Jaggar et al., 2000; Perez et al., 2001). Sparks can occur spontaneously or be triggered by TRPV4 sparklets in the form of a CICR mechanism. Structurally, plasmalemmal BK channels in VSMCs are formed by four pore forming alpha subunits encoded by the slo gene and regulatory β1 subunits that are not necessary for the formation of a functional channel (Toro et al., 1998). However, the β1 subunits play a significant role in modulating the Ca2+ sensitivity and hence functional coupling to RyRs (Brenner et al., 2000). It has been demonstrated by several groups that ablation of the β1 subunit in mice leads to desensitization to Ca2+ and functional uncoupling of BK channels from Ca2+ sparks, causing membrane depolarization, increases in arterial tone, and eventually hypertension (Brenner et al., 2000; Pluger et al., 2000). Furthermore for ATII-induced hypertension, it has been reported that the β1, but not the pore-forming alpha subunit, is downregulated, which mediates a decrease in the sensitivity of BK channels and thereby contributes to vascular dysfunction (Amberg et al., 2003; Nieves-Cintron et al., 2007). Consistent with these results, associations between gain of function mutations of the β1 subunit and a lower prevalence of diastolic hypertension have been described (Fernandez-Fernandez et al., 2004; Nelson and Bonev, 2004; Senti et al., 2005). Additionally, it has been demonstrated that β1 subunit downregulation in ATII-induced hypertension is mediated by enhanced activity of the transcription factor NFATc3 (Amberg et al., 2004; Nieves-Cintron et al., 2007). However in hypertensive animals, there have also been studies that have found higher expression of the α subunit in VSMCs, suggesting that the BK channel is primarily involved in a compensatory response to increased VSMC tone from enhanced LTCC or decreased Kv activity (reviewed in Cox and Rusch, 2002). Hence BK channels appear to be involved in the pathogenesis in some as well as compensation and protection in other forms of hypertension.
Using different strategies to modulate plasmalemmal K+ channel activity to inhibit β1 downregulation in developing hypertension or to increase β1 expression in VSMCs would appear to be a promising approach for the treatment of hypertension. In addition, a number of BK channel openers are currently in development (Webb et al., 2015); however, the use of BK channel openers for the treatment of hypertension is limited by concerns for off-site effects in other smooth muscle tissues. As the β1 subunit of the BK channel seems to be expressed predominantly in VSMCs (Tanaka et al., 1997), targeting β1 expression through gene therapy or modulation of the NFATc3 pathway represents a possible alternative (reviewed by Joseph et al., 2013).
b. Ca2+ gated Cl− channel-coupled sparks.
In some VSMCs, Ca2+ sparks are coupled to CaCCs, and their activation is followed by “spontaneous transient inward currents” or STICs. The two families of CaCCs that have only recently been identified are called bestrophins and TMEM16A. They are also expressed in renal tubular epithelium as well as the heart and are hence thought to have a multidimensional role in blood pressure regulation (reviewed by Matchkov et al., 2015). Because the activation of these channels results in plasma membrane depolarizing currents, they are thought to have an amplifying effect on vascular contractile stimuli by indirectly causing the opening of LTCCs (Leblanc et al., 2005; Matchkov et al., 2013; Bulley and Jaggar, 2014). Indeed, downregulation or inhibition of TMEM16A led to decreased arterial constriction in a variety of studies (Jensen and Skott, 1996; Bulley et al., 2012; Davis et al., 2013; Dam et al., 2014), and a smooth muscle KO of TMEM16A in mice lead to a decrease in the ability of ATII infusion to produce hypertension. As CaCCs are also permeable to other anions such as HCO3−, it is also possible that some effect may be due to changes in intracellular pH that would affect pH-sensitive enzymes including Rho kinase (Boedtkjer et al., 2011). Although there are a number of substances that can inhibit CaCC activity in vitro, the unknown molecular identity of TMEM16A as well as its expression in various tissues would suggest that there is poor pharmacological specificity in vivo and there would be many off target effects (Greenwood and Leblanc, 2007; Boedtkjer et al., 2008).
3. Ca2+ Waves.
Activation of CaCCs is also often mediated by Ca2+ waves, a Ca2+ signal in which subsequent openings of IP3Rs and in some tissues RyRs on the SR cause a wave of Ca2+ release events across the entire length of the VSMC, usually close to the plasma membrane (Iino et al., 1994; Hill-Eubanks et al., 2011; Amberg and Navedo, 2013). Westcott and colleagues described the contrasting roles of RyRs and IP3Rs for the effects of Ca2+ waves in arterioles and upstream feed arteries. Although arteriolar Ca2+ waves are solely IP3R mediated and therefore not inhibited by ryanodine, RyR inhibitors decreased Ca2+ waves in feed arteries. In both tissues, Ca2+ waves were inhibited with phospholipase C (PLC) inhibitors and IP3R blockers, which led to a decrease in [Ca2+i] and vasodilation. Therefore, IP3Rs contribute to Ca2+ waves in both tissues as part of a positive feedback loop for myogenic tone. In contrast, despite the inhibition of Ca2+ sparks and waves in feed arteries, inhibition of RyRs caused an increase in [Ca2+i] and led to vasoconstriction. This was abolished in the presence of BK-channel blocker paxilline, which supports the hypothesis that RyRs, which are involved in Ca2+ waves, are also coupled to BK channels and part of a negative feedback regulation of myogenic tone (Lamont et al., 2003; Westcott and Jackson, 2011; Stewart et al., 2012; Westcott et al., 2012).
In arterioles, Ca2+ waves are initiated by IP3-dependent opening of an IP3R creating a Ca2+ “blip,” a single IP3R opening (Bootman and Berridge, 1996), or a “puff,” short Ca2+ release from a small cluster of IP3Rs that is biophysically different from a RyR-mediated spark (Bootman and Berridge, 1996; Thomas et al., 1998). Subsequently, clusters of IP3Rs open in response to the Ca2+ released by adjacent IP3Rs (CICR) and are inactivated as the [Ca2+] rises further. The IP3R’s Ca2+-dependent activation/inactivation properties are reflected in the wave-like pattern of IP3R-mediated Ca2+ release (Foskett et al., 2007). Ca2+ wave initiation depends on IP3 synthesis by PLC, which occurs after activation of G-protein-coupled receptors by their respective agonists, including norepinephrine and endothelin-1 (Lamont et al., 2003; Westcott and Jackson, 2011; Stewart et al., 2012; Westcott et al., 2012). However, Ca2+ waves are also seen in the absence of agonists and depend on the spontaneous basal production of IP3 by PLC, which varies in different vascular beds, and thus will affect the frequency of Ca2+ release through RyRs via CICR (Gordienko and Bolton, 2002). In arterioles, the wave leads to VSMC contraction by directly increasing [Ca2+]I, and this effect is amplified by the Ca2+-dependent opening of CaCCs in the plasma membrane that leads to membrane depolarization and increased Ca2+ influx through LTCCs.
4. Store-Operated Calcium Entry.
When SR Ca2+ stores are depleted after release through IP3Rs, the SR Ca2+ sensor STIM (stromal interaction molecule) relocates to the SR-plasmalemmal junction and physically interacts with and activates the selective Ca2+ channel Orai [CRAC (calcium release activated calcium channel); reviewed by Trebak, 2012]. For VSMCs in the normal physiologic contractile phenotype, the expression of STIM/Orai is relatively low, but its expression is upregulated when the VSMC changes its phenotype to the proliferative state (Potier et al., 2009). In a rodent STIM/Orai knockdown model, nuclear factor activated T-cells (NFAT) translocation to the nucleus was decreased and VSMC proliferation in response to vascular injury was impaired (Aubart et al., 2009; Guo et al., 2009; Zhang et al., 2011). In spontaneously hypertensive rats, STIM/Orai is upregulated and depletion of SR stores lead to greater SOCE, which may represent an independent mechanism leading to increased VSMC [Ca2+]i in hypertension (Giachini et al., 2009b). Furthermore, these investigators found evidence suggesting that increased STIM/Orai activity may underlie sex differences in the development of hypertension. They determined that inactivation of the STIM/Orai mechanism with CRAC inhibitors as well as antibodies to STIM or Orai during and after store depletion abolished sex differences in spontaneous contractions of VSMCs (Giachini et al., 2009a). Thus CRACs represent a novel target in the treatment of hypertension.
However, there are a number of studies also suggesting a role of TRPC channels in SOCE (reviewed by Beech, 2012). Both TRPCs and Orai channels can be activated by STIM after store depletion (Zeng et al., 2008; Park et al., 2009); however, their individual contribution to SOCE is variable. Studies have demonstrated a partial suppression of SOCE by Orai and TRPC siRNA, respectively (Li et al., 2008). The nature of TRPC-Orai interaction, or if there is in fact one, is currently unresolved (refer to Earley and Brayden, 2015), but both channels can also be activated independently from store depletion or Ca2+ release and their downstream effects on activation differ. TRPs exhibit multiplicity of gating and hence have been suggested to integrate various cellular signals including store depletion (Albert and Large, 2002). TRPC1 mediates Ca2+ influx after store depletion with thapsigargin (Xu and Beech, 2001; Sweeney et al., 2002; Lin et al., 2004) and is thought to be involved in contractile modulation and regulation of cell proliferation; however, more data are needed to determine its exact function. TRPC6 is a channel that mediates cation movement in a variety of experimental settings. In some tissues, inhibition of TRPC6 leads to a decrease in SOCE, but it also appears to be involved in SR-independent signaling. Studies demonstrated that TRPC6 is store and receptor operated, as well as stretch and osmotically activated in VSMCs. It can associate and form heteromultimers with TRPC3, which leads to tonic channel activation (Dietrich et al., 2003). TRPC3 and TRPC6 are upregulated in idiopathic pulmonary hypertension and an siRNA-induced decrease of TRPC6 expression decreases proliferation of cultured pulmonary artery VSMC isolated from patients with pulmonary hypertension. Furthermore, chronic hypoxia increases TRPC6 expression, whereas the ET-1 antagonist bosentan, a common treatment of PAH, lowers TRPC6 expression in pulmonary VSMCs (Kunichika et al., 2004; Lin et al., 2004). These are merely examples of the various roles TRPC channels occupy in VSMC signaling and a complete discussion of TRPC channels in health and disease has been recently presented in a number of reviews (Beech, 2005, 2012; Earley and Brayden, 2015).
C. Excitation-Transcription Coupling
An important mode in which Ca2+ can regulate VSMC contractility is by regulating the composition of the contractile apparatus, ion channels, and cellular signaling molecules by influencing VSMC gene transcription (reviewed by Kudryavtseva et al., 2013). In certain cytoplasmic locations, high [Ca2+] activates specific kinases or phosphatases that in turn lead to activation and translocation of transcription factors to the nucleus. In the nucleus, Ca2+ can bind helix-loop-helix-loop structural domain or motif (EF hand) containing transcription factors directly (Carrion et al., 1999) or modulate transcription factor binding via Ca2+/CaM-S100 complexes (Hermann et al., 1998). Although in many disease states VSMCs can completely lose their contractile function due to a phenotype switch toward a proliferative ECM-producing phenotype, more subtle changes within the contractile phenotype are also thought to play a role in the increased VSMC contractility observed in hypertension.
NFAT is a major target of calcineurin, and it translocates to the nucleus upon calcineurin-mediated dephosphorylation. Calcineurin activation is enhanced by the activity of the AKAP150-bound LTCC signaling pentad (Oliveria et al., 2007; Nieves-Cintron et al., 2008). Hence, NFAT activity is regulated by the level of persistent sparklet activity, but is also dependent on simultaneous inhibition of its nuclear export (Gomez et al., 2003). Interestingly, although membrane-depolarizing signals such as IP3R-mediated Ca2+ waves are thought to cause an increase in NFATc3 activation via enhanced LTCC activity, RyR-mediated Ca2+ release from the SR decreases NFAT activity by an LTCC independent mechanism (Gomez et al., 2002). SOCE has also been implicated in NFAT activation, and its disruption led to reduced hypoxia-induced NFAT nuclear translocation in pulmonary VSMCs (Bierer et al., 2011; Hou et al., 2013). Although a variety of Ca2+ signals lead to NFAT activation, persistently raised levels of intracellular Ca2+ fail to induce NFAT (Stevenson et al., 2001; Gonzalez Bosc et al., 2004). Therefore it is thought that oscillating Ca2+ signals (Matchkov et al., 2012) and concomitant inhibition of nuclear export (Gomez et al., 2003) leads to nuclear NFAT accumulation. It is well documented that inhibition of the calcineurin/NFAT pathway reduces VSMC proliferation, neointima formation, and vascular remodeling in response to injury (Liu et al., 2005; Nilsson et al., 2008; Esteban et al., 2011; Hou et al., 2013). However there are also studies indicating a role for NFAT within the contractile phenotype, altering the expression of plasmalemmal ion channels including BK (Nieves-Cintron et al., 2007) and Kv channels (Amberg et al., 2004) and thereby increasing VSMC contractility and ultimately arterial tone.
In contrast to NFAT, CREB is regulated by the Ca2+ dependent kinases CaMKII and CaMKIV (Cartin et al., 2000). Ca2+ influx through LTCCs is important for activated phospho-CREB to accumulate in the nucleus (Stevenson et al., 2001). Signals that increase LTCC activity including IP3R-mediated Ca2+ waves (Barlow et al., 2006) and SOCE (Pulver et al., 2004; Takahashi et al., 2007) lead to increased CREB-induced transcription, whereas Ca2+ sparks counteract CREB activity by hyperpolarizing the plasmalemma and reducing LTCC flux (Cartin et al., 2000; Wellman and Nelson, 2003). Because CREB activates genes involved in the contractile, as well as the proliferative phenotype, the ultimate effect of CREB activation on VSMC phenotype has not yet been determined. However in contrast to NFAT, CREB is induced by any signal that causes a sustained increase in Ca2+ entry through LTCCs.
In addition to controlling transcription indirectly through CREB, LTCCs have also been found to directly influence gene expression in VSMCs. In a study by Bannister et al. (2013) it was determined that when the C-terminal end of the LTCC (CCt) is cleaved, it either reassociates with LTCCs and reduces LTCC sparklet activity or it relocates to the nucleus and inhibits the transcription of LTCCs. The CCt thus acts as a bimodal vasodilator by decreasing LTCC transcription and reducing voltage-dependent LTCC opening. However, the enzyme responsible for CCt cleavage and the mechanism(s) for regulation have yet to be determined. Potentiating the effects of CCt through increased cleavage or possibly stimulation of CCt promoter sequences may offer another novel approach to controlling vascular contractility.
D. Conclusion
There are a variety of Ca2+-mediated mechanisms that increase VSMC contractility and are possible targets in antihypertensive therapy, some of which are well understood and can be specifically inhibited in vitro. However the development of novel treatments is often limited by the expression of the targets in nonvascular smooth muscle tissues and thus the many off site effects. A possible solution to this issue could be targeting specific therapies to VSMCs using viral vectors. There are a number of successful proof of concept studies using this technique that were recently reviewed by Joseph et al. (2013). Another issue that limits progress in the effort to find novel pharmacologic therapies in Ca2+ signaling is that the composition of Ca2+ signaling microdomains differs in various vascular beds, and hence results cannot always be generalized for VSMCs. This problem again highlights the importance of genetic KO and knockdown studies as a tool to explore targets for gene therapy for the treatment of hypertension.
III. Vascular Smooth Muscle Signal Transduction
A. Signaling Pathways—Overview
Many potential therapeutic strategies are designed to activate or inhibit specific signaling pathways in the vascular smooth muscle cell. It is clear that multiple vascular signaling pathways coexist as spatially separate signaling compartments in individual differentiated vascular smooth muscle (dVSM) cells and coordinated by a multitude of scaffolding proteins. However, these pathways are often overlapping, multilayered, and tissue specific. The tissue-specific nature of these pathways, even between different vessels or sizes of vessels, has led to much controversy on the relative importance of one pathway versus another. Ultimately, however, the possibility of multiple pathways that could be activated or inhibited in various disease states or as functional compensation to physiologic stress gives the system considerable functional plasticity.
At the simplest level, it is well established that vascular tone can be increased either by increasing activation of myosin (Pathways #1 & #2, Fig. 3) or, in a manner analogous to that in striated muscle, by removal of inhibition of actin (Pathway #3, Fig. 3). Either mechanism will lead to an increase in actomyosin activation and crossbridge cycling. Recently, several laboratories have reported more controversial mechanisms by which agonists or biomechanical forces can regulate both vascular and airway smooth muscle contractility by remodeling cytoskeletal attachments (Walsh and Cole, 2013; Zhang et al., 2015) (Pathway #4, Fig. 3). These four pathways are discussed in more detail below.
Fig. 3.

Overview of pathways regulating vascular tone. See text for details. For additional detailed pathways, see subsequent figures.
1. Major Pathways Leading to Changes in the Activity of Smooth Muscle Myosin.
This has been a very active area of investigation by vascular smooth muscle biologists and, as discussed below, has already identified many potential pharmacologic target molecules and in some cases led to possible drug candidates.
Smooth muscle myosin differs from skeletal and cardiac myosins in that it lacks intrinsic myosin ATPase activity in the pure state. Smooth muscle myosin requires a posttranslational modification, phosphorylation of Ser 19 of the 20-kDa regulatory light chain to display enzymatic activity. This phosphorylation is caused by a dedicated Ser/Thr kinase, myosin light chain kinase (MLCK). (Ito and Hartshorne, 1990)
MLCK is a Ca/CaM-dependent kinase and is most simply activated by increases in cytoplasmic ionized Ca ([Ca2+i]) levels (Pathway #1, Fig. 3) such as occurs with a large number of G-protein coupled receptor-mediated agonists, such as alpha agonists or by depolarization of the cell membrane by channel activity or experimentally by equimolar replacement of NaCl with KCl in physiologic saline solution. It has also been reported that increases in the free CaM level (Hulvershorn et al., 2001) or Ca-independent changes in the kinase activity of MLCK can also occur (Kim et al., 2000) by phosphorylation-mediated events.
Dephosphorylation of myosin by myosin phosphatase (MP) decreases its activity, and conversely, inhibition of MP will increase its activity. A large number of pathways, such as those activated by PGF2a and lysophosphatidic acid (Pathway #2, Fig. 3), have been reported to inhibit MP through either Rho-associated protein kinase (ROCK)-dependent mechanisms or those involving Zipper-interacting protein kinase. These pathways are discussed in detail in section IV.
CaMKinase II is another Ca/CaM-dependent kinase with the interesting property, when activated, of autophosphorylating itself on T287, which leads to a sustained activity after Ca is removed, giving it a chemical “memory” of having been activated (Hudmon and Schulman, 2002; Lisman et al., 2002). Conversely, when S26 in the catalytic domain is autophosphorylated, it can terminate sustained kinase activity, making it “forget” prior activation (Yilmaz et al., 2013).
There are four main isoforms of CaMKII, the alpha, beta, gamma, and delta isoforms. The gamma (especially the G-2 variant) (Kim et al., 2000; Marganski et al., 2005) and delta (especially the d2 variant) (Ginnan et al., 2012) isoforms have been shown to play important roles in smooth muscle, with the gamma isoforms primarily regulating contractility and the delta isoforms regulating proliferation. To a large degree the gamma/delta ratio represents the degree of a phenotype switch between the contractile/proliferative phenotypes displayed by smooth muscle in different settings. Vascular injury reduces gamma isoform expression and upregulates delta expression (Singer, 2012). Conversely, siRNA-mediated knock down of the delta isoform attenuates VSM proliferation and neointimal formation. The conditional smooth muscle knockout of CaMKIIdelta significantly delays the progression of airway smooth muscle hyperresponsiveness to an ovalbumin challenge and this isoform is upregulated in the wild-type mouse in response to the same challenge. Thus the delta isoform may play a role in smooth muscle inflammatory responses (Spinelli et al., 2015).
With respect to the gamma isoform and its regulation of smooth muscle contractility, six smooth variants of the gamma isoform of CaMKII have been described with varying kinetics of Ca/CaM-dependent activation/deactivation (Gangopadhyay et al., 2003). One variant, the G-2 variant, which has unique sequence in the association domain of the kinase (Gangopadhyay et al., 2003), has been shown to have scaffolding properties with respect to ERK (extracellular regulated kinase). Antisense specific for the G-2 variant (Marganski et al., 2005) or generic against the gamma isoform or small molecule inhibitor studies (Kim et al., 2000; Rokolya and Singer, 2000) have all demonstrated roles for gamma CaMKII in the regulation of contractility. The CaMKII gamma G-2 variant is reported to be associated with vimentin intermediate filaments and dense bodies in unstimulated vascular smooth muscle cells and on activation by depolarization-mediated increases in cytosolic Ca2+ levels the G-2 variant translocates to the cortical focal adhesions (Marganski et al., 2005; Gangopadhyay et al., 2008). This variant also has been shown to be specifically dephosphorylated by an SCP3 phosphatase. SCP3 is a PP2C typed protein phosphatase, primarily expressed in vascular tissues and specifically binds to the unique association domain sequence in CaMKII gamma G-2. G-2 is bound to this phosphatase in unstimulated vascular smooth muscle tissue but is released upon depolarization-mediated Ca2+ influx. This phosphatase does not appear to regulate kinase activity but rather is thought to result in the exposure of a SH3 domain targeting of the kinase, which leads to targeting to focal adhesions (Gangopadhyay et al., 2008).
Although CaMKIIgamma is known to be activated by stimuli that increase the free Ca2+ level in dVSM, and antisense or inhibitors to CaMKII decrease the amplitude of the contraction to KCl PSS, the exact pathways that regulate contractility are still being confirmed. It has been shown that knock down of the gamma isoform or the G-2 variant specifically, as well as small molecule inhibitors of the kinase, lead to an inhibition of ERK activation and an inhibition of MLCK (Kim et al., 2000; Rokolya and Singer, 2000; Marganski et al., 2005). ERK has been shown to be capable of activating MLCK in other systems (Morrison et al., 1996; Nguyen et al., 1999), but whether this is the link in smooth muscle has not been definitively shown. Additionally in cultured vascular cells, CaMKII is rapidly activated after upon adherence of the cells upon plating onto ECM or poly-lysine. Adherence led to CaMKII-dependent tyrosine phosphorylation of paxillin and ERK activation (Lu et al., 2005). The CaMKII delta 2 variant has also been shown to regulate vascular smooth muscle cell motility in culture through a Src-family tyrosine kinase, Fyn (Ginnan et al., 2013). Because focal adhesions are known to serve as ERK scaffolds in contractile smooth muscle, this is an appealing possible link. Thus, at the present time, although CaMKII is clearly an important regulator of Ca2+-dependent vascular contractility, the complete molecular details of the CaMKII pathway used by contractile vascular smooth muscle to regulate contractility are not yet resolved. It is clear, however, that these details represent considerable untapped potential as future therapeutic targets for the modulation of Ca2+-dependent vascular contractility and hence blood pressure. Additionally, the wealth of information on isoform specific effects of CaMKII, especially the gamma G-2 variant, offers the potential of considerable tissue and smooth muscle phenotype specificity of such therapeutics.
2. Pathways Leading to Changes in Actin Availability for Interaction with Myosin.
In contrast to the pathways described above, ex vivo studies (Walsh et al., 1994; Horowitz et al., 1996a; Dessy et al., 1998) have demonstrated that phorbol esters, or alpha agonists, by activating PKC can trigger increases in contractile force that in some tissues are either Ca2+ independent or cause leftward shifts in the [Ca2+i]-force relationship. The Ca2+ dependence of phorbol ester contractions varies in different smooth muscle tissues, dependent on the isoforms of PKC present in those tissues. The alpha, beta, and gamma isoforms are calcium dependent, but delta and epsilon are calcium independent. Thus, phorbol ester and alpha agonist-induced contractions have been described as being Ca2+ independent in experiments using the aorta of the ferret, which contains significant amounts of the epsilon isoform of PKC, but tissues containing more PKC alpha, such as the portal vein of the ferret, show an increased Ca2+ sensitivity but are still Ca2+ sensitive (Lee et al., 1999) and lead to the activation of Pathway #3 in Fig. 3.
Pathway 3 can be activated by diacylglycerol (DAG) release, generated by the activation of GPCRs in vascular smooth muscle, or by myometrial stretch, RU486, and labor in the rat and human myometrium (Li et al., 2003, 2004; Morgan, 2014). Interestingly, in the presence of extracellular Ca2+ (Cae), phenylephrine (PE) will activate pathways 1, 3, and 4 but in the absence of Cae and in the absence of changes in 20kda light chain phosphorylation, a contraction persists in aorta of the ferret (Dessy et al., 1998). Phorbol esters give a maximal tonic contraction in the absence of changes in 20kda light chain phosphorylation, even in the presence of Cae in this system (Jiang and Morgan, 1989).
When activated, Pathway #3 leads, indirectly, to the PKC-dependent activation of MEK, a dual activity kinase that phosphorylates ERK on a tyrosine and threonine, resulting in activation of ERK. ERK activation can have multiple downstream effects, largely controlled by output-specific scaffolding proteins (see below). In contractile smooth muscle, these downstream effects include phosphorylation of the actin binding protein caldesmon. Caldesmon has been described as being functionally analogous to the troponin complex in striated muscle in that it blocks the access of myosin to actin and hence impairs crossbridge cycling. The C-terminal end of caldesmon is responsible for the direct inhibition of myosin ATPase activity (Sobue et al., 1982; Bryan et al., 1990; Wang et al., 1991). Investigators have demonstrated that the interaction of the actin-binding domain of caldesmon with actin is responsible for the inhibition the actomyosin ATPase (AMATPase) (Velaz et al., 1990) by decreasing the rate of Pi release by 80% (Alahyan et al., 2006).
Caldesmon has an NH2-terminal myosin-binding domain, in addition to the COOH-terminal actin-binding domain, and thus, in theory, could crosslink actin and myosin (Goncharova et al., 2001). However, Lee et al. (2000b) also observed a tethering effect of the N-terminal region of caldesmon to myosin that has the proposed agonist-dependent functional effect of positioning caldesmon so that its C-terminal end no longer inhibits myosin activity. The binding of caldesmon to myosin is regulated by Ca2+-calmodulin, whereas the interaction with actin is regulated by ERK phosphorylation at Ser789 on caldesmon (Hemric et al., 1993; Patchell et al., 2002).
In general, in most systems it appears that phosphorylation of caldesmon on Ser789 by ERK, PAK, or other serine kinases can reverse caldesmon-mediated inhibition of myosin ATPase activity (Childs et al., 1992; Foster et al., 2000; Kim et al., 2008a) (Pathway #3, Fig. 3). However, results from mechanical experiments examining caldesmon function are variable. In smooth muscle from caldesmon KO mice, compared with WT controls, both the rate of force activation and the steady-state force in response to depolarization, phorbol esters, and carbachol were similar, but the rate of force relaxation was reduced (Guo et al., 2013). In contrast to these results, an siRNA-induced decrease in caldesmon expression lowered both isometric force and muscle shortening velocity (Smolock et al., 2009).
In cultured smooth muscle cells, p42/44 MAPK has been clearly demonstrated to phosphorylate caldesmon at Ser789 (Hedges et al., 2000), but for agonist activation of intact smooth muscle, the kinase responsible for caldesmon phosphorylation remains a matter of controversy or may involve different kinases in different settings (Wang, 2008). In skinned smooth muscle strips, ERK-induced phosphorylation of caldesmon did not alter the force-Ca2+ relationship (Nixon et al., 1995). Porcine carotid artery preparations did not display detectable phosphorylation of caldesmon at the ERK sites during phorbol ester stimulation, (D'Angelo et al., 1999), but PAK phosphorylation at Thr627, Ser631, Ser635, and Ser642 was demonstrated to reduce caldesmon’s inhibition of the AMATPase (Hamden et al., 2010). On the other hand, ERK-mediated phosphorylation of caldesmon at 789 has been clearly shown in ferret aorta preparations as well as mouse aorta and rat myometrium. Furthermore, although an increase in caldesmon phosphorylation was observed by Katoch and Moreland (1995) in porcine carotid artery during both depolarization and histamine stimulation, experiments using inhibitors suggested that a second kinase in addition to ERK also phosphorylates caldesmon (Gorenne et al., 2004).
In contrast to the myosin regulatory pathways, this is a relatively untapped area of investigation for the discovery of new target molecules with therapeutic potential. The relative importance of these pathways are definitely tissue and species specific. Interestingly, the strongest evidence of the importance of these pathways appears to have come from myometrial smooth muscle in the setting of preterm labor (Li et al., 2003, 2004, 2007, 2009). Thus, the potential is there for novel and possibly quite specific therapeutic targets within these pathways.
3. Tyrosine Phosphorylation of Smooth Muscle Proteins.
The vast majority of known protein phosphorylation events in the contractile, differentiated smooth muscle cell are serine/threonine events. Where phosphotyrosine screening with immunoblots of contractile vascular as well as myometrial (Li et al., 2007, 2009; Min et al., 2012) smooth muscle tissue has been performed, the reactive bands have been almost exclusively focal adhesion-associated proteins. These tyrosine phosphorylations are largely sensitive to Src inhibitors, pointing to the presence of focal adhesion remodeling in nonproliferating, nonmigrating smooth muscle (Poythress et al., 2013; Ohanian et al., 2014; Zhang et al., 2015). These mechanisms have been especially studied in vascular and airway smooth muscles, resulting in pathways extending from Pathway #4 (Fig. 3). These mechanisms will be discussed in further detail in section V below.
4. Calcium Sensitization of the Contractile Apparatus.
When our group first (Bradley and Morgan, 1982, 1985) measured intracellular Ca levels ([Ca2+]i) in dVSM with the photoprotein aequorin, we noticed that agonists often cause tonic contractions with only transient increases in [Ca2+]i or differing magnitudes of [Ca2+]i, reflecting apparent changes in “Ca2+ sensitivity” of the contractile apparatus (Bradley and Morgan, 1985). This dissociation between [Ca2+]i and force has been confirmed with many agonists and many different Ca2+ indicators in contractile smooth muscle tissues and with permeabilized smooth muscle preparations where leftward shifts in the Ca2+-force relationship in response to agonists and various agents are seen (Ruegg and Pfitzer, 1985; Somlyo et al., 1999). Mechanistically, we now have molecular explanations for this phenomenology. Changes in the apparent Ca2+ sensitivity of the contractile apparatus have been partially explained by the ability of agonists to regulate the activity of myosin phosphatase (MP) (Somlyo and Somlyo, 2003) (Pathway #2, Fig. 3), partially by the ability of ERK to regulate the action of caldesmon (CaD) to inhibit acto-myosin interactions (Kordowska et al., 2006) (Pathway #3, Fig. 3) and clearly also by yet to be defined pathways.
B. Subcellular Spatial Organization of Signaling Pathways
The complexity of signaling pathways in the smooth muscle cell raises the issue of how kinases connect with their complex specific upstream activators and downstream substrates in an agonist-specific manner within the three-dimensional space of the interior of a cell. Scaffold proteins are now recognized to play important roles in coordinating mammalian signal transduction (Morrison and Davis, 2003; Kolch, 2005). Protein scaffolds are defined as docking platforms that colocalize components of kinase cascades and facilitate activation of the kinases (McKay and Morrison, 2007). The scaffolds themselves generally lack enzymatic activity but promote specific outcomes of the pathway. Protein scaffolds can be thought of as “traffic cops” in what would otherwise be the chaos of multiple competing intracellular signaling pathways. Because scaffold proteins add specificity to the cellular pathways, they also present very attractive targets for drug discovery programs. Two major types of scaffolds relevant for the smooth muscle cell are ERK scaffolds and scaffolds for regulators of myosin phosphatase.
1. Extracellular Regulated Kinase Scaffolds (Calponin, SmAV, Paxillin, Caveolin, FAK, IQGAP).
ERK is known to often be targeted to the intranuclear space in proliferative cells and to regulate nuclear signaling, especially to transcription factors (Dhanasekaran et al., 2007). In the smooth muscle cell these pathways can lead to a proliferative phenotype for the smooth muscle cell. These pathways will not be discussed here, but rather we will focus on those most relevant for the fully differentiated contractile cell. Even so, much of this work has been performed using cell culture models and no doubt needs further work in specific contractile smooth muscle tissue systems.
Calponin is a bit of an enigma and its function in smooth muscle is still debated. It has been reported to serve both cytoskeletal and signaling functions (Winder and Walsh, 1990; Birukov et al., 1991; Menice et al., 1997; Leinweber et al., 1999a,b, 2000; Appel et al., 2010). Both PKC and CAM kinase II phosphorylate calponin at Ser175 (Winder and Walsh, 1990), and after phosphorylation, calponin loses its ability both to bind actin and inhibit the AMATPase (Winder et al., 1993). Calponin has been reported directly to regulate contractility (el-Mezgueldi and Marston, 1996; Obara et al., 1996; Winder et al., 1998; Takahashi et al., 2000; Je et al., 2001; Szymanski et al., 2003) but others have reported negative results (Matthew et al., 2000). Calponin phosphorylation increases during carbachol stimulation of smooth muscle (Winder et al., 1993). Consistent with a physiologic role for calponin in the regulation of contractility are results in skinned smooth muscle; the addition of exogenous calponin reduces both Ca2+ activated force (Horowitz et al., 1996b; Obara et al., 1996) and maximal shortening velocity (Jaworowski et al., 1995). In the smooth muscle isolated from calponin KO mice, compared with WT controls, muscle-shortening velocity is significantly higher, but there is no difference in the force produced by Ca2+, carbachol, or phorbol esters (Matthew et al., 2000). However, the addition of exogenous calponin reduces force in skinned single smooth muscle cells, and the Ser175Ala calponin mutant has no effect on force (Horowitz et al., 1996a). In intact smooth muscle during agonist-induced activation, calponin redistributes from the contractile filaments to the cell surface, which is attenuated with the inhibition of PKC (Parker et al., 1994, 1998; Gallant et al., 2011). Thus, these results are consistent with a role for calponin in the regulation of smooth muscle contraction; agonist stimulation leads to the activation of PKC, which phosphorylates calponin at Ser-175 to decrease calponin’s interaction with actin to relieve calponin’s inhibition of the AMATPase.
Three isoforms exist for calponin. h1CaP/CNN1/basic calponin is one of the most specific and rigorous markers for the differentiated smooth muscle phenotype. h2CaP/CNN2/neutral calponin and h3/acidic CaP/CNN3/aCaP are more widely distributed but appear to also be expressed in some smooth muscles (Takahashi et al., 1988; Strasser et al., 1993; Applegate et al., 1994). Work in our group has led us to propose that calponin is an adaptor protein for ERK (Leinweber et al., 1999a,b; Appel et al., 2010). Antisense knock down of calponin (Je et al., 2001) led to decreased ERK activity and contractile force after alpha agonist activation but not after a depolarizing stimulus. Also, protein chemistry studies and cellular immunoprecipitation studies demonstrated that CaP directly binds both PKC and ERK and in intact vascular smooth muscle cells (Leinweber et al., 2000) and is bound to the thin filaments but translocates to the cortex of the cell in response to alpha agonists (Parker et al., 1998). A detailed model has been suggested where agonists activate PKC, which phosphorylates CaP, releasing it from the thin filaments (Kim et al., 2008a). Colocalization of ERK and CaP is seen in unstimulated vascular smooth muscle cells and agonist-activation leads to the translocation of a PKC/CaP/ERK complex to the cell cortex, likely meeting up with SmaV (see below), Raf, and MEK, which leads to the activation of ERK, at which point it is seen to return to the contractile filaments and CaD phosphorylation of the ERK sites is observed (Khalil and Morgan, 1993).
SmAV is the smooth muscle isoform of a major scaffolding protein supervillin (Pestonjamasp et al., 1997). SmAV was initially cloned and identified SmAV as a CaP binding partner in a two-hybrid assay with CaP as bait (Gangopadhyay et al., 2004), and it was found that SmAV acts as an ERK scaffold, leading to the regulation of CaD phosphorylation (Gangopadhyay et al., 2009). Data have been published indicating that CaP (Menice et al., 1997), SmAV (Gangopadhyay et al., 2004) and CaV (Je et al., 2001) function as scaffolds coordinating Pathway #3.
Paxillin, better known as a focal adhesion protein, is also known to bind the classic ERK “signaling module” of Raf, MEK, and ERK (McKay and Morrison, 2007). In quiescent cultured cells, paxillin is constitutively associated with MEK, but Ishibe et al. (2003) showed that when cells are stimulated with HGF, Src-mediated phosphorylation of paxillin at Y118 leads to the recruitment of ERK, followed by Raf, which leads to ERK phosphorylation and activation. Shortly thereafter focal adhesion kinase (FAK) is recruited to the complex, leading to FA remodeling in both cultured cell systems and airway smooth muscle (Zhang et al., 2015). Thus, paxillin provides a signaling hub in the vicinity of focal adhesions that can have specific cytoskeletal outcomes.
Caveolin is an extensively studied protein but there are still many mysteries regarding its function. A caveolin-associated protein has also discovered and named cavin (Liu and Pilch, 2008; Ding et al., 2014; Kovtun et al., 2015). The exact way in which caveolin and cavin interact and the role of cavin specifically in smooth muscle is not yet clear; however, a cavin knockout mouse has been produced (Sward et al., 2014). In this knockout animal not only were arterial expression of cavin-1, cavin-2, and cavin-3 reduced but also all isoforms of caveolin were reduced. As a result, caveolae were absent from both smooth muscle and endothelial cells. An enhanced contractile response to an alpha 1 adrenergic agent was seen, but was likely to be due to the increased thickness of the vascular wall. In contrast, myogenic tone was essentially absent. Surprisingly, blood pressure of the knockout mouse was well maintained, presumably due to opposing influences from smooth muscle and endothelial effects.
Inhibition of caveolin function by a caveolin decoy peptide or by methyl-beta-cyclodextrin has been shown to disrupt ERK activation in vascular smooth muscle (Je et al., 2004). Work using cultured vascular smooth muscle cell models has suggested that caveolin-mediated scaffolding of ERK leads to different functional outputs than actin/calponin-mediated scaffolding (Vetterkind et al., 2013). This concept has not yet been pursued in contractile smooth muscle but illustrates the idea of scaffold proteins regulating the output of kinase cascades toward separate purposes and serving as traffic cops for complex cellular signaling pathways.
IQGAP (IQ motif containing GTPase activating protein) is an ERK-binding and actin-binding protein that has been extensively studied in nonmuscle systems but little studied in smooth muscle systems. In cultured vascular smooth muscle cells, knockdown of IQGAP prevents the phosphorylation and activation of an actin-associated pool of ERK in response to PKC activation. Proximity ligation assays demonstrated direct tethering of ERK1/2 to actin by IQGAP. Interestingly caveolin is also required for activation of this pathway unless ERK is already associated with actin. Caveolin appears to be required specifically for upstream C-raf activation (Vetterkind et al., 2013).
2. Myosin Phosphatase Scaffolds.
Myosin regulation is discussed in detail in section IV below, but multiple pathways have been suggested to coordinate signaling associated with myosin phosphatase (MP), and hence, myosin activity (Pathway #2, Fig. 1), and it seems likely that scaffolds play a role to regulate/facilitate these pathways. One MP putative scaffold, M-RIP, also called p116RIP, is thought to link active Rho/ROCK to the inhibition of MP (Surks and Mendelsohn, 2003; Mulder et al., 2004; Koga and Ikebe, 2005). Vetterkind and Morgan (2009) reported that another scaffold/adaptor protein, Par-4, also regulates myosin phosphatase activity in contractile smooth muscle. We have described a “padlock” model to explain the actions of Par-4, whereby binding of Par-4 to MYPT1 activates MP. This is postulated to occur by the physical blockade by Par-4 of the MYPT1 inhibitory phosphorylation sites. Conversely, this model indicates that inhibitory phosphorylation of MYPT1 by Zipper-interacting protein kinase requires “unlocking” of the blockade by phosphorylation and displacement of Par-4 (Vetterkind et al., 2010). Whether M-RIP and Par-4 facilitate or antagonize each other’s actions is not known.
The complexity of this system is impressive, but it is expected that the multiple scaffolding proteins and signaling molecules involved in regulating myosin phosphorylation will lead to the development of rational and selective therapeutic approaches to cardiovascular disease.
C. Link to Hypertension
We describe here a number of pathways by which vascular smooth muscle contraction and stiffness are directly regulated and hence will affect blood pressure. It should be mentioned that many other indirect pathways are also involved, with a major mechanism being the development of inflammation and subsequent reduction-oxidation reaction (REDOX) signaling pathways (Sorescu et al., 2001; Loirand and Pacaud, 2014). These pathways are triggered by angiotensin-induced signaling, and as a result, inhibitors of the effects of and production of angiotensin are major ways of regulating blood pressure, including blood vessel contraction. For further details, we refer you to Mehta and Griendling (2007) for a review of this topic.
D. Potential Novel Therapeutic Targets/Approaches/Critical Analysis of Pathway-Specific Inhibitors
1. Rho Kinase Inhibitors.
Y27632, the first ROCK inhibitor described, decreases blood pressure in 11-Deoxycorticosterone acetate (DOCA)-salt rat model of hypertension. A similar effect was obtained with the newer ROCK inhibitors fasudil, SAR07899, in other animal models of hypertension, including the spontaneously hypertensive rat (SHR), angiotensin II-induced hypertension in several animals, and L-NG-Nitroarginine Methyl Ester (L-NAME)-induced hypertension (Uehata et al., 1997; Mukai et al., 2001; Kumai et al., 2007; Lohn et al., 2009). Of note, this class of inhibitors also has a major part of their effect on hypertension through inhibition of inflammatory pathways and cardiovascular remodeling. For more details we refer you to a recent review by Loirand and Pacaud (2014).
2. Endothelin Inhibitors.
The endothelin pathway, linked to PLC and ERK signaling, has been identified as an effective antihypertensive target (Sandoval et al., 2014).
3. Beta Adrenergic Receptor Mediated Inhibition.
Of interest is the fact that beta receptor mediated relaxation of vascular smooth muscle has been reported to decline with age in both the human and animal models. In aortas from Fischer 344 rats, an increase in the level of G-protein receptor kinase-2, which desensitizes the beta adrenergic receptor by phosphorylation of the receptor has been reported to increase with age (Schutzer et al., 2005), and thus inhibitors of G-protein receptor kinase-2 may promote beneficial restoration of beta receptor mediated vasodilation.
IV. Regulation of Smooth Muscle Myosin
A. Overview of Regulation of the Smooth Muscle Actomyosin ATPase and 20kda light chain Phosphorylation/Smooth Muscle Activation
The crossbridge cycle describes the development of force through a series of complexes between actin (A), myosin (M), ATP, and its hydrolysis products, ADP and Pi (Sweeney and Houdusse, 2010) (Fig. 4 and the termination of Pathway #1; Fig. 3). Beginning in the rigor state (AM), ATP binding to AM results in rapid dissociation of AM, forming an A+M-ATP state, and then ATP is hydrolyzed by myosin. After hydrolysis, the crossbridge enters a weakly attached, pre-powerstroke AM-ADP-Pi state, and then transitions to a strongly bound, force producing AM-ADP-Pi state. After Pi release from the AM-ADP-Pi state, the crossbridge enters a AM-ADP state, which then isomerizes to a high force generating state (AM-ADP) followed by ADP release and returning to the rigor state (AM). MgATP subsequently binds to the AM state, causing rapid crossbridge detachment, and then another crossbridge cycle commences. The duty cycle is defined as the proportion of time crossbridges spend in strongly attached states divided by the time for the total crossbridge cycle (De La Cruz and Ostap, 2004); high duty cycle motors are capable of processive movement (i.e., dynein, myosin V), whereas skeletal muscle myosin has a low duty cycle that prevents the development of an internal load from strongly bound crossbridges, which would decrease shortening velocity. Although the crossbridge cycle for all types of myosin is frequently described in this generic manner, differences exist between the kinetics of skeletal, cardiac, and smooth muscle and even within different smooth muscle tissues, requiring changes in the crossbridge cycle to explain the differences in AMATPase rates (Rosenfeld et al., 2000).
Fig. 4.

AMATPase: Actomyosin ATPase cycle; ATP is hydrolyzed by myosin (M) and the subsequent interaction of myosin with actin (A) produces force and/or displacement (see text for details).
The smooth muscle AMATPase is similar to that of striated muscle, albeit the kinetics are slower. The kinetics and individual rate constants of the steps in the actomyosin ATPase have been defined in a number of studies (Rosenfeld et al., 2000; Baker et al., 2003; Haldeman et al., 2014), and similar to other myosin IIs, the ATPase is limited by phosphate release or the transition from weak to strong binding states (Haldeman et al., 2014). Both cardiac and skeletal muscle myosin is functionally on, i.e., myosin will hydrolyze ATP in the presence of actin. Smooth muscle (SM) myosin will hydrolyze ATP in the presence of actin, albeit very slowly; however, after phosphorylation of the 20-kDa regulatory myosin light chain (RLC), the rate of hydrolysis is increased (Chacko et al., 1977; Ikebe and Hartshorne, 1985; Ikebe and Morita, 1991; Ellison et al., 2000) due to an ∼1000-fold increase in the rate of product release (Sellers and Adelstein, 1985). Thus, changes in RLC phosphorylation regulate smooth muscle activation and relaxation.
In smooth muscle, in addition to RLC phosphorylation regulating the AMATPase, it also controls the structure of SM myosin and filament formation (Ikebe and Hartshorne, 1985). In the absence of RLC phosphorylation, myosin is in the 10S conformation [high sedimentation velocity and low ATPase (Ikebe and Hartshorne, 1985), with the tail of myosin bending back over the head neck junction interacting with the regulatory light chain (Jung et al., 2011; Salzameda et al., 2006)]. After RLC phosphorylation, the interaction of the myosin tail with the RLC is perturbed (Jung et al., 2011), and myosin exists in an extended conformation [6S, low sedimentation velocity, high ATPase (Ikebe and Hartshorne, 1985)] and also forms filaments (Applegate and Pardee, 1992). Other investigators have suggested that in the absence of RLC phosphorylation, the interaction of the NH2-terminal region of caldesmon (see Fig. 3 and section III.A.1) with the myosin crossbridge disrupts the interaction of the myosin head with its neck-tail region to promote a transition from the 10S to the active, 6S conformation (Wang, 2008). Nonetheless, in human smooth muscle, there is significant pool of 10S myosin that can be converted by changes in cellular conditions to 6S myosin that can then assemble into side polar thick filaments (Milton et al., 2011). These data could suggest that in addition to the regulation of the AMATPase, changes in RLC phosphorylation could regulate the formation of myosin filaments within the smooth muscle during activation and relaxation (Pratusevich et al., 1995; Ali et al., 2007; Liu et al., 2013; Seow, 2013; Lan et al., 2015); however in vivo, the ability of RLC phosphorylation to regulate filament formation is controversial (Seow, 2015; Somlyo, 2015).
The level of RLC phosphorylation is defined by the relative activities of myosin light chain kinase (MLCK) and MLC phosphatase (Gong et al., 1992), i.e., RLC phosphorylation is related to MLCK/(MLCK+MLC phosphatase). Thus, changes in the activity of either MLCK or MLC phosphatase will change RLC phosphorylation and force or vascular tone. MLCK is regulated by Ca2+-calmodulin (Ikebe and Hartshorne, 1985), whereas MLC phosphatase activity is regulated by a number of signaling pathways (Hartshorne et al., 1998). At a constant [Ca2+], a decrease in MLC phosphatase activity increases SM RLC phosphorylation and force to produce Ca2+ sensitization (Somlyo and Somlyo, 2003), whereas an increase in MLC phosphatase activity decreases SM RLC phosphorylation and force to produce Ca2+ desensitization (Somlyo and Somlyo, 2003).
MLC phosphatase is a holoenzyme (Hartshorne et al., 1998) consisting of a catalytic subunit, a 20-kDa subunit of unknown function, and a myosin targeting subunit (MYPT1). Alternative splicing of a 123-bp central exon results in MYPT1 isoforms that differ by a 41-aa central insert (CI), referred to as M130 and M133 (Hartshorne et al., 1998). Additionally, alternative splicing of the 3′ exon (Khatri et al., 2001) is responsible for generating MYPT1 isoforms that differ by the presence or absence of a carboxy-terminal leucine zipper (LZ). Thus in humans and other species, alternative splicing generates four MYPT1 isoforms that differ by the presence or absence of a CI and LZ: CI+LZ+, CI-LZ+, CI+LZ−, CI-LZ− (Hartshorne et al., 1998).
B. Guanine Nucleotide Exchange Factor Signaling, Rac/Rho, and Analysis of Inhibitors
The Rho GTPases are within the RAS superfamily of small G proteins (Jaffe and Hall, 2005), which exist as either active GTP-bound and inactive GDP-bound forms. The conversion between active and inactive forms is controlled by guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), and guanine dissociation inhibitors. The role of RhoA for the regulation of smooth muscle tone has been well described (Fig. 5; Ca2+ sensitization and Pathway #2, Fig. 3). RhoA/Rho kinase signaling is activated by G protein-coupled receptors, and the role of this pathway for the inhibition of MLC phosphatase and Ca2+ sensitization has been the subject of a number of reviews (Arner and Pfitzer, 1999; Somlyo and Somlyo, 2003; Puetz et al., 2009). The activation of Rho kinase has been demonstrated to phosphorylate CPI-17 at Thr38 (Eto et al., 1995; Kitazawa et al., 2000), PHI-1 at Thr57 (El-Touhky et al., 2005, 2006), and MYPT1 (Trinkle-Mulcahy et al., 1995) at both T696 and T850 (Muranyi et al., 2005). MYPT1 phosphorylation at T696 has been demonstrated to decrease MLC phosphatase activity to increase force at a constant Ca2+ (Kitazawa et al., 1991a), whereas the phosphorylation of MYPT1 at T850 dissociates the holoenzyme, which results in a decrease in MLC phosphatase activity (Velasco et al., 2002), which produces Ca2+ sensitization. Similarly, agonist activation of G-protein-coupled receptors has been demonstrated to lead to the activation of PKC, which phosphorylates both CPI-17 at Thr38 (Eto et al., 1995; Kitizawa et al., 1991b) PHI-1 at Thr57 (Eto et al., 1999). When phosphorylated, these proteins will bind to the catalytic core of the catalytic subunit of MLC phosphatase to decrease phosphatase activity (El-Toukhy et al., 2006; Eto et al., 2007). In addition to Rho kinase, Zip kinase will phosphorylate MYPT1 (MacDonald et al., 2001a) as well as CPI-17 (MacDonald et al., 2001b), and integrin-linked kinase will phosphorylate MYPT1 (Kiss et al., 2002; Muranyi et al., 2002) as well as CPI-17 and PHI-1 (Deng et al., 2002).
Fig. 5.

Ca2+ sensitization: Agonist activation of G-protein coupled receptors activates several signaling pathways (IP3, RhoA/Rho kinase, PKC, RAC1) that modulate Ca2+ release from the SR and/or lead to Ca2+ sensitization of the contractile filaments (see text for details).
There are a number of studies demonstrating Rho kinase signaling mediates both MYPT1 phosphorylation and Ca2+ sensitization (Somlyo and Somlyo, 2003; Puetz et al., 2009). However, in smooth muscle of the guinea pig ilium, although inhibition of Rho kinase decreased the carbachol-induced increase in Ca2+ sensitivity, it had no effect on MYPT1 phosphorylation (Pfitzer, 2001). In this study, although staurosporin prevented MYPT1 phosphorylation, specific PKC inhibitors had no effect on the phosphorylation of MYPT1, which could suggest in this tissue Zip kinase is involved in a physiologically important signaling pathway for Ca2+ sensitization. In bladder smooth muscle, others have demonstrated that there is constitutive phosphorylation of MYPT1 at T696, which was unaffected by inhibition of either Rho kinase or PKC, whereas MYPT1 phosphorylation at T850 was primarily mediated by Rho kinase (Chen et al., 2015). These investigators generated T696A and T850A MYPT1 mutant mice to demonstrate that the MYPT1 phosphorylation at T696, but not T850, is important for increasing RLC phosphorylation and Ca2+ sensitization during the sustained phase of force maintenance. These results demonstrate that during activation of smooth muscle, the physiologically important signaling pathways mediating both MYPT1 phosphorylation as well as Ca2+ sensitization are agonist as well as tissue specific.
Although the role of RhoA in the regulation of Ca2+ sensitization of smooth muscle is established, the role of other Rho GTPases for force regulation in smooth muscle is not well defined. Pak1, a downstream target of Rac1, has been demonstrated to inhibit MLCK and relax permeabilized intestinal smooth muscle (Wirth et al., 2003). However in airway smooth muscle, both the knockout or inhibition of Pak reduces tone (Hoover et al., 2012), and Pak3 has been shown to induce a Ca2+-independent contraction of permeabilized smooth muscle (Van Eyk et al., 1998; McFawn et al., 2003). Furthermore, Pak also has been demonstrated to phosphorylate CPI-17 (Takizawa et al., 2002). Recently, the role of Rac1 in force regulation has been further delineated (Rahman et al., 2014). Using a smooth muscle-specific, conditional Rac1 KO, these investigators demonstrated that the decrease in Rac1 reduced force in both bladder and saphenous arterial smooth muscle. Furthermore, the inhibition of Rac1 with EHT1864, which affects nucleotide binding, decreased the Ca2+ transient and force produced in response to depolarization, agonist activation, and activation of PKC. However, inhibition of Rac1 with NSC23766, which blocks the interaction of Rac1 with GEFs, decreased force for phenylephrine (α-agonist) activation but increased the force produced by stimulation with both prostaglandin F2α and thromboxane (U46619). These results suggest that Rac1 signaling is agonist dependent and can result in either an enhancement of the Ca2+ transient and force or an inhibition of Ca2+ sensitization.
The well documented role of RhoA/Rho kinase signaling for the inhibition of MLC phosphatase and Ca2+ sensitization (Somlyo and Somlyo, 2003) could suggest that Ca2+ sensitization of vascular smooth muscle cells contributes to the molecular mechanism for the increase in vascular tone and/or systemic vascular resistance that produces hypertension. Consistent with this hypothesis are the results demonstrating that in SHR compared with control rats, both the sensitivity to G-protein-coupled agonists and the magnitude and sensitivity of Ca2+ sensitization are increased (Satoh et al., 1994) as well as studies defining the role of G12-G13-induced activation of Rho kinase-mediated Ca2+ sensitization for the development of DOCA salt-sensitive hypertension (Wirth et al., 2008). Furthermore, the infusion of the Rho kinase inhibitor Y-27632 reduced blood pressure in normal Wistar rats, as well as several animal models of hypertension including the SHR and DOCA salt-sensitive rat models of hypertension (Uehata et al., 1997), as well as L-NG-Nitroarginine Methyl Ester (L-NAME)-induced hypertension (Seko et al., 2003). Additionally, the expression and activity of Rho kinase are higher in vascular smooth muscle isolated from the SHR compared with controls (Mukai et al., 2001), and the specific Rho kinase inhibitor fasudil (Uehata et al., 1997) reduced blood pressure in the SHR model of essential hypertension (Mukai et al., 2001).
Rho/Rho kinase signaling has also been implicated as an important contributor for the regulation of vascular tone in other mammals; the Rho kinase inhibitor HA1077 has been shown to dilate canine coronary arteries (Asano et al., 1989). Furthermore, Rho kinase is upregulated in a porcine model of coronary vasospasm, and in this model, inhibition of Rho kinase with Y-27632 decreases coronary vasospasm (Kandabashi et al., 2000). In humans, fasudil has been demonstrated to be effective in treating the cerebral vasospasm associated with subarachnoid hemorrhage (Tanaka et al., 2005; Kim et al., 2006), and fasudil is approved for treating patients with pulmonary hypertension (Archer et al., 2010). In humans, Masumoto et al. (2001) demonstrated that brachial artery infusion of fasudil did not decrease systemic blood pressure, but did produce a dose-dependent increase in forearm blood flow in hypertensive patients, but not controls without hypertension. Sodium nitroprusside, on the other hand, resulted in comparable increases in forearm blood flow in both hypertensive and normal humans. These results suggest that an increase in RhoA/Aho kinase signaling may be involved in the molecular mechanism that produces essential hypertension, and development of drugs that target this pathway could represent an effective, novel class of therapeutic agents.
C. Phenotypic Switching of Contractile Proteins during Development and Disease: Role of MYPT1 in Ca2+ Sensitization/Desensitization
1. Smooth Muscle Myosin Heavy Chain.
Four isoforms of the smooth muscle myosin heavy chain (SM MHC) are produced by alternative splicing of a single gene. Alternative splicing of a 5′-site produces two isoforms that express a unique aa sequence of either 43 (SM1, 204 kDa) or 9 (SM2, 200 kDa) residues at the carboxy terminus of the SM MHC tail (Babij and Periasamy, 1989; Nagai et al., 1989), whereas alternative splicing of a 21-bp insert produces a difference of 7 aa near the ATP binding site of the SM MHC (Kelley et al., 1993). Although SM1 or SM2 homodimers are more common than herterodimers in a single myosin rod, SM1 and SM2 homodimers will copolymerize and assemble into side polar thick filaments (Rovner et al., 2002). Estrogen has been demonstrated to increase the ratio of SM1/SM2 expression, and this shift in the expression of SM1 MHC isoform has been suggested to contribute to changes in the sensitivity to the agonist norepinephrine and KCl depolarization (Paul et al., 2007). However, others have demonstrated in the motility assay, there is no difference in the ability of SM1 and SM2 to translate actin (Rovner et al., 2002). These investigators also demonstrated that although there is no difference in the length of SM1 and SM2 filaments, the differences in SM1 and SM2 at the carboxy terminus of the myosin tail influenced filament packing and stability; SM1 filaments have greater stability (Rovner et al., 2002). Consistent with these results, others have demonstrated that the smooth muscle thick filaments of the SM2 KO are similar in length to that of WT mice, but the SM1 thick filaments isolated from SM2 KO mice are smaller in diameter and there are fewer thick filaments per high-powered field (Chi et al., 2008). However, despite the decrease in the number of smooth muscle myosin thick filaments and a concomitant decrease in the expression of SM1, the force produced by both KCl depolarization and carbachol activation was higher in the SM2 KO (Chi et al., 2008).
The other SM MHC isoform is due to a 7-aa insert at the amino terminus, near the ATP binding site of the SM MHC (Kelley et al., 1993). The 7-aa insert is expressed in both SM1 and SM2 SM MHCs, and the SM MHC with the 7-aa insert has been referred to as SMB, whereas SM MHC lacking the insert is SMA (Kelley et al., 1993). SMB, when compared with SMA, has been demonstrated to have a twofold higher AMATPase activity and a 2.5-fold faster velocity of actin translation in the in vitro motility assay (Kelley et al., 1993). Whether the presence of the insert confers a functional difference to the mechanical properties of smooth muscle was assessed using bladder smooth muscle isolated from a transgenic mouse line; the maximum velocity of muscle shortening of bladder smooth muscle strips from WT (SMB+/+) is higher than that of either of heterozygous (SMB+/−) animals or the SMB KO mice (Babu et al., 2001; Karagiannis et al., 2004). Further analysis of the mechanical responses of Ca2+-activated skinned bladder strips to elevated Pi and ADP suggested that the lower shortening velocity of the SMA isoform is due to a slower rate of ADP dissociation or an additional force producing isomerization of the AM-ADP state in the crossbridge cycle (Karagiannis et al., 2003). These results demonstrate that the insert near the SM MHC ATP binding site will alter the mechanical properties of smooth muscle, and the duty cycle of SMA should be higher than SMB, which would be predicted to increase vascular tone and/or vascular resistance and thus blood pressure. Consistent with this prediction are the results demonstrating that compared with WT mice, the isometric force for mesenteric vessels of SMB KO mice was increased (Babu et al., 2004). However, the blood pressure of SMB KO animals compared with WT controls has not been reported, and whether SMA/SMB isoform expression is altered during hypertension has not been investigated.
2. ELC17.
For the 17-kDa essential myosin light chain (ELC17), alternative splicing of exon six, which encodes 44 bp, also produces two isoforms, which differ in the expression of 9 aa at the carboxy terminus of the ELC17 (Nabeshima et al., 1987; Lenz et al., 1989; Hasegawa and Morita, 1992). Exon 6 is excluded in the nonmuscle, more basic isoform (ELC17a), whereas exon 6 is included in the smooth muscle, more acidic isoform (ELC17b) (Hasegawa and Morita, 1992; Kelley et al.,1993). Changes in the ELC17 could affect the stiffness of the SM MHC lever arm, and thus myosin step size and or unitary force. However for ELC17 isoforms, the motility assay does not show any difference in the velocity of actin translocation (Kelley et al., 1993; Quevillon-Cheruel et al., 2000). Nonetheless in fast smooth muscle, the expression of ELC17a and SMA is higher than in slow smooth muscle (Malmqvist and Arner, 1991).
3. MYPT1.
Alternative mRNA splicing produces four splice variant MYPT1 isoforms, formed by the presence or absence of a 43 aa central insert (CI+/−) and carboxy-terminal leucine zipper (LZ+/−). The expression of the CI (residues 512–552) (Shimizu et al., 1994) is both developmentally regulated and tissue specific (Dirksen et al., 2000), and phosphorylation of MYPT1 at Thr696 has been demonstrated to inhibit phosphatase activity (Ichikawa et al., 1996). Although there is evidence that the CI+ MYPT1 isoform may be preferentially phosphorylated during Ca2+ sensitization (Richards et al., 2002), a role for the CI for the regulation of smooth muscle contractile properties has not been established. The MYPT1 LZ+ isoform is generated by the exclusion of a 3′- to 31-bp exon, whereas exon inclusion generates a LZ− MYPT1 isoform (Khatri et al., 2001). The aa sequence of the MYPT1 LZ domain is identical from avians to mammals and 75% identical in mammals and worms (Khatri et al., 2001), which could suggest that this domain is important for the regulation of MLC phosphatase activity. Surks et al. (1999) were the first to demonstrate an important functional role for the MYPT1 LZ domain. These investigators demonstrated that the LZ MYPT1 domain was important for the interaction of MYPT1 and PKG (Surks et al., 1999; Surks and Mendelsohn, 2003). Subsequently it has been demonstrated that PKG interacts with the LZ domain (Lee et al., 2007; Sharma et al., 2008) as well as a MYPT1 domain between aa 888 and 928 (Given et al., 2007; Sharma et al., 2008), but the LZ domain is necessary for the PKG-mediated activation of the MLC phosphatase during Ca2+ desensitization (Fig. 6,; Ca2+ desensitization) and/or nitric oxide (NO)-mediated vasodilatation (Huang et al., 2004). PKG phosphorylates MYPT1 at Ser695 and Ser849, which excludes the Rho kinase-mediated MYPT1 phosphorylation at Thr696 and Thr850, to prevent a Rho kinase-mediated decrease in MLC phosphatase activity (Wooldridge et al., 2004; Nakamura et al., 2007), but MYPT1 phosphorylation at these sites by PKG does not increase MLC phosphatase activity (Nakamura et al., 2007). The mechanism for the increase in MLC phosphatase activity during NO/cGMP-mediated Ca2+ desensitization was recently defined; PKG phosphorylates only LZ+ MYPT1 isoforms at Ser668 (Yuen et al., 2011, 2014), and the Ser668 MYPT1 phosphorylation increases the activity of MLC phosphatase (Yuen et al., 2011; Yuen et al., 2014). MYPT1 LZ+/− isoform expression is developmentally regulated, tissue specific (Khatri et al., 2001; Payne et al., 2006), and modulated in animal models of heart failure (Karim et al., 2004; Chen et al., 2006; Ararat and Brozovich, 2009; Han and Brozovich, 2013), pre-eclampsia (Lu et al., 2008), portal hypertension (Payne et al., 2004; Lu et al., 2008), pulmonary hypertension (Konik et al., 2013), nitrate tolerance (Dou et al., 2010), and sepsis (Reho et al., 2015). Furthermore, a decrease in LZ+ expression decreases the sensitivity to NO-mediated vasodilatation (Huang et al., 2004; Yuen et al., 2011, 2014). Thus, the molecular mechanism that underlies the differential response of the vasculature to NO and NO-based vasodilators is in part due to differential expression of LZ+/− MYPT1 isoforms, and furthermore, changes in the relative LZ+/− expression could tune the vasculature between a low-resistance vascular bed (relatively vasodilated), which is NO responsive and Rho kinase/PKC resistant, and a high-resistance vascular bed (relatively vasoconstricted) that is resistant to NO but responsive to Rho kinase/PKC.
Fig. 6.

Ca2+ desensitization: ACh stimulation of muscarinic receptors on the vascular endothelium leads to the production of NO, and NO diffuses into smooth muscle cells to activate guanylate cyclase. The NO/cGMP signaling pathway relaxes smooth muscle by both decreasing intracellular Ca2+ and activating MLC phosphatase, which results in Ca2+ desensitization of the contractile filaments (see text for details).
The importance of the LZ domain of MYPT1 for the regulation of vascular tone has been established using two different transgenic mice. Michael et al. (2008) produced a transgenic mouse with mutations in the LZ domain of PKG, which disrupts the interaction of PKG with MYPT1. Compared with control littermates, the vascular smooth muscle isolated from these mice were less sensitive to NO-mediated vasodilatation, and thus these mice were hypertensive. In addition and also consistent with an important role for LZ+ MYPT1 isoform expression for the regulation of vascular tone are the results demonstrating that transgenic mice expressing only the LZ+ MYPT1 isoform are more sensitive to NO-mediated relaxation and hypotensive compared with WT littermates (Reho et al., 2015).
Because NO-mediated vasodilation is a fundamental property of the vasculature (Furchgott and Zawadzki, 1980; Furchgott, 1999), preservation of the normal response to NO by maintaining the normal LZ+ MYPT1 isoform expression could improve the outcome for the treatment of diseases of the vasculature. For the treatment of heart failure, both ACE inhibitors (Chen et al., 2006; Chen and Brozovich, 2008) and ARBs (Ararat and Brozovich, 2009) have been demonstrated to preserve both LZ+ MYPT1 expression and the sensitivity to NO-mediated vasodilatation, which could underlie the beneficial effects of inhibition of angiotensin II signaling in the treatment of heart failure compared with other vasodilators (Pfeffer et al., 1992; Yusuf et al., 1992, 2000; Pitt et al., 2000; Granger et al., 2003). Additionally, differential expression of LZ+/− MYPT1 isoforms in patients with heart failure could underlie the mortality benefit of ACE inhibitors in white compared with black patients. ACE inhibitors reduced mortality in white patients, whereas in black patients, although ACE inhibitors did not show a benefit, treatment with the combination of hydralazine and isosorbide dinitrate reduced mortality (Carson et al., 1999).
The regulation for LZ+/− MYPT1 expression is unknown, but LZ+/− MYPT1 expression is both developmental regulated and tissue specific (Khatri et al., 2001; Payne et al., 2006), as well as modulated during disease states (Karim et al., 2004; Payne et al., 2004; Lu et al., 2008; Fisher, 2010). During heart failure (HF), p42/44 MAP kinase is activated and the expression of LZ+ MYPT1 decreases (Ararat and Brozovich, 2009), and further losartan therapy prevents the activation of p42/44 MAP kinase and preserves LZ+ MYPT1 expression (Ararat and Brozovich, 2009). Additionally, the expression of LZ+ MYPT1 isoforms as well as Tra-2β, an atypical member of RNA binding proteins, are higher in fast (phasic) compared with slow (tonic) smooth muscle, and in an animal model of portal hypertension, Tra-2β is downregulated coincident with the decrease in LZ+ MYPT1 expression (E23 exon exclusion) and the shift to the expression of LZ− MYPT1 isoforms (Shukla and Fisher, 2008). These investigators demonstrated both that Tra-2β binds to E23 and transactivates E23 splicing (exon inclusion) to generate a LZ− MYPT1 isoform and deletion or mutation of the Tra2β binding site abolished E23 splicing (exon exclusion) to produce a LZ+ MYPT1. These investigators also showed that an siRNA-induced decrease of Tra-2β decreased E23 splicing to produce an increase in LZ+ MYPT1 expression, which is consistent with Tra-2β expression regulating E23 splicing and LZ+/− MYPT1 expression (Shukla and Fisher, 2008). Further investigation of the signaling pathway for the regulation of LZ+/− MYPT1 isoform expression could reveal novel targets in this pathway. Small molecules could be designed to modulate either total MYPT1 or LZ+ MYPT1 expression, which would both improve both the vascular response to endogenous NO and pharmacological response during the treatment of hypertension as well as a number of other diseases of the vasculature.
D. Implications for Disease and Treatment
1. Pressurized Resistance Vessels, Implications of the Myogenic Response for Hypertension, and Critical Analysis of Inhibitors.
In most systems, flow and pressure are linearly related; as pressure increases, so does flow. However, small arteries vasoconstrict in response to an increase in pressure and vasodilate in response to a decrease in pressure, and the change in vessel diameter in response to changes in intravascular pressure is referred to as the myogenic response (Bayliss, 1902; Lassen, 1959; Davis and Hill, 1999). The myogenic response is an intrinsic property of the small resistance vessels and does not require flow (Davis and Hill, 1999). Furthermore, because of the intrinsic myogenic response as well as the modulatory actions of vasoactive substances (Bayliss, 1902; Lassen, 1959; Davis and Hill, 1999; Walsh and Cole, 2013), the resistance vessels maintain a constant blood flow over a wide range of perfusion pressures, and this property is termed the autoregulation of blood flow (Bayliss, 1902; Lassen, 1959; Davis and Hill, 1999).
Hypertension is associated with abnormalities of the myogenic response (Immink et al., 2004; Jarajapu and Knot, 2005; Kim et al., 2008b), and thus, the mechanism underlying the myogenic response is important in both health and disease. There are significant regional as well as vessel dimension differences in the magnitude and mechanism governing the autoregulation of blood flow (Jones et al., 1995; Davis and Hill, 1999). Further complicating investigation of the mechanism(s) responsible for the myogenic response are the small size (<200 μm diameter) of the resistance vessels; until recently, investigation of the signaling pathways regulating the myogenic response of the small resistance arteries was not possible. But, recent improvements in the sensitivity of protein phosphorylation levels have allowed for direct investigation of signaling pathways that regulate the myogenic response (Takeya et al., 2008; Johnson et al., 2009; El-Yazbi et al., 2010; Moreno-Dominguez et al., 2013).
Although the signaling pathway for the myogenic response is still being investigated, the first element is the mechanosensor that responds to the changes in intraluminal pressure. Martinez-Lemus et al. demonstrated that blocking integrin function with either anti-integrin antibodies (Martinez-Lemus et al., 2005) or integrin-specific peptides (Martinez-Lemus et al., 2003) results in a significant inhibition of the myogenic response. Furthermore, both the activation of integrins and the myogenic response are associated with tyrosine phosphorylation and the activation of focal adhesion kinase (FAK) and Src family tyrosine kinases (Murphy et al., 2001; 2002). Additionally, agonist stimulation of smooth muscle lead to a tyrosine phosphorylation of the protein paxillin (Pavalko et al., 1995) as well as activation of p42/44 MAPK (Spurrell et al., 2003) and the L-type Ca2+ channel (Chan et al., 2010). These data suggest that because of their ability to link the extracellular matrix to the cytoskeleton of the smooth muscle cell, integrins participate in the sensing and transmission of changes in intravascular pressure.
There are a number of studies that have demonstrated an important role for TRP channels Earley and Brayden (2015), specifically TRPC6 and TRPM4 in the myogenic response. Gonzales et al. (2014) demonstrated that selective inhibition of TRPM4 decreased the transient inward cation current induced by membrane stretch. These investigators demonstrated that the generation of IP3 by PLCγ1 and the subsequent Ca2+ release from internal stores is required for both TRPM4 activity and myogenic tone. Furthermore, both TRPC6 inhibitors and antibodies that bind to an extracellular epitope of TRPC6, which block TRPC6 currents, attenuated the stretch-induced activation of TRPM4 current. These investigators also demonstrated that the inhibition of Src nonreceptor tyrosine kinases, which signal through PLCγ, decreased myogenic tone. These results suggest that Src tyrosine kinase activity is important in the stretch-induced increases PLCγ, which generates IP3. Subsequently, IP3 binds to the IP3 receptor, which stimulates Ca2+ release from the SR. Thus, in response to stretch, a local increase in Ca2+ generated by both TRPC6 and IP3 are important for the activation of TRPM4, which changes membrane potential and increases the conductance of voltage-dependent Ca2+ channels, which results in smooth muscle cell contraction to generate the myogenic response.
Additionally, both Ca2+ release and Ca2+ sensitization of the contractile filaments contribute to the myogenic response. The pressure-induced increase in wall tension leads to depolarization of the smooth muscle cells, which results in opening of voltage-gated Ca2+ channels (Knot and Nelson, 1998; Davis and Hill, 1999) and an increase in intracellular Ca2+. However, in addition to the pressure-induced increase in Ca2+, other mechanisms contribute to the myogenic response (Worley et al., 1991; Osol et al., 2002). Using a highly sensitive biochemical technique, El-Yazbi et al. (2010) demonstrated that only in the presence of myogenic tone, serotonin stimulation produced a Rho kinase-mediated phosphorylation of MYPT1, which induced a Ca2+ sensitization of the contractile filaments. Furthermore, this group has also demonstrated that both a Rho kinase-mediated phosphorylation of MYPT1, as well as a Rho kinase- and PKC-mediated increase in actin polymerization are important determinants of the myogenic response (Moreno-Dominguez et al., 2013; El-Yazbi et al., 2015). The activation of the Ca2+-dependent tyrosine kinase, Pyk2, has also been demonstrated to occur with KCl depolarization, and although the inhibition of Pyk2 did not change the rapid increase in force and RLC phosphorylation in response to KCl depolarization, Pyk2 inhibition did depress RLC phosphorylation, Thr-696, and Thr-850 MYPT1 phosphorylation and force during the sustained phase of the contraction (Mills et al., 2015). These results suggest that a Ca2+-induced activation of Pyk2 leads to an activation of RhoA/Rho kinase and Ca2+ sensitization, which is important for force maintenance, or the tonic phase of smooth muscle contraction.
Because hypertension is associated with abnormalities of the myogenic response (Immink et al., 2004; Jarajapu and Knot, 2005; Kim et al., 2008b), antihypertensive agents aimed at the signaling pathway for the myogenic response should be effective for the control of blood pressure. Drugs that decrease Ca2+ influx as well as agents that decrease and/or block the activation of Rho kinase signaling such as ACE inhibitors, ARBs, Rho kinase inhibitors should be and are effective antihypertensives. However, novel agents designed to decrease the sensitivity of the mechanosensor such as blocking the activation of integrins or decreasing tyrosine kinase activation could reduce blood pressure. However, tyrosine kinase inhibitors used for the treatment of carcinomas are known to be cardiotoxic (Chu et al., 2007; Force et al., 2007), and hypertension is the most common cardiovascular side effect (Chu et al., 2007). The mechanism by which tyrosine kinase inhibition produces hypertension is not well understood, but hypothesized to be due to fluid retention, endothelial dysfunction, and an inhibition of NO (Cabanillas et al., 2011). These data demonstrate the complex interplay between the myogenic response, circulating vasoactive substances, and the kidney in the regulation of blood pressure.
2. Smooth Muscle Myosin versus Nonmuscle Myosin, Implications for Force Maintenance and Vascular Tone.
There are three classes of nonmuscle (NM) myosin (Golomb et al., 2004), NMIIA, NMIIB, and NMIIC, and both NMIIA and NMIIB are expressed in smooth muscle (Gaylinn et al., 1989; Morano et al., 2000; Lofgren et al., 2003; Eddinger et al., 2007; Yuen et al., 2009; Guvenc et al., 2010; El-Yazbi et al., 2015), whereas NMIIC is only expressed in neuronal tissue (Golomb et al., 2004; Jana et al., 2009). Both NMIIA and NMIIB are able to form bipolar thick filaments (Billington et al., 2013), and similar to SM, myosin RLC phosphorylation promotes filament formation (Ikebe and Hartshorne, 1985; Applegate and Pardee, 1992). In smooth muscle, NM myosin expression represents ∼10–15% of total myosin (Yuen et al., 2009; Konik et al., 2013). Furthermore, NMIIA has been demonstrated to form mixed bipolar filaments with myosin 18A (Billington et al., 2015), and SM1 and SM2 will copolymerize to form filaments (Rovner et al., 2002). However, it is unclear if NM myosin and SM myosin copolymerize to form mixed filaments or whether two distinct pools of SM and NM myosin thick filaments exist within the smooth muscle cells.
The kinetics of the individual steps of the AMATPase of NMIIB (Wang et al., 2003) and NMIIA (Kovacs et al., 2003) are slower than other types of class II myosin. NMIIA and NMIIB have a high ADP affinity, and for NMIIB, the rate of ADP release is similar to that of the steady-state ATPase. Additionally for NMIIB, actin augments ADP binding rather than accelerating ADP release (Wang et al., 2003), which results in NMIIB spending the majority of its kinetic cycle in states that are strongly bound to actin (Rosenfeld et al., 2003; Wang et al., 2003). On the other hand, the rate of ADP release for NMIIA is one order of magnitude faster than the ATPase, which results in NMIIA spending much less of its ATPase cycle in strongly actin bound states (Kovacs et al., 2003). Although the slow kinetics of NMIIA suggest that NMIIA could contribute to force maintenance, the high duty ratio make NMIIB an ideal candidate for the so called “latch crossbridge” (Dillon et al., 1981; Dillon and Murphy, 1982; Hai and Murphy, 1989), and NM myosin may be an important component for the sustained phase of smooth muscle contraction.
Kovacs et al. (2007) recently extended his kinetic studies and demonstrated that ADP release from NMIIB is slow and strain dependent; positive strain increases the rate of ADP release by a factor of fourfold, whereas negative strain decreases ADP release by 12-fold. Load dependence of ADP release prevents NMIIB from slowing either shortening or the rate of force generation by smooth muscle myosin. However for NMIIB, negative strain increases the duty ratio, which would contribute to force maintenance (Kovacs et al., 2007); i.e., for NMIIB, rapid ADP binding and load-dependent ADP release prolongs the attachment of NMIIB to actin at 10–100 μM ADP (at normal MgATP), which would decrease the rate of ATP usage to <0.01 ATP per head per second during force maintenance (Kovacs et al., 2007). Furthermore, during force maintenance both heads of the NMIIB would be attached to actin, which is ideal for a crossbridge that maintains tone.
Similar to NM myosin, the kinetics of SM myosin have been demonstrated to depend on load (Veigel et al., 2003). Using optical tweezers, these investigators demonstrated that the displacement of the SM crossbridge occurs in successive steps of 4 and 2 nm. The duration of the first phase (4-nm displacement) is strain dependent, increasing by twofold with a negative strain and decreasing by twofold with positive strain. These results could suggest that the increase in attachment time of SM myosin due to the negative strain on the SM crossbridge during force maintenance could contribute to the latch state. Furthermore, recent data from optical trap experiments demonstrated that the attachment time of smooth muscle myosin to actin varies with SM RLC phosphorylation (Tanaka et al., 2008); when only one of the two heads of smooth muscle myosin is phosphorylated, the dwell time was fit with a double exponential with rates of 24 second−1 and 1 second−1, compared with a single rate of 29 second−1 when both heads were phosphorylated. These investigators suggested that the long attachment time of the singly phosphorylated myosin could explain the latch state, or force maintenance, in smooth muscle. However, experiments in the optical trap are performed at low ionic strength to both promote actin-myosin interaction and increase interaction times; at physiologic conditions, rates are much faster. Tanaka et al. (2008) also demonstrated that the ATP turnover rate of singly phosphorylated myosin was 30% of that compared with doubly phosphorylated myosin, which contrasts with the results of Rovner et al. (2006) who demonstrated that the ATP turnover rate of myosin with a single phosphorylated head was over 50% of that when both heads were phosphorylated. The reason for the discrepancy between the results for the ATP turnover is unclear. However, it is likely that the single molecule mechanics of NM myosin with the RLC of one or both heads phosphorylated would be similar to smooth muscle myosin, although much slower, making NM myosin a more attractive candidate for a latch crossbridge.
The regulation of NM myosin has been studied in nonmuscle cells (Kolega, 2003), and similar to smooth muscle myosin, NM myosin is regulated by phosphorylation of its regulatory light chain (NM RLC); NM RLC phosphorylation promotes NM myosin filament assembly (Kolega, 2003) and also results in a 10-fold increase in the Vmax of the NM myosin AMATPase (Cremo et al., 2001). In nonmuscle cells, Rho kinase phosphorylates the NM RLC at Ser19 (Kolega, 2003), and in epithelial cells, results are consistent with both MLCK and Rho kinase as important for the phosphorylation of NM RLC (Connell and Helfman, 2006). In epithelial carcinoma cell lines, Rho kinase increases the phosphorylation of the NM RLC at Ser19 phosphorylation of both NMIIA and NMIIB (Sandquist et al., 2006). During KCl depolarization of smooth muscle, both SM RLC and NM RLC phosphorylation increase, and the increase in RLC phosphorylation is not dependent on either Rho kinase or PKC (Yuen et al., 2009). However for angiotensin II (Ang II) activation, inhibition of either Rho kinase or PKC blunted SM RLC phosphorylation, whereas only a Rho kinase-dependent pathway regulated NM RLC phosphorylation (Yuen et al., 2009). These results demonstrate that similar to SM myosin, NM myosin is regulated in smooth muscle, and both MLCK and Rho kinase regulate the activation of NM myosin (Fig. 7).
Fig. 7.

Force maintenance involving either nonmuscle myosin or smooth muscle myosin: There are several proposed mechanisms for force maintenance in smooth muscle including the interaction of actin with either smooth muscle or NM myosin (latch crossbridge) as well as changes in the cytoskeleton (see text for details and see also Fig. 3).
Recent evidence from mechanical studies also suggests that NM myosin contributes to the force maintenance phase of smooth muscle tissue contraction. Morano et al. (2000) showed that bladder smooth muscle from transgenic mice lacking smooth muscle myosin still contract, albeit with a very slow tonic response, as opposed to the rapid phasic contraction (with transient peaks in force and Vmax) characteristic of wild-type bladder smooth muscle (Morano et al., 2000; Lofgren et al., 2003). The force produced by NM myosin in KO tissues is low (Lofgren et al., 2003), which would suggest that NM myosin will not participate in the rapid phase of force activation, but rather the kinetics of NM myosin are tuned for force maintenance (Kovacs et al., 2007). Consistent with this hypothesis are results demonstrating that the inhibition of the NM AMATPase with blebbistatin reduced force maintenance (Rhee et al., 2006); for phasic smooth muscle, blebbistatin did not affect the rapid rise in force, but decreased maintained force, and for tonic smooth muscle, blebbistatin decreased force maintenance. However, although blebbistatin is thought to be specific for the inhibition of the NM myosin AMATPase (Straight et al., 2003), the specificity of blebbistatin for the NM versus SM AMATPase has been questioned (Eddinger et al., 2007). Nonetheless, consistent with a role of NM myosin for force maintenance are the results with heterozygous NMIIB KO mice; when compared with WT control mice, force maintenance is depressed by 25% in smooth muscle of heterozygous NMIIB KO (Yuen et al., 2009). Interestingly, Sward et al. (2000) demonstrated for carbachol activation of guinea pig ileum that inhibition of Rho kinase did not affect peak force but decreased force maintenance, and Rho/Rho kinase signaling regulates NM RLC phosphorylation (Yuen et al., 2009). These results are consistent with NM myosin participating in force maintenance (Morano et al., 2000; Lofgren et al., 2003; Rhee et al., 2006; Yuen et al., 2009; Guvenc et al., 2010), and the inhibition of Rho kinase would lead to a decrease in the activation of NM myosin (Yuen et al., 2009), which results in a reduction in both the force maintenance phase of smooth muscle contraction and vascular tone (SVR). It is also interesting to speculate on the contributions of other pathways, which have been demonstrated to be important contributors for changes in vascular tone for the regulation of NM myosin including tyrosine kinase (Moreno-Dominguez et al., 2013; El-Yazbi et al., 2015; Mills et al., 2015).
3. Force Maintenance/Latch and the Regulation of Vascular Tone: The Tonic versus Phasic Contractile Phenotype and Contributions to Pathogenesis of Hypertension.
Smooth muscle contractile properties have been classified as phasic or tonic (Somlyo and Somlyo, 1968); after activation, for phasic smooth muscle, force rises rapidly to a peak before falling to a lower steady-state level, whereas for tonic smooth muscle, force slowly increases to a sustained steady state. Smooth muscle has also been termed “fast” and “slow” because of the differences in Vmax. The molecular mechanism that governs tonic and/or phasic contractile properties has yet to be elucidated, although emerging evidence suggests that there are tonic and phasic contractile phenotypes (see Fisher, 2010). In general, the fast isoforms of the SM MHC (SMB) and ECL17 (ECL17a) are expressed in phasic smooth muscle while slow isoforms (SMA and ECL17b) expression predominates in tonic smooth muscle (Malmqvist and Arner, 1991). Similarly, the expression of NM myosin is significantly higher in tonic smooth muscle (Lofgren et al., 2003; Rhee et al., 2006). In addition, there are differences in the expression of the regulatory proteins in phasic and tonic smooth muscle. The expression of MLCK (Gong et al., 1992), MYPT1 (Woodsome et al., 2001), and the LZ− MYPT1 isoform (Dirksen et al., 2000; Khatri et al., 2001) is higher in phasic compared with tonic smooth muscle. Furthermore, in phasic versus tonic smooth muscle, the expression of CPI-17 is lower (Woodsome et al., 2001), whereas telokin expression is higher (Gallagher et al., 1991; Wu et al., 1998; Herring et al., 2001). These results could suggest that a gene program exists that governs the differential expression of contractile proteins in fast (phasic) versus slow (tonic) smooth muscle (Fisher, 2010), and the contractile phenotype regulates systemic vascular resistance.
Resistance is inversely related to the vessel radius to the forth power (1/r4), and thus, SVR is predominantly regulated at the level of the small resistance arteries with a diameter of 50–300 μm. Because of their small size, the molecular contractile phenotype of the resistance vessels have not been fully characterized, but the small resistance vessels express a mixture of fast and slow contractile proteins (Fisher, 2010) and exhibit a mixture of tonic and phasic contractile activity, which is referred to as vasomotion (Peng et al., 2001; Haddock and Hill, 2005). The molecular basis of essential hypertension is unknown, but the molecular contractile phenotype is known to be modulated during disease in both the large conduit vessels (Karim et al., 2004; Chen et al., 2006; Ararat and Brozovich, 2009), as well smaller resistance vessels (Zhang and Fisher, 2007; Han and Brozovich, 2013). In the small resistance vessels, an increase in the relative expression of protein isoforms associated with the tonic contractile phenotype (i.e., an increase in NM myosin expression) or decrease in LZ+ MYPT1 isoform expression would produce an increase in vascular tone and/or SVR, which would produce hypertension.
4. Autoregulation of Vascular Resistance/Flow-Mediated Vasodilatation and Nitric Oxide Signaling with Analysis of Current Inhibitors.
Flow is governed by the simple equation, flow = pressure/resistance, and the ability of NO, or flow, to mediate changes in vascular tone is considered a fundamental property of the vasculature (Furchgott, 1999). The autoregulation of blood flow to a vascular bed maintains a constant flow over a wide range of pressures to ensure that perfusion is maintained despite either hypo- or hypertension. The mechanism governing the autoregulation of blood flow has yet to be fully elucidated but is known to be dependent on the intrinsic myogenic response as well as the modulatory actions of vasoactive substances (Lassen, 1959; Walsh and Cole, 2013), which importantly includes the vascular response to NO. NO is produced by the vascular endothelium in response to shear stress. The NO produced by the endothelium diffuses into the smooth muscle cells where it activates the soluble pool of guanylate cyclase and results in an increase in cGMP. cGMP activates protein kinase G (PKGI), which has a number of targets that produce smooth muscle relaxation (Lincoln, 1989; Schmidt et al., 1993; Alioua et al., 1998; Fukao et al., 1999; Lincoln et al., 2001), including myosin light chain (MLC) phosphatase (Surks et al., 1999). As perfusion pressure increases, the subsequent increase in blood flow will increase shear stress on the endothelial cells, which will stimulate NO production and a subsequent vasodilatation to decrease flow. Conversely, a decrease in perfusion pressure will decrease endothelial shear stress and NO production, and the resulting vasoconstriction will increase flow. Therefore, the NO-induced changes in vascular resistance can be viewed as part of a negative feedback loop that blunts the myogenic response; the myogenic response generates a vasoconstriction with an increase in pressure and vasodilatation with a decrease in pressure (Bayliss, 1902; Lassen, 1959; Davis and Hill, 1999; Walsh and Cole, 2013). Thus, NO production regulates vascular resistance and is essential for the normal regulation of blood flow.
The sensitivity and response of the vasculature to NO and NO-based vasodilators are well known to be heterogenous, and the molecular basis for this variable response to NO is controversial. Nonetheless, during NO signaling, activation of the MLC phosphatase requires the expression of a LZ+ MYPT1 isoform (Surks et al., 1999, 2003; Huang et al., 2004; Yuen et al., 2011, 2014) and in both health (Khatri et al., 2001; Huang et al., 2004; Payne et al., 2006) and disease (Karim et al., 2004; Payne et al., 2004; Zhang and Fisher, 2007; Lu et al., 2008; Dou et al., 2010; Han and Brozovich, 2013; Konik et al., 2013). The sensitivity of the vasculature to NO-mediated vasodilation is regulated by the relative expression of LZ+/LZ− MYPT1 isoforms; i.e., an increase and/or decrease in LZ+ MYPT1 expression will produce an increase and/or decrease in the sensitivity to NO, respectively. Nitrates and nitrate-based vasodilators are a well-known class of antihypertensive agents that will decrease blood pressure in the acute setting. However, tolerance to nitrates is a well-known phenomenon, which may limit the efficacy of nitrates for the treatment of hypertension, and a decrease in LZ+ MYPT1 expression has been demonstrated to contribute to the molecular mechanism of nitrate tolerance (Dou et al., 2010). In the treatment of heart failure, both ACE inhibitors (Chen et al., 2006; Chen and Brozovich, 2008) and ARBs (Ararat and Brozovich, 2009) have been demonstrated to maintain the normal expression of LZ+ MYPT1 expression and sensitivity to NO-mediated vasodilatation. In essential hypertension, both whether changes in LZ+ MYPT1 expression contribute to the molecular mechanism producing this disease and strategies to improve LZ+ MYPT1 expression and the sensitivity to NO-based vasodilators for the treatment of hypertension have not been investigated.
5. Mouse Models (Contractile Protein Knockout) and Implications for Hypertension.
Several strains of genetically modified mice have been produced to evaluate the contribution of abnormalities in the regulation of vascular tone and/or vascular dysfunction to hypertension (Pfeifer et al., 1998; Brenner et al., 2000; Chutkow et al., 2002; Zhu et al., 2002; Tang et al., 2003; Michael et al., 2008; Wirth et al., 2008; Qiao et al., 2014), and experimental results and their implications will be discussed in this section.
Ca2+ signaling is well known to be important for the regulation of vascular tone (Arner and Pfitzer, 1999); the activation of voltage-gated Ca2+ channels increases cytoplasmic Ca2+ and results in vasoconstriction. However, the increase in Ca2+ also activates Ca2+-activated potassium channels (BK channels), and Ca2+ activation of these channels results in a membrane hyperpolarizing current that opposes vasoconstriction (Nelson et al., 1995) and K+ channel openers, such as nicorandil, hyperpolarize smooth muscle to produce vasodilatation (Nelson et al., 1990). Brenner et al. (2003) deleted the β1 subunit of the BK channel in mice and demonstrated that, compared with controls, the open probability of BK channels was 100-fold lower and the BK current in response to Ca2+ sparks was impaired in vascular smooth muscle from the β1 KO animals. Furthermore, myography demonstrated that cerebral arteries from the β1 KO animals mice were more constricted at all pressures, which resulted in hypertension in these animals. Additionally, BK channels have been demonstrated to increase their conductance in response to NO/cGMP signaling (Alioua et al., 1998).
In smooth muscle, contractile agonists have been demonstrated to activate the G-proteins Gq and G11 and stimulate phospholipase C, which leads to an increase intracellular Ca2+ and activation of MLCK to increase RLC phosphorylation and force (Somlyo and Somlyo, 2003). However, many vasoconstrictors also couple with G12 and G13 to activate Rho kinase-mediated signaling, which inhibits MLC phosphatase to produce a Ca2+-independent increase in force (Somlyo and Somlyo, 2003). Wirth et al. (2008) produced mice with smooth muscle-specific ablation of Gq-G11 or G12-G13 to investigate the relative contributions of Gq-G11 versus G12-G13 on vascular tone and the development of hypertension. In aortic smooth muscle isolated from the Gq-G11 KO mice, the contractile response to both phenylephrine and Ang II was completely blocked, whereas the response to serotonin, endothelin, vasopressin, and the thromboxane analog U46619 was inhibited. Ablation of G12-G13 had no effect on the dose-response relationship for phenylephrine or serotonin, but it reduced the steady-state force produced by Ang II, endothelin, vasopressin, and U46619. Furthermore, compared with WT mice, blood pressure was no different in the mice with ablation of G12-G13, but significantly lower in the Gq-G11 KO mice, indicating that the normal regulation of blood pressure requires Gq-G11 signaling. Additionally, ablation of either Gq-G11 or G12-G13 attenuated the increase in blood pressure produced by DOCA-salt treatment, which suggests that although G12-G13 signaling and the subsequent activation of a Rho kinase pathway leading to Ca2+ sensitization is not required for maintenance of normotension, it contributes to the development of DOCA-salt-dependent hypertension. These investigators also demonstrated that the RhoGEF protein LARG is important for the G12-G13-mediated activation of Rho kinase and DOCA-induced hypertension.
The importance of the NO/cGMP signaling pathways for the maintenance of a normal blood pressure has been established with several different models. In mice, the disruption eNOS has been demonstrated to eliminate ACh-mediated relaxation of aortic smooth muscle rings, and as would be predicted, the relaxation produced by sodium nitroprusside was no different in eNOS KO and WT animals. This defect in vascular reactivity resulted in hypertension in the eNOS KO mice (Huang et al., 1995). Similarly, PKGI KO mice were hypertensive compared with WT littermates (Pfeifer et al., 1998). These investigators demonstrated compared with WT, that although the response to contractile agonist was not different in aortic rings from the PKGI KO, ACh- and 8Br-cGMP-mediated relaxation was significantly attenuated. Further preincubation of aortic cells with 8Br-cGMP attenuated the Ca2+ transient in response to NE in WT aortic smooth muscle cells, but had no effect on the Ca2+ transient in the PKGI KO aortic smooth muscle cells.
Most contractile agonists activate Gq-coupled receptors, resulting in the activation of phospholipase C, the generation of IP3, Ca2+ release, and the activation of MLCK as well as Rho/Rho kinase and a resulting inhibition of the MLC phosphatase (Somlyo and Somlyo, 1994; Davis and Hill, 1999). Mendelsohn’s group (Tang et al., 2003) demonstrated that the dose-dependent increase in IP3 production by thrombin was inhibited by both S-nitrosocysteine and 8Br-cGMP. Because NO and cGMP inhibit thromboxane signaling via a PKG-mediated phosphorylation of the cytoplasmic tail of the thromboxane receptor (Wang et al., 1998) and RGS-2 terminates G-protein receptor signaling by accelerating the GTP hydrolysis rate by Gα subunits (Watson et al., 1996), these investigators (Tang et al., 2003) examined the function of RGS-2 in NO/cGMP signaling cascade. They demonstrated that phosphorylation of RGS-2 by PKG resulted in a translocation of RGS-2 from the cytosolic to particulate fractions and decreased Gαq GTPase activity. These data show that NO/cGMP-mediated activation of PKG results in a phosphorylation of RGS-2, which increases the activation of Gq-coupled receptors. Consistent with these data are the results that show agonist-induced force production was increased and the vasodilatory response to NO was reduced in aortic smooth muscle from RGS-2 KO mice, and the resulting increase in vascular tone (relative vasoconstriction) was responsible for the increase in blood pressure in RGS-2 KO mice compared with WT littermates (Tang et al., 2003). The importance of the NO/cGMP signaling cascade for the regulation of vascular tone and blood pressure has also been demonstrated in three recent studies (Michael et al., 2008; Qiao et al., 2014; Reho et al., 2015). Michael et al. (2008) produced a knock-in mouse in which the initial four leucine/isoluecine residues of PKGIα were replaced by alanine. This PKGIα LZ mutant does not interact with MYPT1 (Surks and Mendelsohn, 2003), and as would be predicted, when compared with WT, aortic rings isolated from the PKGIα LZ mutant were less sensitive to ACh- and 8Br-cGMP-mediated relaxation, which produced hypertension. Qiao et al. (2014) produced a conditional MYPT1 KO, which were hypertensive compared with WT controls. The lack of MYPT1, which would disrupt the ability of the MLC phosphatase to target to its substrate, the RLC, resulted in an increase in the phosphorylation of the RLC as well as force in response to both KCl depolarization and contractile agonists. However, surprisingly, the sensitivity of relaxation produced by NO/cGMP signaling was similar in the control and MYPT1 KO mice. The lack of a decrease in the sensitivity to NO of secondary branches of the mesenteric arteries from MYPT1 KO animals could be explained if the conditional KO of MYPT1 increased RGS-2 expression or decreased NO/cGMP-mediated PKG phosphorylation of RGS-2 or the functional replacement of MYPT1 by MBS85, which is a member of the MYPT family that is also expressed in smooth muscle (Hartshorne et al., 2004). Reho et al. (2015) generated a conditional deletion of MYPT1 E24 to produce a LZ+ MYPT1 KI mouse. Vascular tissue isolated for the LZ+ MYPT1 mice was more sensitive to NO/cGMP-mediated relaxation, and as would be expected, the mice were hypotensive compared with controls. Of course, it must be kept in mind that NO, cGMP, and PKG also can dilate vascular smooth muscle by more than one mechanism, thus pathways not involving MYPT1 are also possible but we are not aware of appropriate animal models yet available to test those possibilities.
Mendelsohn’s group has also demonstrated that NO signaling is regulated by estrogens; Zhu et al. (2002) compared the vascular responses in WT, iNOS KO, and estrogen receptor β KO mice. Estrogen was found to decrease PE-induced contraction of aortic smooth muscle in WT, but not iNOS KO mice. Furthermore, iNOS is upregulated by estrogen, as well as transfection with estrogen receptor β. In vascular rings from estrogen receptor β KO mice, compared with WT mice, the contractile response to PE was reduced, which could be the result of a decrease in iNOS in the estrogen receptor β KO tissues. The sensitivity of smooth muscle relaxation to sodium nitroprusside was similar in estrogen receptor β KO and WT animals. Nonetheless, in the estrogen receptor β KO animals, the abnormalities in vascular tone produced hypertension.
The data presented in this section demonstrate the importance of the regulation of vascular tone for the regulation of blood pressure and consistently demonstrate that disruption in the NO/cGMP signaling pathway reduces the sensitivity to NO-mediated vasodilatation, and this decrease in the vascular response to NO and/or NO-based vasodilators produces hypertension.
E. Summary of Contractile Phenotype and Contributions to Pathogenesis of Hypertension with Analysis of Current Therapies for Hypertension
Diuretics reduce blood pressure by decreasing intravascular volume, which mechanistically fits with a Guytonian view for the regulation of blood pressure (Guyton, 1991), and diuretics, as a class, are useful for treating essential hypertension (ALLHAT and Coordinators for the ALLHAT Collaborative Research Group, 2002; Rosendorff et al., 2007). Given the importance of Ca2+ signaling for the regulation of vascular tone (Arner and Pfitzer, 1999; Somlyo and Somlyo, 2003), the benefit of CCB for treating humans with essential hypertension is not surprising (ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group, 2002). Similarly, ACE inhibitors and ARBs will decrease the activation of the Ang II G-protein-coupled receptor and inhibit Ang II-stimulated increase in intracellular Ca2+ and activation of Rho kinase signaling to decrease vascular tone (Somlyo, 1997). Crowley et al. (2005) demonstrated that the KO of the AT1 receptor in only the peripheral vasculature decreases SVR and blood pressure, and thus, these agents would be expected to and have been demonstrated to be effective therapies of essential hypertension (Guyton, 1991; ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group, 2002). In fact, drugs that block any G-protein-coupled receptor, i.e., α-receptor blockers (Koshy et al., 1977), would be expected to decrease vascular tone and lower blood pressure. β-Blockers decrease the activation of the cardiac β-adrenergic receptor, and the subsequent decrease in cAMP and PKA signaling produce negative chronotropic and inotropic response, which decreases blood pressure (Rosendorff et al., 2007). However, in the vascular smooth muscle, an increase PKA activity produces a vasodilatation ((Nakamura et al., 2007; Wooldridge et al., 2004), and β-blocker therapy could potentially result in an increase in vascular tone, but because of the inhibition of central sympathetic output, SVR decreases with β-blocker therapy (Man in't Veld et al., 1988). Drugs designed to increase K+ channel conductance produce smooth muscle hyperpolarization and a resulting vasodilatation of vascular smooth muscle (Nelson et al., 1990; Sobey, 2001; Barbato, 2005), and these agents have been demonstrated to both decrease blood pressure (Wang et al., 2005) and be effective treating angina (Group, 2002). Although NO and NO-based vasodilators will result in a decrease in vascular tone (Furchgott, 1999) and are effective for treating angina (Rosendorff et al., 2007), they have not been demonstrated to be useful in treating essential hypertension (Rosendorff et al., 2007), possibly because of the phenomenon of nitrate tolerance (Munzel et al., 2005).
Although there are a number of effective classes of antihypertensives (Rosendorff et al., 2007), when treating patients with essential hypertension, the selection of an antihypertensive agent does not consider the possible changes in the smooth muscle contractile phenotype that may contribute to the molecular mechanism for essential hypertension. This could explain the variable racial and individual response to drug classes (Cushman et al., 2000; Johnson, 2008; Gupta, 2010). Thus, a critical need for a personalized approach for the treatment of essential hypertension exists.
F. Potential Novel Targets for Treatment of Essential Hypertension
Despite the effectiveness of current antihypertensive (Rosendorff et al., 2007), many patients with essential hypertension either do not respond to one or more therapeutic agents or have a resistant hypertension that requires treatment with multiple classes of antihypertensives for adequate control of blood pressure. Other than the well-known racial and regional differences in response to various classes of antihypertensives (Cushman et al., 2000; Johnson, 2008; Gupta, 2010), there is no known method to determine for any individual drug class whether a patient with essential hypertension will have a therapeutic response or the magnitude of the response to therapy. Because the molecular mechanism that produces essential hypertension is unknown, the diversity in the response to treatment is not unexpected and demonstrates the importance of identifying the mechanism producing hypertension in each individual. As outlined in the preceding sections, there are a number of changes in the contractile phenotype that could contribute to hypertension, and the contribution of the contractile phenotype could be variable among patients.
NO-mediated vasodilatation is a fundamental property of the vasculature (Furchgott, 1999), and studies of transgenic mice have demonstrated the importance of NO/cGMP signaling for maintenance of normal SVR and blood pressure (Huang et al., 1995; Pfeifer et al., 1998; Brenner et al., 2000; Zhu et al., 2002; Michael et al., 2008; Qiao et al., 2014; Reho et al., 2015). However, nitrate-based vasodilators have not been demonstrated to be effective for the treatment of essential hypertension (Rosendorff et al., 2007), possibly because of nitrate tolerance (Munzel et al., 2005). A number of mechanisms contribute to this phenomenon, but recently a decrease in LZ+ MYPT1 isoform has been suggested to play a role in the molecular mechanism for nitrate tolerance (Dou et al., 2010). Because PKG-induced phosphorylation of LZ+ MYPT1 isoforms and subsequent activation of the MLC phosphatase (Yuen et al., 2011; Yuen et al., 2014) is a key component in the NO/cGMP signaling pathway leading to vasodilatation (Lincoln, 1989), increasing MYPT1 LZ+ expression should be effective for both for reversing nitrate tolerance and the treatment of essential hypertension. The regulation for LZ+/LZ− MYPT1 expression is unknown, but investigators have demonstrated that Tra-2β appears to be an important regulator for LZ+/LZ− MYPT1 expression (Shukla and Fisher, 2008). Furthermore, both ACE-inhibitors (Chen et al., 2006) and ARBs (Ararat and Brozovich, 2009) were demonstrated to preserve both the normal LZ+ MYPT1 expression and sensitivity to NO-mediated vasodilation in heart failure, which may explain the benefit of these agents in treating heart failure (Pfeffer et al., 1992; Yusuf et al., 1992; Pitt et al., 2000; Yusuf et al., 2000, 2003; Granger et al., 2003) compared with other vasodilators (Awan et al., 1977), as well as the mortality differences for treatment of heart failure with hydralazine and isosorbide dinitrate versus enalapril in white and black patients (Carson et al., 1999). Investigation of the signaling pathway for the regulation of total MYPT1 as well as LZ+/− MYPT1 isoform expression could reveal novel targets in this pathway that would increase LZ+ MYPT1 expression. Small molecules could be designed to modulate either total MYPT1 or LZ+ MYPT1 expression in vascular smooth muscle, and therapies designed to increase LZ+ MYPT1 expression in vascular smooth muscle would improve both the vascular response to endogenous NO and pharmacological response during the treatment of hypertension. Other targets in the NO signaling pathway could also be exploited for the treatment of essential hypertension; i.e., novel activators of RGS-2 would be expected to decrease the activation of Gq-coupled receptors (Tang et al., 2003; Watson et al., 1996) and ultimately decrease vascular tone and blood pressure.
NM myosin could represent another novel target for drug development for the treatment of hypertension. The inhibition (Rhee et al., 2006), as well as the KO (Yuen et al., 2009) of NM myosin has been demonstrated to decrease force during the tonic phase of smooth muscle contraction, and thus inhibition of NM myosin would be expected to decrease both vascular tone and blood pressure. As outlined above, there are significant differences in the expression of contractile proteins between tonic and phasic smooth muscle (Fisher, 2010), and resistance vessels express a mixture of fast and slow contractile proteins (Fisher, 2010), as well as tonic and phasic contractile properties (Peng et al., 2001; Haddock and Hill, 2005). The contractile phenotype is known to be modulated during disease in both the large conduit vessels (Ararat and Brozovich, 2009) and smaller resistance vessels (Zhang and Fisher, 2007; Han and Brozovich, 2013), and thus essential hypertension could be due to changes in the gene program governing the contractile phenotype and a resulting change in the contractile properties of the resistance vessels from a phasic to more tonic phenotype. This could suggest that targeting the gene program to enhance the expression of phasic contractile proteins would decrease blood pressure and represent a novel therapy for essential hypertension.
V. Cytoskeletal Regulation
Regulation of smooth muscle function by myosin isoforms, particularly nonmuscle myosin isoforms is discussed above in section IV. Here we will discuss the cytoskeletal proteins that until recently were assumed to serve a primarily structural role in smooth muscle. There are three types of cytoskeletal proteins in this category: intermediate filaments, microtubules, and actin filaments.
A. Intermediate Filaments, Dystrophin, Utrophin, and Microtubules
Intermediate filaments have been studied relatively little in contractile smooth muscle but of note is literature that suggests that intermediate filaments may be the glue that sticks together different functional domains of the smooth muscle cellular cytoskeleton. This is based on anatomic studies of several types of smooth muscle (Devine and Somlyo, 1971; Ashton et al., 1975; Small et al., 1986; Siegman, 2014) showing that a population of actin filaments that are not directly contacting myosin surround the actomyosin filament bundles and insert into cytoplasmic “dense bodies.” These cytoplasmic dense bodies in turn are connected to “dense bodies” at the surface of the cell (focal adhesions) by intermediate filaments. Studies on airway smooth muscle have also indicated that intermediate filaments form cable-like structures that connect dense bodies and that these structures have a functional plasticity due to mechanisms, yet to be defined, by which the cable length can be adjusted in a regulatory manner (Zhang et al., 2010). Intermediate filaments in contractile vascular smooth muscle have also been suggested to serve functional roles that are not simply structural. For example, these filaments bind CaMKII and are phosphorylated by this kinase (Marganski et al., 2005), but whether the intermediate filaments are serving a signaling scaffolding role or whether CaMKII is somehow changing the function of the intermediate filaments is not known. Similarly, in airway smooth muscle, PAK has been shown to phosphorylate intermediate filaments (Wang et al., 2006, 2007). Interestingly, antisense knockdown of vimentin in smooth muscle inhibits agonist-induced force development (Tang, 2008), but, again, the exact molecular mechanism involved is not clear.
1. Dystrophin/Utrophin.
Of interest is the fact that smooth muscle contains large quantities of dystrophin and utrophin. These proteins have been little studied in smooth muscle, but in lung and vascular smooth muscle they and the dystroglycan complexes they form connect with caveolin and cavin in caveoli as well as the actin cytoskeleton (Palma-Flores et al., 2014; Sharma et al., 2014). Knockout of dystrophin in a mouse model decreases contractility of tracheal rings (Sharma et al., 2014).
With respect to microtubules, as expected, the microtubular network is sparse in nonproliferative, nondividing smooth muscle (Somlyo, 1980). They could serve a transport purpose, but we are unaware of such functions yet being demonstrated in contractile smooth muscle.
B. Actin
A far more extensive literature has developed with respect to actin in smooth muscle. The actin in contractile smooth muscle can be divided into that associated with smooth muscle myosin, generally called the thin filaments, and the nonmuscle actin cytoskeleton. The mechanisms of regulation of actomyosin have been discussed above. Here we will focus on the emerging role of the nonmuscle cytoskeleton in the regulation of vascular function.
Several groups have demonstrated that vasoconstrictors and myogenic contractions (Cipolla et al., 2002; Rembold et al., 2007; Kim et al., 2008a, 2010; Tejani et al., 2011) regulate the structure of the actin cytoskeleton and its connection to focal adhesions (FAs) (Poythress et al., 2013; Saphirstein et al., 2013, 2015) (Pathway #4, Fig. 3). These smooth muscle FAs appear to be essentially identical FAs in cultured cells except for their less dynamic nature (Poythress et al., 2013). Increasing evidence (Hill et al., 2001; Gunst and Zhang, 2008; Sun et al., 2008; Saphirstein et al., 2015) indicates that tension modulates the function of signaling pathways in the smooth muscle tissue/cell and that these biomechanical functions are mediated by remodeling of the smooth muscle focal adhesions and its nonmuscle actin cytoskeletal connections. It is important to emphasize that smooth muscles generally lack tendons for the transmission and summation of contractile forces generated by the muscles. In contrast, in smooth muscles the extracellular matrix (ECM) forms a sort of “intramuscular tendon” to which the integrins attach. It is one function of the focal adhesions, then, to channel, somehow, all contractile force generated by the contractile apparatus through the integrins and to the ECM. The exact mechanisms involved are now an active area of investigation in smooth muscle cells.
Moreno-Dominguez et al. (2014) have reported that a decline in G-actin content occurs in response to pressurization or agonist activation of cerebral resistance arteries, resulting in an increase in contractility in the absence of detectable myosin or actin phosphorylation that could be blocked by Rho kinase and PKC inhibitors. These results pointed to cytoskeletal remodeling contributing to the contractile responses observed. Smooth muscle actin exists as four different isoforms. Alpha smooth muscle actin and gamma smooth muscle actin are the dominant actins associated with myosin in the contractile filaments, what has been called “mini sarcomers,” (Fatigati and Murphy, 1984; Kargacin et al., 1989; Herrera et al., 2005) in vascular and gastrointestinal smooth muscle, respectively. Beta nonmuscle and gamma nonmuscle actins are thought to be present in all smooth muscles (Fatigati and Murphy, 1984; Drew et al., 1991; Kim et al., 2008a). These four isoforms are separate gene products and clearly have different functions and different expression patterns, but interestingly they have highly similar protein sequences with no isoform sharing less than 93% identity with any other isoform (Perrin and Ervasti, 2010). Kim et al. (2008a) mapped the individual actin isoforms in vascular smooth muscle during contraction in response to an alpha agonist and found that the overall F/G ratio increases by about twofold in response to the alpha agonist phenylephrine, but only gamma nonmuscle actin displays a statistically significant increase in polymerization. Interesting, in response to a phorbol ester, 12-deoxyphorbol 13-isobutyrate 20-acetate, only beta nonmuscle actin showed significant evidence of remodeling.
The actin isoforms also appear to define distinct subcellular domains within the geography of the cell. It has been found that alpha smooth muscle actin is associated with contractile filaments, beta nonmuscle actin is associated with cytoplasmic dense bodies and focal adhesions and gamma nonmuscle actin describes in interesting submembranous cortical domain in vascular smooth muscle (Parker et al., 1994, 1998; Gallant et al., 2011). Truly specific gamma nonmuscle antibodies have only recently become available, but earlier studies described qualitatively similar organizations in gut smooth muscle (Furst et al., 1986; Small et al., 1986; North et al., 1994a,b).
C. Focal Adhesion Remodeling
The smooth muscle FA, by connecting with the nonmuscle actin and intermediate filament cytoskeleton, is thought to serve the purpose of transmitting force generated in the contractile filaments through the integrins to the extracellular matrix and the tissue as a whole. FAs in contractile smooth muscle, like those in cultured, migrating cells, are thought to undergo considerable plasticity and remodeling in response to biomechanical forces, agonists, and hormones. When this has been directly investigated in contractile VSMCs, some, but not all, proteins have been shown to dissociate from the bulk of the FA during vasoconstrictor stimulation. Zyxin and vasodilator-stimulated phosphoprotein (VASP), but notably not FAK, undergo Src-dependent endosomal processing and regulate, through processes not yet fully understood, vascular contractility and stiffness (Poythress et al., 2013). In contrast, in airway smooth muscle, FAK has been shown to be quite mobile, shuttling between FAs at the plasmalemma and the cell interior (Opazo Saez et al., 2004). This highlights differences not only in organ function but also in cellular mechanisms between different types of smooth muscle.
Zyxin and vasodilator-stimulated phosphoprotein (VASP) are FA proteins associated with regulation of actin polymerization. This is consistent with a model whereby in VSMCs, only proteins at the edge of the focal adhesion, furthest from the plasmalemma, are capable of breaking off and undergoing remodeling in smooth muscle. This model is consistent with recent nanoscale super-resolution imaging of cultured fibroblast focal adhesion function described by Kanchanawong et al. (2010). In the Kanchanawong model, their data describe three sublayers within individual FAs: 1) the “integrin signaling layer,” containing integrin and its direct connections such as FAK closest to the plasmalemma; 2) the “force transduction layer,” including vinculin and talin, which are more interior; and 3) the “actin regulatory layer,” containing alpha actinin and zyxin, which is deepest into the cytoplasm.
D. Link to Hypertension
An interesting scenario occurs immediately after birth when there is a drastic demand for a decrease in pulmonary vascular resistance. This triggers a transition from the intrauterine pulmonary vascular requirement for only 8–10% of cardiac output, whereas the placenta performs the function of the primary site of gas exchange to the independent support of the newborn’s lung function. As a result, in the first days to weeks of the newborn’s life, dramatic vascular remodeling is required. If this fails it is associated with the disorder of persistent pulmonary hypertension of the newborn. Interestingly, the remodeling is not just of the cells of the vascular tissue but also a subcellular remodeling of the actin isoforms within the vascular cells with a persistence of excessive gamma actin being a marker of persistent pulmonary hypertension of the newborn (Fediuk and Dakshinamurti, 2015). Cortical gamma actin polymerization is reported to prevent nuclear translocation of the transcription factor YY1, which downregulates SM22, important for smooth muscle specific differentiation (Sotiropoulos et al., 1999).
A similar link between actin polymerization at a subcellular level within the vascular smooth muscle cell and blood pressure has been suggested by the effect of vasoconstrictor agonists to cause both actin polymerization and inward remodeling of resistance vessels associated with many forms of hypertension. Furthermore, inhibition of actin polymerization prevented the agonist-induced inward remodeling of the resistance vessels (Staiculescu et al., 2013). It is well known that increased intraluminal pressure elicits a myogenic vasoconstriction from resistance vessels and a further increase in blood pressure. This myogenic response has been associated with increased Ca2+ entry to the cell, activation of myosin light chain kinase, as well as increased activation of ROCK and protein kinase C pathways that regulate actin polymerization (Moreno-Dominguez et al., 2013).
Further investigation of the signaling pathway for the myogenic response has implicated integrins as the link between the extracellular matrix and the cytoskeleton that senses changes in pressure (Martinez-Lemus et al., 2003, 2005). Because the activation of integrins and the myogenic response is associated with tyrosine phosphorylation and the activation of focal adhesion kinase (FAK) and Src family tyrosine kinases (Murphy et al., 2001, 2002), blocking the activation of integrins or decreasing the activation of tyrosine kinase could be novel targets for treating essential hypertension. Although nonreceptor tyrosine kinases can phosphorylate paxillin and induce actin polymerization (Ohanian et al., 2005) and a PKC-mediated pathway will lead to the phosphorylation, or activation, of PYK2 and Src tyrosine kinases (Hodges et al., 2007; Chang et al., 2012), there is also evidence that receptor tyrosine kinases participate in the regulation of vascular tone. ephrin (EPH) kinases are the largest family of receptor tyrosine kinases, and the ligand for EPH tyrosine kinases are ephrins. Ephrins are also cell surface molecules that transduce signals into cells, and multiple EPHs will interact with multiple ephrins (Pasquale, 2008). Recently, the smooth muscle-specific deletion of the tyrosine kinase EPHB4 was demonstrated to result in decreased contractile responses to the agonist phenylephrine and hypotension in male, but not female, mice (Wang et al., 2015). Contrasting with these results are those demonstrating that blood pressure was unaffected by deletion of the tyrosine kinase EPHB6 in male and female mice, but blood pressure was elevated and the contractile response to phenylephrine was enhanced in castrated male EPHB6 KO mice (Luo et al., 2012; Wu et al., 2012). These results demonstrate that there is interplay between the various members of the non-receptor and receptor tyrosine kinase families as well as other factors that ultimately contribute to the regulation of blood pressure that may provide untapped targets for the development of novel antihypertensive therapeutics.
VI. Identifying Therapeutic Targets in Vascular Smooth Muscle through Biomechanical Studies
A. Arterial Stiffness as a Predictor of Negative Cardiovascular Events with Aging
Increased arterial stiffness is a prominent concept in studies of cardiovascular disease (CVD), especially in the context of aging, as an independent biomarker and predictor of hypertension and atherosclerosis, which are associated with increased mortality rates and severe damage to organ systems via stroke, renal failure, and heart disease (Ross, 1993, 1999; Dustan et al., 1996; Blacher et al., 1999; Guerin et al., 2001; Laurent et al., 2001; Cruickshank et al., 2002; Mattace-Raso et al., 2006; Laurent and Boutouyrie, 2007). Given the overwhelming predominance and persistence of CVD as the leading cause of human death worldwide, there is great interest and urgency to develop a thorough understanding of arterial stiffness as a possible cause of, and thus a potential therapeutic target for preventing or treating, CVD in humans.
Assessment of arterial stiffness exists on multiple scale levels. Macroscale techniques that characterize the hemodynamics of pulsatile blood flow are available clinically in humans and more easily interpretable, whereas microscale techniques better elucidate the underlying mechanisms causative of changes in the physical properties of blood vessels (Kohn et al., 2015). In studying arterial stiffness, the predominant challenge moving forward is to establish an integrated model of increased arterial stiffness across macro- and microscopic scales that enable the development of treatments to alleviate or prevent later-onset CVD. Recent studies suggest that vascular smooth muscle represents an attractive therapeutic target for such designs.
1. Pulse Wave Velocity: The Clinical Standard
In vivo arterial stiffness can be measured representatively and noninvasively as pulse wave velocity (PWV). This measurement of the pulse wave generated by cardiac systole is the ratio of the distance it travels along the vascular wall to the time delay between its arrivals at different points along the circulatory pathway. Most commonly, PWV is measured via ultrasound between the carotid and femoral arteries as representative of aortic stiffness. As the current “gold standard” of arterial stiffness measurements, increased PWV is strongly correlated to both aging and CVD (Mitchell et al., 2007; Willum-Hansen et al., 2006; Ben-Shlomo et al., 2014).
The validity of PWV as an in vivo approximation of aortic stiffness is described by the Moens-Korteweg equation:
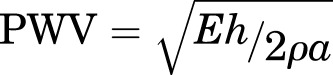 |
where E is the incremental Young’s modulus of the vessel, h and a are the thickness and radius of the aortic wall, respectively, and ρ is blood density. The value of E represents a material stiffness of the wall that is more precisely descriptive of its biomechanical properties. The Moens-Korteweg equation is itself dependent upon key assumptions: that the aortic wall is isotropic and homogeneous and that the aorta undergoes small isovolumetric changes in response to pulse wave propagation. Unfortunately, these assumptions do not hold, as demonstrated by previous studies (Clark and Glagov, 1985; Gosling and Budge, 2003); therefore, whereas PWV is an undoubtedly useful prognosticative tool, its accuracy as a biomechanical indicator is unclear.
2. The Importance of Ex Vivo Material Stiffness.
It is appropriate here to distinguish between “functional stiffness”—a relation between pulse pressure applied to a vessel and its resultant deformation, or strain—and “material stiffness,” the normalization of functional stiffness to vessel geometry, most notably vessel diameter and wall thickness (Greenwald, 2007). Material stiffness corresponds most directly to the value of E in the Moens-Korteweg relation and is the most independent representation of an intrinsic property to resist deformation in response to an applied force (O'Rourke, 1990; Gamble et al., 1994).
Therefore, PWV can more accurately be described as a standard for functional but not material stiffness. Functional stiffness is derived from material stiffness, but the opposite is not true. In understanding this subtle difference, the diverse terminology used in reference to aortic biomechanical properties may present significant difficulties, compounded by the prevalence of terms reciprocal to stiffness such as “compliance” and “distensibility,” which are sometimes used interchangeably among studies in the context of independence from vessel geometry. The most concise discussion of this complicated canvas is provided by Bank and Kaiser (1998), who cite conflicting results attributable to imprecise terminology and summarize their own study, showing that changes to PWV in brachial arteries, in response to smooth muscle relaxation, are not necessarily predictive of any corresponding change in material stiffness.
Analysis of arterial mechanical properties is integral to understanding more broadly the epidemiologic link between arterial stiffness and negative cardiovascular outcomes. Although arterial PWV is an easily demonstrable and a clinically valuable measure of functional stiffness, there is also a strong impetus to evaluate material stiffness of the aortic wall directly to explain how different components of the aortic wall, a highly complex and layered network of interconnected cellular and extracellular elements, contribute to altered or defective mechanisms in disease models. This is most conveniently and commonly done ex vivo by subjecting arterial tissue to mechanical stretch.
B. Regulation of Arterial Stiffness by Vascular Smooth Muscle
Although numerous studies and quantitative models have highlighted the extracellular matrix as the primary effector of dramatic changes in arterial stiffness due to aging or other cardiovascular defects (Greenwald, 2007; Zulliger and Stergiopulos, 2007; Fleenor et al., 2010, 2012; Holzapfel and Ogden, 2010; Valentin et al., 2011; Wagenseil and Mecham, 2012), vascular smooth muscle cells (VSMCs) are implicated to be major regulatory factors of arterial stiffness overall.
1. Homeostatic Interactions between Cellular and Extracellular Components of the Arterial Wall.
All major biologic components of the arterial wall play integral roles in maintaining normal cardiovascular function. Abnormalities in cellular mechanotransduction, which is regulated by cytoskeletal structures, transmembrane receptors, matrix proteins, and cell-cell and cell-matrix adhesions, have been linked to changes in cell mechanics or sensory machinery, as well as structural alterations in extracellular matrix, which are commonly featured in a wide range of diseases (Ingber, 2003, 2006). Indeed, there is a critical codependence between medial elastin and VSMCs in maintaining structure and function in the arterial wall, with studies showing both that elastin regulates VSMC proliferation (Brooke et al., 2003) and that VSMC knockout phenotypes result in severe degradation of elastin (Fry et al., 2015). VSMCs have been implicated in recruitment of both elastin (Dobrin, 1978) and collagen (Bank and Kaiser, 1998) during their contractile response.
Physiologic factors such as aging and hypertension induce observable physical changes in the wall due to VSMC remodeling (Glagov et al., 1993; Intengan and Schiffrin, 2000). With aging in particular, vascular smooth muscle cells (VSMCs) exhibit increased stiffness (Qiu et al., 2010) and are capable of switching their phenotype from contractile to synthetic, resulting in fundamental changes to cellular function as well as wall structure, because remodeled VSMCs migrate and proliferate in concert with alterations to other wall components, most notably elastin and collagen (Greenwald, 2007; Avolio et al., 2011).
The endothelium has also become an increasingly visible nexus of aging-induced changes, with widespread effects on cellular and extracellular processes throughout the arterial wall and especially those linked to VSM function. Nitric oxide (NO) produced by endothelial cells induces vasodilation and relaxation in VSM (Russell and Watts, 2000). NO bioavailability is reduced with aging, which attenuates this benefit, increasing VSM stiffness as a result. This downstream effect may characterize a powerful positive feedback loop where the increased stiffness leads to further reduction of NO (Cannon, 1998; Avolio et al., 2011).
2. The Focal Adhesion and Actin Cytoskeleton as Regulatory Sites of Arterial Material Stiffness.
Recently, attempts to quantify the constituency of total arterial stiffness attributable to VSMC activity have become more prevalent. Ex vivo studies have shown that VSMCs contribute significantly to total stiffness, potentially in conjunction with recruitment of collagen (Barra et al., 1993; Bank et al., 1996). Under optimally physiologic conditions for smooth muscle viability, up to 50% of maximal ex vivo aortic stiffness is observable only after activating VSMCs by alpha agonist and eliminating NO-mediated vasodilation (Gao et al., 2014b), quantifiable as shown in Fig. 8.
Fig. 8.

Separability of contractile components to the generation of aortic stiffness. Modified from Gao et al. (2014b).
Therefore, it is of great interest to probe the cellular mechanisms that produce this effect to identify underlying origins of dysfunction and degeneration. A series of studies highlighting stimulated VSMCs have found that focal adhesions connecting the cortical cytoskeleton to the matrix play a critical role in the regulation of signaling cascades triggered by smooth muscle activation, independently of actomyosin crossbridges (Kim et al., 2008a, 2010; Min et al., 2012; Poythress et al., 2013). In particular, disruption of the FAK-Src signaling complex reduces contractility and stiffness, as well as biochemical markers of focal adhesion signaling, induced by smooth muscle activation (Saphirstein et al., 2013; Saphirstein and Morgan, 2014). Interestingly, these mechanisms are defective with aging (Gao et al., 2014b), and may not be limited to the focal adhesions, but are also processes inherent in the cortical actin cytoskeleton (Saphirstein et al., 2015).The normal plasticity of the cytoskeleton may be an important sort of “shock absorber” for the aortic wall that is lost with aging. A model for the focal adhesion and actin cytoskeleton in the VSMC as core machinery regulating arterial stiffness, which is compromised with aging, has emerged from these findings, as shown in Fig. 9.
Fig. 9.

A diagrammatic model of how cytoskeletal remodeling could provide plasticity and an important “shock absorber” for the cardiovascular system (modified from Gao et al., 2014b.)
It has become increasingly clear that VSM may play a major role in the regulation of arterial stiffness.
VII. Regulation of Vascular Smooth Muscle Cell Function by Epigenetic Mechanisms
Epigenetic changes refer to the heritable alterations in gene expression that occur without changes in genome sequence (Jaenisch and Bird, 2003). The conventional epigenetic mechanisms include DNA methylation and histone modification, which alter the accessibility of transcription factors at DNA regulatory regions, such as promoters or enhancers (Kouzarides, 2007). Over the last decade, RNA-based modifications, which alter the translation of genetic information, have emerged as important regulators of development and disease. Collectively, these mechanisms likely play a role in the remarkable plasticity exhibited by vascular smooth muscle cells (VSMCs). This section will discuss the role of epigenetics in vascular smooth muscle (VSM) biology and describe ways by which these changes may influence the risk of cardiovascular diseases, such as hypertension.
A. DNA Methylation
DNA methylation involves the covalent addition of a methyl group to the 5′-position of the pyrimidine ring of cytosine residues to form 5-methylcytosine (Lorenzen et al., 2012). This process, catalyzed by DNA methyltransferases, leads to transcriptional repression by blocking the access of transcription factors to response elements in the promoter region of genes. DNA methylation had long been considered irreversible until the recent discovery of the members of the ten-eleven translocation (TET 1-3) family of proteins. TET proteins oxidize 5-methylcytosine to 5-hydroxymethyl cytosine, which ultimately revert to cytosine via the demethylation pathway (Boedtkjer et al., 2011; Lorenzen et al., 2012).
Variable methylation patterns are observed in the genes of VSMCs originating from different tissues (Zhang et al., 2012). Abnormalities in DNA methylation patterns have been observed to associate with vascular diseases and have been studied extensively in atherosclerosis (Dong et al., 2002; Castro et al., 2003; Hiltunen and Yla-Herttuala, 2003). SMCs of atherosclerotic lesions in humans and animal models exhibit reduced levels of 5-methylcytosine. The resulting DNA hypomethylation enhanced switching of VSMCs from a contractile to synthetic phenotype (Laukkanen et al., 1999; Ying et al., 2000; Hiltunen et al., 2002).
B. Histone Modifications
The modifications at protruding N-terminal tails of histones act as an integral epigenetic tag for chromatin remodeling. The histone tails can be modified by acetylation, methylation, ubiquitylation, and phosphorylation (Cheung et al., 2000; Kouzarides, 2007). The unique pattern of histone modifications in SMCs can alter chromatin packaging, which leads to the differential expression of crucial VSMC genes (Alexander and Owens, 2012). For example, the chromatin structure of the CArG-box [CC(A/T)6GG] motif, essential for tissue-specific expression of SMC marker genes, exists in an easily accessible euchromatin-like form in SMCs but not in non-SMCs (Alexander and Owens, 2012). Various histone modifications, such as dimethylation of H3K4 and H3K79 and acetylation of H4 and H3K9, have been observed in the CArG elements of SM α-actin and SM MHC in VSMC gene loci (McDonald et al., 2006). In contrast, these histone modifications are associated with transcriptional silencing in non-SMCs, such as endothelial cells (ECs), embryonic stem cells, and fibroblasts (McDonald et al., 2006). ECs exhibit high levels of H4 acetylation and H3K4 dimethylation at the EC-selective vascular endothelial cadherin gene locus. Increased levels of H4K20 dimethylation of this locus in VSMCs, however, are associated with transcriptional silencing (McDonald et al., 2006). These studies implicate histone modifications as important epigenetic tags in the regulation of SMC determination and differentiation.
1. Histone Acetylases and Histone Deacetylases.
Histone acetylation is characterized by the posttranslational modification of histones, which lead to chromatin remodeling by loosening histone-DNA contacts (Lorenzen et al., 2012). These modifications affect many SMC processes, such as differentiation, phenotypic switching, proliferation/apoptosis, and migration. Acetylation and deacetylation of histones are catalyzed by enzymes known as histone acetylases (HATs) and histone deacetylases (HDACs), respectively. HATs are classified into three families, including GNAT, MYST, and CBP/p300 [cAMP response element-binding protein (CREB)]. HDACs are categorized into four classes in mammals: class I (HDAC1-3, HDAC8), class II (HDAC4-7, HDAC9-10), class III sirtuins (SIRT1-7), and class IV (HDAC11) (Lorenzen et al., 2012). The class I, II, and IV HDACs are Zn2+-dependent deacetylases, whereas the class III HDACs, sirtuins, possess nicotinamide adenine dinucleotide (NAD)-dependent deacetylase activity (Lorenzen et al., 2012).
HATs and HDACs work in a dynamic manner to regulate cellular gene expression. Histone acetylation by p300 HAT allows binding of serum response factor to previously inaccessible CArG-containing regions of SMC-specific genes enabling the recruitment of coactivators, such as myocardin, to stimulate expression of α-actin (Gabbiani et al., 1981), γ-actin, calponin (Duband et al., 1993), SM22α (Duband et al., 1993), h-caldesmon (Frid et al., 1992), MLC, and SM MHCs (SM1 and SM2) (Nagai et al., 1988). Retinoic acid treatment of A404 cells, an SMC embryonic cell line, enhances acetylation of histones H3 and H4 at SM α-actin and SM MHC CArG-containing promoter regions (Manabe and Owens, 2001). Unlike myocardin, KLF4 (Kruppel-like factor 4) is a transcription factor associated with myogenic repression that regulates SMC phenotypic switching both in vitro and in vivo after vascular injury. KLF4 acts through epigenetic mechanisms to decrease histone H4 deacetylase by recruiting HDAC2 and HDAC5, which block the binding of serum response factor to methylated histones and CArG box chromatin during repression of SMC gene expression (McDonald and Owens, 2007; Yoshida et al., 2008).
Although the studies identifying the role of HATs and HDACs in the regulation of SMC functions warrant further in vivo studies, they highlight the relevance of inhibitors as epigenetic therapeutics in the treatment of vascular diseases. One such study demonstrated that inhibition of p300 HAT activity by Lys-CoA-TAT decreases the effects of retinoic acid on the differentiation of A404 cells toward an SMC lineage (Spin et al., 2010). In a separate study in A404 cells, overexpression of HDACs increased CREB-CArG-dependent SM22 promoter activity, whereas the HDAC inhibitor trichostatin A had the opposite effect (Qiu and Li, 2002). In contrast, HDAC inhibition after tributyrin treatment inhibited migration of cultured VSMCs after hyperacetylation of histone H3 and reduced expression of HDAC7 (Qiu and Li, 2002). Pharmacological inhibition of HDAC activity or knockdown of HDAC expression with small interfering RNAs (siRNA) prevents platelet-derived growth factor-induced VSMC proliferation (Findeisen et al., 2011).
Recently, HATs and HDACs have been found to act on the lysine (K) residues of a wide range of nonhistone proteins and are therefore also denoted as KATs and KDACs, respectively (Europe-Finner et al., 2015). Nonhistone targets for KATs and KDACs include large cellular macromolecular complexes involved in the actin nucleation complex (Choudhary et al., 2009) and other proteins participating in microfilament formation and dynamics (Posern et al., 2004). A majority of proteins involved in smooth and striated muscle contraction have been shown as substrates undergoing acetylation (Karolczak-Bayatti et al., 2011; Lundby et al., 2012). KDAC8 regulates the differentiation and contraction of SMCs by interacting with α-actin (Waltregny et al., 2005; Chen et al., 2013), tropomyosin, and cortactin proteins (Chen et al., 2013; Li et al., 2014a). KDAC6 is involved in the modulation of the cytoskeleton, cell migration, and cell-cell interactions by interacting with proteins such as SIRT2 (Valenzuela-Fernandez et al., 2008). KDAC inhibition by trichostatin A and hydroxamic acid was associated with increased protein acetylation and reduction of agonist-stimulated contractions in VSM tissues (Krennhrubec et al., 2007; Karolczak-Bayatti et al., 2011; Chen et al., 2013). In a separate study, during contraction, the cytoskeletal proteins were dynamically regulated through HDAC inhibition and activation, resulting in the regulation of cytoskeletal structure (Kim et al., 2006).
a. Histone deacetylases and link to hypertension.
Epigenetic mechanisms influence the changes in vascular structure that occur during hypertension (Lacolley et al., 2012). Angiotensin II (Ang II) at least partially influences vascular remodeling via HDACs in the pathogenesis of hypertension (Xu et al., 2007). Specifically, HDAC5 and HDAC4, after phosphorylation by Ang II, export from the nucleus where they activate myocyte enhancer factor-2, leading to SMC hypertrophy (Xu et al., 2007; Li et al., 2010). Treatment of spontaneously hypertensive rats with HDAC inhibitors, such as trichostatin A or valproic acid, reduced SMC hypertrophy, blood pressure, and vascular inflammation (Cardinale et al., 2010; Bogaard et al., 2011; Usui et al., 2012). All these studies highlight an important therapeutic potential for HDAC inhibitors in the treatment of vascular diseases.
2. Sirtuins.
Sirtuins are a class of proteins that have multiple functions including NAD+-dependent deacetylation. Currently, there are seven known mammalian sirtuins (SIRT1-7), located in the nucleus (SIRT1, 6, and 7) (Michan and Sinclair, 2007), cytoplasm sirtuin (SIRT2), or mitochondria sirtuins (SIRT3, 4, and 5) (Michishita et al., 2008). Because SIRTs are multifunctional, they have a plethora of protein targets, including p53, nuclear factor κB, Ku70, FOXO, tubulin, eNOS, BAD, CytoC, Ndufa9, GDH, ACS2, and ISDH2 (Haigis and Sinclair, 2010; Corbi et al., 2013). Extensive reviews are available on the role of sirtuins in aging, calorie restriction, mitochondrial biogenesis, and neurodegenerative diseases (Gagnon et al., 2008; Pfister et al., 2008; Haigis and Sinclair, 2010; Guarente, 2011),
Sirtuins have largely been shown to have protective effects on the vasculature. For instance, SIRT1 modulates vascular biology during hypoxia-induced redox stress by deacetylating the transcription factor, HIF (Hypoxia-inducible factor)-2α, which modulates vascular tone and enhances cell survival through induction of antioxidant enzymes to promote angiogenesis (Dioum et al., 2009). The deacetylase activity of SIRT1 has also been shown to regulate the proliferation and migration of VSMCs via the suppression of cellular senescence mediator, p21 protein, and enhancement of senescence-resistant cell replication (Stein and Matter, 2011). Overexpression of SIRT1 increases the activity of metalloproteinase-3 inhibitor, which inhibits cell migration. In addition, SIRT1 also deacetylates and activates eNOS, which enhances NO-induced vasodilation (Mattagajasingh et al., 2007).
Interestingly, recent studies implicated SIRT1 in blood pressure regulation through targeting VSMCs. Overexpression of SIRT1 in VSMCs has been found to modulate blood pressure by downregulation of the Ang II type 1a receptor gene (Miyazaki et al., 2008). Moreover, resveratrol treatment activated SIRT1, resulting in the repression of Ang II type 1a receptor gene transcription to counteract Ang II-induced hypertension in mice (Miyazaki et al., 2008). SIRT1 expression in human VSMCs was demonstrated to correlate inversely with donor age (Thompson et al., 2014). This was associated with functional defects, such as reduced migratory capacity and increased senescence (Thompson et al., 2014). Interestingly, Ang II infusion significantly decreased the expression of SIRT1 in mouse aorta, which was associated with increased blood pressure and elevated vascular remodeling. Importantly, a VSMC-specific SIRT1 transgene attenuated both Ang II-induced increases in blood pressure and vascular remodeling (Gao et al., 2014a). These recent studies point to the potential of therapeutically improving SIRT1 function in VSMCs to treat hypertension.
C. Noncoding RNA
Noncoding RNAs comprise many functional RNA transcripts that are not transcribed into proteins, but regulate the transcription, stability, or translation of protein-encoding genes (Ling et al., 2013). Noncoding RNAs were once assumed only to regulate generic functions of cells, such as transcription, translation, and splicing. It is now recognized, however, that a wide variety of noncoding RNA transcripts are transcribed and have diverse biologic activity. The complex nature of this RNA-based regulatory network may partially explain the vast diversity of the characteristics of mammals, despite possessing relatively similar proteomes (Mattick and Makunin, 2006). In addition, epigenetic mechanisms, such as DNA methylation, can regulate the expression of noncoding RNAs, adding a further level of complexity (Liang et al., 2009). This section discusses microRNAs and long noncoding RNAs, which function in the broadly termed RNA silencing machinery.
1. MicroRNAs.
MicroRNAs (miRs) are a family of short (21–25 nucleotide) RNAs, which typically negatively regulate protein translation by direct binding to the 3′ untranslated (3′ UTR) region of mRNA targets. The first miR, lin-4, was discovered to have a role in Caenorhabditis elegans larval development more than 20 years ago (Lee et al., 1993). To date, over 30,000 miRs have been discovered in 206 species, including 2578 in humans (van Rooij and Kauppinen, 2014). It is now certain that miRs play an important role in many cellular and developmental processes. Crucially, miRs are dysregulated in many pathophysiological conditions, including cardiovascular disease.
MicroRNAs are transcribed in the nucleus by RNA polymerase II to long primary transcripts (Lee et al., 2002) (Fig. 10). They are subsequently processed by the RNAse III enzyme Drosha (Lee et al., 2003) in the nucleus to a 70 nucleotide hairpin structure termed preliminary miRNAs (pre-miRNA). After export into the cytoplasm, pre-miRNAs are further processed by a separate RNAse III enzyme, Dicer (Hutvagner et al., 2001), into a 21–25 nucleotide duplex. The mature miR strand is then incorporated onto Argonaute protein to form the RNA-induced silencing complex (RISC) (Hammond et al., 2001; Hutvagner et al., 2001), leading to unraveling and degradation of the complimentary strand (Khvorova et al., 2003; Schwarz et al., 2003). The miR then guides the RISC to complimentary sequences in the 3′ UTR of target mRNA (Lee et al., 1993). In plants, the near-perfect complementarity between the miR and target sequence promotes mRNA cleavage of the target, akin to the mechanism for siRNA-induced silencing (Hake, 2003). In mammals, however, this level of complementarity is rare. In contrast, association of a 6–8 nucleotide ("seed") sequence with the 3′ UTR of target mRNA (Lewis et al., 2005) leads to repression of mRNA translation through inhibition of translation initiation and/or promotion of mRNA decay (reviewed by (Ameres and Zamore, 2013)).
Fig. 10.

Summary of the main mechanisms of miR biogenesis and function and avenues for pharmacological intervention. (A) MiRs are first transcribed in the nucleus, primarily from introns located in both coding and noncoding DNA, into long primary transcripts termed pri-miRNAs, then are processed in the nucleus by the RNAse III enzyme Drosha into a shorter (∼70 nucleotide) hairpin duplex termed pre-miRs. (B) After export to the cytoplasm by the dsRNA-binding protein Exportin 5, pre-microRNAs are processed by an RNAse III enzyme, Dicer, into a short (21–25 nucleotide) duplex. (C) The miR duplex is unwound, primarily leading to preferential incorporation of a single strand onto Argonaute protein to form the RNA-induced silencing complex (RISC). A short sequence (6–8 nucleotides) at the 5′ end of the miR, known as the "seed" sequence, targets the RISC to complimentary sequences in the 3′ UTR of target mRNAs. (D) Translational repression, through the blocking of translation initiation or recruitment of translational blocking proteins, and/or (E) mRNA decay, via 5′ to 3′ decay and deadenylation of the poly (A) tail. (F) miR mimics are designed as RNA duplexes composed of a passenger strand, chemically modified (e.g., phosphate addition) to permit entry to the cell and subsequent unwinding after entry (i.e., by containing several nucleotide mismatches), and a guide strand, which consists of an identical sequence to the endogenous miR. (G) An anti-miR is designed to be complimentary to the endogenous miR of interest, thus inhibiting target mRNA binding, which therefore increases translation of the target mRNA. (H) A lipid nanoparticle delivery system can induce expression of a miR in specific cell types [e.g., in smooth muscle cells by expressing the miR under the influence of a smooth muscle cell (SMC) promoter].
a. Dicer knockout mice.
There are several excellent reviews that discuss the role of miRs in vascular structure, function, and disease (Yu et al., 2014; Gupta and Li, 2015; Maegdefessel et al., 2015a; Marques et al., 2015). The current review will mainly focus on miRs that have a role in regulating vascular smooth muscle cell (VSMC) contractility, thus influencing the risk of hypertension. The importance of miRs in vascular smooth muscle was demonstrated in experiments using smooth muscle-specific Dicer KO mice. Knockout of Dicer in vascular smooth muscle (VSM) induced embryonic lethality, which was associated with widespread hemorrhaging, loss of contractile function, and reduced VSMC proliferation (Albinsson et al., 2010). A further study using a tamoxifen-inducible SMC-specific knockout of Dicer, suggested miRs are necessary for blood pressure regulation and contractile function (Albinsson et al., 2011). Blood pressure was reduced after Dicer knockdown, with no change in cardiac dimensions. This was associated with impairment of both receptor- and calcium-mediated contraction of small mesenteric arteries and reduction of expression of contractile proteins (Albinsson et al., 2011). Furthermore, the stretch-dependent increase in contractile gene expression in the portal veins of mice was dependent on Dicer expression (Turczynska et al., 2013). Loss of the miR-143/-145 cluster is thought to be at least partially responsible for this reduced vascular contractility (Boettger et al., 2009; Norata et al., 2012; Dahan et al., 2014). Pathways that promote the vascular smooth muscle contractile phenotype, such as Notch and bone morphogenic protein signaling, function partially through promoting the expression of miR-143/-145 (Boettger et al., 2009; Albinsson et al., 2010; Boucher et al., 2011; Davis-Dusenbery et al., 2011; Norata et al., 2012; Turczynska et al., 2013; Dahan et al., 2014). In addition to miR-143/-145, the expression of miR-21 (Kang et al., 2012) and miR-24 (Chan et al., 2010) is similarly induced by bone morphologic protein (BMP) signaling, promoting the contractile phenotype by targeting Programmed Cell Death 4 and Tribbles-like protein-3, respectively.
The Dicer KO model has also demonstrated an important role for miRs in the regulation of small arterial myogenic tone, therefore modulating peripheral arterial resistance, which is raised in hypertension. Myogenic tone of mesenteric arteries was abolished from Dicer KO mice, which was associated with a loss of calcium influx through the L-type calcium channel (Bhattachariya et al., 2014). It is therefore possible that the aberrant expression of miRs may increase the development of myogenic constriction of small arteries, thereby increasing peripheral resistance and systemic blood pressure.
b. Regulation of vascular smooth muscle cell contractility.
Several miRs have been found to posttranscriptionally modulate the expression of VSMC contractile proteins. Overexpression of miR-143/-145 in VSMCs increases the expression of contractile proteins, such as SM-MHC, calponin, and SM22α, through the targeted downregulation of KLF4 and 5 (Albinsson et al., 2010, 2011; Boucher et al., 2011; Davis-Dusenbery et al., 2011; Norata et al., 2012; Turczynska et al., 2013; Chettimada et al., 2014; Dahan et al., 2014; Riches et al., 2014). VSMCs from Type 2 diabetic rats (Chettimada et al., 2014) and humans (Riches et al., 2014) display an increased expression of miR-145, which contributes to the enhanced expression of vascular contractile proteins in Type 2 diabetes. Myocardin increases the expression of miR-1 in human aortic SMCs, which blocks the expression of the contractile proteins α-SMA and SM22, possibly acting as a negative feedback mechanism to counter myocardin-induced increase in contractility (Jiang et al., 2010). Additionally, although miR-21 may promote the contractile phenotype, it may also negatively regulate VSMC contraction through the targeted downregulation of myosin phosphatase, Rho-interacting protein, and cofilin-2 (Kotlo et al., 2011).
c. Regulation of vascular smooth muscle cell ion channels.
MicroRNAs have also been demonstrated to posttranscriptionally regulate the expression of VSMC ion channels, thereby influencing vascular contractility and thus blood pressure regulation. For example, miR-145 indirectly increases VSM contractility by targeted downregulation of CamKIIδ, which results in increased expression of the α1C subunit of the L-type calcium channel (Turczynska et al., 2012). Similarly, miR-328 has been demonstrated to directly target the α1C subunit of the L-type calcium channel (Guo et al., 2012). Overexpression of the miR-424/322 resulted in decrease in cyclin D1 and calcium-regulating proteins calumenin and stromal-interacting molecule 1, which reduces store-operated calcium entry in human and rat VSMCs (Merlet et al., 2013). Further studies have elucidated a role for miR-9a-3p (Li et al., 2015) and miR-190 (Li et al., 2014b) in the downregulation of the VSMC KATP and potassium channel Kv7.5, respectively, causing an increase in contractility.
d. Regulation of the extracellular regulated kinase pathway.
MicroRNAs have also been implicated in the downregulation of proteins involved in the ERK 1/2 signaling pathway in proliferating, but not contractile, VSMCs. The miRs-21 (Stein et al., 2014), -31 (Liu et al., 2011), -132 (Choe et al., 2013), and -155 (Yang et al., 2015) all affect ERK activation. It remains to be determined if those miRs similarly affect ERK activation in fully differentiated VSMCs.
2. Long Noncoding RNAs.
Long noncoding RNAs (LncRNAs), arbitrarily classified as longer than 200 nucleotides, have a role in regulating the expression of neighboring genes. LncRNAs have a much more widespread mode of action than miRs, and their function cannot currently be inferred from sequence or structure (Mercer et al., 2009; Rinn and Chang, 2012). LncRNAs can recruit chromatin-remodeling complexes to epigenetically regulate specific genomic loci. They can also influence transcriptional regulation of genes by recruiting RNA binding proteins to gene promoters, acting as cofactors to modulate transcription factor and RNA polymerase II activity. Finally, LncRNAs can also regulate posttranscriptional processing events, by recognizing complimentary sequences of RNA. Like miRs, they have the ability to bind to mRNA targets, affecting their translation and degradation, but they can also influence other posttranscriptional processes such as splicing, editing, and transport. For a deeper description of LncRNA function the reader may refer to the following references (Mercer et al., 2009; Rinn and Chang, 2012).
Although the field is newer than that for miRs, LncRNAs are emerging as potentially important players in cardiovascular disease (recently reviewed by Uchida and Dimmeler, 2015 and Miano and Long, 2015). Through RNA sequencing approaches in human coronary arteries, VSMCs have been demonstrated to express several LncRNAs, including smooth muscle and endothelial cell-enriched migration/differentiation-associated long noncoding RNA (Bell et al., 2014). Depletion of endothelial cell-enriched migration/differentiation-associated long noncoding RNA in VSMCs was associated with decreased expression of myocardin and other contractile genes and increased expression of the promigratory proteins MDK and PTN, suggesting a role in maintenance of the contractile phenotype (Bell et al., 2014). The Lnc-Ang362 was increased in Ang II-treated rats, which was associated with an increased VSMC proliferation. Interestingly, Lnc-Ang362 was cotranscribed with miR-221 and -222 and was required for their expression (Leung et al., 2013). This study provided evidence for the regulation of LncRNAs by Ang II, which may have important implications in the pathogenesis of Ang II-associated cardiovascular diseases, such as hypertension (Leung et al., 2013).
Recent evidence suggested the expression of LncRNAs may affect VSMC proliferation and apoptosis, therefore influencing the risk of aortic aneurysm (He et al., 2015) and atherosclerosis (Congrains et al., 2012a; Bayoglu et al., 2014; Li et al., 2014c; Vigetti et al., 2014; Wu et al., 2014). For example, LncRNA-p21 was demonstrated to inhibit cell proliferation and neointimal hyperplasia by releasing the mouse double minute 2 repression of p53 (Wu et al., 2014). Importantly, this LncRNA was downregulated in atherosclerotic plaques from ApoE−/− mice (Wu et al., 2014). Furthermore, the expression of the LncRNA ANRIL was influenced by several atherosclerosis-associated single nucleotide polymorphisms (SNPs) in the 9p21 locus (Holdt et al., 2011; Congrains et al., 2012a,b; Bayoglu et al., 2014). These data alert one to the possibility that previous genome-wide association studies (GWAS) may have misinterpreted SNPs in regions that encode nonprotein coding RNA as without effect on disease risk.
3. Strategies to Regulate microRNAs in Vascular Disease.
MicroRNAs continue to provide great therapeutic potential, and the strategies to alter miR function will be discussed below. Because the functions of LncRNAs in VSM biology are still being determined, approaches to modify their function have not yet been extensively examined in this setting.
One of the benefits of miRs is that they are strongly conserved in species (albeit potentially targeting differing mRNA targets), thus aiding the design of preclinical studies to determine their efficacy and safety. In addition, miRs have the potential to target many members of the same molecular pathway and as a result having a greater combined effect than siRNAs, which typically target only a single gene. For example, the miR-200 family has been implicated in regulating many targets that control actin filament organization and dynamics (Bracken et al., 2014). Furthermore, miR therapy will not completely knock down their target protein’s expression. Rather, they will result in a "fine-tuning" effect on target protein expression, making them an attractive proposition for therapeutic intervention.
There are two main techniques to modulate miR activity in vivo (reviewed in Ling et al., 2013; van Rooij and Kauppinen, 2014) (Fig. 10): 1) restoring the expression of a downregulated miR by use of a miR-mimic and 2) inhibiting the activity of an abnormally expressed miR by use of an anti-miR. A miR-mimic is a synthetically designed RNA duplex, containing a passenger strand, chemically modified to enhance uptake and disengage once inside the cell, and a guide strand, which is identical to a miR of interest. Conversely, an anti-miR is designed to be complementary to the endogenous miR and therefore binds and inhibits it from acting on its mRNA target. Several modifications, such as locked nucleic acid and phosphodiester additions, have improved the in vivo stability of anti-miRs.
The great strength of miRs, their ability to target multiple mRNAs, is also their greatest weakness because they could potentially result in many off-target effects. In addition, miR mimics often increase the miR concentration to supraphysiologic levels, again increasing the risk of off-target actions. To lower such risk, viral constructs have been developed to re-express the miRs to endogenous levels. Recently, lipid nanoparticle delivery has proved promising to deliver miR regulators to target cells and reduced delivery to nontarget cells (Fig. 10). For instance, anti-miR-145 therapy in rats with Sugen-5416/hypoxia-induced pulmonary arterial hypertension (PAH) using the Star:Star-mPEG delivery system, reduced the severity of pulmonary hypertension without significant off-target effects (McLendon et al., 2015).
Extracellular vesicles, such as exosomes, microvesicles, and apoptotic bodies, allow long-distance delivery of cellular information, including noncoding RNA. They recently emerged as important regulators of cardiovascular diseases, such as atherosclerosis (recently reviewed by Das and Halushka, 2015) and may serve as important diagnostic and prognostic biomarkers for these conditions.
a. Pulmonary hypertension.
Several miRs have been found to contribute to clinical and experimental pulmonary hypertension, which is characterized by enhanced proliferation and constriction of pulmonary artery smooth muscle cells (PASMCs). For example, miR-130a promotes PASMC proliferation through the negative regulation of its targets CDKN1A and growth arrest-specific homeobox (Wu et al., 2011; Brock et al., 2015). Interestingly, the miR-130/-301 family also regulates the expression of peroxisome proliferation receptor gamma, which influences the expression of many vasoactive factors, such as endothelin-1 (Bertero et al., 2015). The expression of miR-190 was augmented in PASMCs of the hypoxic rat, which acts as a model for pulmonary hypertension. Vasoconstriction to both potassium chloride and phenylephrine was enhanced after transfection of pulmonary arterial rings with a miR-190 mimic. The same study demonstrated that miR-190 downregulates the protein expression of the potassium channel Kv7.5, which results in raised intracellular calcium in PASMCs (Li et al., 2014b). The miR-328 has been demonstrated to directly target the α1C subunit of the L-type calcium channel and was reduced in the pulmonary artery from patients with pulmonary hypertension, thus enabling an increase in vasoconstriction (Guo et al., 2012).
b. Systemic hypertension.
There have been fewer animal studies focusing on the role of miRs in the development of essential hypertension. Clinical studies, however, have highlighted the possibility of therapeutic intervention to prevent or treat systemic hypertension. A study from Greece found that the expression of miR-145, -143, and -133 was upregulated, and miR-21 and -1 were downregulated in the peripheral blood of patients presenting with essential hypertension (Kontaraki et al., 2014). These miRs are all important regulators of VSMC phenotype plasticity. A further study in a Chinese population identified the SNP rs12731181 (A to G) in the prostaglandin F2α receptor that was more prevalent in hypertensive individuals (Xiao et al., 2015). This polymorphism reduces the likelihood of miR-590-3p binding, thus increasing prostaglandin F2α receptor expression and enhancing prostaglandin-mediated contractility of VSMCs (Xiao et al., 2015).
c. Other vascular diseases.
Several studies have demonstrated the feasibility of miR therapies to slow the development of abdominal aortic aneurysms. In particular, the miR-29 family has been heavily implicated in aneurysm development in mouse models. Murine in vivo delivery of miR-29 was found to decrease elastin mRNA and increase MMP activity and aneurysm development (Boon et al., 2011; Jones et al., 2011; Maegdefessel et al., 2012; Merk et al., 2012). Importantly, blockade of miR-29 reduced aneurysm development in Ang II-infused mice (Boon et al., 2011), a mouse model for Marfan syndrome (Merk et al., 2012), and ApoE−/− mice (Maegdefessel et al., 2012), signifying its therapeutic potential. In addition, miR-21 (Maegdefessel et al., 2012), -712/-205, and -195 (Zampetaki et al., 2014) have all been demonstrated to promote aneurysm development, whereas miR-24 (Maegdefessel et al., 2015b) slows aneurysm progression in mouse models.
Interestingly, miR-29 was found to increase with aging in the aorta from mice, which correlated with aneurysm development (Boon et al., 2011). The targets affected by miR-29 are strongly implicated in aortic stiffness, which is also known to increase with age. Few studies have focused on the role of miRs in vascular stiffness. A clinical study found that two SNPs, rs978906 (A allele) and rs9808232 (C allele), were associated with high pulse wave velocity in 856 individuals in a Chinese population (Liao et al., 2015). These polymorphisms rendered the expression of the aortic stiffness-associated ROCK2 (Noma et al., 2007) less responsive to changes in miR-1183 expression, thus enhancing ROCK2 expression (Liao et al., 2015). In addition, miR-145 was strongly reduced in ApoE−/− mice, which was associated with an increase in collagen and stiffer arteries (Kothapalli et al., 2012).
These studies demonstrate the capacity of miR therapeutics to improve treatments for vascular complications such as hypertension, abdominal aortic aneurysm, and arterial stiffness. Indeed, anti-miR-33 (atherosclerosis, Regulus Therapeutics, (Carlsbad, CA)), anti-miR-92 (peripheral artery disease, miRagen Therapeutics Boulder, CO), and anti-miR-145 (vascular occlusion, miRagen Therapeutics) have already reached preclinical trials. This has clearly been a rapidly expanding field and we expect to see many further developments, including approaches to target LncRNAs, in the coming years.
VIII. Vascular Smooth Muscle Diseases and Their Treatments
A. Review of Current Therapies and Their Targets
Excess vasoconstriction and the resulting increase in vascular tone and SVR are an important contributing factor to the pathogenesis of essential hypertension. CCBs, ACE inhibitors, ARBs, and direct vasodilators (i.e., hydralazine) all target the smooth muscle cell to decrease vascular tone and blood pressure. CCBs decrease the activation of SM myosin by decreasing intracellular Ca2+, whereas ACE inhibitors and ARBs will decrease the activation of the RhoA/Rho kinase signaling pathway to reduce Ca2+ sensitization. K+ channel openers produce vasodilatation; nicorandil acts on BK channels and hyperpolarizes smooth muscle cells (Nelson et al., 1990), whereas minoxidil opens ATP-sensitive K+ channels and hyperpolarizes the smooth muscle cells (Wickenden et al., 1991). The mechanism of action for hydralazine, on the other hand, is unclear, but evidence suggests that hydralazine both activates K+ channels and also stimulates NO production by the vascular endothelium (Knowles et al., 2004).
In addition to essential hypertension, there are a number of other diseases of the vasculature and diseases with associated abnormal vascular reactivity. These defects in vascular reactivity contribute to morbidity and mortality. In this section, we will outline these diseases, their mechanism, current therapies, and suggest novel targets for both the treatment of essential hypertension as well as these other diseases of the vasculature (Table 1).
TABLE 1.
Disease-associated vascular abnormalities
In addition to hypertension, there are a number of diseases with abnormalities of the vasculature that contribute to patient morbidity and mortality. The vascular abnormality, possible mechanisms as well as therapies and potential new, novel targets for therapy are listed and discussed in detail in the text.
| Disease | Abnormality | Possible Mechanisms | Treatments |
|---|---|---|---|
| Heart Failure | Resting Vasoconstriction, | Defect in NO/cGMP mediated vasodilatation; ?Decrease in LZ+ MYPT1 | Vasodilators (ACE inhibitors, ARBs, hydralazine, nitrates) |
| Decrease in sensitivity to NO | |||
| Idiopathic Pulmonary Hypertension | Pulmonary Vascular Vasoconstriction & | Proliferation of pulmonary SMCs, Defect in NO/cGMP mediated vasodilatation. ?Decrease in LZ+ MYPT1 and increase in NM myosin | Prostaglandins (Epoprostenol), Phosphodiesterase inhibitors (Sildenafil), Guanylate cyclase stimulators (Riocigaut), Endothelin antagonists (Bosentan), NO inhalation & Rho kinase inhibitors (Fasudil) |
| Decrease in sensitivity to NO | |||
| Portal Hypertension | Sensitivity to vasodilators increased & to vasoconstrictors decreased | ?Changes in isoform expression of contractile proteins which influence both smooth muscle activation and relaxation | Management of fluid status |
| Raynaud’s Phenomenon | Transient vasospasm of digital vessels | Altered reactivity of vascular smooth muscle | Keeping digits warm, CCBs |
| Pre-eclampsia/Pregnancy Induced Hypertension | Increase in vascular tone | Altered reactivity to RhoA, PKC & Ca2+, decrease NO | Antihypertensives (non-fetotoxic) |
B. Other Major Vascular Diseases Including Analysis of Current Therapies and Novel Targets
1. Heart failure.
Considerable evidence from both human subjects and animal models have shown that HF is associated with an impaired vasodilatory response to acetylcholine, via an endothelium-dependent mechanism, as well as to nitroglycerin, via an endothelial-independent mechanism (Kaiser et al., 1989; Kubo et al., 1991; Katz et al., 1993). Both MYPT1 LZ+ expression and the vasodilatory response to NO/cGMP has been demonstrated to decrease in HF (Karim et al., 2004; Chen et al., 2006; Ararat and Brozovich, 2009; Han and Brozovich, 2013), which demonstrates that the fall in LZ+ MYPT1 expression contributes to the decrease in sensitivity to NO-mediated vasodilatation associated with HF. Additionally, treatment of HF with either ACE inhibitors (Chen et al., 2006) or ARBs (Ararat and Brozovich, 2009) has been demonstrated to preserve the normal expression of the LZ+ MYPT1 isoform and sensitivity to NO/cGMP-mediated vasodilatation. These results are similar to those of Abassi et al., 1997; this group demonstrated that both endothelium-dependent and -independent responses in renal blood flow were impaired in HF rats compared with control, and Ang II receptor blockade normalized these vasodilatory responses.
The mechanism for the regulation of LZ+/LZ− MYPT1 expression by Ang II-induced activation of the AT1 receptor is unknown, but evidence suggests that both the activation of p42/44 MAPK (Ararat and Brozovich, 2009) and Tra2β (Shukla and Fisher, 2008) are important. Ang II produces vasoconstriction through its effect on both the endothelium and vascular smooth muscle (Nickenig and Harrison, 2002). Additionally, Ang II activates nuclear factor κB to produce a proinflammatory state by inducing expression of interleukin (IL)-6 and tumor necrosis factor (TNF)-α (Dzau, 2001; Nickenig and Harrison, 2002) and also activates membrane NADH/NADPH oxidases, generating reactive superoxide anions, which increase NO catabolism and therefore decrease NO bioavailability (Griendling et al., 1994). In heart failure, inhibition of Ang II signaling counter these deleterious effects and could contribute to the reduction in cardiovascular morbidity and mortality observed with ACE inhibitors and ARBs (Pfeffer et al., 1992; Yusuf et al., 1992). Furthermore, similar beneficial effects on survival have not been observed with other vasodilators such as prazosin, although prazosin has also been shown to improve cardiac index and fractional shortening for heart failure patients (Awan et al., 1977; Miller et al., 1977), which suggests that ACE inhibitor have other beneficial effects beyond reducing afterload and improving left ventricular remodeling.
In the setting of advanced heart failure, marked activation of the renin-angiotensin system and worsening functional status have been associated with a progressive elevation of the proinflammatory marker TNF-α (Levine et al., 1990; Torre-Amione et al., 1996), which has been demonstrated to contribute to endothelial dysfunction and also an increase in vascular tone that is produced by a RhoA/Rho kinase-mediated inhibition of MLC phosphatase (Parris et al., 1999; Mann, 2002). Additionally, the inflammatory marker IL-1β, which is also elevated in heart failure, increases Ser850 MYPT1 phosphorylation to inhibit MLC phosphatase (Kandabashi et al., 2000; Mann, 2002). It has been demonstrated that impaired endothelial and vascular smooth muscle dysfunction are associated with a poor prognosis in heart failure (Mann, 2002). Potentially IL-1β and TNF-α could be involved in the regulation of MYPT1 isoform expression, and defining the role of inflammatory cytokines for the regulation of MYPT1 isoform expression could reveal a novel therapy to reverse the vascular dysfunction and improve prognosis in heart failure.
In addition to inhibiting the RhoA/Rho kinase pathway (Arner and Pfitzer, 1999; Somlyo and Somlyo, 2003), ACE inhibitors and ARBs also preserve the LZ+ MYPT1 isoform expression (Chen et al., 2006; Ararat and Brozovich, 2009). The variability in LZ+ MYTP1 expression among different vascular smooth muscle cells and the changes that occur during heart failure could provide an explanation as to why racial differences play an important role in predicting responses to ACE inhibitor therapy compared with the combination of vasodilators and nitrates (Carson et al., 1999). There are racial variations in plasma norepinephrine and renin (Carson et al., 1999), and this could influence both MYPT1 isoform expression as well as MLC phosphatase activity. These factors could explain the more significant blood pressure and mortality reduction seen in white compared with black patients (Carson et al., 1999). MYPT1 polymorphisms may also exist, which could contribute to the diversity of symptoms in patients with similar reductions in LVEF. Hence, the ability of ACE inhibitors and ARBs to alter the vascular smooth muscle cell phenotype could contribute to the improvement in survival in patients with heart failure (Pfeffer et al., 1992; Yusuf et al., 1992; Granger et al., 2003), and thus enhancing LZ+ MYPT1 expression and normalizing the vascular response to NO may represent another novel target for the treatment of heart failure.
2. Pulmonary hypertension.
Pulmonary arterial hypertension (PAH) is a rare, incurable disease with a poor prognosis (Geraci et al., 2001a,b; Archer et al., 2010). In patients with PAH, the pulmonary vasculature is characterized by a resting vasoconstriction and an abnormal response to NO, and in the majority of patients, NO-mediated vasodilatation is severely blunted (Sitbon et al., 1998; McGoon et al., 2004). Clinically, the initial assessment of patients with PAH includes the evaluation of the pulmonary response to NO (Badesch et al., 2004; Barst et al., 2004; McGoon et al., 2004) prognosis is improved if NO decreases pulmonary arterial pressure and PVR by 20% (Sitbon et al., 1998; Barst et al., 2004). However, less than 10% of patients with PAH demonstrate a significant vasodilatory response to NO (Barst et al., 2004), and although there are a number of proposed mechanisms for the progressive increase in PVR (Stenmark et al., 2006; Archer et al., 2010; Farkas et al., 2011), the molecular mechanism that results in the lack of vasodilatation to NO in PAH is unknown.
There are two components that produce PAH: the extent of the structural changes (smooth muscle cell proliferation, smooth muscle cell hypertrophy, and the deposition of matrix proteins within the media of pulmonary arterial vessels (Stenmark et al., 2006; Archer et al., 2010; Farkas et al., 2011) and excess vasoconstriction (Oka et al., 2007; Archer et al., 2010). Structural changes have been documented in animal models of PAH, which have demonstrated that chronic hypoxia leads to pulmonary vascular remodeling that increases PVR and decreases the vasodilatory response to NO (reviewed in Stenmark et al., 2006; Archer et al., 2010; Farkas et al., 2011). However, despite the fact that for PAH the target and aim of all current therapeutic agents [prostaglandins (epoprostenol), phosphodiesterase inhibitors (sildenafil), guanylate cyclase stimulators (riocigaut), endothelin antagonists (bosentan), NO inhalation and Rho kinase inhibitors (fasudil)] is the smooth muscle cell to reduce pulmonary vascular resistance, the fundamental changes in the pulmonary smooth muscle contractile phenotype that contribute to the excess vasoconstriction, or the increase in PVR, as well as the decrease in vasodilatory response to NO, are poorly characterized. Evidence supports a contribution of changes in the pulmonary smooth muscle contractile phenotype in PAH; an increase in the expression and activity of Rho kinase has been demonstrated to contribute to the pathogenesis of PAH (Connolly and Aaronson, 2011), and Rho kinase inhibition reduces pulmonary pressure in some animal models of PAH (Oka et al., 2007; Archer et al., 2010). The importance of the NO/cGMP signaling pathway for the pathogenesis of PAH is highlighted by data demonstrating that PKG KO mice develop pulmonary hypertension (Zhao et al., 2012), and mice with mutations in the PKG LZ domain, which disrupts PKG interaction with MYPT1 (Surks et al., 1999, 2003; Huang et al., 2004; Yuen et al., 2011; Yuen et al., 2014), develop progressive increases in pulmonary pressures and right ventricular hypertrophy (Ramchandran et al., 2014). Furthermore, activation of guanylate cyclase has been demonstrated to improve exercise capacity and reduce pulmonary pressures in patients with PAH (Zhao et al., 2012; Ghofrani et al., 2013a,b; Ramchandran et al., 2014). All these studies are consistent with a defect in NO/cGMP signaling contributing to the pathogenesis of pulmonary hypertension, but do not isolate the step in the NO signaling pathway that produces PAH.
In smooth muscle, investigators have demonstrated that NM myosin (Fig. 7) contributes to the sustained force response (Morano et al., 2000; Lofgren et al., 2003) and a decrease in NM myosin expression will result in a decrease in vascular tone (Yuen et al., 2009). Additionally, changes in the sensitivity to NO-mediated vasodilatation are due to changes in the expression of LZ+/LZ− MYPT1 (Huang et al., 2004). Thus, an increase in NM myosin and decrease in LZ+ MYPT1 expression will produce abnormalities of pulmonary vascular reactivity and may participate in the molecular mechanism that produces PAH (Konik et al., 2013). Konik et al. (2013) demonstrated that with the development of severe PAH, there is a 3.5-fold increase in NM myosin expression, which could contribute to the prolongation in the rates of both force activation and relaxation, as well as the increase in force maintenance. Similarly, an increase in NM myosin expression has also been reported for hypoxia induced PAH (Packer et al., 1998), and therefore drugs that target NM myosin expression or activation could represent a new class of therapeutic agents for the treatment of PAH.
Konik’s data also demonstrated that a decrease in LZ+ MYPT1 expression is associated with severe PAH (Konik et al., 2013). The sensitivity of smooth muscle to NO is regulated, in part, by relative LZ+ MYPT1 expression (Huang et al., 2004). In severe PAH, these data would suggest that the decrease in LZ+ MYPT1 expression would result in a decrease in the sensitivity of the pulmonary vasculature to NO. Similarly, a decrease in LZ+ MYPT1 expression has been reported for cultured pulmonary SMC exposed to hypoxia (Singh et al., 2011). Additionally, the transition from a phasic to tonic SM contractile phenotype has been suggested to be associated with a decrease in the ratio of LZ+ MYPT1/MYPT1 expression (Fisher, 2010). Tonic contractile properties would produce an increase in vascular tone, similar to that observed in patients with PAH. These results are consistent with a decrease in LZ+ MYPT1 expression contributing to the pathogenesis of PAH; a decrease in LZ+ MYPT1 expression would reduce the sensitivity to cGMP, and riociguat treatment (Ghofrani et al., 2013a,b) would increase cGMP to counteract and overcome the decrease in sensitivity to NO to reduce PVR and pulmonary pressures. If a decrease in LZ+ MYPT1 expression contributes to the molecular mechanism for PAH, these data could provide a unifying mechanism to explain the variable response to drugs that act on the NO/cGMP signaling pathway (NO, PDE5 inhibitors, and riociguat), i.e., patients with a mild decrease in LZ+ MYPT1 expression would represent “responders,” whereas patients in which LZ+ MYPT1 expression is severely depressed would not respond to this class of therapeutics. Developing therapeutics that increase LZ+ MYPT1 expression should normalize the pulmonary vascular response to NO and decrease PVR, which would represent a novel target for treating patients with PAH.
3. Portal hypertension.
Vascular reactivity is known to be altered in portal hypertension, with increased sensitivity to dilatation and decrease sensitivity to constriction (Benoit et al., 1984; Sikuler et al., 1985; Bomzon and Blendis, 1994; Wu and Benoit, 1994; Payne et al., 2004). The mechanism that contributes to this altered reactivity is unclear, but evidence suggests that splice variant expression of the contractile proteins is modulated by flow and/or pressure (Zhang and Fisher, 2007). Payne et al. (2004) examined the expression of SM MHC, ELC17, and MYPT1 in the portal vein stenosis model of portal hypertension (Benoit et al., 1984). The ligature of the portal vein produces dynamic changes in flow and pressure; there is an initial increase in both splanchnic pressure and flow (Benoit et al., 1984), which fall with the development of porto-systemic shunting. Similar to these dynamic changes in flow, these investigators (Payne et al., 2004) demonstrated that in both the portal veins and mesenteric arteries within 1 day of the increase in portal pressure, although total MYPT1 expression decreased, the expression of the LZ+ MYPT1 isoform increased. These changes in MYPT1 expression were maintained for ∼7 days before returning to preportal hypertension levels by 14 days after the ligature of the portal vein. Similar to MYPT1 isoform expression, ELC17 and SM MHC expression were modulated with an increase in the expression of the slow isoforms (ELC17b and SMA) 3 days after ligature, but SMA expression returned to baseline at day 14.
Others have altered flow in the mesenteric circulation by ligating alternative pairs of second order mesenteric arteries to produce either low or high flow in the upstream first order mesenteric arteries (Zhang and Fisher, 2007). These investigators demonstrated that there was an initial increase in LZ+ MYPT1 expression in both low and high flow vessels, but by 1 month LZ+ MYPT1 expression returned toward baseline in the low flow vessels, whereas in the high flow vessels, LZ+ MYPT1 expression continued to increase. As would be predicted, the sensitivity to NO/cGMP-mediated relaxation paralleled LZ+ MYPT1 expression. Furthermore, total MYPT1 expression rapidly fell in both the low and high flow mesenteric vessels, and pretreatment with an inhibitor of the proteosome attenuated the decrease in MYPT1. These data suggest that the rapid decrease in total MYPT1 is due to degradation by the proteosome (Zhang and Fisher, 2007), which is similar to the results in the mesenteric arterioles in heart failure (Han and Brozovich, 2013). These data demonstrate that flow and/or pressure is also important in regulating the smooth muscle contractile phenotype, and thus vascular reactivity.
4. Raynaud's phenomenon.
Raynaud’s phenomenon is described as a transient cessation of blood flow in the fingers and toes and can be triggered by either cold or emotional stress. The phenomenon is the result of a transient vasospasm of the digital arteries in the hands and/or feet that leads to a cessation of blood flow, which produces pallor, followed by cyanosis and pain due ischemia of the sensory nerves. As the vasospastic attack subsides, blood flow to the digits is restored, and the resulting hyperemia produces a red phase. The estimates of the prevalence of this disease vary, but it is more common in women, and the onset and severity of the disease peaks between menarche and menopause. Treatment of Raynaud’s involves keeping the digits warm and pharmacologic therapy with CCBs can be helpful; however the usefulness of CCBs is often limited by hypotension (Cooke and Marshall, 2005).
There are a number of mechanisms proposed for the transient vasoconstriction of the digital vessels; these have been reviewed (Cooke and Marshall, 2005) and there is evidence supporting roles for hyperactivity of adrenergic nervous system, α2 adrenoceptors, central stress responses, 5-HT, endothelin, oxidative stress, cyclo-oxygenase, and NO. However, it is interesting that the vasodilation of the digital vessels produced by both sodium nitroprusside and ACh are reduced in patients with Raynaud’s (Morris and Shore, 1996; Cooke and Marshall, 2005), which suggest that the defect lies within the cGMP/PKG signaling cascade. Consistent with this hypothesis are the results that show l-arginine does not restore the normal vasodilatory response to ACh (Khan et al., 1997), which indicates that the vasodilatory response to NO is depressed in Raynaud’s disease, and the mechanism lies beyond the bioavailability of NO within the cGMP/PKG signaling cascade. These data could suggest that a decrease in LZ+ MYPT1 expression in the digital vessels could contribute to the abnormal vascular reactivity.
5. Pre-eclampsia/pregnancy-induced hypertension.
During a normal pregnancy, vascular resistance falls, which is thought to be the results of increased synthesis and activity of NO and prostacyclin (Bird et al., 2003; James and Nelson-Piercy, 2004). However, approximately 5–15% of pregnancies are complicated by hypertension, which increases perinatal morbidity and mortality of both the mother and fetus (Lindheimer, 1993; James and Nelson-Piercy, 2004). Antihypertensive therapy is limited due to the effects on the fetus, i.e., ACE inhibitors and ARBs are fetotoxic (James and Nelson-Piercy, 2004). A number of mechanisms have been proposed for the increase in PVR and blood pressure, including both a decrease in NO produced by endothelial dysfunction and an increase in circulating vasoconstrictors including Ang II, thromboxane, and endothelin (Khalil and Granger, 2002; Bird et al., 2003; Granger et al., 2003). NOS inhibition is commonly used to produce a model of pregnancy-induced hypertension, and studies have suggested that the increase in blood pressure is due to altered vascular reactivity produced by RhoA, PKC, and Ca2+ signaling (Khalil and Granger, 2002; Carter and Kanagy, 2003).
Lu et al. (2008) recently examined MYPT1 isoform expression in pregnancy-induced hypertension. These investigators demonstrated that during a normal pregnancy total MYPT1 expression increased, but there was no change in MYPT1 isoform expression. However, during pregnancy-induced hypertension, although there were no changes in MYPT1 expression in mesenteric arteries or the aorta, in uterine arteries, pregnancy-induced hypertension was associated with an increase in LZ+ MYPT1 expression but an ∼50% decrease in total MYPT1 expression. As would be predicted, the increase in LZ+ MYPT1 expression was associated with an increase in the sensitivity of uterine artery relaxation to both sodium nitroprusside and cGMP. The increase in LZ+ MYPT1 expression did not occur when the blood pressure of the pregnant hypertensive rats was normalized with hydralazine. It is interesting to speculate that during pregnancy-induced hypertension the changes in MYPT1 expression are compensatory, and the resulting reduction in uterine vascular resistance maintains blood flow despite the inward remodeling of the arteries (Lu et al., 2008).
C. Personalized Medicine
A number of studies demonstrate that blood pressure is a genetically determined trait with heritability estimates of 31–68% (Padmanabhan et al., 2012). There are several, rare monogenic syndromes, which are due to abnormalities in renal fluid balance that result in hypertension (Lifton et al., 2001; Ji et al., 2008). However, because the regulation of blood pressure is due to complex interactions of fluid balance, cardiac contractility, and vascular tone, the genetic basis of essential hypertension has yet to be elucidated (Oparil et al., 2003; Padmanabhan et al., 2012). Recently, a number of genome wide-association studies (GWAS) have identified SNPs in a number of genes that could contribute to the development of essential hypertension (Adeyemo et al., 2009; Johnson et al., 2011; Kato et al., 2011; Wain et al., 2011; Havulinna et al., 2013; Tragante et al., 2014). These studies have focused on populations of European decent, but GWAS have examined East Asia (Kato et al., 2011) as well as African American cohorts (Adeyemo et al., 2009). A complete discussion of GWAS methods as well as the results of these studies are beyond the scope of the present review, and we would refer readers to recent reviews on this subject (Ehret, 2010; Padmanabhan et al., 2012). In addition to SNPs, gene expression is also regulated by miR and DNA methylation. The importance of miRs in the regulation of blood pressure is highlighted by a recent study demonstrating that mice lacking miR-142 and miR-145 have reduced vascular tone and blood pressure (Xin et al., 2009). Additionally, hcmv-miR-UL122 is highly expressed in hypertensive patients (Cheng et al., 2009), a number of miRs are increased in hypertensive nephrosclerosis (Wang et al., 2010), and furthermore, renin expression appears to be regulated by mi-RNA-181a and miR-663 renin (Marques et al., 2011). These factors illustrate that SNPs, miRs, and posttranslational modifications all play a role in the development of hypertension.
In addition to revealing candidate genes important in the pathogenesis of essential hypertension, pharmacogenomic GWAS have the potential to reveal the likelihood of a patient to respond to therapy or even to develop a rare adverse drug reaction (Crowley et al., 2009; Johnson et al., 2008). There are several reports of an association between polymorphisms in the β1-adrenergic receptor gene and the lowering of blood pressure (reviewed in Shin and Johnson, 2007). An association of Ser49Gly and Arg389Gly polymorphisms has been demonstrated to be associated with a significant reduction in blood pressure with β-blocker therapy (Johnson et al., 2003), and furthermore, one or both of these SNPs were carried by 54% of Chinese and 44% of whites (Johnson et al., 2003), perhaps suggesting an etiology of the ethnic differences in response to β-blocker therapy. The PEAR study (Gong et al., 2012) evaluated the association of 39 SNPs known to be associated with hypertension from GWAS studies with a response to monotherapy with atenolol or HCTZ in 768 hypertensive patients (60% white and 40% black). The response to atenolol therapy was greater in the white patients; six SNPs were associated with a response to atenolol therapy, with greater responses with in those with all six BP lowering alleles. HCTZ lowered blood pressure less in white compared with black patients. In the white patients, three alleles were associated with BP lowering for HCTZ monotherapy, and similarly, the association was strongest for those with all three alleles, but none of these were associated with BP lowering in blacks. Interestingly for HCTZ therapy, one SNP was associated with a BP reduction in white hypertensive patients, whereas it was associated with an increase in BP in the black patients. A G825C polymorphism in the gene encoding the G-protein β3-subunit has been associated with a response to HCTZ therapy, and this genotype was a stronger predictor of response to diuretics than ethnicity (Turner et al., 2001). There have been two large trials examining polymorphisms in the genes encoding ACE (Arnett et al., 2005) and the AT1 receptor (Brunner et al., 2007) and the response to ACE inhibition. No association was found for the insertion/deletion polymorphism in the ACE gene and the response to lisinopril therapy (Arnett et al., 2005), and similarly for all ethnic groups examined, no genetic association was present for the 1166A-C genotype of the AT1 receptor and response to trandolapril. These data suggest that pharmacogenomic GWAS have the potential to identify genotypes that not only contribute to the development of hypertension but will also predict a positive response to antihypertensive therapy. An individual’s genotype could be then used for a personalized approach for selecting an effective antihypertensive with minimal chance of the patient developing an adverse reaction.
D. Summary of Novel Targets and Potential for Improved Therapies
There is no question that essential hypertension is due, in part, to changes at the level of vascular smooth muscle that lead to excess vasoconstriction and a concomitant increase in SVR and blood pressure. In this review, we suggested have several novel targets for the treatment of essential hypertension. Details and the strategy for the each target can be found in the individual sections. These targets include those that affect Ca2+, including 1) downregulation of AKAP150, 2) increasing the expression of the β1 subunit of the BK channel, 3) novel BK channel openers, 4) inhibiting or decreasing the expression of STIM/Orai (Ca2+ release-activated Ca2+ channels), and 5) increasing cleavage of the C terminal of LTCC or increasing CCt expression. One could also consider inhibition of MLCK as a novel target; the soybean isoform of calmodulin (SCaM-4) has been demonstrated to bind Ca2+ but not activate MLCK and thus inhibit the activation of smooth muscle (Lee et al., 2000a; Van Lierop et al., 2002). Thus, targeted expression of SCaM-4 or a CaM fragment in vascular smooth muscle should reduce SVR and blood pressure. There are also a number of scaffolds including those for ERK (calponin, SmAV, and paxillin) and MLC phosphatase (M-RIP and Par-4) that could be examined as potential therapeutic targets, which could result in a reduction of vascular tone. Inhibition of the signaling pathways for Ca2+ sensitization including Rho kinase and GEF signaling could represent another area for rational drug design, and similarly, activation of pathways leading to Ca2+ desensitization including guanylate cyclase and increasing the expression of LZ+ MYPT1 would enhance NO/flow mediated vasodilatation, which should decrease blood pressure. Other attractive targets would be pathways leading to remodeling of the actin cytoskeleton including integrins, focal adhesion proteins, as well as the activation of tyrosine kinases. Targeted delivery of miRs to the vasculature also may prove to be an effective strategy for the treatment of hypertension; miRs could be used as a strategy to change vascular smooth muscle gene program from high force (tonic) and NO unresponsive to a lower force (phasic) and NO responsive and hence produce a reduction in vascular tone and blood pressure.
These novel targets could form the basis for rationale drug design, and could be exploited for development of therapeutic agents that are effective for the treatment of hypertension. For some targets, small molecule inhibitors/activators would appear to be a reasonable choice, whereas for other targets, i.e., to reverse the decreases in MYPT1 expression, adenoviral delivery of MYPT1 to the vasculature could also be used as a strategy to increase the sensitivity to NO or flow-mediated vasodilation, which would decrease in vascular tone and blood pressure.
The contribution of alteration in the vascular phenotype from Ca2+ signaling, regulation of the cytoskeleton and contractility, and biomechanics to the pathogenesis of essential hypertension may vary among ethnic groups and/or individuals. Currently, antihypertensive therapy is generally approached by a method that could be best summarized as trial and error: an agent is selected, usually without consideration of the patient’s ethnic background, and if control is not adequate, either the dose is increased or another agent is added.
In this review, we discussed the importance of the vascular phenotype in the mechanism that leads to essential hypertension. Therefore, mechanisms to identify the etiology of the increase in blood pressure in each patient are important for ultimately selecting an individualized and effective therapeutic agent. These future studies will hopefully lead to a more personalized approach to antihypertensive therapy.
Abbreviations
- ACE
angiotensin converting enzyme
- AM
rigor state
- Ang II
angiotensin II
- ARB
angiotensin receptor blocker
- AT1
angiotensin type 1
- BK
Ca2+-dependent K+ channels
- BP
blood pressure
- CaCC
Ca2+ gated Cl− channel
- CaD
caldesmon
- Cae
extracellular Ca2+
- CaP
calponin
- CCB
calcium channel blocker
- CCt
C-terminal end of the LTCC
- CI
central insert
- CICR
Ca2+-induced Ca2+ release
- CO
cardiac output
- CRAC
calcium release activated calcium channel
- CREB
cAMP response element-binding protein
- CVD
cardiovascular disease
- EC
endothelial cells
- ECM
extracellular matrix
- ELC17
17-kDa essential myosin light chain
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular regulated kinase
- FA
focal adhesion
- FAK
focal adhesion kinase
- GAP
GTPase activating protein
- GEF
guanine nucleotide exchange factor
- GWAS
genome wide-association studies
- HAT
histone acetylase
- HDAC
histone deacetylase
- HF
heart failure
- IL
interleukin
- KLF
Kruppel-like factor
- KO
knockout
- LTCC
L-type Ca2+ channels
- LncRNA
long noncoding RNA
- LZ
leucine zipper
- MHC
muscle myosin heavy chain
- miR
microRNAs
- MLCK
myosin light chain kinase
- MP
myosin phosphatase
- NAD
nicotinamide adenine dinucleotide
- NM
nonmuscle
- NO
nitric oxide
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary artery smooth muscle cells
- PKGI
protein kinase G
- pre-miRNA
preliminary miRNA
- PWV
pulse wave velocity
- RISC
RNA-induced silencing complex
- RLC
regulatory myosin light chain
- RyR
ryanodine receptors
- SHR
spontaneously hypertensive rat
- SIRT
sirtuin
- SM
smooth muscle
- SMA
slow isoform of smooth muscle myosin heavy chain
- SMB
fast isoform of smooth muscle myosin heavy chain
- SNP
single nucleotide polymorphism
- SOCE
store-operated calcium entry
- SR
sarcoplasmic reticulum
- STIM
stromal interaction molecule
- SVR
systemic vascular resistance
- TNF
tumor necrosis factor
- 3′ UTR
3′ untranslated
- VSMC
vascular smooth muscle cells
- WT
wild type
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Brozovich, Nicholson, Degen, Gao, Aggarwal, and Morgan.
Footnotes
References
- Abassi ZA, Gurbanov K, Mulroney SE, Potlog C, Opgenorth TJ, Hoffman A, Haramati A, Winaver J. (1997) Impaired nitric oxide-mediated renal vasodilation in rats with experimental heart failure: role of angiotensin II. Circulation 96:3655–3664. [DOI] [PubMed] [Google Scholar]
- Adeyemo A, Gerry N, Chen G, Herbert A, Doumatey A, Huang H, Zhou J, Lashley K, Chen Y, Christman M, et al. (2009) A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet 5:e1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alahyan M, Webb MR, Marston SB, El-Mezgueldi M. (2006) The mechanism of smooth muscle caldesmon-tropomyosin inhibition of the elementary steps of the actomyosin ATPase. J Biol Chem 281:19433–19448. [DOI] [PubMed] [Google Scholar]
- Albert AP, Large WA. (2002) A Ca2+-permeable non-selective cation channel activated by depletion of internal Ca2+ stores in single rabbit portal vein myocytes. J Physiol 538:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albinsson S, Skoura A, Yu J, DiLorenzo A, Fernández-Hernando C, Offermanns S, Miano JM, Sessa WC. (2011) Smooth muscle miRNAs are critical for post-natal regulation of blood pressure and vascular function. PLoS One 6:e18869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC. (2010) MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol 30:1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MR, Owens GK. (2012) Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74:13–40. [DOI] [PubMed] [Google Scholar]
- Ali F, Chin L, Pare PD, Seow CY. (2007) Mechanism of partial adaptation in airway smooth muscle after a step change in length. J Appl Physiol 103:569–577. [DOI] [PubMed] [Google Scholar]
- Alioua A, Tanaka Y, Wallner M, Hofmann F, Ruth P, Meera P, Toro L. (1998) The large conductance, voltage-dependent, and calcium-sensitive K+ channel, Hslo, is a target of cGMP-dependent protein kinase phosphorylation in vivo. J Biol Chem 273:32950–32956. [DOI] [PubMed] [Google Scholar]
- ALLHAT and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (2002) Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). JAMA 288:2998–3007. [DOI] [PubMed] [Google Scholar]
- Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. (2003) Modulation of the molecular composition of large conductance, Ca(2+) activated K(+) channels in vascular smooth muscle during hypertension. J Clin Invest 112:717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC, Navedo MF. (2013) Calcium dynamics in vascular smooth muscle. Microcirculation 20:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC, Navedo MF, Nieves-Cintrón M, Molkentin JD, Santana LF. (2007) Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J Physiol 579:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC, Rossow CF, Navedo MF, Santana LF. (2004) NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem 279:47326–47334. [DOI] [PubMed] [Google Scholar]
- Ameres SL, Zamore PD. (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14:475–488. [DOI] [PubMed] [Google Scholar]
- Appel S, Allen PG, Vetterkind S, Jin JP, Morgan KG. (2010) h3/Acidic calponin: an actin-binding protein that controls extracellular signal-regulated kinase 1/2 activity in nonmuscle cells. Mol Biol Cell 21:1409–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate D, Feng W, Green RS, Taubman MB. (1994) Cloning and expression of a novel acidic calponin isoform from rat aortic vascular smooth muscle. J Biol Chem 269:10683–10690. [PubMed] [Google Scholar]
- Applegate D, Pardee JD. (1992) Actin-facilitated assembly of smooth muscle myosin induces formation of actomyosin fibrils. J Cell Biol 117:1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ararat E, Brozovich FV. (2009) Losartan decreases p42/44 MAPK signaling and preserves LZ+ MYPT1 expression. PLoS One 4:e5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Weir EK, Wilkins MR. (2010) Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121:2045–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner A, Pfitzer G. (1999) Regulation of cross-bridge cycling by Ca2+ in smooth muscle. Rev Physiol Biochem Pharmacol 134:63–146. [DOI] [PubMed] [Google Scholar]
- Arnett DK, Davis BR, Ford CE, Boerwinkle E, Leiendecker-Foster C, Miller MB, Black H, Eckfeldt JH. (2005) Pharmacogenetic association of the angiotensin-converting enzyme insertion/deletion polymorphism on blood pressure and cardiovascular risk in relation to antihypertensive treatment: the Genetics of Hypertension-Associated Treatment (GenHAT) study. Circulation 111:3374–3383. [DOI] [PubMed] [Google Scholar]
- Asano T, Suzuki T, Tsuchiya M, Satoh S, Ikegaki I, Shibuya M, Suzuki Y, Hidaka H. (1989) Vasodilator actions of HA1077 in vitro and in vivo putatively mediated by the inhibition of protein kinase. Br J Pharmacol 98:1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton FT, Somlyo AV, Somlyo AP. (1975) The contractile apparatus of vascular smooth muscle: intermediate high voltage stereo electron microscopy. J Mol Biol 98:17–29. [DOI] [PubMed] [Google Scholar]
- Aubart FC, Sassi Y, Coulombe A, Mougenot N, Vrignaud C, Leprince P, Lechat P, Lompré AM, Hulot JS. (2009) RNA interference targeting STIM1 suppresses vascular smooth muscle cell proliferation and neointima formation in the rat. Mol Ther 17:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avolio A, Butlin M, Liu Y-Y, Viegas K, Avadhanam B, Lindesay G. (2011) Regulation of arterial stiffness: Cellular, molecular and neurogenic mechanisms. Artery Res 5:122–127. [Google Scholar]
- Awan NA, Miller RR, DeMaria AN, Maxwell KS, Neumann A, Mason DT. (1977) Efficacy of ambulatory systemic vasodilator therapy with oral prazosin in chronic refractory heart failure. Concomitant relief of pulmonary congestion and elevation of pump output demonstrated by improvements in symptomatology, exercise tolerance, hemodynamics and echocardiography. Circulation 56:346–354. [DOI] [PubMed] [Google Scholar]
- Babij P, Periasamy M. (1989) Myosin heavy chain isoform diversity in smooth muscle is produced by differential RNA processing. J Mol Biol 210:673–679. [DOI] [PubMed] [Google Scholar]
- Babu GJ, Loukianov E, Loukianova T, Pyne GJ, Huke S, Osol G, Low RB, Paul RJ, Periasamy M. (2001) Loss of SM-B myosin affects muscle shortening velocity and maximal force development. Nat Cell Biol 3:1025–1029. [DOI] [PubMed] [Google Scholar]
- Babu GJ, Pyne GJ, Zhou Y, Okwuchukuasanya C, Brayden JE, Osol G, Paul RJ, Low RB, Periasamy M. (2004) Isoform switching from SM-B to SM-A myosin results in decreased contractility and altered expression of thin filament regulatory proteins. Am J Physiol Cell Physiol 287:C723–C729. [DOI] [PubMed] [Google Scholar]
- Badesch DB, Abman SH, Ahearn GS, Barst RJ, McCrory DC, Simonneau G, McLaughlin VV, American College of Chest Physicians (2004) Medical therapy for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126(1, Suppl)35S–62S. [DOI] [PubMed] [Google Scholar]
- Baker JE, Brosseau C, Fagnant P, Warshaw DM. (2003) The unique properties of tonic smooth muscle emerge from intrinsic as well as intermolecular behaviors of Myosin molecules. J Biol Chem 278:28533–28539. [DOI] [PubMed] [Google Scholar]
- Bank AJ, Kaiser DR. (1998) Smooth muscle relaxation: effects on arterial compliance, distensibility, elastic modulus, and pulse wave velocity. Hypertension 32:356–359. [DOI] [PubMed] [Google Scholar]
- Bank AJ, Wang H, Holte JE, Mullen K, Shammas R, Kubo SH. (1996) Contribution of collagen, elastin, and smooth muscle to in vivo human brachial artery wall stress and elastic modulus. Circulation 94:3263–3270. [DOI] [PubMed] [Google Scholar]
- Bannister JP, Leo MD, Narayanan D, Jangsangthong W, Nair A, Evanson KW, Pachuau J, Gabrick KS, Boop FA, Jaggar JH. (2013) The voltage-dependent L-type Ca2+ (CaV1.2) channel C-terminus fragment is a bi-modal vasodilator. J Physiol 591:2987–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbato JC. (2005) Nicorandil: the drug that keeps on giving. Hypertension 46:647–648. [DOI] [PubMed] [Google Scholar]
- Barlow CA, Rose P, Pulver-Kaste RA, Lounsbury KM. (2006) Excitation-transcription coupling in smooth muscle. J Physiol 570:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barra JG, Armentano RL, Levenson J, Fischer EI, Pichel RH, Simon A. (1993) Assessment of smooth muscle contribution to descending thoracic aortic elastic mechanics in conscious dogs. Circ Res 73:1040–1050. [DOI] [PubMed] [Google Scholar]
- Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, Gaine S. (2004) Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 43(12, Suppl S)40S–47S. [DOI] [PubMed] [Google Scholar]
- Bayliss WM. (1902) On the local reactions of the arterial wall to changes of internal pressure. J Physiol 28:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayoglu B, Arslan C, Gode S, Kaya Dagistanli F, Arapi B, Burc Deser S, Dirican A, Cengiz M. (2014) The severity of internal carotid artery stenosis is associated with the cyclin-dependent kinase inhibitor 2A gene expression. J Atheroscler Thromb 21:659–671. [DOI] [PubMed] [Google Scholar]
- Beech DJ. (2005) Emerging functions of 10 types of TRP cationic channel in vascular smooth muscle. Clin Exp Pharmacol Physiol 32:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. (2012) Integration of transient receptor potential canonical channels with lipids. Acta Physiol (Oxf) 204:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. (2013) Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ J 77:570–579. [DOI] [PubMed] [Google Scholar]
- Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL, Zhou Q, Han Y, Spector DL, Zheng D, et al. (2014) Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol 34:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, Boutouyrie P, Cameron J, Chen C-H, Cruickshank JK, et al. (2014) Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol 63:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit JN, Barrowman JA, Harper SL, Kvietys PR, Granger DN. (1984) Role of humoral factors in the intestinal hyperemia associated with chronic portal hypertension. Am J Physiol 247:G486–G493. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. (2006) Calcium microdomains: organization and function. Cell Calcium 40:405–412. [DOI] [PubMed] [Google Scholar]
- Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM, et al. (2015) The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem 290:2069–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattachariya A, Dahan D, Turczyńska KM, Swärd K, Hellstrand P, Albinsson S. (2014) Expression of microRNAs is essential for arterial myogenic tone and pressure-induced activation of the PI3-kinase/Akt pathway. Cardiovasc Res 101:288–296. [DOI] [PubMed] [Google Scholar]
- Bierer R, Nitta CH, Friedman J, Codianni S, de Frutos S, Dominguez-Bautista JA, Howard TA, Resta TC, Bosc LV. (2011) NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am J Physiol Lung Cell Mol Physiol 301:L872–L880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE. (2014) Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol Rev 66:513–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington N, Beach JR, Heissler SM, Remmert K, Guzik-Lendrum S, Nagy A, Takagi Y, Shao L, Li D, Yang Y, et al. (2015) Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr Biol 25:942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington N, Wang A, Mao J, Adelstein RS, Sellers JR. (2013) Characterization of three full-length human nonmuscle myosin II paralogs. J Biol Chem 288:33398–33410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird IM, Zhang L, Magness RR. (2003) Possible mechanisms underlying pregnancy-induced changes in uterine artery endothelial function. Am J Physiol Regul Integr Comp Physiol 284:R245–R258. [DOI] [PubMed] [Google Scholar]
- Birukov KG, Stepanova OV, Nanaev AK, Shirinsky VP. (1991) Expression of calponin in rabbit and human aortic smooth muscle cells. Cell Tissue Res 266:579–584. [DOI] [PubMed] [Google Scholar]
- Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. (1999) Impact of aortic stiffness on survival in end-stage renal disease. Circulation 99:2434–2439. [DOI] [PubMed] [Google Scholar]
- Boedtkjer DM, Matchkov VV, Boedtkjer E, Nilsson H, Aalkjaer C. (2008) Vasomotion has chloride-dependency in rat mesenteric small arteries. Pflugers Arch 457:389–404. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Füchtbauer AC, Simonsen U, Füchtbauer EM, Aalkjaer C. (2011) Disruption of Na+,HCO₃⁻ cotransporter NBCn1 (slc4a7) inhibits NO-mediated vasorelaxation, smooth muscle Ca²⁺ sensitivity, and hypertension development in mice. Circulation 124:1819–1829. [DOI] [PubMed] [Google Scholar]
- Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T. (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119:2634–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. (2011) Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 183:1402–1410. [DOI] [PubMed] [Google Scholar]
- Bomzon A, Blendis LM. (1994) The nitric oxide hypothesis and the hyperdynamic circulation in cirrhosis. Hepatology 20:1343–1350. [PubMed] [Google Scholar]
- Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, et al. (2011) MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109:1115–1119. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ. (1996) Subcellular Ca2+ signals underlying waves and graded responses in HeLa cells. Curr Biol 6:855–865. [DOI] [PubMed] [Google Scholar]
- Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. (2011) The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. J Biol Chem 286:28312–28321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken CP, Li X, Wright JA, Lawrence DM, Pillman KA, Salmanidis M, Anderson MA, Dredge BK, Gregory PA, Tsykin A, et al. (2014) Genome-wide identification of miR-200 targets reveals a regulatory network controlling cell invasion. EMBO J 33:2040–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley AB, Morgan KG. (1985) Cellular Ca2+ monitored by aequorin in adenosine-mediated smooth muscle relaxation. Am J Physiol 248:H109–H117. [DOI] [PubMed] [Google Scholar]
- Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. (2000) Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407:870–876. [DOI] [PubMed] [Google Scholar]
- Brock M, Haider TJ, Vogel J, Gassmann M, Speich R, Trenkmann M, Ulrich S, Kohler M, Huber LC. (2015) The hypoxia-induced microRNA-130a controls pulmonary smooth muscle cell proliferation by directly targeting CDKN1A. Int J Biochem Cell Biol 61:129–137. [DOI] [PubMed] [Google Scholar]
- Brooke BS, Karnik SK, Li DY. (2003) Extracellular matrix in vascular morphogenesis and disease: structure versus signal. Trends Cell Biol 13:51–56. [DOI] [PubMed] [Google Scholar]
- Brown DI, Griendling KK. (2015) Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116:531–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M, Cooper-DeHoff RM, Gong Y, Karnes JH, Langaee TY, Pepine CJ, Johnson JA, Investigators I, INVEST Investigators (2007) Factors influencing blood pressure response to trandolapril add-on therapy in patients taking verapamil SR (from the International Verapamil SR/Trandolapril [INVEST] Study). Am J Cardiol 99:1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan J. (1990) Caldesmon: fragments, sequence, and domain mapping. Ann N Y Acad Sci 599:100–110. [DOI] [PubMed] [Google Scholar]
- Bulley S, Jaggar JH. (2014) Cl⁻ channels in smooth muscle cells. Pflugers Arch 466:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W, Jaggar JH. (2012) TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res 111:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas ME, Hu MI, Durand JB, Busaidy NL. (2011) Challenges associated with tyrosine kinase inhibitor therapy for metastatic thyroid cancer. J Thyroid Res 2011:985780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon RO., 3rd (1998) Role of nitric oxide in cardiovascular disease: focus on the endothelium. Clin Chem 44:1809–1819. [PubMed] [Google Scholar]
- Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM, Francis J. (2010) HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension 56:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrión AM, Link WA, Ledo F, Mellström B, Naranjo JR. (1999) DREAM is a Ca2+-regulated transcriptional repressor. Nature 398:80–84. [DOI] [PubMed] [Google Scholar]
- Carson P, Ziesche S, Johnson G, Cohn JN, Vasodilator-Heart Failure Trial Study Group (1999) Racial differences in response to therapy for heart failure: analysis of the vasodilator-heart failure trials. J Card Fail 5:178–187. [DOI] [PubMed] [Google Scholar]
- Carter RW, Kanagy NL. (2003) Mechanism of enhanced calcium sensitivity and alpha 2-AR vasoreactivity in chronic NOS inhibition hypertension. Am J Physiol Heart Circ Physiol 284:H309–H316. [DOI] [PubMed] [Google Scholar]
- Cartin L, Lounsbury KM, Nelson MT. (2000) Coupling of Ca(2+) to CREB activation and gene expression in intact cerebral arteries from mouse : roles of ryanodine receptors and voltage-dependent Ca(2+) channels. Circ Res 86:760–767. [DOI] [PubMed] [Google Scholar]
- Castro R, Rivera I, Struys EA, Jansen EE, Ravasco P, Camilo ME, Blom HJ, Jakobs C, Tavares de Almeida I. (2003) Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem 49:1292–1296. [DOI] [PubMed] [Google Scholar]
- Chacko S, Conti MA, Adelstein RS. (1977) Effect of phosphorylation of smooth muscle myosin on actin activation and Ca2+ regulation. Proc Natl Acad Sci USA 74:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, Lieberman J, Lagna G, Hata A. (2010) Molecular basis for antagonism between PDGF and the TGFbeta family of signalling pathways by control of miR-24 expression. EMBO J 29:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MY, Huang DY, Ho FM, Huang KC, Lin WW. (2012) PKC-dependent human monocyte adhesion requires AMPK and Syk activation. PLoS One 7:e40999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Karolczak-Bayatti M, Sweeney M, Treumann A, Morrissey K, Ulrich SM, Europe-Finner GN, Taggart MJ. (2013) Lysine deacetylase inhibition promotes relaxation of arterial tone and C-terminal acetylation of HSPB6 (Hsp20) in vascular smooth muscle cells. Physiol Rep 1:e00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Chen X, Qiao YN, Wang P, He WQ, Zhang CH, Zhao W, Gao YQ, Chen C, Tao T, et al. (2015) In vivo roles for myosin phosphatase targeting subunit-1 phosphorylation sites T694 and T852 in bladder smooth muscle contraction. J Physiol 593:681–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FC, Brozovich FV. (2008) Gene expression profiles of vascular smooth muscle show differential expression of mitogen-activated protein kinase pathways during captopril therapy of heart failure. J Vasc Res 45:445–454. [DOI] [PubMed] [Google Scholar]
- Chen FC, Ogut O, Rhee AY, Hoit BD, Brozovich FV. (2006) Captopril prevents myosin light chain phosphatase isoform switching to preserve normal cGMP-mediated vasodilatation. J Mol Cell Cardiol 41:488–495. [DOI] [PubMed] [Google Scholar]
- Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves-Cintrón M, Scott JD, Santana LF. (2011) Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ Res 109:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Ke Q, Jin Z, Wang H, Kocher O, Morgan JP, Zhang J, Crumpacker CS. (2009) Cytomegalovirus infection causes an increase of arterial blood pressure. PLoS Pathog 5:e1000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG, Gupte SA. (2014) Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol 306:H214–H224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Allis CD, Sassone-Corsi P. (2000) Signaling to chromatin through histone modifications. Cell 103:263–271. [DOI] [PubMed] [Google Scholar]
- Chi M, Zhou Y, Vedamoorthyrao S, Babu GJ, Periasamy M. (2008) Ablation of smooth muscle myosin heavy chain SM2 increases smooth muscle contraction and results in postnatal death in mice. Proc Natl Acad Sci USA 105:18614–18618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs TJ, Watson MH, Sanghera JS, Campbell DL, Pelech SL, Mak AS. (1992) Phosphorylation of smooth muscle caldesmon by mitogen-activated protein (MAP) kinase and expression of MAP kinase in differentiated smooth muscle cells. J Biol Chem 267:22853–22859. [PubMed] [Google Scholar]
- Choe N, Kwon JS, Kim JR, Eom GH, Kim Y, Nam KI, Ahn Y, Kee HJ, Kook H. (2013) The microRNA miR-132 targets Lrrfip1 to block vascular smooth muscle cell proliferation and neointimal hyperplasia. Atherosclerosis 229:348–355. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. [DOI] [PubMed] [Google Scholar]
- Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, Woulfe K, Pravda E, Cassiola F, Desai J, et al. (2007) Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370:2011–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, McNally EM. (2002) Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 K(ATP) channels. J Clin Invest 110:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Gokina NI, Osol G. (2002) Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J 16:72–76. [DOI] [PubMed] [Google Scholar]
- Clark JM, Glagov S. (1985) Transmural organization of the arterial media. The lamellar unit revisited. Arteriosclerosis 5:19–34. [DOI] [PubMed] [Google Scholar]
- Coffman TM, Crowley SD. (2008) Kidney in hypertension: guyton redux. Hypertension 51:811–816. [DOI] [PubMed] [Google Scholar]
- Collier ML, Ji G, Wang Y, Kotlikoff MI. (2000) Calcium-induced calcium release in smooth muscle: loose coupling between the action potential and calcium release. J Gen Physiol 115:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congrains A, Kamide K, Katsuya T, Yasuda O, Oguro R, Yamamoto K, Ohishi M, Rakugi H. (2012a) CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem Biophys Res Commun 419:612–616. [DOI] [PubMed] [Google Scholar]
- Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, Kawai T, Kusunoki H, Yamamoto H, Takeya Y, et al. (2012b) Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 220:449–455. [DOI] [PubMed] [Google Scholar]
- Connell LE, Helfman DM. (2006) Myosin light chain kinase plays a role in the regulation of epithelial cell survival. J Cell Sci 119:2269–2281. [DOI] [PubMed] [Google Scholar]
- Connolly MJ, Aaronson PI. (2011) Key role of the RhoA/Rho kinase system in pulmonary hypertension. Pulm Pharmacol Ther 24:1–14. [DOI] [PubMed] [Google Scholar]
- Cooke JP, Marshall JM. (2005) Mechanisms of Raynaud’s disease. Vasc Med 10:293–307. [DOI] [PubMed] [Google Scholar]
- Corbi G, Conti V, Russomanno G, Longobardi G, Furgi G, Filippelli A, Ferrara N. (2013) Adrenergic signaling and oxidative stress: a role for sirtuins? Front Physiol 4:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RH, Rusch NJ. (2002) New expression profiles of voltage-gated ion channels in arteries exposed to high blood pressure. Microcirculation 9:243–257. [DOI] [PubMed] [Google Scholar]
- Cremo CR, Wang F, Facemyer K, Sellers JR. (2001) Phosphorylation-dependent regulation is absent in a nonmuscle heavy meromyosin construct with one complete head and one head lacking the motor domain. J Biol Chem 276:41465–41472. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Sullivan PF, McLeod HL. (2009) Pharmacogenomic genome-wide association studies: lessons learned thus far. Pharmacogenomics 10:161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, et al. (2005) Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 115:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, Gosling RG. (2002) Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation 106:2085–2090. [DOI] [PubMed] [Google Scholar]
- Cushman WC, Reda DJ, Perry HM, Williams D, Abdellatif M, Materson BJ, Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents (2000) Regional and racial differences in response to antihypertensive medication use in a randomized controlled trial of men with hypertension in the United States. Arch Intern Med 160:825–831. [DOI] [PubMed] [Google Scholar]
- Dahan D, Ekman M, Larsson-Callerfelt AK, Turczyńska K, Boettger T, Braun T, Swärd K, Albinsson S. (2014) Induction of angiotensin-converting enzyme after miR-143/145 deletion is critical for impaired smooth muscle contractility. Am J Physiol Cell Physiol 307:C1093–C1101. [DOI] [PubMed] [Google Scholar]
- Dam VS, Boedtkjer DM, Nyvad J, Aalkjaer C, Matchkov V. (2014) TMEM16A knockdown abrogates two different Ca(2+)-activated Cl (-) currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch 466:1391–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo G, Graceffa P, Wang CA, Wrangle J, Adam LP. (1999) Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Quantitation using novel anti-phosphopeptide antibodies. J Biol Chem 274:30115–30121. [DOI] [PubMed] [Google Scholar]
- Das S, Halushka MK. (2015) Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc Pathol 24:199–206. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Shi J, Pritchard HA, Chadha PS, Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP, Greenwood IA. (2013) Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh) -A01. Br J Pharmacol 168:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. (1999) Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79:387–423. [DOI] [PubMed] [Google Scholar]
- Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A. (2011) down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem 286:28097–28110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz EM, Ostap EM. (2004) Relating biochemistry and function in the myosin superfamily. Curr Opin Cell Biol 16:61–67. [DOI] [PubMed] [Google Scholar]
- Deng JT, Sutherland C, Brautigan DL, Eto M, Walsh MP. (2002) Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem J 367:517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. (1998) A role for MAP kinase in differentiated smooth muscle contraction evoked by alpha-adrenoceptor stimulation. Am J Physiol 275:C1081–C1086. [DOI] [PubMed] [Google Scholar]
- Devine CE, Somlyo AP. (1971) Thick filaments in vascular smooth muscle. J Cell Biol 49:636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. (2007) Scaffold proteins of MAP-kinase modules. Oncogene 26:3185–3202. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. (2003) N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem 278:47842–47852. [DOI] [PubMed] [Google Scholar]
- Dillon PF, Aksoy MO, Driska SP, Murphy RA. (1981) Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science 211:495–497. [DOI] [PubMed] [Google Scholar]
- Dillon PF, Murphy RA. (1982) Tonic force maintenance with reduced shortening velocity in arterial smooth muscle. Am J Physiol 242:C102–C108. [DOI] [PubMed] [Google Scholar]
- Ding SY, Lee MJ, Summer R, Liu L, Fried SK, Pilch PF. (2014) Pleiotropic effects of cavin-1 deficiency on lipid metabolism. J Biol Chem 289:8473–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, Garcia JA. (2009) Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324:1289–1293. [DOI] [PubMed] [Google Scholar]
- Dirksen WP, Vladic F, Fisher SA. (2000) A myosin phosphatase targeting subunit isoform transition defines a smooth muscle developmental phenotypic switch. Am J Physiol Cell Physiol 278:C589–C600. [DOI] [PubMed] [Google Scholar]
- Dobrin PB. (1978) Mechanical properties of arterises. Physiol Rev 58:397–460. [DOI] [PubMed] [Google Scholar]
- Dong C, Yoon W, Goldschmidt-Clermont PJ. (2002) DNA methylation and atherosclerosis. J Nutr 132(8, Suppl)2406S–2409S. [DOI] [PubMed] [Google Scholar]
- Dou D, Ma H, Zheng X, Ying L, Guo Y, Yu X, Gao Y. (2010) Degradation of leucine zipper-positive isoform of MYPT1 may contribute to development of nitrate tolerance. Cardiovasc Res 86:151–159. [DOI] [PubMed] [Google Scholar]
- Drew JS, Moos C, Murphy RA. (1991) Localization of isoactins in isolated smooth muscle thin filaments by double gold immunolabeling. Am J Physiol 260:C1332–C1340. [DOI] [PubMed] [Google Scholar]
- Duband JL, Gimona M, Scatena M, Sartore S, Small JV. (1993) Calponin and SM 22 as differentiation markers of smooth muscle: spatiotemporal distribution during avian embryonic development. Differentiation 55:1–11. [DOI] [PubMed] [Google Scholar]
- Dustan HP, Roccella EJ, Garrison HH. (1996) Controlling hypertension. A research success story. Arch Intern Med 156:1926–1935. [PubMed] [Google Scholar]
- Dzau VJ. (2001) Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 37:1047–1052. [DOI] [PubMed] [Google Scholar]
- Earley S, Brayden JE. (2015) Transient receptor potential channels in the vasculature. Physiol Rev 95:645–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. (2009) TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol 297:H1096–H1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddinger TJ, Meer DP, Miner AS, Meehl J, Rovner AS, Ratz PH. (2007) Potent inhibition of arterial smooth muscle tonic contractions by the selective myosin II inhibitor, blebbistatin. J Pharmacol Exp Ther 320:865–870. [DOI] [PubMed] [Google Scholar]
- Ehret GB. (2010) Genome-wide association studies: contribution of genomics to understanding blood pressure and essential hypertension. Curr Hypertens Rep 12:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-Mezgueldi M, Marston SB. (1996) The effects of smooth muscle calponin on the strong and weak myosin binding sites of F-actin. J Biol Chem 271:28161–28167. [DOI] [PubMed] [Google Scholar]
- El-Touhky A, Given AM, Cochard A, Brozovich FV. (2005) PHI-1 induced enhancement of myosin phosphorylation in chicken smooth muscle. FEBS Lett 579:4271–4277. [DOI] [PubMed] [Google Scholar]
- El-Toukhy A, Given AM, Ogut O, Brozovich FV. (2006) PHI-1 interacts with the catalytic subunit of myosin light chain phosphatase to produce a Ca(2+) independent increase in MLC(20) phosphorylation and force in avian smooth muscle. FEBS Lett 580:5779–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Yazbi AF, Abd-Elrahman KS, Moreno-Dominguez A. (2015) PKC-mediated cerebral vasoconstriction: Role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem Pharmacol 95:263–278. [DOI] [PubMed] [Google Scholar]
- El-Yazbi AF, Johnson RP, Walsh EJ, Takeya K, Walsh MP, Cole WC. (2010) Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol 588:1747–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison PA, Sellers JR, Cremo CR. (2000) Kinetics of smooth muscle heavy meromyosin with one thiophosphorylated head. J Biol Chem 275:15142–15151. [DOI] [PubMed] [Google Scholar]
- Essin K, Welling A, Hofmann F, Luft FC, Gollasch M, Moosmang S. (2007) Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol 584:205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban V, Méndez-Barbero N, Jiménez-Borreguero LJ, Roqué M, Novensá L, García-Redondo AB, Salaices M, Vila L, Arbonés ML, Campanero MR, et al. (2011) Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med 208:2125–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Karginov A, Brautigan DL. (1999) A novel phosphoprotein inhibitor of protein type-1 phosphatase holoenzymes. Biochemistry 38:16952–16957. [DOI] [PubMed] [Google Scholar]
- Eto M, Kitazawa T, Matsuzawa F, Aikawa S, Kirkbride JA, Isozumi N, Nishimura Y, Brautigan DL, Ohki SY. (2007) Phosphorylation-induced conformational switching of CPI-17 produces a potent myosin phosphatase inhibitor. Structure 15:1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. (1995) A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J Biochem 118:1104–1107. [DOI] [PubMed] [Google Scholar]
- Europe-Finner GN, Karolczak-Bayatti M, Taggart MJ. (2015) The multifaceted KDAC8: a smooth muscle contractile regulator. Trends Pharmacol Sci 36:493. [DOI] [PubMed] [Google Scholar]
- Farkas L, Gauldie J, Voelkel NF, Kolb M. (2011) Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol 45:1–15. [DOI] [PubMed] [Google Scholar]
- Fatigati V, Murphy RA. (1984) Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem 259:14383–14388. [PubMed] [Google Scholar]
- Fediuk J, Dakshinamurti S. (2015) A role for actin polymerization in persistent pulmonary hypertension of the newborn. Can J Physiol Pharmacol 93:185–194. [DOI] [PubMed] [Google Scholar]
- Fernández-Fernández JM, Tomás M, Vázquez E, Orio P, Latorre R, Sentí M, Marrugat J, Valverde MA. (2004) Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J Clin Invest 113:1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D. (2011) Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol 31:851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SA. (2010) Vascular smooth muscle phenotypic diversity and function. Physiol Genomics 42A:169–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Marshall KD, Durrant JR, Lesniewski LA, Seals DR. (2010) Arterial stiffening with ageing is associated with transforming growth factor-β1-related changes in adventitial collagen: reversal by aerobic exercise. J Physiol 588:3971–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Eng JS, Nair DP, Dodson RB, Seals DR. (2012) Sodium nitrite de-stiffening of large elastic arteries with aging: role of normalization of advanced glycation end-products. Exp Gerontol 47:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force T, Krause DS, Van Etten RA. (2007) Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 7:332–344. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87:593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DB, Shen LH, Kelly J, Thibault P, Van Eyk JE, Mak AS. (2000) Phosphorylation of caldesmon by p21-activated kinase. Implications for the Ca(2+) sensitivity of smooth muscle contraction. J Biol Chem 275:1959–1965. [DOI] [PubMed] [Google Scholar]
- Frid MG, Shekhonin BV, Koteliansky VE, Glukhova MA. (1992) Phenotypic changes of human smooth muscle cells during development: late expression of heavy caldesmon and calponin. Dev Biol 153:185–193. [DOI] [PubMed] [Google Scholar]
- Fry JL, Shiraishi Y, Turcotte R, Yu X, Gao YZ, Akiki R, Bachschmid M, Zhang Y, Morgan KG, Cohen RA, et al. (2015) Vascular Smooth Muscle Sirtuin-1 Protects Against Aortic Dissection During Angiotensin II-Induced Hypertension. J Am Heart Assoc 4:e002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao M, Mason HS, Britton FC, Kenyon JL, Horowitz B, Keef KD. (1999) Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J Biol Chem 274:10927–10935. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. (1999) Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide. Biosci Rep 19:235–251. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. (1980) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288:373–376. [DOI] [PubMed] [Google Scholar]
- Fürst DO, Cross RA, De Mey J, Small JV. (1986) Caldesmon is an elongated, flexible molecule localized in the actomyosin domains of smooth muscle. EMBO J 5:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G, Schmid E, Winter S, Chaponnier C, de Ckhastonay C, Vandekerckhove J, Weber K, Franke WW. (1981) Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci USA 78:298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon A, Wilson RD, Audibert F, Allen VM, Blight C, Brock JA, Désilets VA, Johnson JA, Langlois S, Summers A, et al. Society of Obstetricians and Gynaecologists of Canada Genetics Committee (2008) Obstetrical complications associated with abnormal maternal serum markers analytes. J Obstet Gynaecol Can 30:918–949. [DOI] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Griffin SA, Stull JT. (1991) Molecular characterization of a mammalian smooth muscle myosin light chain kinase. J Biol Chem 266:23936–23944. [PMC free article] [PubMed] [Google Scholar]
- Gallant C, Appel S, Graceffa P, Leavis P, Lin JJ, Gunning PW, Schevzov G, Chaponnier C, DeGnore J, Lehman W, et al. (2011) Tropomyosin variants describe distinct functional subcellular domains in differentiated vascular smooth muscle cells. Am J Physiol Cell Physiol 300:C1356–C1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble G, Zorn J, Sanders G, MacMahon S, Sharpe N. (1994) Estimation of arterial stiffness, compliance, and distensibility from M-mode ultrasound measurements of the common carotid artery. Stroke 25:11–16. [DOI] [PubMed] [Google Scholar]
- Gangopadhyay SS, Barber AL, Gallant C, Grabarek Z, Smith JL, Morgan KG. (2003) Differential functional properties of calmodulin-dependent protein kinase IIgamma variants isolated from smooth muscle. Biochem J 372:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangopadhyay SS, Gallant C, Sundberg EJ, Lane WS, Morgan KG. (2008) Regulation of Ca2+/calmodulin kinase II by a small C-terminal domain phosphatase. Biochem J 412:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangopadhyay SS, Kengni E, Appel S, Gallant C, Kim HR, Leavis P, DeGnore J, Morgan KG. (2009) Smooth muscle archvillin is an ERK scaffolding protein. J Biol Chem 284:17607–17615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangopadhyay SS, Takizawa N, Gallant C, Barber AL, Je HD, Smith TC, Luna EJ, Morgan KG. (2004) Smooth muscle archvillin: a novel regulator of signaling and contractility in vascular smooth muscle. J Cell Sci 117:5043–5057. [DOI] [PubMed] [Google Scholar]
- Gao P, Xu TT, Lu J, Li L, Xu J, Hao DL, Chen HZ, Liu DP. (2014a) Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J Mol Med (Berl) 92:347–357. [DOI] [PubMed] [Google Scholar]
- Gao YZ, Saphirstein RJ, Yamin RY, Suki B, Morgan KG. (2014b) Aging impairs smooth muscle mediated regulation of aortic stiffness: a defect in shock absorption function? Am J Physiol Heart Circ Physiol 307:H1252–H1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaylinn BD, Eddinger TJ, Martino PA, Monical PL, Hunt DF, Murphy RA. (1989) Expression of nonmuscle myosin heavy and light chains in smooth muscle. Am J Physiol 257:C997–C1004. [DOI] [PubMed] [Google Scholar]
- Geraci MW, Gao B, Hoshikawa Y, Yeager ME, Tuder RM, Voelkel NF. (2001a) Genomic approaches to research in pulmonary hypertension. Respir Res 2:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, Gao B, Loyd JE, Tuder RM, Voelkel NF. (2001b) Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res 88:555–562. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, Mayer E, Simonneau G, Wilkins MR, Fritsch A, et al. CHEST-1 Study Group (2013a) Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 369:319–329. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, et al. PATENT-1 Study Group (2013b) Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369:330–340. [DOI] [PubMed] [Google Scholar]
- Giachini FR, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, Tostes RC, Webb RC. (2009a) Sex differences in vascular expression and activation of STIM-1/Orai-1 during hypertension: focus on calcium regulation. FASEB J 23:Suppl 1, 781–15. [Google Scholar]
- Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, Tostes RC, Webb RC. (2009b) Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension 53:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginnan R, Sun LY, Schwarz JJ, Singer HA. (2012) MEF2 is regulated by CaMKIIδ2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem J 444:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginnan R, Zou X, Pfleiderer PJ, Mercure MZ, Barroso M, Singer HA. (2013) Vascular smooth muscle cell motility is mediated by a physical and functional interaction of Ca2+/calmodulin-dependent protein kinase IIδ2 and Fyn. J Biol Chem 288:29703–29712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Given AM, Ogut O, Brozovich FV. (2007) MYPT1 mutants demonstrate the importance of aa 888-928 for the interaction with PKGIalpha. Am J Physiol Cell Physiol 292:C432–C439. [DOI] [PubMed] [Google Scholar]
- Glagov S, Zarins CK, Masawa N, Xu CP, Bassiouny H, Giddens DP. (1993) Mechanical functional role of non-atherosclerotic intimal thickening. Front Med Biol Eng 5:37–43. [PubMed] [Google Scholar]
- Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. (2004) Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J Biol Chem 279:2800–2808. [DOI] [PubMed] [Google Scholar]
- Gomez MF, Bosc LV, Stevenson AS, Wilkerson MK, Hill-Eubanks DC, Nelson MT. (2003) Constitutively elevated nuclear export activity opposes Ca2+-dependent NFATc3 nuclear accumulation in vascular smooth muscle: role of JNK2 and Crm-1. J Biol Chem 278:46847–46853. [DOI] [PubMed] [Google Scholar]
- Gomez MF, Stevenson AS, Bonev AD, Hill-Eubanks DC, Nelson MT. (2002) Opposing actions of inositol 1,4,5-trisphosphate and ryanodine receptors on nuclear factor of activated T-cells regulation in smooth muscle. J Biol Chem 277:37756–37764. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Shirinsky VP, Shevelev AY, Marston SB, Vorotnikov AV. (2001) Actomyosin cross-linking by caldesmon in non-muscle cells. FEBS Lett 497:113–117. [DOI] [PubMed] [Google Scholar]
- Gong MC, Cohen P, Kitazawa T, Ikebe M, Masuo M, Somlyo AP, Somlyo AV. (1992) Myosin light chain phosphatase activities and the effects of phosphatase inhibitors in tonic and phasic smooth muscle. J Biol Chem 267:14662–14668. [PubMed] [Google Scholar]
- Gong Y, McDonough CW, Wang Z, Hou W, Cooper-DeHoff RM, Langaee TY, Beitelshees AL, Chapman AB, Gums JG, Bailey KR, et al. (2012) Hypertension susceptibility loci and blood pressure response to antihypertensives: results from the pharmacogenomic evaluation of antihypertensive responses study. Circ Cardiovasc Genet 5:686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, Hill-Eubanks DC, Nelson MT, Earley S. (2014) A PLCγ1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal 7:ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. (2004) Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation: role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J Biol Chem 279:10702–10709. [DOI] [PubMed] [Google Scholar]
- Gordienko DV, Bolton TB. (2002) Crosstalk between ryanodine receptors and IP(3) receptors as a factor shaping spontaneous Ca(2+)-release events in rabbit portal vein myocytes. J Physiol 542:743–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorenne I, Su X, Moreland RS. (2004) Caldesmon phosphorylation is catalyzed by two kinases in permeabilized and intact vascular smooth muscle. J Cell Physiol 198:461–469. [DOI] [PubMed] [Google Scholar]
- Gosling RG, Budge MM. (2003) Terminology for describing the elastic behavior of arteries. Hypertension 41:1180–1182. [DOI] [PubMed] [Google Scholar]
- Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B, Ostergren J, Pfeffer MA, Swedberg K, CHARM Investigators and Committees (2003) Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet 362:772–776. [DOI] [PubMed] [Google Scholar]
- Greenwald SE. (2007) Ageing of the conduit arteries. J Pathol 211:157–172.17200940 [Google Scholar]
- Greenwood IA, Leblanc N. (2007) Overlapping pharmacology of Ca2+-activated Cl- and K+ channels. Trends Pharmacol Sci 28:1–5. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Lassègue B, Murphy TJ, Alexander RW. (1994) Angiotensin II receptor pharmacology. Adv Pharmacol 28:269–306. [DOI] [PubMed] [Google Scholar]
- Group IS, IONA Study Group (2002) Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet 359:1269–1275. [DOI] [PubMed] [Google Scholar]
- Guarente L. (2011) Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med 364:2235–2244. [DOI] [PubMed] [Google Scholar]
- Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, London GM. (2001) Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation 103:987–992. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Zhang W. (2008) Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295:C576–C587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Huang R, Semba S, Kordowska J, Huh YH, Khalina-Stackpole Y, Mabuchi K, Kitazawa T, Wang CL. (2013) Ablation of smooth muscle caldesmon affects the relaxation kinetics of arterial muscle. Pflugers Arch 465:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S, Tian H, Jiang C, Zhu D. (2012) The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-α1C. Hypertension 59:1006–1013. [DOI] [PubMed] [Google Scholar]
- Guo RW, Wang H, Gao P, Li MQ, Zeng CY, Yu Y, Chen JF, Song MB, Shi YK, Huang L. (2009) An essential role for stromal interaction molecule 1 in neointima formation following arterial injury. Cardiovasc Res 81:660–668. [DOI] [PubMed] [Google Scholar]
- Gupta AK. (2010) Racial differences in response to antihypertensive therapy: does one size fits all? Int J Prev Med 1:217–219. [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Li L. (2015) Modulation of miRNAs in Pulmonary Hypertension. Int J Hypertens 2015:169069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvenc B, Ustunel C, Ozturk N, Brozovich FV. (2010) A dynamic approach reveals non-muscle myosin influences the overall smooth muscle cross-bridge cycling rate. FEBS Lett 584:2862–2866. [DOI] [PubMed] [Google Scholar]
- Guyton AC. (1991) Blood pressure control--special role of the kidneys and body fluids. Science 252:1813–1816. [DOI] [PubMed] [Google Scholar]
- Haddock RE, Hill CE. (2005) Rhythmicity in arterial smooth muscle. J Physiol 566:645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai CM, Murphy RA. (1989) Cross-bridge dephosphorylation and relaxation of vascular smooth muscle. Am J Physiol 256:C282–C287. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar I, Kotchen JM, Kotchen TA. (2006) Hypertension: trends in prevalence, incidence, and control. Annu Rev Public Health 27:465–490. [DOI] [PubMed] [Google Scholar]
- Hake S. (2003) MicroRNAs: a role in plant development. Curr Biol 13:R851–R852. [DOI] [PubMed] [Google Scholar]
- Haldeman BD, Brizendine RK, Facemyer KC, Baker JE, Cremo CR. (2014) The kinetics underlying the velocity of smooth muscle myosin filament sliding on actin filaments in vitro. J Biol Chem 289:21055–21070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamden SS, Schroeter MM, Chalovich JM. (2010) Phosphorylation of caldesmon at sites between residues 627 and 642 attenuates inhibitory activity and contributes to a reduction in Ca2+-calmodulin affinity. Biophys J 99:1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. (2001) Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293:1146–1150. [DOI] [PubMed] [Google Scholar]
- Han YS, Brozovich FV. (2013) Altered reactivity of tertiary mesenteric arteries following acute myocardial ischemia. J Vasc Res 50:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne DJ, Ito M, Erdödi F. (1998) Myosin light chain phosphatase: subunit composition, interactions and regulation. J Muscle Res Cell Motil 19:325–341. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ, Ito M, Erdödi F. (2004) Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J Biol Chem 279:37211–37214. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Morita F. (1992) Role of 17-kDa essential light chain isoforms of aorta smooth muscle myosin. J Biochem 111:804–809. [DOI] [PubMed] [Google Scholar]
- Havulinna AS, Kettunen J, Ukkola O, Osmond C, Eriksson JG, Kesäniemi YA, Jula A, Peltonen L, Kontula K, Salomaa V, et al. (2013) A blood pressure genetic risk score is a significant predictor of incident cardiovascular events in 32,669 individuals. Hypertension 61:987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Tan J, Yu B, Shi W, Liang K. (2015) Long noncoding RNA HIF1A-AS1A reduces apoptosis of vascular smooth muscle cells: implications for the pathogenesis of thoracoabdominal aorta aneurysm. Pharmazie 70:310–315. [PubMed] [Google Scholar]
- Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT. (2000) Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am J Physiol Cell Physiol 278:C718–C726. [DOI] [PubMed] [Google Scholar]
- Hemric ME, Lu FW, Shrager R, Carey J, Chalovich JM. (1993) Reversal of caldesmon binding to myosin with calcium-calmodulin or by phosphorylating caldesmon. J Biol Chem 268:15305–15311. [PMC free article] [PubMed] [Google Scholar]
- Hermann S, Saarikettu J, Onions J, Hughes K, Grundström T. (1998) Calcium regulation of basic helix-loop-helix transcription factors. Cell Calcium 23:135–142. [DOI] [PubMed] [Google Scholar]
- Herrera AM, McParland BE, Bienkowska A, Tait R, Paré PD, Seow CY. (2005) ‘Sarcomeres’ of smooth muscle: functional characteristics and ultrastructural evidence. J Cell Sci 118:2381–2392. [DOI] [PubMed] [Google Scholar]
- Herring BP, Lyons GE, Hoggatt AM, Gallagher PJ. (2001) Telokin expression is restricted to smooth muscle tissues during mouse development. Am J Physiol Cell Physiol 280:C12–C21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L. (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383:1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Falcone JC, Meininger GA. (1990) Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am J Physiol 259:H1586–H1594. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. (2001) Invited review: arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J Appl Physiol (1985) 91:973–983. [DOI] [PubMed] [Google Scholar]
- Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. (2011) Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol 3:a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen MO, Turunen MP, Häkkinen TP, Rutanen J, Hedman M, Mäkinen K, Turunen AM, Aalto-Setälä K, Ylä-Herttuala S. (2002) DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med 7:5–11. [DOI] [PubMed] [Google Scholar]
- Hiltunen MO, Ylä-Herttuala S. (2003) DNA methylation, smooth muscle cells, and atherogenesis. Arterioscler Thromb Vasc Biol 23:1750–1753. [DOI] [PubMed] [Google Scholar]
- Hodges RR, Horikawa Y, Rios JD, Shatos MA, Dartt DA. (2007) Effect of protein kinase C and Ca(2+) on p42/p44 MAPK, Pyk2, and Src activation in rat conjunctival goblet cells. Exp Eye Res 85:836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdt LM, Sass K, Gäbel G, Bergert H, Thiery J, Teupser D. (2011) Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis 214:264–270. [DOI] [PubMed] [Google Scholar]
- Holzapfel GA, Ogden RW. (2010) Constitutive modelling of arteries. Proceedings of the Royal Society 466:1551–1597. [Google Scholar]
- Hoover WC, Zhang W, Xue Z, Gao H, Chernoff J, Clapp DW, Gunst SJ, Tepper RS. (2012) Inhibition of p21 activated kinase (PAK) reduces airway responsiveness in vivo and in vitro in murine and human airways. PLoS One 7:e42601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz A, Clément-Chomienne O, Walsh MP, Morgan KG. (1996a) Epsilon-isoenzyme of protein kinase C induces a Ca(2+)-independent contraction in vascular smooth muscle. Am J Physiol 271:C589–C594. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Clément-Chomienne O, Walsh MP, Tao T, Katsuyama H, Morgan KG. (1996b) Effects of calponin on force generation by single smooth muscle cells. Am J Physiol 270:H1858–H1863. [DOI] [PubMed] [Google Scholar]
- Hou X, Chen J, Luo Y, Liu F, Xu G, Gao Y. (2013) Silencing of STIM1 attenuates hypoxia-induced PASMCs proliferation via inhibition of the SOC/Ca2+/NFAT pathway. Respir Res 14:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. (1995) Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377:239–242. [DOI] [PubMed] [Google Scholar]
- Huang QQ, Fisher SA, Brozovich FV. (2004) Unzipping the role of myosin light chain phosphatase in smooth muscle cell relaxation. J Biol Chem 279:597–603. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. (2002) Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem 71:473–510. [DOI] [PubMed] [Google Scholar]
- Hulvershorn J, Gallant C, Wang CA, Dessy C, Morgan KG. (2001) Calmodulin levels are dynamically regulated in living vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 280:H1422–H1426. [DOI] [PubMed] [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293:834–838. [DOI] [PubMed] [Google Scholar]
- Ichikawa K, Ito M, Hartshorne DJ. (1996) Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J Biol Chem 271:4733–4740. [DOI] [PubMed] [Google Scholar]
- Iino M, Kasai H, Yamazawa T. (1994) Visualization of neural control of intracellular Ca2+ concentration in single vascular smooth muscle cells in situ. EMBO J 13:5026–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikebe M, Hartshorne DJ. (1985) Effects of Ca2+ on the conformation and enzymatic activity of smooth muscle myosin. J Biol Chem 260:13146–13153. [PubMed] [Google Scholar]
- Ikebe M, Morita J. (1991) Identification of the sequence of the regulatory light chain required for the phosphorylation-dependent regulation of actomyosin. J Biol Chem 266:21339–21342. [PubMed] [Google Scholar]
- Immink RV, van den Born BJ, van Montfrans GA, Koopmans RP, Karemaker JM, van Lieshout JJ. (2004) Impaired cerebral autoregulation in patients with malignant hypertension. Circulation 110:2241–2245. [DOI] [PubMed] [Google Scholar]
- Ingber DE. (2003) Mechanobiology and diseases of mechanotransduction. Ann Med 35:564–577. [DOI] [PubMed] [Google Scholar]
- Ingber DE. (2006) Cellular mechanotransduction: putting all the pieces together again. FASEB J 20:811–827. [DOI] [PubMed] [Google Scholar]
- Intengan HD, Schiffrin EL. (2000) Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants. Hypertension 36:312–318. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Zhu X, Cantley LG. (2003) Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell 12:1275–1285. [DOI] [PubMed] [Google Scholar]
- Ito I, Jarajapu YP, Grant MB, Knot HJ. (2007) Characteristics of myogenic tone in the rat ophthalmic artery. Am J Physiol Heart Circ Physiol 292:H360–H368. [DOI] [PubMed] [Google Scholar]
- Ito M, Hartshorne DJ. (1990) Phosphorylation of myosin as a regulatory mechanism in smooth muscle. Prog Clin Biol Res 327:57–72. [PubMed] [Google Scholar]
- Jaenisch R, Bird A. (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 (Suppl):245–254. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21:247–269. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Porter VA, Lederer WJ, Nelson MT. (2000) Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278:C235–C256. [DOI] [PubMed] [Google Scholar]
- James PR, Nelson-Piercy C. (2004) Management of hypertension before, during, and after pregnancy. Heart 90:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana SS, Kim KY, Mao J, Kawamoto S, Sellers JR, Adelstein RS. (2009) An alternatively spliced isoform of non-muscle myosin II-C is not regulated by myosin light chain phosphorylation. J Biol Chem 284:11563–11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarajapu YP, Knot HJ. (2005) Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am J Physiol Heart Circ Physiol 289:H1917–H1922. [DOI] [PubMed] [Google Scholar]
- Jaworowski A, Anderson KI, Arner A, Engström M, Gimona M, Strasser P, Small JV. (1995) Calponin reduces shortening velocity in skinned taenia coli smooth muscle fibres. FEBS Lett 365:167–171. [DOI] [PubMed] [Google Scholar]
- Je HD, Gallant C, Leavis PC, Morgan KG. (2004) Caveolin-1 regulates contractility in differentiated vascular smooth muscle. Am J Physiol Heart Circ Physiol 286:H91–H98. [DOI] [PubMed] [Google Scholar]
- Je HD, Gangopadhyay SS, Ashworth TD, Morgan KG. (2001) Calponin is required for agonist-induced signal transduction--evidence from an antisense approach in ferret smooth muscle. J Physiol 537:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BL, Skøtt O. (1996) Blockade of chloride channels by DIDS stimulates renin release and inhibits contraction of afferent arterioles. Am J Physiol 270:F718–F727. [DOI] [PubMed] [Google Scholar]
- Ji H, Menini S, Zheng W, Pesce C, Wu X, Sandberg K. (2008) Role of angiotensin-converting enzyme 2 and angiotensin(1-7) in 17beta-oestradiol regulation of renal pathology in renal wrap hypertension in rats. Exp Physiol 93:648–657. [DOI] [PubMed] [Google Scholar]
- Jiang MJ, Morgan KG. (1989) Agonist-specific myosin phosphorylation and intracellular calcium during isometric contractions of arterial smooth muscle. Pflugers Arch 413:637–643. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Yin H, Zheng XL. (2010) MicroRNA-1 inhibits myocardin-induced contractility of human vascular smooth muscle cells. J Cell Physiol 225:506–511. [DOI] [PubMed] [Google Scholar]
- Johnson AD, Newton-Cheh C, Chasman DI, Ehret GB, Johnson T, Rose L, Rice K, Verwoert GC, Launer LJ, Gudnason V, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. Global BPgen Consortium. Women’s Genome Health Study (2011) Association of hypertension drug target genes with blood pressure and hypertension in 86,588 individuals. Hypertension 57:903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA. (2008) Ethnic differences in cardiovascular drug response: potential contribution of pharmacogenetics. Circulation 118:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF. (2003) Beta 1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin Pharmacol Ther 74:44–52. [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Feig DI, Nakagawa T, Sanchez-Lozada LG, Rodriguez-Iturbe B. (2008) Pathogenesis of essential hypertension: historical paradigms and modern insights. J Hypertens 26:381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. (2009) Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol 587:2537–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CJ, Kuo L, Davis MJ, Chilian WM. (1995) Regulation of coronary blood flow: coordination of heterogeneous control mechanisms in vascular microdomains. Cardiovasc Res 29:585–596. [PubMed] [Google Scholar]
- Jones JA, Stroud RE, O’Quinn EC, Black LE, Barth JL, Elefteriades JA, Bavaria JE, Gorman JH, 3rd, Gorman RC, Spinale FG, et al. (2011) Selective microRNA suppression in human thoracic aneurysms: relationship of miR-29a to aortic size and proteolytic induction. Circ Cardiovasc Genet 4:605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph BK, Thakali KM, Moore CL, Rhee SW. (2013) Ion channel remodeling in vascular smooth muscle during hypertension: Implications for novel therapeutic approaches. Pharmacol Res 70:126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Billington N, Thirumurugan K, Salzameda B, Cremo CR, Chalovich JM, Chantler PD, Knight PJ. (2011) Role of the tail in the regulated state of myosin 2. J Mol Biol 408:863–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser L, Spickard RC, Olivier NB. (1989) Heart failure depresses endothelium-dependent responses in canine femoral artery. Am J Physiol 256:H962–H967. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. (2010) Nanoscale architecture of integrin-based cell adhesions. Nature 468:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandabashi T, Shimokawa H, Miyata K, Kunihiro I, Kawano Y, Fukata Y, Higo T, Egashira K, Takahashi S, Kaibuchi K, et al. (2000) Inhibition of myosin phosphatase by upregulated rho-kinase plays a key role for coronary artery spasm in a porcine model with interleukin-1beta. Circulation 101:1319–1323. [DOI] [PubMed] [Google Scholar]
- Kang H, Davis-Dusenbery BN, Nguyen PH, Lal A, Lieberman J, Van Aelst L, Lagna G, Hata A. (2012) Bone morphogenetic protein 4 promotes vascular smooth muscle contractility by activating microRNA-21 (miR-21), which down-regulates expression of family of dedicator of cytokinesis (DOCK) proteins. J Biol Chem 287:3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis P, Babu GJ, Periasamy M, Brozovich FV. (2003) The smooth muscle myosin seven amino acid heavy chain insert’s kinetic role in the crossbridge cycle for mouse bladder. J Physiol 547:463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis P, Babu GJ, Periasamy M, Brozovich FV. (2004) Myosin heavy chain isoform expression regulates shortening velocity in smooth muscle: studies using an SMB KO mouse line. J Muscle Res Cell Motil 25:149–158. [DOI] [PubMed] [Google Scholar]
- Karaki H. (1989) Ca2+ localization and sensitivity in vascular smooth muscle. Trends Pharmacol Sci 10:320–325. [DOI] [PubMed] [Google Scholar]
- Kargacin GJ. (1994) Calcium signaling in restricted diffusion spaces. Biophys J 67:262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargacin GJ, Cooke PH, Abramson SB, Fay FS. (1989) Periodic organization of the contractile apparatus in smooth muscle revealed by the motion of dense bodies in single cells. J Cell Biol 108:1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim SM, Rhee AY, Given AM, Faulx MD, Hoit BD, Brozovich FV. (2004) Vascular reactivity in heart failure: role of myosin light chain phosphatase. Circ Res 95:612–618. [DOI] [PubMed] [Google Scholar]
- Karolczak-Bayatti M, Sweeney M, Cheng J, Edey L, Robson SC, Ulrich SM, Treumann A, Taggart MJ, Europe-Finner GN. (2011) Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J Biol Chem 286:34346–34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, Tay WT, Chen CH, Zhang Y, Yamamoto K, et al. (2011) Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 43:531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoch SS, Moreland RS. (1995) Agonist and membrane depolarization induced activation of MAP kinase in the swine carotid artery. Am J Physiol 269:H222–H229. [DOI] [PubMed] [Google Scholar]
- Katz SD, Schwarz M, Yuen J, LeJemtel TH. (1993) Impaired acetylcholine-mediated vasodilation in patients with congestive heart failure. Role of endothelium-derived vasodilating and vasoconstricting factors. Circulation 88:55–61. [DOI] [PubMed] [Google Scholar]
- Kelley CA, Takahashi M, Yu JH, Adelstein RS. (1993) An insert of seven amino acids confers functional differences between smooth muscle myosins from the intestines and vasculature. J Biol Chem 268:12848–12854. [PubMed] [Google Scholar]
- Khalil RA, Granger JP. (2002) Vascular mechanisms of increased arterial pressure in preeclampsia: lessons from animal models. Am J Physiol Regul Integr Comp Physiol 283:R29–R45. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. (1993) PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle cell activation. Am J Physiol 265:C406–C411. [DOI] [PubMed] [Google Scholar]
- Khan F, Litchfield SJ, McLaren M, Veale DJ, Littleford RC, Belch JJ. (1997) Oral L-arginine supplementation and cutaneous vascular responses in patients with primary Raynaud’s phenomenon. Arthritis Rheum 40:352–357. [DOI] [PubMed] [Google Scholar]
- Khatri JJ, Joyce KM, Brozovich FV, Fisher SA. (2001) Role of myosin phosphatase isoforms in cGMP-mediated smooth muscle relaxation. J Biol Chem 276:37250–37257. [DOI] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD. (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115:209–216. [DOI] [PubMed] [Google Scholar]
- Kim HR, Gallant C, Leavis PC, Gunst SJ, Morgan KG. (2008a) Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol 295:C768–C778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R, Morgan KG. (2010) Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol 298:C559–C571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Je HD, Gallant C, Zhan Q, Riper DV, Badwey JA, Singer HA, Morgan KG. (2000) Ca2+-calmodulin-dependent protein kinase II-dependent activation of contractility in ferret aorta. J Physiol 526:367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23:607–618. [DOI] [PubMed] [Google Scholar]
- Kim YS, Immink RV, Stok WJ, Karemaker JM, Secher NH, van Lieshout JJ. (2008b) Dynamic cerebral autoregulatory capacity is affected early in Type 2 diabetes. Clin Sci (Lond) 115:255–262. [DOI] [PubMed] [Google Scholar]
- Kiss E, Murányi A, Csortos C, Gergely P, Ito M, Hartshorne DJ, Erdodi F. (2002) Integrin-linked kinase phosphorylates the myosin phosphatase target subunit at the inhibitory site in platelet cytoskeleton. Biochem J 365:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Yamazaki J. (2001) Chloride channels and their functional roles in smooth muscle tone in the vasculature. Jpn J Pharmacol 85:351–357. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. (2000) Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem 275:9897–9900. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. (1991a) G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem 266:1708–1715. [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. (1991b) G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc Natl Acad Sci USA 88:9307–9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. (1998) Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles HJ, Tian YM, Mole DR, Harris AL. (2004) Novel mechanism of action for hydralazine: induction of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and angiogenesis by inhibition of prolyl hydroxylases. Circ Res 95:162–169. [DOI] [PubMed] [Google Scholar]
- Koga Y, Ikebe M. (2005) p116Rip decreases myosin II phosphorylation by activating myosin light chain phosphatase and by inactivating RhoA. J Biol Chem 280:4983–4991. [DOI] [PubMed] [Google Scholar]
- Kohn JC, Lampi MC, Reinhart-King CA. (2015) Age-related vascular stiffening: causes and consequences. Front Genet 6:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W. (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 6:827–837. [DOI] [PubMed] [Google Scholar]
- Kolega J. (2003) Asymmetric distribution of myosin IIB in migrating endothelial cells is regulated by a rho-dependent kinase and contributes to tail retraction. Mol Biol Cell 14:4745–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konik EA, Han YS, Brozovich FV. (2013) The role of pulmonary vascular contractile protein expression in pulmonary arterial hypertension. J Mol Cell Cardiol 65:147–155. [DOI] [PubMed] [Google Scholar]
- Kontaraki JE, Marketou ME, Zacharis EA, Parthenakis FI, Vardas PE. (2014) Differential expression of vascular smooth muscle-modulating microRNAs in human peripheral blood mononuclear cells: novel targets in essential hypertension. J Hum Hypertens 28:510–516. [DOI] [PubMed] [Google Scholar]
- Kordowska J, Huang R, Wang CL. (2006) Phosphorylation of caldesmon during smooth muscle contraction and cell migration or proliferation. J Biomed Sci 13:159–172. [DOI] [PubMed] [Google Scholar]
- Koshy MC, Mickley D, Bourgiognie J, Blaufox MD. (1977) Physiologic evaluation of a new antihypertensive agent: prazosin HCl. Circulation 55:533–537. [DOI] [PubMed] [Google Scholar]
- Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, et al. (2012) Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Reports 2:1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlo KU, Hesabi B, Danziger RS. (2011) Implication of microRNAs in atrial natriuretic peptide and nitric oxide signaling in vascular smooth muscle cells. Am J Physiol Cell Physiol 301:C929–C937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2007) Chromatin modifications and their function. Cell 128:693–705. [DOI] [PubMed] [Google Scholar]
- Kovács M, Thirumurugan K, Knight PJ, Sellers JR. (2007) Load-dependent mechanism of nonmuscle myosin 2. Proc Natl Acad Sci USA 104:9994–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács M, Wang F, Hu A, Zhang Y, Sellers JR. (2003) Functional divergence of human cytoplasmic myosin II: kinetic characterization of the non-muscle IIA isoform. J Biol Chem 278:38132–38140. [DOI] [PubMed] [Google Scholar]
- Kovtun O, Tillu VA, Ariotti N, Parton RG, Collins BM. (2015) Cavin family proteins and the assembly of caveolae. J Cell Sci 128:1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krennhrubec K, Marshall BL, Hedglin M, Verdin E, Ulrich SM. (2007) Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg Med Chem Lett 17:2874–2878. [DOI] [PubMed] [Google Scholar]
- Kubo SH, Rector TS, Bank AJ, Williams RE, Heifetz SM. (1991) Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation 84:1589–1596. [DOI] [PubMed] [Google Scholar]
- Kudryavtseva O, Aalkjaer C, Matchkov VV. (2013) Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J 280:5488–5499. [DOI] [PubMed] [Google Scholar]
- Kumai T, Takeba Y, Matsumoto N, Nakaya S, Tsuzuki Y, Yanagida Y, Hayashi M, Kobayashi S. (2007) Fasudil attenuates sympathetic nervous activity in the adrenal medulla of spontaneously hypertensive rats. Life Sci 81:1193–1198. [DOI] [PubMed] [Google Scholar]
- Kunichika N, Landsberg JW, Yu Y, Kunichika H, Thistlethwaite PA, Rubin LJ, Yuan JX. (2004) Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med 170:1101–1107. [DOI] [PubMed] [Google Scholar]
- Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. (2012) The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 95:194–204. [DOI] [PubMed] [Google Scholar]
- Lamont C, Vainorius E, Wier WG. (2003) Purinergic and adrenergic Ca2+ transients during neurogenic contractions of rat mesenteric small arteries. J Physiol 549:801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan B, Deng L, Donovan GM, Chin LY, Syyong HT, Wang L, Zhang J, Pascoe CD, Norris BA, Liu JC, et al. (2015) Force maintenance and myosin filament assembly regulated by Rho-kinase in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 308:L1–L10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen NA. (1959) Cerebral blood flow and oxygen consumption in man. Physiol Rev 39:183–238. [DOI] [PubMed] [Google Scholar]
- Laukkanen MO, Mannermaa S, Hiltunen MO, Aittomäki S, Airenne K, Jänne J, Ylä-Herttuala S. (1999) Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol 19:2171–2178. [DOI] [PubMed] [Google Scholar]
- Laurent S, Boutouyrie P. (2007) Recent advances in arterial stiffness and wave reflection in human hypertension. Hypertension 49:1202–1206. [DOI] [PubMed] [Google Scholar]
- Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. (2001) Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37:1236–1241. [DOI] [PubMed] [Google Scholar]
- Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O’Driscoll K, Britton F, Perrino BA, Greenwood IA. (2005) Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol 83:541–556. [DOI] [PubMed] [Google Scholar]
- Lee E, Hayes DB, Langsetmo K, Sundberg EJ, Tao TC. (2007) Interactions between the leucine-zipper motif of cGMP-dependent protein kinase and the C-terminal region of the targeting subunit of myosin light chain phosphatase. J Mol Biol 373:1198–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854. [DOI] [PubMed] [Google Scholar]
- Lee SH, Johnson JD, Walsh MP, Van Lierop JE, Sutherland C, Xu A, Snedden WA, Kosk-Kosicka D, Fromm H, Narayanan N, et al. (2000a) Differential regulation of Ca2+/calmodulin-dependent enzymes by plant calmodulin isoforms and free Ca2+ concentration. Biochem J 350:299–306. [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, et al. (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21:4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Gallant C, Guo H, Li Y, Wang CA, Morgan KG. (2000b) Regulation of vascular smooth muscle tone by N-terminal region of caldesmon. Possible role of tethering actin to myosin. J Biol Chem 275:3213–3220. [DOI] [PubMed] [Google Scholar]
- Lee YH, Kim I, Laporte R, Walsh MP, Morgan KG. (1999) Isozyme-specific inhibitors of protein kinase C translocation: effects on contractility of single permeabilized vascular muscle cells of the ferret. J Physiol 517:709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinweber B, Parissenti AM, Gallant C, Gangopadhyay SS, Kirwan-Rhude A, Leavis PC, Morgan KG. (2000) Regulation of protein kinase C by the cytoskeletal protein calponin. J Biol Chem 275:40329–40336. [DOI] [PubMed] [Google Scholar]
- Leinweber BD, Leavis PC, Grabarek Z, Wang CL, Morgan KG. (1999a) Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem J 344:117–123. [PMC free article] [PubMed] [Google Scholar]
- Leinweber BD, Parissenti A, Leavis PC, Morgan KG. (1999b) Calponin binds the regulatory domain of PKCe and activates kinase activity. abstract Mol Biol Cell 10:247a. [Google Scholar]
- Lenz S, Lohse P, Seidel U, Arnold HH. (1989) The alkali light chains of human smooth and nonmuscle myosins are encoded by a single gene. Tissue-specific expression by alternative splicing pathways. J Biol Chem 264:9009–9015. [PubMed] [Google Scholar]
- Leung A, Trac C, Jin W, Lanting L, Akbany A, Sætrom P, Schones DE, Natarajan R. (2013) Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ Res 113:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kalman J, Mayer L, Fillit HM, Packer M. (1990) Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 323:236–241. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. [DOI] [PubMed] [Google Scholar]
- Li H, Li W, Gupta AK, Mohler PJ, Anderson ME, Grumbach IM. (2010) Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol 298:H688–H698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen S, Cleary RA, Wang R, Gannon OJ, Seto E, Tang DD. (2014a) Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am J Physiol Cell Physiol 307:C288–C295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zheng L, Lin P, Danielpour D, Pan Z, Ma J. (2008) Overexpression of Bax induces down-regulation of store-operated calcium entry in prostate cancer cells. J Cell Physiol 216:172–179. [DOI] [PubMed] [Google Scholar]
- Li SS, Ran YJ, Zhang DD, Li SZ, Zhu D. (2014b) MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K⁺ channel in arterial smooth muscle cells. J Cell Biochem 115:1196–1205. [DOI] [PubMed] [Google Scholar]
- Li SS, Wu Y, Jin X, Jiang C. (2015) The SUR2B subunit of rat vascular KATP channel is targeted by miR-9a-3p induced by prolonged exposure to methylglyoxal. Am J Physiol Cell Physiol 308:C139–C145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gallant C, Malek S, Morgan KG. (2007) Focal adhesion signaling is required for myometrial ERK activation and contractile phenotype switch before labor. J Cell Biochem 100:129–140. [DOI] [PubMed] [Google Scholar]
- Li Y, Je HD, Malek S, Morgan KG. (2003) ERK1/2-mediated phosphorylation of myometrial caldesmon during pregnancy and labor. Am J Physiol Regul Integr Comp Physiol 284:R192–R199. [DOI] [PubMed] [Google Scholar]
- Li Y, Je HD, Malek S, Morgan KG. (2004) Role of ERK1/2 in uterine contractility and preterm labor in rats. Am J Physiol Regul Integr Comp Physiol 287:R328–R335. [DOI] [PubMed] [Google Scholar]
- Li Y, Reznichenko M, Tribe RM, Hess PE, Taggart M, Kim H, DeGnore JP, Gangopadhyay S, Morgan KG. (2009) Stretch activates human myometrium via ERK, caldesmon and focal adhesion signaling. PLoS One 4:e7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yan J, Wu C, Wang Z, Yuan W, Wang D. (2014c) CD137-CD137L interaction regulates atherosclerosis via cyclophilin A in apolipoprotein E-deficient mice. PLoS One 9:e88563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Bates DJ, Wang E. (2009) Epigenetic control of microRNA expression and aging. Curr Genomics 10:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YC, Liu PY, Lin HF, Lin WY, Liao JK, Juo SH. (2015) Two functional polymorphisms of ROCK2 enhance arterial stiffening through inhibiting its activity and expression. J Mol Cell Cardiol 79:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifton RP, Gharavi AG, Geller DS. (2001) Molecular mechanisms of human hypertension. Cell 104:545–556. [DOI] [PubMed] [Google Scholar]
- Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. (2004) Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95:496–505. [DOI] [PubMed] [Google Scholar]
- Lincoln TM. (1989) Cyclic GMP and mechanisms of vasodilation. Pharmacol Ther 41:479–502. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Dey N, Sellak H. (2001) Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 91:1421–1430. [DOI] [PubMed] [Google Scholar]
- Lindheimer MD. (1993) Hypertension in pregnancy. Hypertension 22:127–137. [DOI] [PubMed] [Google Scholar]
- Ling H, Fabbri M, Calin GA. (2013) MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov 12:847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190. [DOI] [PubMed] [Google Scholar]
- Liu JC, Rottler J, Wang L, Zhang J, Pascoe CD, Lan B, Norris BA, Herrera AM, Paré PD, Seow CY. (2013) Myosin filaments in smooth muscle cells do not have a constant length. J Physiol 591:5867–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Pilch PF. (2008) A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem 283:4314–4322. [DOI] [PubMed] [Google Scholar]
- Liu X, Cheng Y, Chen X, Yang J, Xu L, Zhang C. (2011) MicroRNA-31 regulated by the extracellular regulated kinase is involved in vascular smooth muscle cell growth via large tumor suppressor homolog 2. J Biol Chem 286:42371–42380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhang C, Dronadula N, Li Q, Rao GN. (2005) Blockade of nuclear factor of activated T cells activation signaling suppresses balloon injury-induced neointima formation in a rat carotid artery model. J Biol Chem 280:14700–14708. [DOI] [PubMed] [Google Scholar]
- Löfgren M, Ekblad E, Morano I, Arner A. (2003) Nonmuscle myosin motor of smooth muscle. J Gen Physiol 121:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhn M, Plettenburg O, Ivashchenko Y, Kannt A, Hofmeister A, Kadereit D, Schaefer M, Linz W, Kohlmann M, Herbert JM, et al. (2009) Pharmacological characterization of SAR407899, a novel rho-kinase inhibitor. Hypertension 54:676–683. [DOI] [PubMed] [Google Scholar]
- Loirand G, Pacaud P. (2014) Involvement of Rho GTPases and their regulators in the pathogenesis of hypertension. Small GTPases 5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen JM, Martino F, Thum T. (2012) Epigenetic modifications in cardiovascular disease. Basic Res Cardiol 107:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KK, Armstrong SE, Ginnan R, Singer HA. (2005) Adhesion-dependent activation of CaMKII and regulation of ERK activation in vascular smooth muscle. Am J Physiol Cell Physiol 289:C1343–C1350. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zhang H, Gokina N, Mandala M, Sato O, Ikebe M, Osol G, Fisher SA. (2008) Uterine artery myosin phosphatase isoform switching and increased sensitivity to SNP in a rat L-NAME model of hypertension of pregnancy. Am J Physiol Cell Physiol 294:C564–C571. [DOI] [PubMed] [Google Scholar]
- Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, et al. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Reports 2:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Wu Z, Tremblay J, Thorin E, Peng J, Lavoie JL, Hu B, Stoyanova E, Cloutier G, Qi S, et al. (2012) Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J Biol Chem 287:6819–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JA, Borman MA, Murányi A, Somlyo AV, Hartshorne DJ, Haystead TA. (2001a) Identification of the endogenous smooth muscle myosin phosphatase-associated kinase. Proc Natl Acad Sci USA 98:2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JA, Eto M, Borman MA, Brautigan DL, Haystead TA. (2001b) Dual Ser and Thr phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by MYPT-associated kinase. FEBS Lett 493:91–94. [DOI] [PubMed] [Google Scholar]
- Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, et al. (2012) Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest 122:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegdefessel L, Rayner KJ, Leeper NJ. (2015a) MicroRNA regulation of vascular smooth muscle function and phenotype: early career committee contribution. Arterioscler Thromb Vasc Biol 35:2–6. [DOI] [PubMed] [Google Scholar]
- Maegdefessel L, Spin JM, Raaz U, Eken SM, Toh R, Azuma J, Adam M, Nakagami F, Heymann HM, Chernogubova E, et al. (2015b) Erratum: miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat Commun 6:6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmqvist U, Arner A. (1991) Correlation between isoform composition of the 17 kDa myosin light chain and maximal shortening velocity in smooth muscle. Pflugers Arch 418:523–530. [DOI] [PubMed] [Google Scholar]
- Manabe I, Owens GK. (2001) Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res 88:1127–1134. [DOI] [PubMed] [Google Scholar]
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. (1988) Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1:91–96. [DOI] [PubMed] [Google Scholar]
- Mann DL. (2002) Angiotensin II as an inflammatory mediator: evolving concepts in the role of the renin angiotensin system in the failing heart. Cardiovasc Drugs Ther 16:7–9. [DOI] [PubMed] [Google Scholar]
- Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. (2005) Targeting of a novel Ca+2/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res 97:541–549. [DOI] [PubMed] [Google Scholar]
- Marques FZ, Booth SA, Charchar FJ. (2015) The emerging role of non-coding RNA in essential hypertension and blood pressure regulation. J Hum Hypertens 29:459–467. [DOI] [PubMed] [Google Scholar]
- Marques FZ, Campain AE, Tomaszewski M, Zukowska-Szczechowska E, Yang YH, Charchar FJ, Morris BJ. (2011) Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension 58:1093–1098. [DOI] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Crow T, Davis MJ, Meininger GA. (2005) alphavbeta3- and alpha5beta1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol Heart Circ Physiol 289:H322–H329. [DOI] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. (2003) Integrins as unique receptors for vascular control. J Vasc Res 40:211–233. [DOI] [PubMed] [Google Scholar]
- Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. (2001) Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 38:1307–1310. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Boedtkjer DM, Aalkjaer C. (2015) The role of Ca(2+) activated Cl(-) channels in blood pressure control. Curr Opin Pharmacol 21:127–137. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Kudryavtseva O, Aalkjaer C. (2012) Intracellular Ca²⁺ signalling and phenotype of vascular smooth muscle cells. Basic Clin Pharmacol Toxicol 110:42–48. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Secher Dam V, Bødtkjer DM, Aalkjær C. (2013) Transport and function of chloride in vascular smooth muscles. J Vasc Res 50:69–87. [DOI] [PubMed] [Google Scholar]
- Mattace-Raso FUS, van der Cammen TJM, Hofman A, van Popele NM, Bos ML, Schalekamp MADH, Asmar R, Reneman RS, Hoeks APG, Breteler MMB, et al. (2006) Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation 113:657–663. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. (2007) SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA 104:14855–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthew JD, Khromov AS, McDuffie MJ, Somlyo AV, Somlyo AP, Taniguchi S, Takahashi K. (2000) Contractile properties and proteins of smooth muscles of a calponin knockout mouse. J Physiol 529:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV. (2006) Non-coding RNA. Hum Mol Genet 15:R17–R29. [DOI] [PubMed] [Google Scholar]
- McDonald OG, Owens GK. (2007) Programming smooth muscle plasticity with chromatin dynamics. Circ Res 100:1428–1441. [DOI] [PubMed] [Google Scholar]
- McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. (2006) Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest 116:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFawn PK, Shen L, Vincent SG, Mak A, Van Eyk JE, Fisher JT. (2003) Calcium-independent contraction and sensitization of airway smooth muscle by p21-activated protein kinase. Am J Physiol Lung Cell Mol Physiol 284:L863–L870. [DOI] [PubMed] [Google Scholar]
- McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, Loyd JE, American College of Chest Physicians (2004) Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126(1, Suppl)14S–34S. [DOI] [PubMed] [Google Scholar]
- McKay MM, Morrison DK. (2007) Integrating signals from RTKs to ERK/MAPK. Oncogene 26:3113–3121. [DOI] [PubMed] [Google Scholar]
- McLendon JM, Joshi SR, Sparks J, Matar M, Fewell JG, Abe K, Oka M, McMurtry IF, Gerthoffer WT. (2015) Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J Control Release 210:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292:C82–C97. [DOI] [PubMed] [Google Scholar]
- Menice CB, Hulvershorn J, Adam LP, Wang CA, Morgan KG. (1997) Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. J Biol Chem 272:25157–25161. [DOI] [PubMed] [Google Scholar]
- Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, Brayden JE, Santana LF. (2014) Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J Gen Physiol 143:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159. [DOI] [PubMed] [Google Scholar]
- Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, et al. (2012) miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res 110:312–324. [DOI] [PubMed] [Google Scholar]
- Merlet E, Atassi F, Motiani RK, Mougenot N, Jacquet A, Nadaud S, Capiod T, Trebak M, Lompré AM, Marchand A. (2013) miR-424/322 regulates vascular smooth muscle cell phenotype and neointimal formation in the rat. Cardiovasc Res 98:458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM, Long X. (2015) The short and long of noncoding sequences in the control of vascular cell phenotypes. Cell Mol Life Sci 72:3457–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael SK, Surks HK, Wang Y, Zhu Y, Blanton R, Jamnongjit M, Aronovitz M, Baur W, Ohtani K, Wilkerson MK, et al. (2008) High blood pressure arising from a defect in vascular function. Proc Natl Acad Sci USA 105:6702–6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452:492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ED, Jr, Ackerly JA, Vaughan ED, Jr, Peach MJ, Epstein RM. (1977) The renin-angiotensin system during controlled hypotension with sodium nitroprusside. Anesthesiology 47:257–262. [PubMed] [Google Scholar]
- Mills RD, Mita M, Nakagawa J, Shoji M, Sutherland C, Walsh MP. (2015) A role for the tyrosine kinase Pyk2 in depolarization-induced contraction of vascular smooth muscle. J Biol Chem 290:8677–8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton DL, Schneck AN, Ziech DA, Ba M, Facemyer KC, Halayko AJ, Baker JE, Gerthoffer WT, Cremo CR. (2011) Direct evidence for functional smooth muscle myosin II in the 10S self-inhibited monomeric conformation in airway smooth muscle cells. Proc Natl Acad Sci USA 108:1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Reznichenko M, Poythress RH, Gallant CM, Vetterkind S, Li Y, Morgan KG. (2012) Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol 227:3585–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell GF, Guo CY, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, Vasan RS, Levy D. (2007) Cross-sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation 115:2628–2636. [DOI] [PubMed] [Google Scholar]
- Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, Sunagawa K. (2008) SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 28:1263–1269. [DOI] [PubMed] [Google Scholar]
- Morano I, Chai GX, Baltas LG, Lamounier-Zepter V, Lutsch G, Kott M, Haase H, Bader M. (2000) Smooth-muscle contraction without smooth-muscle myosin. Nat Cell Biol 2:371–375. [DOI] [PubMed] [Google Scholar]
- Moreno-Domínguez A, Colinas O, El-Yazbi A, Walsh EJ, Hill MA, Walsh MP, Cole WC. (2013) Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol 591:1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Domínguez A, El-Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, Cole DM, Kargacin GJ, Walsh MP, Cole WC. (2014) Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem 289:20939–20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JP, Morgan KG. (1982) Vascular smooth muscle: the first recorded Ca2+ transients. Pflugers Arch 395:75–77. [DOI] [PubMed] [Google Scholar]
- Morgan KG. (2014) The importance of the smooth muscle cytoskeleton to preterm labour. Exp Physiol 99:525–529. [DOI] [PubMed] [Google Scholar]
- Morris SJ, Shore AC. (1996) Skin blood flow responses to the iontophoresis of acetylcholine and sodium nitroprusside in man: possible mechanisms. J Physiol 496:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. (2003) Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol 19:91–118. [DOI] [PubMed] [Google Scholar]
- Morrison DL, Sanghera JS, Stewart J, Sutherland C, Walsh MP, Pelech SL. (1996) Phosphorylation and activation of smooth muscle myosin light chain kinase by MAP kinase and cyclin-dependent kinase-1. Biochem Cell Biol 74:549–557. [DOI] [PubMed] [Google Scholar]
- Mukai Y, Shimokawa H, Matoba T, Kandabashi T, Satoh S, Hiroki J, Kaibuchi K, Takeshita A. (2001) Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. FASEB 15:1062–1064. [DOI] [PubMed] [Google Scholar]
- Mulder J, Ariaens A, van den Boomen D, Moolenaar WH. (2004) p116Rip targets myosin phosphatase to the actin cytoskeleton and is essential for RhoA/ROCK-regulated neuritogenesis. Mol Biol Cell 15:5516–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Mülsch A. (2005) Explaining the phenomenon of nitrate tolerance. Circ Res 97:618–628. [DOI] [PubMed] [Google Scholar]
- Murányi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ. (2005) Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 579:6611–6615. [DOI] [PubMed] [Google Scholar]
- Murányi A, MacDonald JA, Deng JT, Wilson DP, Haystead TA, Walsh MP, Erdodi F, Kiss E, Wu Y, Hartshorne DJ. (2002) Phosphorylation of the myosin phosphatase target subunit by integrin-linked kinase. Biochem J 366:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TV, Spurrell BE, Hill MA. (2001) Tyrosine phosphorylation following alterations in arteriolar intraluminal pressure and wall tension. Am J Physiol Heart Circ Physiol 281:H1047–H1056. [DOI] [PubMed] [Google Scholar]
- Murphy TV, Spurrell BE, Hill MA. (2002) Cellular signalling in arteriolar myogenic constriction: involvement of tyrosine phosphorylation pathways. Clin Exp Pharmacol Physiol 29:612–619. [DOI] [PubMed] [Google Scholar]
- Nabeshima Y, Nabeshima Y, Nonomura Y, Fujii-Kuriyama Y. (1987) Nonmuscle and smooth muscle myosin light chain mRNAs are generated from a single gene by the tissue-specific alternative RNA splicing. J Biol Chem 262:10608–10612. [PubMed] [Google Scholar]
- Nagai R, Kuro-o M, Babij P, Periasamy M. (1989) Identification of two types of smooth muscle myosin heavy chain isoforms by cDNA cloning and immunoblot analysis. J Biol Chem 264:9734–9737. [PubMed] [Google Scholar]
- Nagai R, Larson DM, Periasamy M. (1988) Characterization of a mammalian smooth muscle myosin heavy chain cDNA clone and its expression in various smooth muscle types. Proc Natl Acad Sci USA 85:1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Koga Y, Sakai H, Homma K, Ikebe M. (2007) cGMP-dependent relaxation of smooth muscle is coupled with the change in the phosphorylation of myosin phosphatase. Circ Res 101:712–722. [DOI] [PubMed] [Google Scholar]
- Navedo MF, Amberg GC. (2013) Local regulation of L-type Ca²⁺ channel sparklets in arterial smooth muscle. Microcirculation 20:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Amberg GC, Votaw VS, Santana LF. (2005) Constitutively active L-type Ca2+ channels. Proc Natl Acad Sci USA 102:11112–11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. (2010) Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res 106:748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Nieves-Cintrón M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. (2008) AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res 102:e1–e11. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Bonev AD. (2004) The beta1 subunit of the Ca2+-sensitive K+ channel protects against hypertension. J Clin Invest 113:955–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. (1995) Relaxation of arterial smooth muscle by calcium sparks. Science 270:633–637. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. (1990) Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol 259:C3–C18. [DOI] [PubMed] [Google Scholar]
- Nguyen DHD, Catling AD, Webb DJ, Sankovic M, Walker LA, Somlyo AV, Weber MJ, Gonias SL. (1999) Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J Cell Biol 146:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickenig G, Harrison DG. (2002) The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis. Circulation 105:393–396. [DOI] [PubMed] [Google Scholar]
- Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. (2008) The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci USA 105:15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves-Cintrón M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. (2007) Activation of NFATc3 down-regulates the beta1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem 282:3231–3240. [DOI] [PubMed] [Google Scholar]
- Nilsson LM, Nilsson-Ohman J, Zetterqvist AV, Gomez MF. (2008) Nuclear factor of activated T-cells transcription factors in the vasculature: the good guys or the bad guys? Curr Opin Lipidol 19:483–490. [DOI] [PubMed] [Google Scholar]
- Nixon GF, Iizuka K, Haystead CM, Haystead TA, Somlyo AP, Somlyo AV. (1995) Phosphorylation of caldesmon by mitogen-activated protein kinase with no effect on Ca2+ sensitivity in rabbit smooth muscle. J Physiol 487:283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, Kimura M, Nakagawa K, Oshima T, Chayama K, et al. (2007) Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J Am Coll Cardiol 49:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norata GD, Pinna C, Zappella F, Elia L, Sala A, Condorelli G, Catapano AL. (2012) MicroRNA 143-145 deficiency impairs vascular function. Int J Immunopathol Pharmacol 25:467–474. [DOI] [PubMed] [Google Scholar]
- North AJ, Gimona M, Cross RA, Small JV. (1994a) Calponin is localised in both the contractile apparatus and the cytoskeleton of smooth muscle cells. J Cell Sci 107:437–444. [DOI] [PubMed] [Google Scholar]
- North AJ, Gimona M, Lando Z, Small JV. (1994b) Actin isoform compartments in chicken gizzard smooth muscle cells. J Cell Sci 107:445–455. [DOI] [PubMed] [Google Scholar]
- Obara K, Szymanski PT, Tao T, Paul RJ. (1996) Effects of calponin on isometric force and shortening velocity in permeabilized taenia coli smooth muscle. Am J Physiol 270:C481–C487. [DOI] [PubMed] [Google Scholar]
- Ohanian J, Gatfield KM, Ward DT, Ohanian V. (2005) Evidence for a functional calcium-sensing receptor that modulates myogenic tone in rat subcutaneous small arteries. Am J Physiol Heart Circ Physiol 288:H1756–H1762. [DOI] [PubMed] [Google Scholar]
- Ohanian J, Pieri M, Ohanian V. (2014) Non-receptor tyrosine kinases and the actin cytoskeleton in contractile vascular smooth muscle. J Physiol 593:3807–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. (2007) Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100:923–929. [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell’Acqua ML, Sather WA. (2007) AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oparil S, Zaman MA, Calhoun DA. (2003) Pathogenesis of hypertension. Ann Intern Med 139:761–776. [DOI] [PubMed] [Google Scholar]
- Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. (2004) Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol 286:C433–C447. [DOI] [PubMed] [Google Scholar]
- O’Rourke M. (1990) Arterial stiffness, systolic blood pressure, and logical treatment of arterial hypertension. Hypertension 15:339–347. [DOI] [PubMed] [Google Scholar]
- Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. (2002) Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol 283:H2260–H2267. [DOI] [PubMed] [Google Scholar]
- Packer CS, Roepke JE, Oberlies NH, Rhoades RA. (1998) Myosin isoform shifts and decreased reactivity in hypoxia-induced hypertensive pulmonary arterial muscle. Am J Physiol 274:L775–L785. [DOI] [PubMed] [Google Scholar]
- Padmanabhan S, Newton-Cheh C, Dominiczak AF. (2012) Genetic basis of blood pressure and hypertension. Trends Genet 28:397–408. [DOI] [PubMed] [Google Scholar]
- Palma-Flores C, Ramírez-Sánchez I, Rosas-Vargas H, Canto P, Coral-Vázquez RM. (2014) Description of a utrophin associated protein complex in lipid raft domains of human artery smooth muscle cells. Biochim Biophys Acta 1838:1047–1054. [DOI] [PubMed] [Google Scholar]
- Park SY, Park SU, Sohn UD. (2009) Regulators involved in the electrically stimulated response of feline esophageal smooth muscle. Pharmacology 84:346–355. [DOI] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tang JX, Tao T, Morgan KG. (1998) Cytoskeletal targeting of calponin in differentiated, contractile smooth muscle cells of the ferret. J Physiol 508:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tao T, Morgan KG. (1994) Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. Am J Physiol 267:C1262–C1270. [DOI] [PubMed] [Google Scholar]
- Parris JR, Cobban HJ, Littlejohn AF, MacEwan DJ, Nixon GF. (1999) Tumour necrosis factor-alpha activates a calcium sensitization pathway in guinea-pig bronchial smooth muscle. J Physiol 518:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale EB. (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133:38–52. [DOI] [PubMed] [Google Scholar]
- Patchell VB, Vorotnikov AV, Gao Y, Low DG, Evans JS, Fattoum A, El-Mezgueldi M, Marston SB, Levine BA. (2002) Phosphorylation of the minimal inhibitory region at the C-terminus of caldesmon alters its structural and actin binding properties. Biochim Biophys Acta 1596:121–130. [DOI] [PubMed] [Google Scholar]
- Paul RJ, Bowman PS, Johnson J, Martin AF. (2007) Effects of sex and estrogen on myosin COOH-terminal isoforms and contractility in rat aorta. Am J Physiol Regul Integr Comp Physiol 292:R751–R757. [DOI] [PubMed] [Google Scholar]
- Pavalko FM, Adam LP, Wu MF, Walker TL, Gunst SJ. (1995) Phosphorylation of dense-plaque proteins talin and paxillin during tracheal smooth muscle contraction. Am J Physiol 268:C563–C571. [DOI] [PubMed] [Google Scholar]
- Payne MC, Zhang HY, Prosdocimo T, Joyce KM, Koga Y, Ikebe M, Fisher SA. (2006) Myosin phosphatase isoform switching in vascular smooth muscle development. J Mol Cell Cardiol 40:274–282. [DOI] [PubMed] [Google Scholar]
- Payne MC, Zhang HY, Shirasawa Y, Koga Y, Ikebe M, Benoit JN, Fisher SA. (2004) Dynamic changes in expression of myosin phosphatase in a model of portal hypertension. Am J Physiol Heart Circ Physiol 286:H1801–H1810. [DOI] [PubMed] [Google Scholar]
- Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H. (2001) Hypothesis for the initiation of vasomotion. Circ Res 88:810–815. [DOI] [PubMed] [Google Scholar]
- Pérez GJ, Bonev AD, Nelson MT. (2001) Micromolar Ca(2+) from sparks activates Ca(2+)-sensitive K(+) channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol 281:C1769–C1775. [DOI] [PubMed] [Google Scholar]
- Perrin BJ, Ervasti JM. (2010) The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 67:630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesic A, Madden JA, Pesic M, Rusch NJ. (2004) High blood pressure upregulates arterial L-type Ca2+ channels: is membrane depolarization the signal? Circ Res 94:e97–e104. [DOI] [PubMed] [Google Scholar]
- Pestonjamasp KN, Pope RK, Wulfkuhle JD, Luna EJ. (1997) Supervillin (p205): A novel membrane-associated, F-actin-binding protein in the villin/gelsolin superfamily. J Cell Biol 139:1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ, Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, et al. The SAVE Investigators (1992) Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. N Engl J Med 327:669–677. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszódi A, Andersson KE, et al. (1998) Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J 17:3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D’Mello SR. (2008) Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One 3:e4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfitzer G. (2001) Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol 91:497–503. [DOI] [PubMed] [Google Scholar]
- Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, et al. (2000) Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial--the Losartan Heart Failure Survival Study ELITE II. Lancet 355:1582–1587. [DOI] [PubMed] [Google Scholar]
- Plüger S, Faulhaber J, Fürstenau M, Löhn M, Waldschütz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. (2000) Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res 87:E53–E60. [DOI] [PubMed] [Google Scholar]
- Poburko D, Kuo KH, Dai J, Lee CH, van Breemen C. (2004) Organellar junctions promote targeted Ca2+ signaling in smooth muscle: why two membranes are better than one. Trends Pharmacol Sci 25:8–15. [DOI] [PubMed] [Google Scholar]
- Posern G, Miralles F, Guettler S, Treisman R. (2004) Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J 23:3973–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M. (2009) Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J 23:2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poythress RH, Gallant C, Vetterkind S, Morgan KG. (2013) Vasoconstrictor-induced endocytic recycling regulates focal adhesion protein localization and function in vascular smooth muscle. Am J Physiol Cell Physiol 305:C215–C227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratusevich VR, Seow CY, Ford LE. (1995) Plasticity in canine airway smooth muscle. J Gen Physiol 105:73–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT, Pfitzer G. (2009) Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 24:342–356. [DOI] [PubMed] [Google Scholar]
- Pulver RA, Rose-Curtis P, Roe MW, Wellman GC, Lounsbury KM. (2004) Store-operated Ca2+ entry activates the CREB transcription factor in vascular smooth muscle. Circ Res 94:1351–1358. [DOI] [PubMed] [Google Scholar]
- Qiao YN, He WQ, Chen CP, Zhang CH, Zhao W, Wang P, Zhang L, Wu YZ, Yang X, Peng YJ, et al. (2014) Myosin phosphatase target subunit 1 (MYPT1) regulates the contraction and relaxation of vascular smooth muscle and maintains blood pressure. J Biol Chem 289:22512–22523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, et al. (2010) Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 107:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Li L. (2002) Histone acetylation and recruitment of serum responsive factor and CREB-binding protein onto SM22 promoter during SM22 gene expression. Circ Res 90:858–865. [DOI] [PubMed] [Google Scholar]
- Quevillon-Chéruel S, Janmot C, Nozais M, Lompré AM, Béchet JJ. (2000) Functional regions in the essential light chain of smooth muscle myosin as revealed by the mutagenesis approach. Eur J Biochem 267:6151–6157. [DOI] [PubMed] [Google Scholar]
- Rahman A, Davis B, Lövdahl C, Hanumaiah VT, Feil R, Brakebusch C, Arner A. (2014) The small GTPase Rac1 is required for smooth muscle contraction. J Physiol 592:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandran R, Raghavan A, Geenen D, Sun M, Bach L, Yang Q, Raj JU. (2014) PKG-1α leucine zipper domain defect increases pulmonary vascular tone: implications in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307:L537–L544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reho JJ, Zheng X, Asico LD, Fisher SA. (2015) Redox signaling and splicing dependent change in myosin phosphatase underlie early versus late changes in NO vasodilator reserve in a mouse LPS model of sepsis. Am J Physiol Heart Circ Physiol 308:H1039–H1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Tejani AD, Ripley ML, Han S. (2007) Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol 293:C993–C1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee AY, Ogut O, Brozovich FV. (2006) Nonmuscle myosin, force maintenance, and the tonic contractile phenotype in smooth muscle. Pflugers Arch 452:766–774. [DOI] [PubMed] [Google Scholar]
- Richards CT, Ogut O, Brozovich FV. (2002) Agonist-induced force enhancement: the role of isoforms and phosphorylation of the myosin-targeting subunit of myosin light chain phosphatase. J Biol Chem 277:4422–4427. [DOI] [PubMed] [Google Scholar]
- Riches K, Alshanwani AR, Warburton P, O’Regan DJ, Ball SG, Wood IC, Turner NA, Porter KE. (2014) Elevated expression levels of miR-143/5 in saphenous vein smooth muscle cells from patients with Type 2 diabetes drive persistent changes in phenotype and function. J Mol Cell Cardiol 74:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokolya A, Singer HA. (2000) Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am J Physiol Cell Physiol 278:C537–C545. [DOI] [PubMed] [Google Scholar]
- Rosendorff C, Black HR, Cannon CP, Gersh BJ, Gore J, Izzo JL, Jr, Kaplan NM, O’Connor CM, O’Gara PT, Oparil S; American Heart Association Council for High Blood Pressure Research; American Heart Association Council on Clinical Cardiology; American Heart Association Council on Epidemiology and Prevention (2007) Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation 115:2761–2788. [DOI] [PubMed] [Google Scholar]
- Rosenfeld SS, Xing J, Chen LQ, Sweeney HL. (2003) Myosin IIb is unconventionally conventional. J Biol Chem 278:27449–27455. [DOI] [PubMed] [Google Scholar]
- Rosenfeld SS, Xing J, Whitaker M, Cheung HC, Brown F, Wells A, Milligan RA, Sweeney HL. (2000) Kinetic and spectroscopic evidence for three actomyosin:ADP states in smooth muscle. J Biol Chem 275:25418–25426. [DOI] [PubMed] [Google Scholar]
- Ross R. (1993) The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362:801–809. [DOI] [PubMed] [Google Scholar]
- Ross R. (1999) Atherosclerosis--an inflammatory disease. N Engl J Med 340:115–126. [DOI] [PubMed] [Google Scholar]
- Rovner AS, Fagnant PM, Lowey S, Trybus KM. (2002) The carboxyl-terminal isoforms of smooth muscle myosin heavy chain determine thick filament assembly properties. J Cell Biol 156:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovner AS, Fagnant PM, Trybus KM. (2006) Phosphorylation of a single head of smooth muscle myosin activates the whole molecule. Biochemistry 45:5280–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüegg JC, Pfitzer G. (1985) Modulation of calcium sensitivity in guinea pig taenia coli: skinned fiber studies. Experientia 41:997–1001. [DOI] [PubMed] [Google Scholar]
- Russell A, Watts S. (2000) Vascular reactivity of isolated thoracic aorta of the C57BL/6J mouse. J Pharmacol Exp Ther 294:598–604. [PubMed] [Google Scholar]
- Salzameda B, Facemyer KC, Beck BW, Cremo CR. (2006) The N-terminal lobes of both regulatory light chains interact with the tail domain in the 10 S-inhibited conformation of smooth muscle myosin. J Biol Chem 281:38801–38811. [DOI] [PubMed] [Google Scholar]
- Sandoval YH, Atef ME, Levesque LO, Li Y, Anand-Srivastava MB. (2014) Endothelin-1 signaling in vascular physiology and pathophysiology. Curr Vasc Pharmacol 12:202–214. [DOI] [PubMed] [Google Scholar]
- Sandquist JC, Swenson KI, Demali KA, Burridge K, Means AR. (2006) Rho kinase differentially regulates phosphorylation of nonmuscle myosin II isoforms A and B during cell rounding and migration. J Biol Chem 281:35873–35883. [DOI] [PubMed] [Google Scholar]
- Saphirstein RJ, Gao YZ, Jensen MH, Gallant CM, Vetterkind S, Moore JR, Morgan KG. (2013) The focal adhesion: a regulated component of aortic stiffness. PLoS One 8:e62461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphirstein RJ, Gao YZ, Lin QQ, Morgan KG. (2015) Cortical actin regulation modulates vascular contractility and compliance in veins. J Physiol 593:3929–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphirstein RJ, Morgan KG. (2014) The contribution of vascular smooth muscle to aortic stiffness across length scales. Microcirculation 21:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Kreutz R, Wilm C, Ganten D, Pfitzer G. (1994) Augmented agonist-induced Ca(2+)-sensitization of coronary artery contraction in genetically hypertensive rats. Evidence for altered signal transduction in the coronary smooth muscle cells. J Clin Invest 94:1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HH, Lohmann SM, Walter U. (1993) The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta 1178:153–175. [DOI] [PubMed] [Google Scholar]
- Schutzer WE, Reed JF, Mader SL. (2005) Decline in caveolin-1 expression and scaffolding of G protein receptor kinase-2 with age in Fischer 344 aortic vascular smooth muscle. Am J Physiol Heart Circ Physiol 288:H2457–H2464. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199–208. [DOI] [PubMed] [Google Scholar]
- Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. (2003) Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res 92:411–418. [DOI] [PubMed] [Google Scholar]
- Sellers JR, Adelstein RS. (1985) The mechanism of regulation of smooth muscle myosin by phosphorylation. Curr Top Cell Regul 27:51–62. [DOI] [PubMed] [Google Scholar]
- Sentí M, Fernández-Fernández JM, Tomás M, Vázquez E, Elosua R, Marrugat J, Valverde MA. (2005) Protective effect of the KCNMB1 E65K genetic polymorphism against diastolic hypertension in aging women and its relevance to cardiovascular risk. Circ Res 97:1360–1365. [DOI] [PubMed] [Google Scholar]
- Seow CY. (2013) Hill’s equation of muscle performance and its hidden insight on molecular mechanisms. J Gen Physiol 142:561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow CY. (2015) Reply from Chun Y. Seow. J Physiol 593:477–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Zhou GP, Kupferman J, Surks HK, Christensen EN, Chou JJ, Mendelsohn ME, Rigby AC. (2008) Probing the interaction between the coiled coil leucine zipper of cGMP-dependent protein kinase Ialpha and the C terminus of the myosin binding subunit of the myosin light chain phosphatase. J Biol Chem 283:32860–32869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Basu S, Mitchell RW, Stelmack GL, Anderson JE, Halayko AJ. (2014) Role of dystrophin in airway smooth muscle phenotype, contraction and lung function. PLoS One 9:e102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Ito M, Miyahara M, Ichikawa K, Okubo S, Konishi T, Naka M, Tanaka T, Hirano K, Hartshorne DJ, et al. (1994) Characterization of the myosin-binding subunit of smooth muscle myosin phosphatase. J Biol Chem 269:30407–30411. [PubMed] [Google Scholar]
- Shin J, Johnson JA. (2007) Pharmacogenetics of beta-blockers. Pharmacotherapy 27:874–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Fisher SA. (2008) Tra2beta as a novel mediator of vascular smooth muscle diversification. Circ Res 103:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegman MJ. (2014) The pathway for force transmission in the rat anococcygeus muscle: a tale of two tendons. Anat Rec (Hoboken) 297:1714–1733. [DOI] [PubMed] [Google Scholar]
- Sikuler E, Kravetz D, Groszmann RJ. (1985) Evolution of portal hypertension and mechanisms involved in its maintenance in a rat model. Am J Physiol 248:G618–G625. [DOI] [PubMed] [Google Scholar]
- Singer HA. (2012) Ca2+/calmodulin-dependent protein kinase II function in vascular remodelling. J Physiol 590:1349–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Sarkar J, Raghavan A, Reddy SP, Raj JU. (2011) Hypoxia modulates the expression of leucine zipper-positive MYPT1 and its interaction with protein kinase G and Rho kinases in pulmonary arterial smooth muscle cells. Pulm Circ 1:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitbon O, Humbert M, Jagot JL, Taravella O, Fartoukh M, Parent F, Herve P, Simonneau G. (1998) Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J 12:265–270. [DOI] [PubMed] [Google Scholar]
- Small JV, Fürst DO, De Mey J. (1986) Localization of filamin in smooth muscle. J Cell Biol 102:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolock EM, Trappanese DM, Chang S, Wang T, Titchenell P, Moreland RS. (2009) siRNA-mediated knockdown of h-caldesmon in vascular smooth muscle. Am J Physiol Heart Circ Physiol 297:H1930–H1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobey CG. (2001) Potassium channel function in vascular disease. Arterioscler Thromb Vasc Biol 21:28–38. [DOI] [PubMed] [Google Scholar]
- Sobue K, Morimoto K, Inui M, Kanda K, Kakiuchi S. (1982) Control of actin-myosin interaction of gizzard smooth muscle by calmodulin and caldesmon-linked flip-flop mechanism. Biomed Res 3:188–196. [Google Scholar]
- Somlyo AP. (1997) Signal transduction. Rhomantic interludes raise blood pressure. Nature 389:908–909, 911. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. (1994) Signal transduction and regulation in smooth muscle. Nature 372:231–236. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83:1325–1358. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Wu X, Walker LA, Somlyo AV. (1999) Pharmacomechanical coupling: the role of calcium, G-proteins, kinases and phosphatases. Rev Physiol Biochem Pharmacol 134:201–234. [DOI] [PubMed] [Google Scholar]
- Somlyo AV. (1980) Ultrastructure of vascular smooth muscle, in Handbook of Physiology, (Geiger SR. ed) pp 33–67, American Physiological Society, Bethesda, MD. [Google Scholar]
- Somlyo AV. (2015) Smooth muscle myosin filament controversy, once again? J Physiol 593:473–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AV, Somlyo AP. (1968) Electromechanical and pharmacomechanical coupling in vascular smooth muscle. J Pharmacol Exp Ther 159:129–145. [PubMed] [Google Scholar]
- Sorescu D, Szöcs K, Griendling KK. (2001) NAD(P)H oxidases and their relevance to atherosclerosis. Trends Cardiovasc Med 11:124–131. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos A, Gineitis D, Copeland J, Treisman R. (1999) Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 98:159–169. [DOI] [PubMed] [Google Scholar]
- Spin JM, Quertermous T, Tsao PS. (2010) Chromatin remodeling pathways in smooth muscle cell differentiation, and evidence for an integral role for p300. PLoS One 5:e14301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli AM, Liu Y, Sun LY, González-Cobos JC, Backs J, Trebak M, Singer HA. (2015) Smooth muscle CaMKIIδ promotes allergen-induced airway hyperresponsiveness and inflammation. Pflugers Arch 467:2541–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurrell BE, Murphy TV, Hill MA. (2003) Intraluminal pressure stimulates MAPK phosphorylation in arterioles: temporal dissociation from myogenic contractile response. Am J Physiol Heart Circ Physiol 285:H1764–H1773. [DOI] [PubMed] [Google Scholar]
- Staiculescu MC, Galiñanes EL, Zhao G, Ulloa U, Jin M, Beig MI, Meininger GA, Martinez-Lemus LA. (2013) Prolonged vasoconstriction of resistance arteries involves vascular smooth muscle actin polymerization leading to inward remodelling. Cardiovasc Res 98:428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JJ, Iwuchukwu C, Maier KG, Gahtan V. (2014) Thrombospondin-1-induced vascular smooth muscle cell migration and proliferation are functionally dependent on microRNA-21. Surgery 155:228–233. [DOI] [PubMed] [Google Scholar]
- Stein S, Matter CM. (2011) Protective roles of SIRT1 in atherosclerosis. Cell Cycle 10:640–647. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA, Frid MG. (2006) Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99:675–691. [DOI] [PubMed] [Google Scholar]
- Stevenson AS, Gomez MF, Hill-Eubanks DC, Nelson MT. (2001) NFAT4 movement in native smooth muscle. A role for differential Ca(2+) signaling. J Biol Chem 276:15018–15024. [DOI] [PubMed] [Google Scholar]
- Stewart M, Needham M, Bankhead P, Gardiner TA, Scholfield CN, Curtis TM, McGeown JG. (2012) Feedback via Ca²⁺-activated ion channels modulates endothelin 1 signaling in retinal arteriolar smooth muscle. Invest Ophthalmol Vis Sci 53:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. (2003) Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299:1743–1747. [DOI] [PubMed] [Google Scholar]
- Strasser P, Gimona M, Moessler H, Herzog M, Small JV. (1993) Mammalian calponin. Identification and expression of genetic variants. FEBS Lett 330:13–18. [DOI] [PubMed] [Google Scholar]
- Sun Z, Martinez-Lemus LA, Hill MA, Meininger GA. (2008) Extracellular matrix-specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. Am J Physiol Cell Physiol 295:C268–C278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surks HK, Mendelsohn ME. (2003) Dimerization of cGMP-dependent protein kinase 1a and the myosin-binding subunit of myosin phosphatase: Role of leucine sipper domains. Cell Signal 15:937–944. [DOI] [PubMed] [Google Scholar]
- Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM, Mendelsohn ME. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286:1583–1587. [DOI] [PubMed] [Google Scholar]
- Surks HK, Richards CT, Mendelsohn ME. (2003) Myosin phosphatase-Rho interacting protein. A new member of the myosin phosphatase complex that directly binds RhoA. J Biol Chem 278:51484–51493. [DOI] [PubMed] [Google Scholar]
- Swärd K, Albinsson S, Rippe C. (2014) Arterial dysfunction but maintained systemic blood pressure in cavin-1-deficient mice. PLoS One 9:e92428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swärd K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. (2000) Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol 522:33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney HL, Houdusse A. (2010) Structural and functional insights into the Myosin motor mechanism. Annu Rev Biophys 39:539–557. [DOI] [PubMed] [Google Scholar]
- Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. (2002) Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283:L144–L155. [DOI] [PubMed] [Google Scholar]
- Szymanski PT, Dickie R, Rogers R, Fredberg JJ. (2003) Extraction and reconstitution of calponin and consequent contractile ability in permeabilized smooth muscle fibers. Anal Biochem 321:8–21. [DOI] [PubMed] [Google Scholar]
- Tabas I, García-Cardeña G, Owens GK. (2015) Recent insights into the cellular biology of atherosclerosis. J Cell Biol 209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajada S, Cidad P, Colinas O, Santana LF, López-López JR, Pérez-García MT. (2013) Down-regulation of CaV1.2 channels during hypertension: how fewer CaV1.2 channels allow more Ca(2+) into hypertensive arterial smooth muscle. J Physiol 591:6175–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Hiwada K, Kokubu T. (1988) Vascular smooth muscle calponin. A novel troponin T-like protein. Hypertension 11:620–626. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yoshimoto R, Fuchibe K, Fujishige A, Mitsui-Saito M, Hori M, Ozaki H, Yamamura H, Awata N, Taniguchi S, et al. (2000) Regulation of shortening velocity by calponin in intact contracting smooth muscles. Biochem Biophys Res Commun 279:150–157. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Watanabe H, Murakami M, Ono K, Munehisa Y, Koyama T, Nobori K, Iijima T, Ito H. (2007) Functional role of stromal interaction molecule 1 (STIM1) in vascular smooth muscle cells. Biochem Biophys Res Commun 361:934–940. [DOI] [PubMed] [Google Scholar]
- Takeya K, Loutzenhiser K, Shiraishi M, Loutzenhiser R, Walsh MP. (2008) A highly sensitive technique to measure myosin regulatory light chain phosphorylation: the first quantification in renal arterioles. Am J Physiol Renal Physiol 294:F1487–F1492. [DOI] [PubMed] [Google Scholar]
- Takizawa N, Koga Y, Ikebe M. (2002) Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem Biophys Res Commun 297:773–778. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Homma K, White HD, Yanagida T, Ikebe M. (2008) Smooth muscle myosin phosphorylated at single head shows sustained mechanical activity. J Biol Chem 283:15611–15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Minami H, Kota M, Kuwamura K, Kohmura E. (2005) Treatment of cerebral vasospasm with intra-arterial fasudil hydrochloride. Neurosurgery 56:214–223, discussion 214–223. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Meera P, Song M, Knaus HG, Toro L. (1997) Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant alpha + beta subunit complexes. J Physiol 502:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DD. (2008) Intermediate filaments in smooth muscle. Am J Physiol Cell Physiol 294:C869–C878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, Kaltenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, et al. (2003) Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med 9:1506–1512. [DOI] [PubMed] [Google Scholar]
- Tejani AD, Walsh MP, Rembold CM. (2011) Tissue length modulates “stimulated actin polymerization,” force augmentation, and the rate of swine carotid arterial contraction. Am J Physiol Cell Physiol 301:C1470–C1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Lipp P, Berridge MJ, Bootman MD. (1998) Hormone-evoked elementary Ca2+ signals are not stereotypic, but reflect activation of different size channel clusters and variable recruitment of channels within a cluster. J Biol Chem 273:27130–27136. [DOI] [PubMed] [Google Scholar]
- Thompson AM, Wagner R, Rzucidlo EM. (2014) Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am J Physiol Heart Circ Physiol 307:H533–H541. [DOI] [PubMed] [Google Scholar]
- Toro L, Wallner M, Meera P, Tanaka Y. (1998) Maxi-K(Ca), a Unique Member of the Voltage-Gated K Channel Superfamily. News Physiol Sci 13:112–117. [DOI] [PubMed] [Google Scholar]
- Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, Mann DL. (1996) Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 93:704–711. [DOI] [PubMed] [Google Scholar]
- Tragante V, Barnes MR, Ganesh SK, Lanktree MB, Guo W, Franceschini N, Smith EN, Johnson T, Holmes MV, Padmanabhan S, et al. (2014) Gene-centric meta-analysis in 87,736 individuals of European ancestry identifies multiple blood-pressure-related loci. Am J Hum Genet 94:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M. (2012) STIM/Orai signalling complexes in vascular smooth muscle. J Physiol 590:4201–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Ichikawa K, Hartshorne DJ, Siegman MJ, Butler TM. (1995) Thiophosphorylation of the 130-kDa subunit is associated with a decreased activity of myosin light chain phosphatase in alpha-toxin-permeabilized smooth muscle. J Biol Chem 270:18191–18194. [DOI] [PubMed] [Google Scholar]
- Turczyńska KM, Bhattachariya A, Säll J, Göransson O, Swärd K, Hellstrand P, Albinsson S. (2013) Stretch-sensitive down-regulation of the miR-144/451 cluster in vascular smooth muscle and its role in AMP-activated protein kinase signaling. PLoS One 8:e65135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turczynska KM, Sadegh MK, Hellstrand P, Swärd K, Albinsson S. (2012) MicroRNAs are essential for stretch-induced vascular smooth muscle contractile differentiation via microRNA (miR)-145-dependent expression of L-type calcium channels. J Biol Chem 287:19199–19206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner ST, Schwartz GL, Chapman AB, Boerwinkle E. (2001) C825T polymorphism of the G protein beta(3)-subunit and antihypertensive response to a thiazide diuretic. Hypertension 37:739–743. [DOI] [PubMed] [Google Scholar]
- Uchida S, Dimmeler S. (2015) Long noncoding RNAs in cardiovascular diseases. Circ Res 116:737–750. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, et al. (1997) Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990–994. [DOI] [PubMed] [Google Scholar]
- Usui T, Okada M, Mizuno W, Oda M, Ide N, Morita T, Hara Y, Yamawaki H. (2012) HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol Heart Circ Physiol 302:H1894–H1904. [DOI] [PubMed] [Google Scholar]
- Valentín A, Humphrey JD, Holzapfel GA. (2011) A multi-layered computational model of coupled elastin degradation, vasoactive dysfunction, and collagenous stiffening in aortic aging. Ann Biomed Eng 39:2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. (2008) HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 18:291–297. [DOI] [PubMed] [Google Scholar]
- Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak-Kazmierczak E, Côté GP, Mak AS. (1998) Different molecular mechanisms for Rho family GTPase-dependent, Ca2+-independent contraction of smooth muscle. J Biol Chem 273:23433–23439. [DOI] [PubMed] [Google Scholar]
- Van Lierop JE, Wilson DP, Davis JP, Tikunova S, Sutherland C, Walsh MP, Johnson JD. (2002) Activation of smooth muscle myosin light chain kinase by calmodulin. Role of LYS(30) and GLY(40). J Biol Chem 277:6550–6558. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Kauppinen S. (2014) Development of microRNA therapeutics is coming of age. EMBO Mol Med 6:851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veigel C, Molloy JE, Schmitz S, Kendrick-Jones J. (2003) Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat Cell Biol 5:980–986. [DOI] [PubMed] [Google Scholar]
- Velasco G, Armstrong C, Morrice N, Frame S, Cohen P. (2002) Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr850 induces its dissociation from myosin. FEBS Lett 527:101–104. [DOI] [PubMed] [Google Scholar]
- Velaz L, Ingraham RH, Chalovich JM. (1990) Dissociation of the effect of caldesmon on the ATPase activity and on the binding of smooth heavy meromyosin to actin by partial digestion of caldesmon. J Biol Chem 265:2929–2934. [PubMed] [Google Scholar]
- Vetterkind S, Lee E, Sundberg E, Poythress RH, Tao TC, Preuss U, Morgan KG. (2010) Par-4: a new activator of myosin phosphatase. Mol Biol Cell 21:1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetterkind S, Morgan KG. (2009) The pro-apoptotic protein Par-4 facilitates vascular contractility by cytoskeletal targeting of ZIPK. J Cell Mol Med 13:887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetterkind S, Poythress RH, Lin QQ, Morgan KG. (2013) Hierarchical scaffolding of an ERK1/2 activation pathway. Cell Commun Signal 11:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigetti D, Deleonibus S, Moretto P, Bowen T, Fischer JW, Grandoch M, Oberhuber A, Love DC, Hanover JA, Cinquetti R, et al. (2014) Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J Biol Chem 289:28816–28826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenseil JE, Mecham RP. (2012) Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5:264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, Bochud M, Rice KM, Henneman P, Smith AV, et al. LifeLines Cohort Study. EchoGen consortium. AortaGen Consortium. CHARGE Consortium Heart Failure Working Group. KidneyGen consortium. CKDGen consortium. Cardiogenics consortium. CardioGram (2011) Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet 43:1005–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MP, Andrea JE, Allen BG, Clément-Chomienne O, Collins EM, Morgan KG. (1994) Smooth muscle protein kinase C. Can J Physiol Pharmacol 72:1392–1399. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Cole WC. (2013) The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 33:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltregny D, Glénisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. (2005) Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J 19:966–968. [DOI] [PubMed] [Google Scholar]
- Wang C-LA, Wang L-WC, Xu SA, Lu RC, Saavedra-Alanis V, Bryan J. (1991) Localization of the calmodulin- and the actin-binding sites of caldesmon. J Biol Chem 266:9166–9172. [PubMed] [Google Scholar]
- Wang CL. (2008) Caldesmon and the regulation of cytoskeletal functions. Adv Exp Med Biol 644:250–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Kovacs M, Hu A, Limouze J, Harvey EV, Sellers JR. (2003) Kinetic mechanism of non-muscle myosin IIB: functional adaptations for tension generation and maintenance. J Biol Chem 278:27439–27448. [DOI] [PubMed] [Google Scholar]
- Wang G, Kwan BC, Lai FM, Choi PC, Chow KM, Li PK, Szeto CC. (2010) Intrarenal expression of microRNAs in patients with IgA nephropathy. Lab Invest 90:98–103. [DOI] [PubMed] [Google Scholar]
- Wang GR, Zhu Y, Halushka PV, Lincoln TM, Mendelsohn ME. (1998) Mechanism of platelet inhibition by nitric oxide: in vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc Natl Acad Sci USA 95:4888–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Long CL, Zhang YL. (2005) A new ATP-sensitive potassium channel opener reduces blood pressure and reverses cardiovascular remodeling in experimental hypertension. J Pharmacol Exp Ther 312:1326–1333. [DOI] [PubMed] [Google Scholar]
- Wang R, Li Q, Tang DD. (2006) Role of vimentin in smooth muscle force development. Am J Physiol Cell Physiol 291:C483–C489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Li QF, Anfinogenova Y, Tang DD. (2007) Dissociation of Crk-associated substrate from the vimentin network is regulated by p21-activated kinase on ACh activation of airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 292:L240–L248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Thorin E, Luo H, Tremblay J, Lavoie JL, Wu Z, Peng J, Qi S, Wu J. (2015) EPHB4 Protein Expression in Vascular Smooth Muscle Cells Regulates Their Contractility, and EPHB4 Deletion Leads to Hypotension in Mice. J Biol Chem 290:14235–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. (1996) RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature 383:172–175. [DOI] [PubMed] [Google Scholar]
- Webb TI, Kshatri AS, Large RJ, Akande AM, Roy S, Sergeant GP, McHale NG, Thornbury KD, Hollywood MA. (2015) Molecular mechanisms underlying the effect of the novel BK channel opener GoSlo: involvement of the S4/S5 linker and the S6 segment. Proc Natl Acad Sci USA 112:2064–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman GC, Nelson MT. (2003) Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium 34:211–229. [DOI] [PubMed] [Google Scholar]
- Westcott EB, Goodwin EL, Segal SS, Jackson WF. (2012) Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol 590:1849–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott EB, Jackson WF. (2011) Heterogeneous function of ryanodine receptors, but not IP3 receptors, in hamster cremaster muscle feed arteries and arterioles. Am J Physiol Heart Circ Physiol 300:H1616–H1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden AD, Grimwood S, Grant TL, Todd MH. (1991) Comparison of the effects of the K(+)-channel openers cromakalim and minoxidil sulphate on vascular smooth muscle. Br J Pharmacol 103:1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. (2006) Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation 113:664–670. [DOI] [PubMed] [Google Scholar]
- Winder SJ, Allen BG, Clément-Chomienne O, Walsh MP. (1998) Regulation of smooth muscle actin-myosin interaction and force by calponin. Acta Physiol Scand 164:415–426. [DOI] [PubMed] [Google Scholar]
- Winder SJ, Allen BG, Fraser ED, Kang HM, Kargacin GJ, Walsh MP. (1993) Calponin phosphorylation in vitro and in intact muscle. Biochem J 296:827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ, Walsh MP. (1990) Smooth muscle calponin. Inhibition of actomyosin MgATPase and regulation by phosphorylation. J Biol Chem 265:10148–10155. [PubMed] [Google Scholar]
- Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, et al. (2008) G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14:64–68. [DOI] [PubMed] [Google Scholar]
- Wirth A, Schroeter M, Kock-Hauser C, Manser E, Chalovich JM, De Lanerolle P, Pfitzer G. (2003) Inhibition of contraction and myosin light chain phosphorylation in guinea-pig smooth muscle by p21-activated kinase 1. J Physiol 549:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. (2001) Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J Physiol 535:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge AA, MacDonald JA, Erdodi F, Ma C, Borman MA, Hartshorne DJ, Haystead TA. (2004) Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J Biol Chem 279:34496–34504. [DOI] [PubMed] [Google Scholar]
- Worley JF, Quayle JM, Standen NB, Nelson MT. (1991) Regulation of single calcium channels in cerebral arteries by voltage, serotonin, and dihydropyridines. Am J Physiol 261:H1951–H1960. [DOI] [PubMed] [Google Scholar]
- Wu C, So J, Davis-Dusenbery BN, Qi HH, Bloch DB, Shi Y, Lagna G, Hata A. (2011) Hypoxia potentiates microRNA-mediated gene silencing through posttranslational modification of Argonaute2. Mol Cell Biol 31:4760–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, et al. (2014) LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130:1452–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Haystead TA, Nakamoto RK, Somlyo AV, Somlyo AP. (1998) Acceleration of myosin light chain dephosphorylation and relaxation of smooth muscle by telokin. Synergism with cyclic nucleotide-activated kinase. J Biol Chem 273:11362–11369. [DOI] [PubMed] [Google Scholar]
- Wu Z, Luo H, Thorin E, Tremblay J, Peng J, Lavoie JL, Wang Y, Qi S, Wu T, Wu J. (2012) Possible role of Efnb1 protein, a ligand of Eph receptor tyrosine kinases, in modulating blood pressure. J Biol Chem 287:15557–15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZY, Benoit JN. (1994) Vascular NE responsiveness in portal hypertension: role of portal pressure and portosystemic shunting. Am J Physiol 266:H1162–H1168. [DOI] [PubMed] [Google Scholar]
- Xiao B, Gu SM, Li MJ, Li J, Tao B, Wang Y, Wang Y, Zuo S, Shen Y, Yu Y, et al. (2015) Rare SNP rs12731181 in the miR-590-3p target site of the prostaglandin F2α receptor gene confers risk for essential hypertension in the Han Chinese population. Arterioscler Thromb Vasc Biol 35:1687–1695. [DOI] [PubMed] [Google Scholar]
- Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. (2009) MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23:2166–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ, Beech DJ. (2001) TrpC1 is a membrane-spanning subunit of store-operated Ca(2+) channels in native vascular smooth muscle cells. Circ Res 88:84–87. [DOI] [PubMed] [Google Scholar]
- Xu X, Ha CH, Wong C, Wang W, Hausser A, Pfizenmaier K, Olson EN, McKinsey TA, Jin ZG. (2007) Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler Thromb Vasc Biol 27:2355–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zheng B, Zhang Y, He M, Zhang XH, Ma D, Zhang RN, Wu XL, Wen JK. (2015) miR-155-dependent regulation of mammalian sterile 20-like kinase 2 (MST2) coordinates inflammation, oxidative stress and proliferation in vascular smooth muscle cells. Biochim Biophys Acta 1852:1477–1489. [DOI] [PubMed] [Google Scholar]
- Yilmaz M, Gangopadhyay SS, Leavis P, Grabarek Z, Morgan KG. (2013) Phosphorylation at Ser²⁶ in the ATP-binding site of Ca²⁺/calmodulin-dependent kinase II as a mechanism for switching off the kinase activity. Biosci Rep 33:e00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying AK, Hassanain HH, Roos CM, Smiraglia DJ, Issa JJ, Michler RE, Caligiuri M, Plass C, Goldschmidt-Clermont PJ. (2000) Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc Res 46:172–179. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Gan Q, Owens GK. (2008) Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol 295:C1175–C1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lu Y, Li Z, Wang Q. (2014) microRNA-133: expression, function and therapeutic potential in muscle diseases and cancer. Curr Drug Targets 15:817–828. [DOI] [PubMed] [Google Scholar]
- Yuen S, Ogut O, Brozovich FV. (2011) MYPT1 protein isoforms are differentially phosphorylated by protein kinase G. J Biol Chem 286:37274–37279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen SL, Ogut O, Brozovich FV. (2009) Nonmuscle myosin is regulated during smooth muscle contraction. Am J Physiol Heart Circ Physiol 297:H191–H199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen SL, Ogut O, Brozovich FV. (2014) Differential phosphorylation of LZ+/LZ- MYPT1 isoforms regulates MLC phosphatase activity. Arch Biochem Biophys 562:37–42. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Pepine CJ, Garces C, Pouleur H, Salem D, Kostis J, Benedict C, Rousseau M, Bourassa M, Pitt B. (1992) Effect of enalapril on myocardial infarction and unstable angina in patients with low ejection fractions. Lancet 340:1173–1178. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, CHARM Investigators and Committees (2003) Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 362:777–781. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G, The Heart Outcomes Prevention Evaluation Study Investigators (2000) Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 342:145–153. [DOI] [PubMed] [Google Scholar]
- Zampetaki A, Attia R, Mayr U, Gomes RS, Phinikaridou A, Yin X, Langley SR, Willeit P, Lu R, Fanshawe B, et al. (2014) Role of miR-195 in aortic aneurysmal disease. Circ Res 115:857–866. [DOI] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell 32:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fisher SA. (2007) Conditioning effect of blood flow on resistance artery smooth muscle myosin phosphatase. Circ Res 100:730–737. [DOI] [PubMed] [Google Scholar]
- Zhang H, Gu S, Al-Sabeq B, Wang S, He J, Tam A, Cifelli C, Mathalone N, Tirgari S, Boyd S, et al. (2012) Origin-specific epigenetic program correlates with vascular bed-specific differences in Rgs5 expression. FASEB J 26:181–191. [DOI] [PubMed] [Google Scholar]
- Zhang J, Herrera AM, Paré PD, Seow CY. (2010) Dense-body aggregates as plastic structures supporting tension in smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 299:L631–L638. [DOI] [PubMed] [Google Scholar]
- Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, et al. (2011) Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ Res 109:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang Y, Wu Y, Gunst SJ. (2015) A novel role for RhoA GTPase in the regulation of airway smooth muscle contraction. Can J Physiol Pharmacol 93:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YD, Cai L, Mirza MK, Huang X, Geenen DL, Hofmann F, Yuan JX, Zhao YY. (2012) Protein kinase G-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation. Am J Pathol 180:2268–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, et al. (2002) Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 295:505–508. [DOI] [PubMed] [Google Scholar]
- Zulliger MA, Stergiopulos N. (2007) Structural strain energy function applied to the ageing of the human aorta. J Biomech 40:3061–3069. [DOI] [PubMed] [Google Scholar]