

Abstract
The isoprenoid biosynthetic pathway (IBP) is critical for providing substrates for the post-translational modification of proteins key in regulating malignant cell properties, including proliferation, invasion, and migration. Inhibitors of the IBP, including statins and nitrogenous bisphosphonates, are used clinically for the treatment of hypercholesterolemia and bone disease respectively. The statins work predominantly in the liver, while the nitrogenous bisphosphonates are highly sequestered to bone. Inhibition of the entire IBP is limited by organ specificity and side effects resulting from depletion of all isoprenoids. We have developed a novel compound, disodium [(6Z,11E,15E)-9-[bis(sodiooxy)phosphoryl]-17-hydroxy-2,6,12,16-tetramethyheptadeca-2,6,11,15-tetraen-9-yl]phosphonate (GGOHBP), which selectively targets geranylgeranyl diphosphate synthase (GGDPS), reducing post-translational protein geranylgeranylation. Intracardiac injection of luciferase-expressing human-derived 22Rv1 PCa cells into SCID mice resulted in tumor development in bone (100%), adrenal glands (72%), mesentery (22%), liver (17%), and the thoracic cavity (6%). Three weeks after tumor inoculation, daily subcutaneous (SQ) injections of 1.5 mg/kg GGOHBP or the vehicle were given for one month. Dissected tumors revealed areduction in adrenal gland tumors corresponding to a 54% (P < 0.005) reduction in total adrenal gland tumor weight of the treated mice as compared to vehicle-treated controls. Western blot analysis of the harvested tissues showed a reduction in Rap1A geranylgeranylation in adrenal glands and mesenteric tumors of the treated mice while non-tumorous tissues and control mice showed no Rap1A alteration. Our findings detail a novel bisphosphonate compound capable of preferentially altering the IBP in tumor-burdened adrenal glands of a murine model of PCa metastasis.
Keywords: Prostate cancer, Metastasis, Adrenal tumor, Geranylgeranyl pyrophosphate, posttranslational modification, isoprenoid, bisphosphonate
Introduction
The isoprenoid biosynthetic pathway (IBP, Figure 1a) is highly conserved across multiple species and is central to many cellular functions.[1] In humans, the IBP has been targeted for therapeutic benefit, most notably the statin family for the treatment of hypercholesterolemia and nitrogenous bisphosphonates for the treatment of osteoporosis.[2] Manipulation of this pathway with nitrogenous bisphosphonates has been beneficial in the treatment of patients with bone metastases, particularly in prostate cancer (PCa) by decreasing skeletal events and minimizing bone pain.[3-5]
Fig 1. Novel compound GGOHBP targets GGDPS.

(a) The isoprenoid biosynthetic pathway with targeted inhibition by statins, nitrogenous bisphosphonates, and novel compounds DGBP and GGOHBP. Compound structures of (b) DGBP and (c) GGOHBP
Nitrogenous bisphosphonates are potent inhibitors of the protein farnesyl pyrophosphate (FPP) synthase (FDPS) which synthesizes the 15 carbon isoprenoid FPP.[6, 7] FPP and the 20 carbon isoprenoid geranylgeranyl pyrophosphate (GGPP) are both important metabolites involved in a host of cellular processes.[1] FPP is crucial for cholesterol synthesis and is also used by the enzyme geranylgeranyl pyrophosphate synthase (GGDPS) to make GGPP. Both FPP and GGPP are used to post-translationally modify small GTPases, including the Ras and Rho family, to facilitate targeting of these proteins to the membrane, a modification central to their proper spatio-temporal activity.[8, 9]
While the statins and nitrogenous bisphosphonates target early steps in the IBP, in vitro work has implicated their reduction of the downstream metabolite GGPP in their function.[10-12] Small GTPases implicated in cancer metastasis and dependent upon geranylgeranylation for proper function include the following: RhoA, Rac1, Cdc42 in cell polarity;[13] RhoA and Rac1 in cell junctions;[14-17] RhoA, Rac, and Cdc42 in cell motility;[18, 19] Rho family in intravasation,[18, 20] and RhoC in motility cell and invasive potential.[21]
The benefit of manipulating the IBP has been demonstrated in multiple epidemiological studies. [22, 23] In a Health Professionals Follow-up study of 34,438 men on statins, they found a 46% reduction in their risk of advanced PCa (p=0.008).[22] In a Finnish study of 49,446 men taking statins, users had a 25% lower risk of advanced PCa as compared to men on other cholesterol lowering agents or on no cholesterol lowering medication.[24-26] Very recently, studies of statin use in cohorts of patients with PCa have shown that more hydrophobic statins show better anti-PCa activity, with men taking statins at the time of diagnosis faring better than those who were prescribed statins post PCa diagnosis.[27, 28]
Both statins and the nitrogenous bisphosphonates have potential drawbacks limiting their application in PCa. Statins act predominantly in the liver where they are subject to first pass metabolism, showing reduced bioavailability outside this organ.[29, 30] Nitrogenous bisphosphonates on the other hand bind extremely tightly to bone, limiting clinical use to skeletal indications.[31] As part of an extensive research program aimed at developing drugs targeting isoprenoid metabolism for the treatment of disease, we have developed a library of compounds which are potent inhibitors of GGDPS.[32-45] These compounds are more lipophilic and are not highly targeted to bone mineral. For this reason they demonstrate more standard pharmcokinetics than the nitrogenous bisphosphonates in current clinical use which are highly bone bound and therefore have extended terminal half-lives.
The present studies use two of these compounds, digeranyl bisphosphonate (DGBP) (Figure 1b) and disodium [(6Z,11E,15E)-9-[bis(sodiooxy)phosphoryl]-17-hydroxy-2,6,12,16-tetramethyheptadeca-2,6,11,15-tetraen-9-yl]phosphonate (GGOHBP, Figure 1c), both inhibiting GGDPS. We hypothesize the selective reduction of geranylgeranylation will reduce the metastatic potential of human PCa cells while further clarifying the role of geranylgeranylation and the IBP in PCa metastasis. We demonstrate for the first time that in vivo inhibition of GGDPS both decreases adrenal gland metastasis and biochemically blocks protein geranylgeranylation in a murine model of human PCa.
Materials and Methods
Compound synthesis
GGOHBP was synthesized as previously described.[46] The concentration was calculated based on the tetra-sodium salt form.
Cell culture
22Rv1 cells were transfected to express luciferase as previously described.[47] Cells were grown at 37°C with 5% CO2 in RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% non-essential amino acids (MEM NEAA), and 1% geneticin.
GGDPS enzyme assays
The GGDPS enzyme was kindly provided by Dr. James E Dunford. The enzyme assay method was modified from Dunford et al.[48] GGDPS was diluted to 2μg/mL (10 mM HEPES, pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM TCEP, and 5 μg/mL BSA) and pre-incubated with inhibitors for 10 min at room temperature in the reaction buffer (50 mM Tris, pH 7.7, 2 mM MgCl2, 0.5 mM TCEP, and 50 μg/mL BSA). Enzyme assay reactions were initiated by the simultaneous addition of 10 μM FPP and 14C-isopentenyl pyrophosphate and allowed to proceed for 15 min at 37°C, at which point no more than 20% of the substrate was used. Reactions were terminated by the addition of 200 μL saturated NaCl, and isoprenoids were extracted with 1 mL saturated butanol. Incorporated 14C was detected by liquid scintillation counting.
MTT assays
Cells were plated at 25,000 cells/well in supplemented RPMI 1640 media lacking phenol red and allowed to adhere overnight. Cells were treated in triplicate with the indicated concentrations of inhibitors and incubated for 44 hrs, at which point 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added (Calbiochem, Billerica, MA). Four hours later MTT stop solution was added and cells incubated at 37°C with a gentle agitation overnight. Absorbance was measured at 540 nm with a reference wavelength at 650 nm.
FPP/GGPP quantification
FPP and GGPP quantification by HPLC was performed as previously described. [49] Briefly, cells were plated at 5 million cells/plate for 48 hrs. Isoprenoid pyrophosphates were extracted and incorporated into fluorescent D*-GCVLA or D*-GCVLL (dansylated peptide substrates) by FPTase or GGPTase I or II and analyzed by HPLC equipped with fluorescent detection.
TX-114 separation
Cells were plated at 3 million cells/plate overnight and treated with the indicated concentrations of inhibitors for 48 hrs. Cells were lysed with TX-114 buffer (1% TX-114, 20 mM Tris HCl, 150 mM NaCl) containing protease inhibitor cocktail and phenylmethanesulfonyle fluoride and incubated at 37°C until lysates became cloudy at which point they were spun for 2 min at 14000 rpm. Aqueous and detergent phases were separated, adding 1% TX-114 buffer to aqueous lysates and diluting detergent lysates to ¼ the volume of aqueous lysates using Buffer B (20 mM Tris HCl, 150 mM NaCl) containing protease inhibitor cocktail and phenylmethanesulfonyle fluoride. Incubation and separation of aqueous and detergent samples was repeated five times, after which detergent lysates were diluted to the total volume of aqueous lysates using Buffer B.
Western blots
Cells were plated at 3 million cells/plate and treated with the indicated concentrations of inhibitors for 48 hrs. Cells were lysed in RIPA (0.15 M NaCl, 0.05 M Tris HCl, 1% w/v sodium deoxycholate, 1% w/v SDS, 1% w/v Triton X-100, 1 mM EDTA) or TX-114 buffer containing protease inhibitor cocktail and phenylmethanesulfonyle fluoride. Protein concentrations were quantified by the BCA method.[50] Equal protein quantities were resolved by SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane overnight. Membranes were blocked for 45 min in 5% non-fat milk followed by incubation with primary antibodies overnight and secondary antibodies for 1 hr. Protein levels were visualized using ECL detection site (GE Healthcare, Buckinghamshire, UK). Primary antibody anti-pan-Ras was obtained from Inter-Biotechnology (Tokyo, Japan), Rap1A (sc-1482), Rab6 (sc-310), β-tubulin (sc-9140), calnexin (sc-23954), and secondary antibody donkey anti-goat IgG-HRP (sc-2033) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and secondary HRP-conjugated anti-rabbit (NA934) and anti-mouse (NXA931) antibodies were obtained from GE Healthcare (Buckinghamshire, UK).
Metastatic murine model
All animal work was performed in accordance with The University of Iowa Animal Care and Use Committee policies. Intracardiac injection of 105 luciferase-expressing 22Rv1 cells into male SCID mice was done as described previously.[47, 51] Weekly bioluminescence imaging with X-ray overlay monitored tumor development. Between weeks three and four after cell injection, mice were randomized into two groups using an algorithm to minimize between group variance in total photon count, at which point daily subcutaneous injections of PBS or 1.5 mg/kg GGOHBP began. After 31 days of treatment, poor body condition, or a greater than 20% loss in body weight, mice were euthanized, ex vivo bioluminescence performed, and all heart, liver, kidney, and any identified tumors were collected.
Tissue harvesting and lysing
Frozen tissues were thawed on ice and weighed. Tissue aliquots of approximately 0.05 g were collected and lysed in RIPA buffer containing protease inhibitor cocktail and phenylmethanesulfonyle fluoride. Tissues were sonicated, incubated with lysing buffer for 30 min with agitation, and centrifuged at 14000 rpm for 20 min at 4°C, collecting the supernatant and repeating centrifugation.
Results
DGBP and GGOHBP inhibit GGDPS and impair protein geranylgeranylation
In enzyme assays both DGBP and GGOHBP inhibit GGDPS with IC50s of 0.2 μM and 0.8 μM respectively (Figure 2a). Intact cell studies were performed using the luciferase-expressing 22Rv1 human-derived prostate cancer cell line. MTT activity of intact cells at 48 hrs was reduced by ~15% at 10 μM treatments of both compounds (Figure 2b). Western blot analysis of Ras, Rap1A, Rab6 was done with antibodies that detect both the unprenylated (upper band) and prenylated (lower band) forms of Ras, only the unprenylated form of Rap1A, and the unprenylated (aqueous bands) and prenylated (detergent bands) of Rab6. As shown in figure 2c, neither compound altered Ras farnesylation at up to 50 μM concentrations at 48 hrs as shown by the lack of an upper band, similar to the no treatment control. Rap1A geranylgeranylation, which is catalyzed by geranylgeranyl protein transferase 1 (GGPTase-I), was impaired at 48 hrs at concentrations of 2.5 μM DGBP and 5 μM GGOHBP. Rab6 geranylgeranylation, which is catalyzed by GGPTase-II, was impaired at 48 hrs at concentrations of at least 1 μM for DGBP and 2.5 μM for GGOHBP. Immune compromised male SCID mice showed no weight loss at 1.5 mg/kg GGOHBP daily for 31 days (Figure 2d) while previous work found the maximum tolerated dose (MTD) of DGBP to be 0.1 mg/kg 3x/week. Due to the comparable inhibition of geranylgeranylation in vitro and apparent improvement in therapeutic index indicated by the increased MTD in vivo, GGOHBP was chosen as the compound of interest for subsequent studies.
Fig 2. DGBP and GGOHBP inhibit GGDPS, reducing protein geranylgeranylation.

(a) Isolated GGDPS enzyme activity following preincubation with the indicated concentrations of DGBP or GGOHBP. (b) MTT activity of DGBP and GGOHBP in luciferase-expressing 22Rv1 cells after 48 hrs of treatment. (c) Representative Western blot analysis of Ras (top upper panel), Rap1A (mid upper panel), and Rab6 (lower panels). Luciferase-expressing 22Rv1 cells were treated with the indicated concentrations of DGBP, GGOHBP, or lovastatin for 48 hrs. The Ras antibody detects the unprenylated (upper band) and prenylated (lower band) forms of Ras. The Rap1A antibody detects the unprenylated form of Rap1A. The Rab6 antibody detects the unprenylated (aqueous bands) and prenylated (detergent bands) forms of Rab6 and requires separation by TX-114 detergent. (d) Average weights of mice treated with the PBS-vehicle, 1.25 mg/kg, or 1.5 mg/kg GGOHBP daily for 31 days. Error bars indicate standard deviation
GGOHBP decreases adrenal gland metastasis from luciferase-expressing 22Rv1 inoculum
The intracardiac injection of luciferase-expressing 22RV1 cells in an in vivo murine model resulted in tumors that distributed to multiple organs. Table 1 lists the sites of metastasis from most to least frequent in affected animals. While all mice had mandibular tumors and 72% had adrenal gland tumors, the distribution of other soft tissue tumors did not follow any discernable pattern. Of the four mice with mesentery tumors, three also presented with adrenal gland tumors. Furthermore, of the three mice with liver tumors and the one mouse with a thoracic tumor, none showed tumors in the adrenal glands. Compared to a larger scale study of the luciferase-expressing 22Rv1 cells inoculated via intracardiac injection by Drake et al, our overall tumor distribution to the bone, adrenal glands, mesenteric, and liver tumors was commonly observed. However, Drake et al additionally observed tumor distribution to the urogenital, brain/dura, and muscle, which were not seen in our study.[47] Between weeks 3 and 4, mice were randomized to afford equal numbers in both control and treatment arms (Table 1) based on photon counts (i.e. total body tumor volume) at which point daily subcutaneous injections of 1.5 mg/kg GGOHBP or the vehicle began.
Table 1.
Distribution of tumors in mice as determined by ex vivo bioluminescence. Mice were randomized between weeks 3 and 4 to afford equal numbers in both control and treatment arms based on photon counts (i.e. total body tumor volume)
| Site of metastasis | Percentage of total mice | Percentage of control mice |
Percentage of treated mice |
|---|---|---|---|
| Mandible | 18/18 (100%) | 9/9 (100%) | 9/9 (100%) |
| Adrenal glands | 13/18 (72.2%) | 6/9 (66.7%) | 7/9 (77.8%) |
| Mesentery | 4/18 (22.2%) | 2/9 (22.2%) | 2/9 (22.2%) |
| Liver | 3/18 (16.6%) | 2/9 (22.2%) | 1/9 (11.1%) |
| Femur | 2/18 (11.1%) | 0/9 (0%) | 2/9 (22.2%) |
| Thoracic cavity | 1/18 (5.5%) | 1/9 (11.1%) | 0/9 (0%) |
Shown in figure 3a is a representative mouse followed weekly with bioluminescence imaging (BLI). Weekly whole body photon counts averaged within the control and treated groups are shown in figure 3b. Despite treatment beginning between weeks 3 and 4, there was not a significant difference between the whole body photon counts of the control and treated mice. However, early in treatment during weeks 4 through 6 the treated mice trended towards maintenance of their whole body photon count while the control mice continued to increase. BLI of the dissected adrenal gland tumors in figure 3c showed a nonsignificant trend towards reduction in the treated mice as compared to the control mice. This corresponded to a significant 54% reduction in the total adrenal gland tumor weights of the treated mice as compared to the control mice (P < 0.005) shown in figure 3d. Weekly BLI analysis of the adrenal gland tumors in vivo found the treated mice to initially plateau in the first few weeks following treatment initiation whereas the control mice continued an increasing trend (Figure 3e). GGOHBP impairs Rap1A geranylgeranylation in adrenal gland and mesenteric metastatic sites-
Fig 3. GGOHBP reduces adrenal gland tumor bioluminescence and weight.

(a) Representative weekly BLI imaging of control mouse 15. BLI exposure times indicated below. (b) Calculated weekly whole body photon count, averaged within the control and GGOHBP treatment groups. (c) Calculated ex vivo photon count of the adrenal gland tumors taken immediately following individual mouse euthanization, averaged within the control and GGOHBP treatment groups. (d) Total adrenal gland tumor weights. Control mice 14, 25, and 28 and treated mice 13 and 29 did not have adrenal gland tumors and thus were not included in the adrenal gland tumor weight analysis. Control mouse 15 and treated mouse 19 had adrenal gland tumors but were sent to pathology and not weighed and consequently were not included in the adrenal gland tumor weight analysis. (e) Weekly adrenal gland tumor photon count, averaged within the control and GGOHBP treatment groups. Statistical significance indicated as ** (P < 0.005) by Student’s t test. Error bars indicate standard deviation
Non-tumorous liver, kidney, and heart tissues were harvested from each mouse. Western blot analysis of Rap1A geranylgeranylation showed no alteration in non-tumor burdened liver (Figure 4a), kidney (Figure 4b), and heart (Figure 4c) tissues of control and treated mice. As identified by ex vivo imaging, soft tissue tumors were collected from each mouse. Analysis of Rap1A geranylgeranylation in the adrenal gland tumors of control and treated mice showed inhibition in all treated mice but not in any control mice (Figure 4d). Within the treated mice there was variability in the inhibition of Rap1A geranylgeranylation. Mouse 17 showed the strongest inhibition of Rap1A geranylgeranylation and had the largest adrenal gland tumor at 38 mg. Conversely, mouse 24 showed the least inhibition of Rap1A geranylgeranylation and had the smallest adrenal gland tumor at 15 mg. Analysis of Rap1A geranylgeranylation in the miscellaneous soft tissue tumors showed inhibition within the mesenteric tumor of a treated mouse with no inhibition evident in the control mice (Figure 4e). FPP, GGPP, Ras, and Rab6 levels are significantly higher in more metastatic PCa cell lines-
Fig 4. Western blot analysis shows a reduction in Rap1A prenylation in the adrenal gland tumors and a mesenteric tumor of treated mice.

Representative Western blot analysis of Rap1A in (a) liver tissues, (b) kidney tissues, (c) heart tissues, (d) adrenal gland tumors, and (e) mesenteric, liver, and thoracic tumors of control and treated mice. Control mice in part e are indicated by C and treated mice in part e are indicated by a T. The Rap1A antibody detects the unprenylated form of Rap1A as indicated by the positive control 10 μM lovastatin treatment in luciferase-expressing 22Rv1 cells
Due to the finding of increased inhibition of Rap1A geranylgeranylation in the larger adrenal gland tumors, we questioned whether there was endogenous variation in levels of the IBP in different prostate cancer cell lines . To address this question, we compared endogenous levels of FPP, GGPP, Ras, and Rab6 in the three distinct prostate cancer cell lines showing various degrees of metastatic behavior. The metastatic cell lines LNCaP and 22Rv1 and the highly metastatic PC3 cell line. Shown in figure 5a, we found significantly higher levels of both FPP and GGPP in the untreated PC3 cell line as compared to the less metastatic LNCaP and 22Rv1 cells when analyzing early cell passages. Correspondingly, we found the PC3 cell line to have higher levels of both farnesylated Ras and geranylgeranylated Rab6 (Figure 5b).
Fig 5. Highly metastatic PC3 cells have significantly more FPP, GGPP, Ras, and Rab6 as compared to the less metastatic LNCaP and 22Rv1 cell lines.
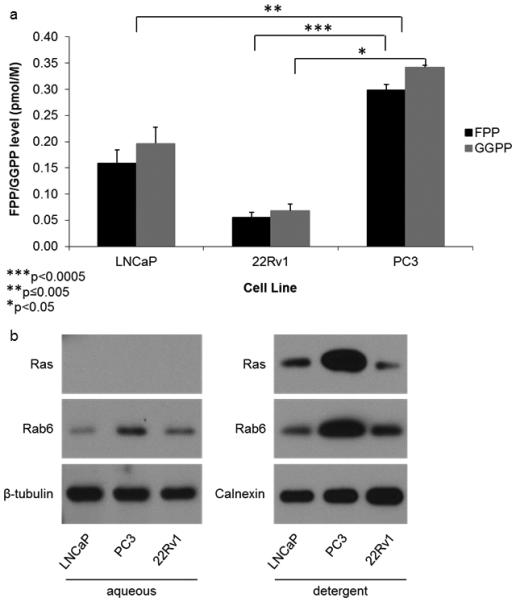
(a) HPLC analysis of FPP and GGPP levels in the non-treated LNCaP, 22Rv1, and PC3 cell lines. (b) Representative Western blot analysis of Ras and Rab6 in the non-treated LNCaP, 22Rv1, and PC3 cell lines. The Ras and Rab6 antibodies detect both the unprenylated (aqueous) and prenylated (detergent) forms of Ras and Rab6 respectively, requiring separation by TX-114. Statistical significance indicated as * (P < 0.05), ** (P < 0.005), and *** (P < 0.0005) as determined by Student’s t Test. Error bars indicate standard deviation
Rap1A geranylgeranylation is not inhibited by GGOHBP in non-tumorous adrenal glands
Daily treatment with 1.5 mg/kg GGOHBP in addition to a high tumor burden caused a significant weight loss in mice. Figure 6a shows the weekly percent change in weight averaged within the control and treated groups following treatment initiation between weeks 3 and 4. A weight loss of 20% signified termination of the mouse in the study. At week 7 (day 25 of treatment), three out of nine treated mice and zero control mice were removed from the study due to weight loss. By week 8 (days 28-31 of treatment), an additional three treated mice and one out of nine control mice were removed from the study due to weight loss. A Kaplan-Meier plot of mice survival is shown in figure 6b. A majority of treated mice were removed from the study due to weight loss, while one treated mouse was removed on day 14 of treatment due to a broken femur at the site of bone metastasis.
Fig 6. GGOHBP reduces mouse weight.
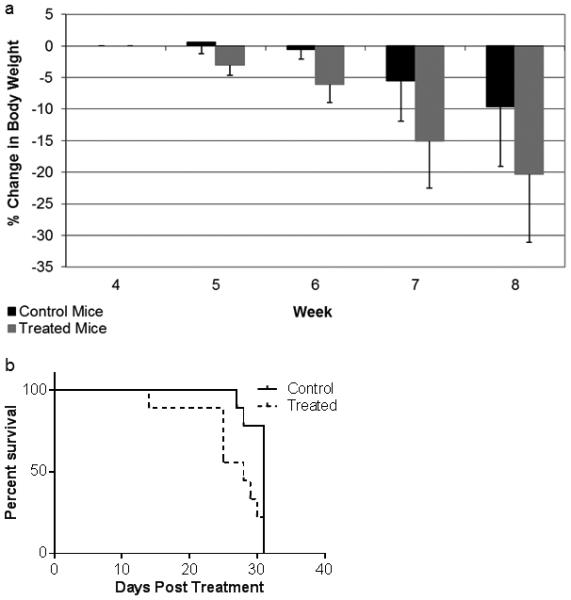
(a) Percent change in body weight from the start of treatment between weeks 3 and 4. Error bars indicate standard deviation. (b) Kaplan-Meier plot of mice survival. Day 0 indicates the beginning of daily subcutaneous treatments of the PBS-vehicle or 1.5 mg/kg GGOHBP
A study of non-tumor bearing mice given 1.25 mg/kg or 1.5 mg/kg GGOHBP subcutaneously over the same period of time did not induce significant weight loss (Figure 2d). A follow-up study on these animals including Western blot analysis of Rap1A geranylgeranylation of the non-tumor burdened mice showed no impairment within the heart (Figure 7a), kidney (Figure 7b), or adrenal gland tissues (Figure 7c).
Fig 7. Daily treatment of GGOHBP does not alter Rap1A prenylation in non-tumor burdened mice.
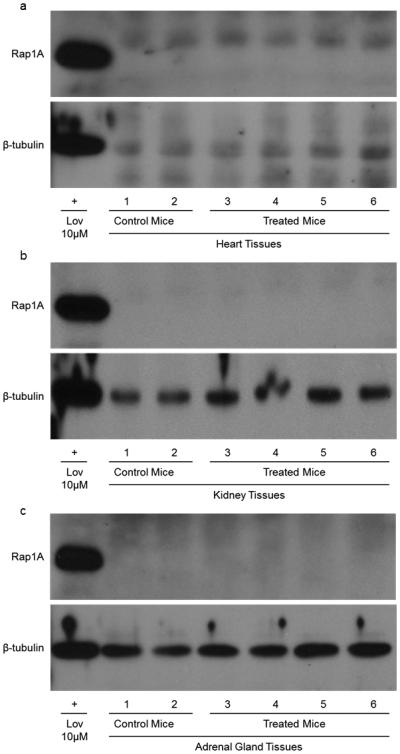
Western blot analysis of Rap1A in the (a) heart, (b) kidney, and (c) adrenal gland tissues of non-tumor bearing mice treated with the vehicle (mice 1 and 2), 1.25 mg/kg (mice 3 and 4) or 1.5 mg/kg (mice 5 and 6) GGOHBP daily for 31 days. The Rap1A antibody detects the unprenylated form of Rap1A as indicated by the positive control 10 μM lovastatin treatment in luciferase-expressing 22Rv1 cells
Discussion
IBP inhibition by statins and nitrogenous bisphosphonates has been shown by multiple epidemiological studies to potentially reduce the risk of PCa advancement. In vitro studies have implicated the reduction of GGPP and geranylgeranylation as the component responsible for reducing the metastatic characteristics of PCa.[52] The most potent of the nitrogen bisphosphonates, such as zoledronate, minodronate, and alendronate have been shown to have direct anti-tumor and anti-metastatic effects in diverse cancer models.[53-60] Clearly the nitrogenous bisphosphonates are having extra-skeletal effects, however their non-standard pharmacokinetic properties and inhibition at an early step in the pathway make them less than desirable to interogate the role of geranylgeranylation in PCa. Our work uses a novel inhibitor of geranylgeranyl pyrophosphate synthase, GGOHBP, to selectively reduce GGPP and geranylgeranylation in vitro and in vivo allowing us to elucidate the specific role of geranylgeranylation in PCa metastasis.
In vitro work using the luciferase-expressing human derived 22Rv1 PCa cell line found GGOHBP to impair Rap1A and Rab6 geranylgeranylation without altering Ras farnesylation at concentrations far below those toxic to the cells. A treatment model of PCa metastasis initiated by intracardiac injection of the 22Rv1 cells showed no significant reduction in whole body photon count in the treated mice as compared to the control mice. However, ex vivo analysis of the adrenal gland tumors, the second most common site of metastasis in our model revealed a significant 54% reduction in adrenal gland tumor weight of the treated mice as compared to the control mice. One possible explanation for the discrepancy between our compounds inability to reduce cell viability in vitro and our observed reduction in adrenal gland tumor burden in vivo is an organ site specific affinity of GGOHBP for the adrenal glands, causing it to accumulate in the adrenal gland tumors at much higher concentrations after 31 days of treatment than were tested at 48 hrs in vitro. In studies of prostate cancer metastasis in an orthotopic model, Tuomela showed reductions in invasion [61] at concentrations of the nitrogen bisphosphonate alendronate that were not toxic to the cell line used. These same authors showed a reduction in lymph node metastasis that was accompanied by reduced vascularization and increased apoptosis, while showing no change in proliferation rate.[59] Thus it is also possible that GGOHBP is reducing the tumor capacity for growth in a manner that is not readily translatable to MTT assay results.
The distribution of tumor sites in our murine model of metastasis was intriguing. In humans prostate cancer commonly metastasizes to bone (83-90%), lymph nodes (10-63%), lung (46-50%), and liver (10-25%).[62-64] Metastasis to the adrenal glands is less common, being found in 13-23% of patients.[62-64] Our model exhibited high rates of metastasis to bone (100%) and adrenal glands (72%), with lower rates to the mesentery (22%), and liver (17%). Due to the presence of mandibular tumors in all mice, the reduction of adrenal gland tumor burden in the treated mice as compared to the control mice is likely being masked by the bone tumor burden when looking at whole body photon counts. GGOHBP appears to have a lower bone affinity than the typical nitrogenous bisphosphonates[65] as evidenced by its activity in soft tissues such as the adrenal glands and mesentery with no notable effect on tumor burden in the bone. We believe the reduced bone affinity of GGOHBP will allow this type of bisphosphonate to be used as a combination therapy treatment for soft tissue tumors of metastatic PCa without being limited to the bone environment.
Western blot analysis of Rap1A geranylgeranylation in the non-tumorous and tumor burdened tissues of the mice showed an impairment in the adrenal gland tumors and in one mesenteric tumor of the treated mice but not in non-tumorous tissues or in the control mice. Interestingly, the extent of Rap1A impairment was higher in treated mice with larger adrenal gland tumor burdens, suggesting GGOHBP activity is dependent on the size of the tumor. In vitro work comparing three metastatic PCa cell lines found the highly metastatic cells to have higher IBP activity as compared to the less metastatic cell lines. This suggests the larger, more aggressive tumors in our study may possess a comparatively larger amount of geranylgeranylated Rap1A, suggesting a higher flux through the IBP, giving our treatments the opportunity to have a greater impact on geranylgeranylation in the larger tumors.
Here we use Rap1A as a convenient biomarker for impairment of post-translational modification by geranylgeranylation of proteins in tissues. There is ample evidence to support the inhibition of protein geranylgeranylation as a strategy to reduce PCa metastasis.[66] Several studies have linked RHO-family proteins such as Rac1,[67] and associated signaling proteins such as guanine nucleotide exchange factors[68, 69] to disease free survival, metastatic spread and poor clinical outcomes.[70] Studies to elucidate the exact proteins responsible for the reduction in adrenal tumor metastasis are ongoing.
Due to the role of the IBP for steroid production in the adrenal glands, we questioned if GGOHBP was targeting the adrenal gland tumor tissues due to the tumor burden or whether GGOHBP also showed effects in the non-tumorous adrenal glands. The production of all steroid hormones is dependent on FPP and reductions in GGPP would have no direct effect on these intermediates.[71, 72] Our previous work has also shown that fatty acid synthesis is regulated by FPP and that GGPP does not seem to play a role in the regulation of this pathway.[73] One possible explanation for the results in the adrenal glands is that the entire pathway is upregulated in this organ due to requirement for FPP for the above processes. In a study of non-tumor bearing mice given GGOHBP, we found no alteration in Rap1A geranylgeranylation in the adrenal glands, suggesting the presence of the tumor was required for GGOHBP to cause detectable changes in Rap1A geranylgeranylation. During this secondary study we tested a mouse cell line to ensure that our antibody was capable of detecting Rap1A of mouse origin. We found that the antibody detected both mouse and human unmodified Rap1A (data not shown).
Unfortunately, weight loss was an issue in the treated mice. This was an interesting and unexpected development because our non-tumor bearing treatment mouse study showed no significant weight loss at this dose. This suggests GGOHBP alone is not solely responsible for the toxic effects to mice but that a combination of altered geranylgeranylation by GGOHBP along with a high tumor burden was required.Further work is being done to define the nature of GGOHBP in the context of high tumor burden.
In conclusion, we found the depletion of geranylgeranylation by the novel inhibitor GGOHBP to significantly reduce adrenal gland tumor metastasis in vivo. This reduction in adrenal gland tumor burden corresponded to a biochemical reduction in geranylgeranylation that was not seen in non-tumor burdened tissues or vehicle-treated control mice. Further work defining the role of geranylgeranylated proteins in the metastatic potential of PCa as well as further refinement of the library of GGDPS inhibitors is currently underway.
Acknowledgments
We would like to thank Nadine Bannick for her support in utilizing the bioluminescence imager and James E Dunford for graciously providing the GGDPS enzyme. This project was supported in part by the Roy J. Carver Charitable Trust, the Roland W. Holden Family Program for Experimental Therapeutics.
References
- 1.Holstein SA, Hohl RJ. Isoprenoids: Remarkable diversity of form and function. Lipids. 2004;39(4):293–309. doi: 10.1007/s11745-004-1233-3. [DOI] [PubMed] [Google Scholar]
- 2.LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease - A meta-analysis of randomized controlled trials. Jama-Journal of the American Medical Association. 1999;282(24):2340–6. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- 3.Saad F, Lipton A. Clinical benefits and considerations of bisphosphonate treatment in metastatic bone disease. Seminars in Oncology. 2007;34(6):S17–S23. doi: 10.1053/j.seminoncol.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Brown JE, et al. Bone turnover markers as predictors of skeletal complications in prostate cancer, lung cancer, and other solid tumors. J Natl Cancer Inst. 2005;97(1):59–69. doi: 10.1093/jnci/dji002. [DOI] [PubMed] [Google Scholar]
- 5.Russell RGG, Rogers MJ. Bisphosphonates: From the laboratory to the clinic and back again. Bone. 1999;25(1):97–106. doi: 10.1016/s8756-3282(99)00116-7. [DOI] [PubMed] [Google Scholar]
- 6.van Beek E, et al. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun. 1999;264(1):108–11. doi: 10.1006/bbrc.1999.1499. [DOI] [PubMed] [Google Scholar]
- 7.Bergstrom JD, et al. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch Biochem Biophys. 2000;373(1):231–41. doi: 10.1006/abbi.1999.1502. [DOI] [PubMed] [Google Scholar]
- 8.Maltese WA. POSTTRANSLATIONAL MODIFICATION OF PROTEINS BY ISOPRENOIDS IN MAMMALIAN-CELLS. Faseb J. 1990;4(15):3319–28. doi: 10.1096/fasebj.4.15.2123808. [DOI] [PubMed] [Google Scholar]
- 9.Clarke S. Protein Isoprenylation and Methylation at Carboxyl-Terminal Cysteine Residues. Annu Rev Biochem. 1992;61:355–86. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]
- 10.Coxon FP, et al. Protein geranylgeranylation is required for osteoclast formation, function, and survival: Inhibition by bisphosphonates and GGTI-298. J Bone Miner Res. 2000;15(8):1467–76. doi: 10.1359/jbmr.2000.15.8.1467. [DOI] [PubMed] [Google Scholar]
- 11.Coxon FP, Rogers MJ. The role of prenylated small GTP-binding proteins in the regulation of osteoclast function. Calcified Tissue International. 2003;72(1):80–4. doi: 10.1007/s00223-002-2017-2. [DOI] [PubMed] [Google Scholar]
- 12.Luckman SP, et al. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res. 1998;13(4):581–9. doi: 10.1359/jbmr.1998.13.4.581. [DOI] [PubMed] [Google Scholar]
- 13.O'Brien LE, et al. Rac1 orientates epithelial apical polarity through effects on basolateral laminin assembly. Nat Cell Biol. 2001;3(9):831–8. doi: 10.1038/ncb0901-831. [DOI] [PubMed] [Google Scholar]
- 14.Quinlan MP. Rac regulates the stability of the adherens junction and its components, thus affecting epithelial cell differentiation and transformation. Oncogene. 1999;18(47):6434–42. doi: 10.1038/sj.onc.1203026. [DOI] [PubMed] [Google Scholar]
- 15.Braga VMM, et al. Activation of the small GTPase Rac is sufficient to disrupt cadherin-dependent cell-cell adhesion in normal human keratinocytes. Molecular Biology of the Cell. 2000;11(11):3703–21. doi: 10.1091/mbc.11.11.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braga VMM, et al. The small GTPases rho and rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol. 1997;137(6):1421–31. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zondag GCM, et al. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J Cell Biol. 2000;149(4):775–81. doi: 10.1083/jcb.149.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Worthylake RA, et al. RhoA is required for monocyte tail retraction during transendothelial migration. J Cell Biol. 2001;154(1):147–60. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumoto Y, et al. Small GTP-binding protein, Rho, both increased and decreased cellular motility, activation of matrix metalloproteinase 2 and invasion of human osteosarcoma cells. Japanese Journal of Cancer Research. 2001;92(4):429–38. doi: 10.1111/j.1349-7006.2001.tb01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adamson P, et al. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a Rho-dependent pathway. Journal of Immunology. 1999;162(5):2964–73. [PubMed] [Google Scholar]
- 21.Clark EA, et al. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 22.Platz EA. Does statin use affect the risk of developing prostate cancer? Nature Clinical Practice Urology. 2009;6(2):70–1. doi: 10.1038/ncpuro1295. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton RJ, et al. Statin Medication Use and the Risk of Biochemical Recurrence After Radical Prostatectomy Results From the Shared Equal Access Regional Cancer Hospital (SEARCH) Database. Cancer. 2010;116(14):3389–98. doi: 10.1002/cncr.25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen SF, Nordestgaard BG, Bojesen SE. Statin Use and Reduced Cancer-Related Mortality. N Engl J Med. 2012;367(19):1792–802. doi: 10.1056/NEJMoa1201735. [DOI] [PubMed] [Google Scholar]
- 25.Murtola TJ, et al. Cholesterol-lowering drugs and prostate cancer risk: A population-based case-control study. Cancer Epidemiology Biomarkers & Prevention. 2007;16(11):2226–32. doi: 10.1158/1055-9965.EPI-07-0599. [DOI] [PubMed] [Google Scholar]
- 26.Murtola TJ, et al. Statins and prostate cancer prevention: where we are now, and future directions. Nature Clinical Practice Urology. 2008;5(7):376–87. doi: 10.1038/ncpuro1146. [DOI] [PubMed] [Google Scholar]
- 27.Lustman A, et al. Statin use and incident prostate cancer risk: does the statin brand matter? A population-based cohort study. Prostate Cancer and Prostatic Diseases. 2014;17(1):6–9. doi: 10.1038/pcan.2013.34. [DOI] [PubMed] [Google Scholar]
- 28.Yu O, et al. Use of Statins and the Risk of Death in Patients With Prostate Cancer. J Clin Oncol. 2014;32(1):5–U77. doi: 10.1200/JCO.2013.49.4757. [DOI] [PubMed] [Google Scholar]
- 29.Corsini A, et al. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacology & Therapeutics. 1999;84(3):413–28. doi: 10.1016/s0163-7258(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 30.Reinoso RF, et al. Preclinical pharmacokinetics of statins. Methods and Findings in Experimental and Clinical Pharmacology. 2002;24(9):593–613. [PubMed] [Google Scholar]
- 31.Sasaki A, et al. Bisphosphonate Risedronate Reduces Metastatic Human Breast-Cancer Burden in Bone in Nude-Mice. Cancer Res. 1995;55(16):3551–7. [PubMed] [Google Scholar]
- 32.Holstein SA, Hohl RJ. Isoprenoid biosynthetic pathway inhibition disrupts monoclonal protein secretion and induces the unfolded protein response pathway in multiple myeloma cells. Leukemia Res. 2011;35(4):551–9. doi: 10.1016/j.leukres.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holstein SA, Tong HX, Hohl RJ. Differential activities of thalidomide and isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Leukemia Res. 2010;34(3):344–51. doi: 10.1016/j.leukres.2009.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wasko BM, Dudakovic A, Hohl RJ. Bisphosphonates Induce Autophagy by Depleting Geranylgeranyl Diphosphate. Journal of Pharmacology and Experimental Therapeutics. 2011;337(2):540–6. doi: 10.1124/jpet.110.175521. [DOI] [PubMed] [Google Scholar]
- 35.Wasko BM, et al. Identification and characterization of novel bisphosphonate inhibitors of squalene synthase. Faseb J. 2010;24 [Google Scholar]
- 36.Weivoda MM, Hohl RJ. The Effects of Direct Inhibition of Geranylgeranyl Pyrophosphate Synthase on Osteoblast Differentiation. J Cell Biochem. 2011;112(6):1506–13. doi: 10.1002/jcb.23087. [DOI] [PubMed] [Google Scholar]
- 37.Wiemer AJ, Hohl RJ, Wiemer DF. The Intermediate Enzymes of Isoprenoid Metabolism as Anticancer Targets. Anti-Cancer Agents in Medicinal Chemistry. 2009;9(5):526–42. doi: 10.2174/187152009788451860. [DOI] [PubMed] [Google Scholar]
- 38.Zhou X, et al. Synthesis of isoprenoid bisphosphonate ethers through C-P bond formations: Potential inhibitors of geranylgeranyl diphosphate synthase. Beilstein Journal of Organic Chemistry. 2014;10:1645–50. doi: 10.3762/bjoc.10.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hohl RJ, et al. Inhibition of hydroxymethylglutaryl coenzyme A reductase activity induces a paradoxical increase in DNA synthesis in myeloid leukemia cells. Blood. 1991;77(5):1064–70. [PubMed] [Google Scholar]
- 40.Hohl RJ, Lewis K. Differential-Effects of Monoterpenes and Lovastatin on Ras Processing. J Biol Chem. 1995;270(29):17508–12. doi: 10.1074/jbc.270.29.17508. [DOI] [PubMed] [Google Scholar]
- 41.Hohl RJ, et al. Stereochemistry-dependent inhibition of RAS farnesylation by farnesyl phosphonic acids. Lipids. 1998;33(1):39–46. doi: 10.1007/s11745-998-0178-x. [DOI] [PubMed] [Google Scholar]
- 42.Hohl RJ, LewisTibesar K. Targeting the isoprenoid pathway for antileukemia therapy in humans. Blood. 1995;86(10):3039. [Google Scholar]
- 43.Hohl RJ, et al. Differential effects of isoprenoid phosphonic acids on RAS farnesylation and cholesterol synthesis. J Invest Med. 1996;44(7):A342–A. [Google Scholar]
- 44.Hohl RJ, et al. Inhibition of RAS farnesylation by isoprenoid phosphonic acids. Clin Pharmacol Ther. 1996;59(2):PII39–PII. [Google Scholar]
- 45.Weivoda MM, Hohl RJ. Geranylgeranyl pyrophosphate stimulates PPAR gamma expression and adipogenesis through the inhibition of osteoblast differentiation. Bone. 2012;50(2):467–76. doi: 10.1016/j.bone.2011.09.056. [DOI] [PubMed] [Google Scholar]
- 46.Maalouf MA. Chemistry. University of Iowa; Iowa City, IA: 2006. Synthesis of isoprenoids incorporating a fluorescent label and evaluation of their effect on the mevalonate pathway. [Google Scholar]
- 47.Drake JM, Gabriel CL, Henry MD. Assessing tumor growth and distribution in a model of prostate cancer metastasis using bioluminescence imaging. Clin Exp Metastasis. 2005;22(8):674–84. doi: 10.1007/s10585-006-9011-4. [DOI] [PubMed] [Google Scholar]
- 48.Dunford JE, et al. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro nd inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. Journal of Pharmacology and Experimental Therapeutics. 2001;296(2):235–42. [PubMed] [Google Scholar]
- 49.Tong H, Holstein SA, Hohl RJ. Simultaneous determination of farnesyl and geranylgeranyl pyrophosphate levels in cultured cells. Anal Biochem. 2005;336(1):51–9. doi: 10.1016/j.ab.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 50.Smith PK, et al. Measurement of Protein Using Bicinchoninic Acid. Anal Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 51.Drake JM, Danke JR, Henry MD. Bone-specific growth inhibition of prostate cancer metastasis by atrasentan. Cancer Biology & Therapy. 2010;9(8):607–14. doi: 10.4161/cbt.9.8.11112. [DOI] [PubMed] [Google Scholar]
- 52.Ghosh PM, et al. Role of RhoA activation in the growth and morphology of a murine prostate tumor cell line. Oncogene. 1999;18(28):4120–30. doi: 10.1038/sj.onc.1202792. [DOI] [PubMed] [Google Scholar]
- 53.Wakchoure S, et al. Bisphosphonates inhibit the growth of mesothelioma cells in vitro and in vivo. Clin Cancer Res. 2006;12(9):2862–8. doi: 10.1158/1078-0432.CCR-05-2766. [DOI] [PubMed] [Google Scholar]
- 54.Clezardin P. Bisphosphonates' antitumor activity: An unravelled side of a multifaceted drug class. Bone. 2011;48(1):71–9. doi: 10.1016/j.bone.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 55.Fournier P, et al. Bisphosphonates inhibit angiogenesis in vitro and testosterone-stimulated vascular regrowth in the ventral prostate in castrated rats. Cancer Res. 2002;62(22):6538–44. [PubMed] [Google Scholar]
- 56.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114(5):623–33. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hiraga T, et al. Zoledronic acid inhibits visceral metastases in the 4T1/luc mouse breast cancer model. Clin Cancer Res. 2004;10(13):4559–67. doi: 10.1158/1078-0432.CCR-03-0325. [DOI] [PubMed] [Google Scholar]
- 58.Ory B, et al. Zoledronic acid suppresses lung metastases and prolongs overall survival of osteosarcoma-bearing mice. Cancer. 2005;104(11):2522–9. doi: 10.1002/cncr.21530. [DOI] [PubMed] [Google Scholar]
- 59.Tuomela JM, et al. Alendronate decreases orthotopic PC-3 prostate tumor growth and metastasis to prostate-draining lymph nodes in nude mice. Bmc Cancer. 2008;8 doi: 10.1186/1471-2407-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamagishi S, et al. Minodronate, a newly developed nitrogen-containing bisphosphonate, suppresses melanoma growth and improves survival in nude mice by blocking vascular endothelial growth factor signaling. American Journal of Pathology. 2004;165(6):1865–74. doi: 10.1016/s0002-9440(10)63239-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Virtanen SS, et al. Alendronate inhibits invasion of PC-3 prostate cancer cells by affecting the mevalonate pathway. Cancer Res. 2002;62(9):2708–14. [PubMed] [Google Scholar]
- 62.Bubendorf L, et al. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Human Pathology. 2000;31(5):578–83. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 63.Gandaglia G, et al. Distribution of Metastatic Sites in Patients With Prostate Cancer: A Population-Based Analysis. Prostate. 2014;74(2):210–6. doi: 10.1002/pros.22742. [DOI] [PubMed] [Google Scholar]
- 64.Shah RB, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004;64(24):9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 65.Nancollas GH, et al. Novel insights into actions of bisphosphonates on bone: differences in interactions with hydroxyapatite. Bone. 2006;38(5):617–27. doi: 10.1016/j.bone.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 66.Lyons LS, et al. Ligand-Independent Activation of Androgen Receptors by Rho GTPase Signaling in Prostate Cancer. Mol Endocrinol. 2008;22(3):597–608. doi: 10.1210/me.2007-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knight-Krajewski S, et al. Deregulation of the Rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21(CIP1) levels in androgen-independent human prostate cancer cells. Oncogene. 2004;23(32):5513–22. doi: 10.1038/sj.onc.1207708. [DOI] [PubMed] [Google Scholar]
- 68.Lin K-T, et al. Vav3-Rac1 Signaling Regulates Prostate Cancer Metastasis with Elevated Vav3 Expression Correlating with Prostate Cancer Progression and Posttreatment Recurrence. Cancer Res. 2012;72(12):3000–9. doi: 10.1158/0008-5472.CAN-11-2502. [DOI] [PubMed] [Google Scholar]
- 69.Qin J, et al. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene. 2009;28(16):1853–63. doi: 10.1038/onc.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Engers R, et al. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr Relat Cancer. 2007;14(2):245–56. doi: 10.1677/ERC-06-0036. [DOI] [PubMed] [Google Scholar]
- 71.Russell DW. CHOLESTEROL-BIOSYNTHESIS AND METABOLISM. Cardiovascular Drugs and Therapy. 1992;6(2):103–10. doi: 10.1007/BF00054556. [DOI] [PubMed] [Google Scholar]
- 72.Schroepfer GJ. STEROL BIOSYNTHESIS. Annu Rev Biochem. 1982;51:555–85. doi: 10.1146/annurev.bi.51.070182.003011. [DOI] [PubMed] [Google Scholar]
- 73.Murthy S, Tong H, Hohl RJ. Regulation of fatty acid synthesis by farnesyl pyrophosphate. J Biol Chem. 2005;280(51):41793–804. doi: 10.1074/jbc.M504101200. [DOI] [PubMed] [Google Scholar]