

Abstract
Huntington’s disease (HD) is a neurodegenerative disorder characterized by massive loss of medium spiny neurons in the striatum. However, the mechanisms by which mutant huntingtin leads to this selective neuronal death remain incompletely understood. Brain-derived neurotrophic factor (BDNF) has been shown to be neuroprotective on HD striatal neurons both in vitro and in vivo. ProBDNF, the precursor of mature BDNF (mBDNF), also can be secreted but promotes apoptosis of neurons expressing p75NTR and sortilin receptors. Although a reduction of total striatal BDNF protein has been reported in HD patients and mouse models, it remains unclear whether conversion of proBDNF to mBDNF is altered in HD, and whether the proBDNF receptors, p75NTR and sortilin are dysregulated, leading to impaired striatal neuron survival. To test these hypotheses, we generated bdnf-HA knock-in (KI) mice on the zQ175 HD background to accurately quantitate the levels of both proBDNF and mBDNF in the HD striatum. In aged zQ175 HD mice, we observed a significant loss of mBDNF and decreased TrkB activation, but no increase of proBDNF or p75NTR levels either in the sensorimotor cortex or the striatum. However, immunoreactivities of p75NTR and sortilin receptor are both increased in immature striatal oligodendrocytes, which associate with significant myelin defects in the HD striatum. Taken together, the present study indicates that diminished mature BDNF trophic signaling through the TrkB receptor, rather than an induction in proBDNF, is a main contributing factor to the vulnerability of striatal neurons in the zQ175 HD mouse model.
Keywords: Huntington’s disease, zQ175 mice, cortex, striatum, proBDNF, mature BDNF, p75NTR, sortilin, oligodendrocyte, myelin
Introduction
Huntington’s disease (HD) is an autosomal dominant disorder associated with progressive neuronal degeneration, especially involving the caudate nucleus and putamen of the basal ganglia and the cortex (Shoulson and Chase, 1975). The behavioral symptoms of HD include a triad of motor dysfunction, psychiatric disturbances and cognitive deficits, as well as peripheral phenotypes that include weight loss and muscle wasting (Walker, 2007). The disease usually manifests in midlife, and is fatal after 10–15 years from the onset (Cattaneo et al., 2005; Crook and Housman, 2011). An expansion of CAG trinucleotide repeats (>35 repeats) in exon I of the interesting transcript 15 (IT15) gene has been identified as the genetic mutation responsible for the disease, which results in an elongated stretch of glutamine in the N-terminal of the huntingtin protein (HDCRG, 1993). However, the underlying mechanisms by which mutant huntingtin leads to selective and progressive neuronal degeneration remain incompletely understood.
Brain-derived neurotrophic factor (BDNF) has been shown to have protective effects on striatal neurons both in vitro (Saudou et al., 1998) and in vivo (Bemelmans et al., 1999; Kells et al., 2004; Kells et al., 2008; Giampa et al., 2013). A decrease in BDNF levels in the cortex and the striatum has been reported in HD patients (Ferrer et al., 2000; Zuccato et al., 2001; Zuccato et al., 2008) and some HD animal models (Supplemental Table. 1). Selective deletion of BDNF in the mouse forebrain leads to loss of striatal neurons and HD-like behavioral phenotypes (Baquet et al., 2004), and these mice exhibit changes in gene expression that are similar to the ones observed in the human HD caudate (Strand et al., 2007). Importantly, BDNF haploinsufficiency results in earlier onset and more severe motor dysfunctions in HD transgenic mice (Canals et al., 2004), while BDNF overexpression ameliorates the symptoms of HD in mouse models (Gharami et al., 2008; Xie et al., 2010). Thus, neurotrophic support from BDNF is a key modulator of disease progression in HD.
The vast majority (~95%) of striatal BDNF is anterogradely transported from the cortex (Altar et al., 1997; Baquet et al., 2004). Current hypotheses underlying reduced levels of striatal BDNF observed in HD patients and animal models include loss of cortical bdnf gene transcription (Zuccato et al., 2001; Zuccato et al., 2003; Conforti et al., 2013) and impaired axonal transport of BDNF protein in cortical neurons (Gauthier et al., 2004). Since BDNF is synthesized as proBDNF (the larger precursor of mature BDNF), and both isoforms can be secreted from neurons (Yang et al., 2009), it remains to be determined whether the conversion of proBDNF to mature BDNF in the HD cortex and the striatum is abnormal, which could not only contribute to HD pathogenesis but also reduce the therapeutic efficacy of BDNF overexpression.
Furthermore, in contrast to mBDNF, proBDNF has been shown to induce cell death (Teng et al., 2005), synaptic long-term depression (Woo et al., 2005) and growth cone collapse (Anastasia et al., 2013) utilizing p75NTR and sortilin receptor family members. Increased p75NTR but reduced TrkB expression has recently been reported in HD patients and some HD transgenic mouse models (Brito et al., 2013), suggesting an imbalance of p75NTR/TrkB signaling in HD (Plotkin et al., 2014).
To test the hypothesis that enhanced proBDNF-p75NTR signaling counteracts the survival signaling mediated by mBDNF-TrkB in the HD striatum, we analyzed the levels of different BDNF isoforms targeted to the striatum and the expression of TrkB and p75NTR receptors in zQ175 KI mouse model of HD. This mouse model carries ~188 CAG repeats contained within a chimeric human/mouse exon 1 of murine huntingtin gene (Menalled et al., 2012). Prior studies document that zQ175 homozygous mice show hypoactivity in the open field (~4 months of age), impaired rotarod activity (~8 months of age), cognitive deficits (~1 year of age), and significantly reduced survival (median ~90 weeks of age). Heterozygous mice exhibit milder behavioral deficits from around 4.5 months of age. (Heikkinen et al., 2012; Menalled et al., 2012). In terms of pathology, homozygous mice have mutant huntingtin inclusions/aggregates (~2–4 months of age), early and significant decrease of striatal gene markers (~3 months of age) and decreased neuronal cell counts. Decreased expression of striatal gene markers are detected in heterozygous mice from ~4.5 months of age (Heikkinen et al., 2012; Menalled et al., 2012; Smith et al., 2014). In addition to these behavioral and pathological phenotypes, zQ175 KI mice also have electrophysiological, histological, and metabolic alterations (Heikkinen et al., 2012; Menalled et al., 2012; Plotkin et al., 2014; Smith et al., 2014; Tong et al., 2014), which suggest it as a robust mouse model to study molecular mechanisms and therapeutic interventions of HD.
To facilitate detection of endogenously expressed BDNF, we utilized previously generated bdnf-HA KI mice, which permits accurate detection and quantification of both proBDNF and mBDNF in the mouse brain by detecting the C-terminal HA epitope tag (Yang et al., 2009). We observe that mBDNF is the predominant BDNF isoform detected in the striatum at all postnatal ages. To assess the impact of mutant huntingtin, we crossed zQ175 KI mice with bdnf-HA mice, and determined levels of different BDNF isoforms in the hippocampus, the striatum and the sensorimotor cortex of +/+ | bdnf-HA, zQ175/+ | bdnf-HA, zQ175/zQ175 | bdnf-HA mice at 2, 6 and 12 months of age. mBDNF is markedly reduced in zQ175 HD mouse striatum at 6 months and 12 months of age, while mBDNF in the sensorimotor cortex of zQ175 mice only decreases significantly at 12 months of age. TrkB phosphorylation is also diminished in the striatum of 12-month-old zQ175 mice. Importantly, no proBDNF upregulation is observed at any time points tested. However, p75NTR- and sortilin-immunoreactivities are both increased in a subset of immature striatal oligodendrocytes in 12-month-old zQ175 mice. Severely impaired myelination is concomitantly observed in the striatum of these mice at 12 months of age.
Taken together, our findings demonstrate a significant decrease of mBDNF-TrkB signaling, but no induction of proBDNF-p75NTR signaling in striatal neurons of zQ175 mice, suggesting that maturation of proBDNF to mBDNF remains intact in this HD mouse model. Nevertheless, local induction of p75NTR and sortilin is found in immature striatal oligodendrocytes and is associated with severe myelin deficits in the striatum of aged zQ175 HD mice.
Material and methods
Mouse strains
bdnf-HA knock-in mice were previously generated (Yang et al., 2009), backcrossed to C57Bl6 for >10 generations, and maintained by intercrossing as bdnf-HA/bdnf-HA homozygous. The zQ175 knock-in mouse line was supplied from The Jackson Laboratory (Stock #370437). zQ175/+ mice were bred with bdnf-HA/bdnf-HA homozygous mice to generate zQ175/+ | bdnf-HA/ bdnf-HA mice and wild type littermate (+/+ | bdnf-HA/ bdnf-HA). zQ175/+ | bdnf-HA/ bdnf-HA mice were then interbred to generate zQ175/zQ175 | bdnf-HA/ bdnf-HA mice. Only male mice were used for all the analyses. To monitor CAG repeat sizes, tail samples from all animals were collected and sent to Laragen, Inc. for CAG sizing. All zQ175 allele carriers have repeat sizes ranging from 171~200.
Immunofluorescence staining and analysis
Mice were injected with sodium pentobarbital (150 mg/kg, I.P.) and transcardially perfused with cold phosphate-buffered saline (PBS, pH7.4), followed by 4% paraformaldehyde (PFA) in PBS. Brains were post-fixed in 4% PFA overnight at 4°C and then cryoprotected with 30% sucrose in PBS. Free-floating coronal brain sections (30μm) were prepared on a freezing microtome. For immunofluorescence detection, brain sections were incubated in blocking buffer (5% normal donkey serum + 0.3% Triton-X 100) and avidin/biotin blocking kit (Vector Laboratories) at room temperature, and then incubated with primary antibodies in blocking buffer for 18 hours at 4°C. The following primary antibodies were used: goat anti-p75NTR (1:100, R&D), rabbit anti-pTrkB Y816 (1:1000, kind gift from Dr. Moses V. Chao), rabbit anti-sortilin (1:1000, Abcam), rabbit anti-Olig2 (1:1000, Millipore), rabbit anti-NG2 (1:200, Millipore), mouse anti-2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) (1:200, Millipore), mouse anti-APC (CC1, 1:100, Millipore), rabbit anti-GFAP (1:1000, DAKO), rabbit anti-Iba1 (1:500, DAKO), rabbit anti-cleaved caspase-3 (1:200, Cell signaling). The secondary antibodies used were biotinylated donkey anti-goat IgG (1:400, Jackson ImmunoResearch), donkey anti-rabbit Alexa 488, donkey anti-mouse Alexa 488 or 647 (1:1000, Jackson ImmunoResearch). Cy3- or Cy5-conjugated streptavidin (1:1000, Jackson ImmunoResearch) was used to visualize biotinylated secondary antibodies. Three animals per genotype were analyzed for each experiment. For quantification, immunofluorescence images were examined on Zeiss Axio Observer.Z1 inverted microscope and captured using the same exposure time / channel / experiment by AxioCam MRC camera. Quantitative analyses (mean pixel intensity measurement) were performed using Image J v1.33 software by Wayne Rasband (National Institutes of Health, Bethesda, MD, USA).
Western blot analysis
Dissected tissues were minced and lysed in lysis buffer (1% NP-40, 1% Triton X-100, 1mM PMSF, 10% glycerol and protease inhibitor cocktail (Sigma) in Tris-buffered saline) for 30 min on ice. Lysates were further titrated using a 30g needle, and supernatants were collected following centrifugation at 14,000 rpm for 5 min. Cleared lysates were then resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following transfer, Western blots were blocked and incubated with primary antibodies, followed by labeling with HRP-conjugated secondary antibodies and developed with the ECL kit (Amersham). The following primary antibodies were used: mouse anti-HA.11 (1:3000, Covance), rabbit anti-TrkB (1:3000, Millipore), rabbit anti-pTrkB Y490 (1:1000, generous gift from Dr. Moses V. Chao), rabbit anti-pERK (1:1000, Cell signaling), rabbit anti-ERK1/2 (1:3000, Cell signaling), rabbit anti-p75 (1:2000, 9992 antisera, kindly provided by Dr. Moses V. Chao), mouse anti-sortilin (1:1000, BD biosciences). For HA immunoblotting, membrane was fixed with 2.5% gluteraldehyde in PBS for 30 min at room temperature after transfer and washed extensively in PBS before blocking and antibody incubation. N=5 animals per genotype were used for each experiment. In Western blot densitometry analysis, values were normalized by loading control protein levels and then averaged to compare with wild type mice.
Enzyme-linked Immunosorbent Assay (ELISA)
To measure levels of total BDNF in brain lysates, mice were euthanized and the cortex, hippocampus and striatum were dissected and rapidly frozen on dry ice. Tissue lysates were prepared as for Western blot analysis. Lysates containing 300μg of hippocampal, 600μg of cortical, or 900μg of striatal proteins were diluted to 100μl using blocking buffer and were analyzed in duplicate. Analysis was performed according to the manufacturer’s instructions (Promega BDNF ELISA kit). Values were calculated as picogram (pg) of BDNF per milligram (mg) of tissue protein and standardized to the average of wild type animals. N=5 animals were used for each genotype at each time point.
Electron microscopy and analysis
Sections were processed for electron microscopy as previously described (Milner et al., 2011). Briefly, 12-month-old wild type or zQ175 homozygous mice were overdosed with sodium pentobarbital (150 mg/kg, i.p.) and perfused transcardially with 2% heparin-saline followed by 3.75% acrolein in 2% paraformaldehyde. The brains were removed from skull, post-fixed in 2% acrolein and 2% paraformaldehyde for 30 minutes, and then placed in 0.1M phosphate buffer (PB; pH7.4). Coronal sections through the brains were cut (40μm thick) using Vibratome, and stored in cryoprotectant at −20°C until use.
For p75NTR immunolabeling, striatal sections were rinsed in PB to remove cryoprotectant and then incubated in 1% sodium borohydride in PB for 30min to remove active aldehydes. Sections were then washed in 8–10 changes of PB until all the gaseous bubbles disappeared and then placed in 0.1 M Tris-buffered saline (TS), pH 7.6. Sections were then incubated sequentially in: (1) 0.5% bovine serum albumin (BSA) in TS; (2) p75NTR antisera (1:800, R&D) in 0.1% BSA + 0.025% Triton X-100 in TS for 1 d at room temperature (~23°C), followed by 3 d cold (~4°C); (3) 1:400 of biotinylated anti-goat IgG (Jackson ImmunoResearch), 30 min; (4) 1:100 peroxidase-avidin complex (Vectastain Elite Kit), 30 min; and (5) 3,3′-diaminobenzidine (DAB; Sigma-Aldrich) and H2O2 in TS, 6 min. All incubations were separated by washes in TS. Sections were rinsed in 0.1M PB and then postfixed in 2% osmium tetroxide in PB for 1 hour, dehydrated and embedded with Epon 812 between two sheets of Aclar plastic. Sections from dorsal striatum were selected, mounted on EMBed chucks and trimmed to 1–1.5mm wide trapezoids. Ultrathin sections (~65 nm thick) within 0.1–0.2 μm to the tissue-plastic interface were cut on a Leica Ultracut ultratome, collected into copper mesh grids, and counterstained with uranyl acetate and Reynold’s lead citrate. Sections were viewed and photographed using a FEI Tecnai Biotwin electron microscope equipped with a digital camera (Advanced Microscopy Techniques, software version 3.2). 10 random but not overlapping EM pictures of p75NTR immunolabeling in the striatum were taken per animal, and three animals of each genotype were analyzed.
G-ratios (calculated as diameter of the axon/outer diameter of the myelinated fiber) of at least 300 myelinated axons per genotype and area were measured. Data are displayed as a scatter-plot against axon diameter. To calculate percentage of unmyelinated fibers, 10 non-overlapping EM pictures from each striatal section containing myelinated axonal bundles were randomly chosen and fibers without myelin sheath (myelin stained black by the osmium stain) were counted in each section. Data are then expressed as number of unmyelinated fibers / total number of fibers analyzed. At least 100 axons within the striatum were quantified per animal. N=3 animals of each genotype were analyzed. Observer was blind to the genotypes during quantitation analysis of all EM images.
RNA isolation and reverse transcription
Total RNA from brain tissues was isolated utilizing TRIZOL reagent (Life Technologies). RNA was treated with DNase I (Life Technologies) to remove DNA contamination. RNA was then reverse transcribed into single-stranded cDNA using SuperScript III First-Strand Synthesis System (Life Technologies) as described by the manufacturer.
Real-time quantitative PCR analysis
Real-time PCR analyses were performed as described previously (Conforti et al., 2013) with minor modifications. Mastercycler Realplex2 real-time PCR machine (Eppendorf) was used, and all reactions were performed in a total volume of 25μl containing 50ng of cDNA, QuantiTect SYBR Green PCR kit (Qiagen) and 0.4μM of forward and reverse primers. Amplification cycles were carried out as follows: an initial denaturing cycle at 95°C for 15min, followed by 50 cycles of 95°C for 30s, 60°C for 30s and 72°C for 30s, for all of the genes analyzed. Ct was quantified during the 60°C annealing step. Amounts of bdnf mRNA were normalized to a reference gene (β-actin). Each sample was repeated in triplicate for statistical analyses. Primer sequences used are as follows: bdnf coding exon forward, 5′-TCGTTCCTTTCGAGTTAGCC-3′; bdnf coding exon reverse, 5′-TTGGTAAACGGCACAAAAC-3′; β-actin forward, 5′-AGTGTGACGTTGACATCCGTA-3′; β-actin reverse, 5′-GCCAGAGCAGTAATCTCCTTCT-3′.
Statistical Analysis
All of the data were analyzed with the program GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA, USA). Values are presented as the mean ± standard error of mean (S.E.M.). Statistical differences between the groups were determined using the unpaired Student’s t test unless indicated otherwise. Statistical differences were considered to be significant for P<0.05 (* P<0.05, ** P<0.01, *** P<0.001, n.s. non-significant).
Results
Mature BDNF is the predominant isoform of BDNF in the wild type striatum and declines with aging
Quantitation of levels of endogenous BDNF proteins in the murine cortex and the striatum is technically challenging, given the extremely low concentration of BDNF isoforms in these regions as well as limitations in antibody specificity and sensitivity. Although reduced trophic support of BDNF has been proposed as an underlying mechanism in HD pathogenesis, prior studies in HD mouse models showed conflicting results regarding the levels of BDNF in the cortex and the striatum (Gines et al., 2003; Seo et al., 2008; Bobrowska et al., 2011; Sontag et al., 2012; Plotkin et al., 2014). Moreover, the ratio of proBDNF to mBDNF in the striatum of wild type and HD mice remains unknown.
To facilitate detection of endogenously expressed BDNF, we utilized bdnf-HA KI mice in which the endogenous bdnf coding exon was replaced with the murine bdnf sequence with a C-terminal HA epitope tag, which permits accurate detection and quantification of both proBDNF and mBDNF in the mouse brain (Yang et al., 2009). The HA tag is not detected in wild type C57Bl/6 mice by Western blot analysis and does not affect the total level of BDNF as quantitated by ELISA (Yang et al., 2009). This bdnf-HA KI mouse line has been extensively evaluated and is indistinguishable from the wild type C57Bl/6 mice, as assessed by neuronal morphology, dendritic spine density, and electrophysiology (Yang et al., 2014).
In mice with two bdnf-HA alleles (bdnf-HA/bdnf-HA), previous studies have shown that levels of both proBDNF and mBDNF in the hippocampus peak around postnatal day 15–21, and then decrease in adulthood (Yang et al., 2014). To evaluate the levels of both BDNF isoforms in the cortex and striatum of wild type mice, we collected sensorimotor cortices and striatum from mice at different ages (Postnatal day 15, 6 months, 12 months), and determined BDNF protein levels using antibodies to detect the HA tag by Western blot analysis. At all ages, mBDNF is the predominant isoform detected in all the three brain regions (the hippocampus, the cortex and the striatum) (Fig. 1A). Notably, proBDNF is detected in the hippocampus and the cortex, but not in the striatum (Fig. 1A, B). mBDNF in the striatum is 49 ± 0.9% lower than mBDNF in the cortex at all ages (Fig. 1C). The ratio of total BDNF levels among these three brain regions is consistent with the quantification of total BDNF levels by ELISA when animals are 6-month-old (Supplemental Fig. 1A). Importantly, unlike hippocampus, both cortical and striatal mBDNF levels drop ~50% from 6 months of age to 12 months of age in wild type animals (Fig. 1C), suggesting that trophic support of BDNF declines with aging in these two brain regions that degenerate early in HD.
Figure 1. mBDNF is the predominant form and its level declines with age.

(A) Representative Western blot showing levels of BDNF isoforms in the hippocampus (HP), the cortex (CTX), and the striatum (STR) of wild type bdnf-HA/bdnf-HA mice at the age of postnatal day 15 (P15), 6 months and 12 months. Note that levels of both BDNF isoforms decrease with age, and intact proBDNF is barely detectable in the striatum at all the time points. Quantitative results of mBDNF level (B) and proBDNF (C) in HP, CTX and STR at the age of P15, 6months and 12 months show that the levels of both BDNF isoforms in CTX and STR are substantially lower than the HP, and both proBDNF and mBDNF levels decline significantly with age in all the three brain regions, except that proBDNF cannot be detected in STR at all the time points. AU, arbitrary units.
Age-dependent reduction of mature BDNF but no increase of proBDNF in the striatum and the cortex of zQ175 mice
To determine whether the total level of BDNF and the ratio between proBDNF and mBDNF are altered in HD, we assessed levels of both proBDNF and mature BDNF in 2-month-old (pre-symptomatic stage), 6-month-old (disease onset), and 12-month-old (symptomatic stage) zQ175 mice with two bdnf-HA alleles and their wild type littermates by Western blot analysis using antibodies to detect the HA tag. At the time point of 2 months, which is before disease onset of zQ175 HD mice (Menalled et al., 2012), no significant changes of either proBDNF or mBDNF are detected in the hippocampus, the cortex or the striatum of zQ175 mice (Fig. 2A, B, C).
Figure 2. Age-dependent loss of mBDNF but no induction of proBDNF in the striatum of zQ175 mice.

(A, B) No significant alterations in either isoform of BDNF are observed in HP, CTX, and STR when animals are 2 months old. (C, D) Decrease of mBDNF without upregulation of proBDNF in the striatum of zQ175 heterozygous and homozygous mice compared with wild type (WT) mice at 6 months of age. proBDNF level decreases while no change of mBDNF level is detected in both HP and CTX. (E, F) Total BDNF levels of both CTX and STR reduce significantly in zQ175 heterozygous and homozygous mice when animals are 12 months old. proBDNF is not induced in either CTX or STR, and there is a decrease of proBDNF level in HP lysates of zQ175 mice compared to WT.
However, we observed a significant reduction of mBDNF in the striatum of zQ175 heterozygous mice (47.7 ± 5.4% reduction, P=0.0076) and zQ175 homozygous mice (47.8 ± 8.6% reduction, P=0.0293) compared to wild type mice at 6 months of age (Fig. 2D, E). The timing of BDNF level reduction in the striatum correlates with the onset of motor behavior deficits in this line of HD mouse model (Smith et al., 2014). Notably, no significant decrease of mBDNF is detected in the hippocampus and the cortex at 6 months of age (about 8.84% and 3.34% reduction, respectively, P>0.05) (Fig. 2D, E). Thus, the selective decrease of BDNF levels in the HD striatum early in disease progression is consistent with the hypothesis that BDNF anterograde transport from the cortex is impaired by mutant huntingtin (Gauthier et al., 2004). Moreover, proBDNF is barely detectable in the WT and HD striatum, and proBDNF levels decrease by approximately 50% in the hippocampus and the cortex of zQ175 heterozygous and homozygous mice as compared to wild type mice (Fig. 2D, F).
In the relatively late stage of the disease, when animals are 12-month-old, we observed a trend towards a decrease of mBDNF in the hippocampus of zQ175 expressing mice as compared to wild type (15.5 ± 5.4% and 24.1 ± 7.2% in zQ175 heterozgyous and homozygous mice respectively), but this did not reach statistical significance. In contrast, we observed a 41.0 ± 6.3% (P=0.0351) and 53.3 ± 5.4% (P=0.0132) reduction in mBDNF in the cortex of mice expressing one or two zQ175 alleles, respectively. In the striatum, mBDNF levels are significantly reduced by approximately 60%, with similar levels observed in zQ175 heterozygous (P=0.0007) and homozygous (P=0.0271) animals (Fig. 2G, H). Given that mBDNF is the predominant isoform of all the three brain regions tested, the alterations of mBDNF levels in the hippocampus, the cortex and the striatum of zQ175 mice at the age of 12 months are in line with the measurement of total BDNF by ELISA (Supplementary Fig. 1B). We also observed a 38.2 ± 5.4% (P=0.0252) and 40.2 ± 9.8% (P=0.0375) reduction of total bdnf mRNA in the cortex of 12-month-old heterozygous and homozygous zQ175 mice respectively (Supplemental Fig. 2), which correlates well with the reduction of mBDNF protein in the cortex of zQ175 mice at 12 months (Fig. 2G, H), and further supports the literature that deficiency in mRNA transcription contributes to the reduced level of BDNF protein (Zuccato et al., 2001). It is noteworthy that proBDNF remains undetectable in the cortex or the striatum of both WT and zQ175 mice at 12 months of age, and hippocampal proBDNF levels further decrease in the zQ175 mice to approximately 30% compared to wild type littermates (Fig. 2G, I).
These results demonstrate that single mutant huntingtin allele is sufficient to induce a reduction of BDNF levels both in the cortex and the striatum of zQ175 mice in an age- and region-dependent manner. However, conversion of proBDNF to mature BDNF is not impaired in this HD animal model.
Diminished TrkB activation in the striatum of zQ175 mice
Striatal neurons express both a full-length, kinase active TrkB receptor (TrkB), which is activated by mBDNF, as well as a truncated, catalytically inactive TrkB receptor (TrkB-T) (Eide et al., 1996). To evaluate whether the reduction in the level of BDNF detected in the striatum results in decreased TrkB activity, we examined levels of both isoforms of TrkB receptor and its phosphorylation in the cortex and the striatum of zQ175 mice at the age of 12 months. We did not observe significant changes of either full-length or truncated TrkB in the cortex (Fig. 3A, C) or in the striatum (Fig. 3B, D) of 1-year-old zQ175 mice. We observed a nonsignificant decrease in phosphorylated TrkB receptor (pTrkB) in the cortex (P=0.1703 and P=0.1321 respectively) (Fig. 3A, E). However, level of pTrkB is markedly reduced to ~50% in the striatal lysates from both zQ175 heterozygous and homozygous animals compared to wild type (P=0.0168 and P=0.0023 respectively) (Fig. 3B, E). This indicates that BDNF neuroprotective signaling via TrkB receptor is impaired in the striatum but not in the cortex, and it precedes depletion of full-length TrkB receptor or increase of truncated TrkB receptor.
Figure 3. Diminished TrkB activation in the striatum of zQ175 mice.

(A) Representative Western blot showing that total TrkB levels (both full-length and truncated TrkB) and phosphorylated TrkB (p-TrkB) levels are modestly reduced in the cortex of zQ175 mice at 12 months of age. (B) Total TrkB levels (both full-length and truncated TrkB) are not changed in the striatum of zQ175 mice, while p-TrkB level is significantly reduced in both 12-month-old zQ175 heterozygous and homozygous animals. (C, D) Densiometric quantification results of full-length TrkB (TrkB) and truncated TrkB (TrkB-T) protein levels in cortical (C) and striatal (D) lysates from wild type and zQ175 mice demonstrate no significant change of total TrkB levels in either the cortex or the striatum. (E) Quantification results of p-TrkB level in the cortex and the striatum of zQ175 mice. TrkB activation is only significantly diminished in the striatum but not the cortex. Values were normalized by total Erk1/2 levels and then averaged to compare with wild type.
To further examine whether pTrkB is decreased in striatal medium spiny neurons, we performed immunofluorescence staining in striatal sections. In the striatum, pTrkB-immunoreactivity (pTrkB-ir) is diffuse. Relatively higher intensities of pTrkB-ir are seen in the cell bodies of medium spiny neurons, as marked by Darpp-32 expression (Supplemental Fig. 3A, C). We found that there is ~30% reduction in pTrkB-ir in the striatum (P=0.0134) but not in the cortex of zQ175 homozygous mice at 12 months of age compared to their wild type littermates (Supplemental Fig. 3B), as quantified by overall intensity in the field and cellular intensity in Darpp-32 labeled cell bodies. The number of Darpp-32 labeled cells per field is similar between wild type and zQ175 mice (Supplemental Fig. 3D, E). This suggests that TrkB phosphorylation is decreased significantly in the HD striatum, including the striatal medium spiny neurons.
Induction of p75NTR and sortilin in immature striatal oligodendrocytes
p75NTR and sortilin receptor family have been shown to mediate apoptosis or degeneration in a variety of cell types responding to proneurotrophins or other ligands (Beattie et al., 2002; Wang et al., 2002; Teng et al., 2005; Charalampopoulos et al., 2012; Takamura et al., 2012). To assess whether p75NTR and sortilin may play a role in the pathogenesis of HD, we examined the expression of these proteins in the striatum of zQ175 mice. In 12-month-old zQ175 mice, we found that the sortilin level is significantly increased by ~30% in the striatum (P=0.0281 and P=0.0155 respectively), but is unchanged in the cortex (P=0.1713 and P=0.0583 respectively), as determined by Western blot analysis (Fig. 4A, B, D). In contrast, there is a significant decrease of p75NTR levels by ~40% in both the cortex and the striatum of zQ175 heterozygous and homozygous animals compared to the wild type (P<0.01 of all values) (Fig. 4A, B, C).
Figure 4. Alterations of p75NTR and sortilin levels in both the cortex and the striatum of zQ175 mice.

(A) At the age of 12 months, p75NTR level is significantly reduced in the cortex of zQ175 mice, while sortilin level is modestly increased. (B) p75NTR level is also significantly reduced in the striatum of 12-month-old zQ175 mice, but sortilin level is significantly increased. Both (C) Quantification results of p75NTR level showing a significant decrease (~40%) of p75NTR in the cortex and the striatum of both zQ175 heterozygous and homozygous animals. (D) Quantification results of sortilin level showing a modest increase of sortilin in the cortex but a significant sortilin induction in the striatum of both zQ175 heterozygous and homozygous animals.
We next evaluated the pattern of p75NTR and sortilin labeling in the wild type and zQ175 mouse striatum. In 12-month-old wild type mice, p75NTR labels patches of processes surrounding 1~3 cell bodies in the striatum (Fig. 5A). Electron microscopy verified that p75NTR immunolabeling is in the axons in the wild type striatum (Supplemental Fig. 5A). The specificity of this labeling is validated using brain sections from p75−/− animals (data not shown). In 12-month-old zQ175 homozygous mice, we found a reduction of patchy labeling of p75NTR in the striatum, but a robust induction of p75NTR signal in the cell bodies of a subpopulation of cells (Fig. 5B). Further analysis reveals that p75NTR is not present in the cell bodies of medium spiny neurons in either WT (Fig. 5C) or zQ175 homozygous mice (Fig. 5D). No co-localization between p75NTR and an astroglial marker (GFAP) or a microglial marker (Iba1) was observed in 12-month-old zQ175 homozygous mice (Supplemental Fig. 4A, B). Instead, the p75NTR-labeled cells in 12-month-old zQ175 homozygous striatum are positive for Olig2 marker (Fig. 6B), suggesting an oligodendrocyte lineage, which is further verified by electron microscopy (Supplemental Fig. 5B). To determine the developmental stage of these oligodendrocytes, we performed immunofluorescence detection of p75NTR with several oligodendrocyte markers (Fig. 6A). The results indicate that the p75NTR-immunoreactive cells are 2′, 3′-Cyclic-nucleotide 3′-phosphodiesterase (CNPase)-immunoreactive (Fig. 6D), but are negative for NG2- or APC (CC1)-labeling (Fig. 6C, E) in the striatum, implying that these cells are not oligodendrocyte precursor cells or mature myelinating oligodendrocytes, but are immature oligodendrocytes. However, the p75NTR-labeled cells are not immunoreactive for cleaved caspase-3 (Fig. 6F), suggesting that p75NTR is not inducing apoptosis of these oligodendrocytes.
Figure 5. Alteration of p75NTR labeling pattern in the striatum of zQ175 mice.
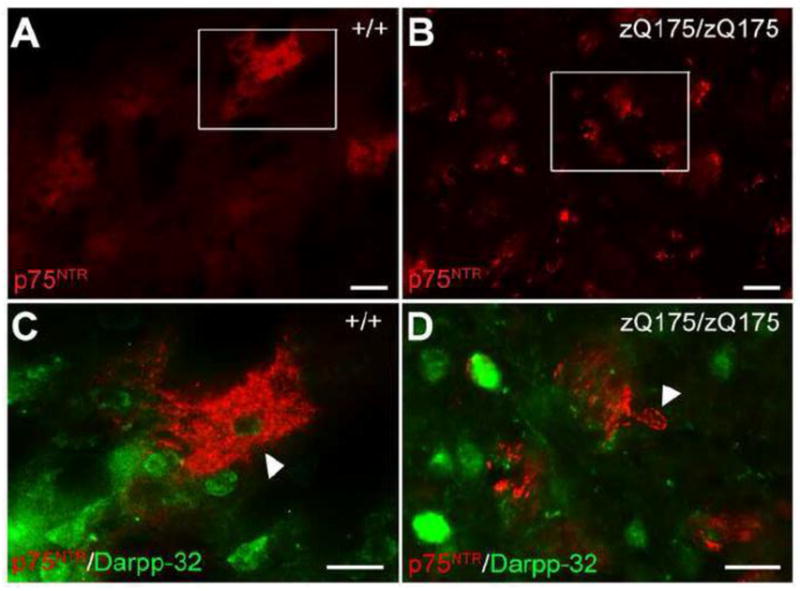
(A, B) Immunofluorescence staining of p75NTR in the striatum of 12-month-old wild type (A) and zQ175 homozygous mice (B). Note the reduction of p75NTR immunoreactivity of the patch-like labeling but induction of p75NTR in the cell bodies in zQ175/zQ175 striatum. Boxes in (A) and (B) are enlarged in (C) and (D), respectively. Double immunofluorescence staining shows no co-localization between p75NTR (arrowhead) and Darpp-32 (medium spiny neuron marker) in the striatum of both wild type (C) and zQ175 homozygous mice (D). Scale bar: 50μm (A, B), 20μm (C, D).
Figure 6. Detection of p75NTR in immature striatal oligodendrocytes of zQ175 mice.

(A) Scheme showing the expression timeline of different oligodendrocyte markers in different stage of oligodendrocyte development. OPC, oligodendrocyte precursor cells. OL, oligodendrocyte. (B–E) Colocalization of p75NTR labeled cells with oligodendrocyte lineage marker Olig2 (B) and CNPase (D), but not oligodendrocyte precursor cell marker NG2 (C) or mature oligodendrocyte marker CC1 (D) in the striatum of zQ175 homozygous mice at the age of 12 months. (F) p75NTR-immunoreactive cells (arrow) in the striatum do not colocalize with cleaved caspase-3 immunoreactivity (arrowhead). Scale bar, 20μm.
Simultaneous induction of sortilin in p75NTR-immunoreactive oligodendrocytes is also observed in the zQ175 homozygous striatum at the age of 12 months (Fig. 7A–D). Importantly, this induction of p75NTR and sortilin in oligodendrocytes was not observed in zQ175 mice at 6 months of age or WT mice at any time points (data not shown).
Figure 7. Simultaneous upregulation of p75NTR and sortilin in the same oligodendrocytes of striatum in zQ175 animals.

Triple immunofluorescence labeling of p75NTR (A), sortilin (B), and CNPase (C) in zQ175/zQ175 striatum at 12 months of age. (D) Overlay of (A–C) shows the presence of both p75NTR and sortilin in the same oligodendrocytes of zQ175 striatum. Scale bar, 10μm.
Impaired myelination in the striatum of aged zQ175 mice
We next assessed whether there is a myelin defect in the HD striatum associated with p75NTR induction in the oligodendrocytes. For this, we analyzed axon clusters of the internal capsule islands in the striatum by electron microscopy. Ultrastructural analysis revealed a significant decrease in myelin sheath thickness in the striatal axons from 12-month-old zQ175 mice (Fig. 8A) as measured by G-ratio (P=0.0005) (Fig. 8B, C). We also found a two-fold increase in percentage of unmyelinated axons (P=0.0202) in the striatum of 12-month-old zQ175 homozygous mice (Fig. 8D). However, no decrease of total number of mature oligodendrocytes as detected by CC1 labeling in the striatum was observed (Fig. 8E). These data suggest that there is deficient myelination correlating with p75NTR induction in immature striatal oligodendrocytes of zQ175 homozygous mice at 12 months of age, and this impairment in myelination is not contributed by a decrease in the number of mature myelinating oligodendrocytes.
Figure 8. Deficient myelination in the striatum of zQ175 homozygous animals.

(A) Representative electron micrographs of myelinated fibers in the striatum of 12-month-old wild type (left panel) and zQ175/zQ175 mice (right panel). Note the thinner myelin sheath in zQ175 homozygous striatum. G-ratio (calculated as axon diameter divided by total fiber diameter) is significantly larger in zQ175/zQ175 striatum compared to WT at 12 months old. Statistics are shown by distribution (B) and average (C). (D) Percentage of unmyelinated axons increases significantly in the striatum of zQ175/zQ175 striatum compared to WT at 12 months old. (E) Cell counts of CC1+ mature oligodendrocytes in the striatum are not different between WT and zQ175 homozygous animals at 12 months of age. Scale bar, 500nm (A).
Discussion
The precise mechanisms that lead to selective striatal neuronal death in HD are unclear. Deficiency in BDNF trophic support has been proposed as an important mechanism underlying HD pathogenesis. However, prior studies have reported inconsistent results regarding levels of BDNF and its receptors in different HD mouse models (Gines et al., 2003; Zuccato et al., 2005; Seo et al., 2008; Bobrowska et al., 2011; Sontag et al., 2012; Plotkin et al., 2014). Furthermore, given that proBDNF and mBDNF have opposing effects on cell survival, the question of whether BDNF maturation is impaired in HD needs to be determined. To clarify this, we evaluated the levels of both proBDNF and mBDNF and alterations of their receptors in the cortex and the striatum of zQ175 mouse model of HD. Our findings demonstrate an age- and region-dependent loss of mBDNF-TrkB signaling, but no induction of proBDNF-p75NTR signaling in striatal neurons of zQ175 mice, suggesting that maturation of proBDNF to mBDNF remains intact in the corticostriatal circuitry of this HD mouse model. However, local induction of p75NTR and sortilin immunoreactivities is found in a subset of immature oligodendrocytes in the striatum and is associated with severe myelin deficits of these fibers in the striatum of aged zQ175 HD mice.
Levels of different BDNF isoforms in the wild type and HD murine striatum
Since the discovery of the potential link between BDNF deficiency and HD pathogenesis, multiple studies have examined BDNF levels in the brain of HD mouse models (summarized in Supplemental Table.1). A variety of factors could contribute to the inconsistencies in previous studies regarding BDNF levels in HD mice: first, different lines of animal models are used, among which the expression levels and structure of mutant huntingtin protein varies (Crook and Housman, 2011). Large differences exist among these genetic HD mouse models regarding the disease progression curve, in which disease onset, as assessed by behavioral tests, can range from 8 weeks to 12 months (Menalled, 2005). Thus, levels of BDNF can vary significantly due to the use of different animal models at different time points. Second, different approaches and reagents have been used to measure the amount of BDNF. In-situ hybridization or RT-PCR provide information on mRNA levels, but may not correlate precisely with protein level, particularly as BDNF can be transported long distances via axonal projections from the cortex to the striatum (Altar et al., 1997). Furthermore, lack of specific and sensitive antibodies to detect BDNF protein, due to its highly conserved sequence across species, results in low sensitivity and specificity, particularly problematic due to the extremely low level of BDNF in the cortex and the striatum of aged animals, using Western blot analysis or ELISA. These factors may contribute to the discrepancy among multiple publications using the same animal model and the same time points.
To quantitatively determine the change of levels of different BDNF isoforms, we utilized bdnf-HA KI mice on zQ175 HD mice background to facilitate the detection of endogenously expressed proBDNF and mBDNF in tissue lysates. At an early stage of disease (6 months), we observed a profound loss of striatal mBDNF but no change of cortical mBDNF, supporting the hypothesis that BDNF anterograde transport from the cortex to the striatum is deficient (Gauthier et al., 2004). This is also in line with a recent publication indicating BDNF mRNA level in the cortex is similar between wild type and zQ175 heterozygous animals (Plotkin et al., 2014). At late stage of disease (12 months), we found that both striatal and cortical mBDNF levels are markedly reduced, which correlate with overt onset of behavioral and cognitive symptoms. The age- and region-dependent reduction of mBDNF is consistent with progressive loss of bdnf gene transcription (Gines et al., 2003; Zuccato et al., 2005; Seo et al., 2008; Bobrowska et al., 2011; Sontag et al., 2012; Plotkin et al., 2014).
ProBDNF can act as a death-promoting factor to neurons. However, no studies have reported the ratio of proBDNF to mBDNF in the wild type murine striatum during aging, and it is unknown whether conversion of proBDNF to mBDNF is altered in HD, which could contribute to HD pathogenesis and hinder the therapeutic potential of increasing BDNF expression. By utilizing bdnf-HA KI animals, we observed that mBDNF is the predominant form present in the striatum during normal postnatal development and with aging.
The lack of striatal proBDNF in wild type mice can be explained by: (1) striatal neurons synthesize relatively low levels of BDNF isoforms, as compared to that transported from other brain regions; (2) efficient conversion of proBDNF to mBDNF during anterograde transport from the cortex. After being synthesized in cortical neurons, proBDNF or the cleaved products of prodomain and mature BDNF targeted to the striatum are transported within dense core vesicles along corticostriatal axons (unpublished observations). Our results suggest that most proBDNF trafficked to the striatum might already be processed to mBDNF in the wild type mice, and the level of intact proBDNF present in the striatum is below the limits of detection by this approach.
It is also noteworthy that no increase in proBDNF levels was observed either in the sensorimotor cortex or the neostriatum of zQ175 HD mice at 12 months, suggesting that the maturation pathway of BDNF is still intact in zQ175 HD cortical neurons at this time point. Given the pathological differences among HD mouse models, utilizing bdnf-HA KI mice on different HD mouse models would be important in the future to clarify the field.
In addition, several publications have shown the therapeutic potentials of overexpressing proBDNF cDNA under the control of the Ca2+/calmodulindependent kinase II (CaMKIIa) promoter in the forebrain of HD transgenic mice (Gharami et al., 2008; Xie et al., 2010). However, it remains to be evaluated whether proBDNF processing is inefficient when there is massive BDNF overexpression in vivo, which would otherwise lead to an imbalance in death-survival signaling.
Alterations of TrkB / p75NTR levels and downstream signaling in HD mouse models
Recent studies suggest that p75NTR/TrkB imbalance is associated with striatal vulnerability in HD (Brito et al., 2013; Plotkin et al., 2014). However, data regarding TrkB / p75NTR levels and their downstream signaling are not consistent among different animal models (Gharami et al., 2008; Xie et al., 2010; Simmons et al., 2013; Plotkin et al., 2014). In aged zQ175 KI HD mice, we observed significant decreased TrkB phosphorylation in the cell bodies of medium spiny neurons using validated antibodies. This result can be explained by the reduction in the amount of the ligand (mBDNF) as well as deficits of BDNF-TrkB retrograde transport in the HD striatum (Liot et al., 2013). These results support the previous hypothesis of diminished mBDNF-TrkB trophic signaling in HD. However, using multiple validated antibodies, we detected little p75NTR-immunoreactivity in the cell bodies of medium spiny neurons in wild type or zQ175 mice, and the total level of p75NTR in the striatum decreases in aged zQ175 HD mice compared with wild type by Western blot analysis. Different p75NTR antibodies and different HD animal models may be responsible for the discrepancy between this result and the previous reports. Nevertheless, we did find an elevation in the JNK/caspase-3 apoptotic signaling cascade (data not shown), which suggests that pro-apoptotic downstream signaling is indeed augmented in the striatum of aged zQ175 HD mice, although p75NTR-mediated mechanisms may not be the only factor contributing to this augmentation.
Role of p75NTR and sortilin in oligodendroglial development and HD pathogenesis
Impaired myelination has been reported in HD patients (Bartzokis et al., 2007) and mouse models (Xiang et al., 2011; Jin et al., 2015). The molecular mechanisms underlying this white matter abnormality are largely unknown, although PGC-1α deficiency (Xiang et al., 2011) and altered proliferation of oligodendrocyte precursor cell (Jin et al., 2015) could be contributing factors. Interestingly, our study revealed that p75NTR- and sortilin-immunoreactivities are both significantly elevated in a subset of immature striatal oligodendrocytes in aged zQ175 HD mice, and this correlates with myelination defect in the HD striatum.
In many neurodegenerative diseases including HD and amyotrophic lateral sclerosis (Bartzokis et al., 2007; Kang et al., 2013), the physiologic response of oligodendrocytes, to repair and remyelinate after myelin breakdown, results in oligodendrocyte progenitor proliferation and differentiation. Failure of newly formed oligodendrocytes to mature leads to progressive demyelination (Kang et al., 2013). It is possible that p75NTR and sortilin upregulation in this subgroup of immature oligodendroglia is relevant to the failed attempt of newly generated oligodendrocyte to mature and remyelinate upon insults, which ultimately leads to deficient myelination.
While p75NTR has been shown to be a positive modulator of remyelination in Schwann cells of peripheral nervous system (Tomita et al., 2007), its role in myelination of central nervous system has not been firmly established. In mature oligodendrocytes, p75NTR is shown to mediate cell apoptosis via JNK and caspase-3 pathway in response to high concentration of mature nerve growth factor (NGF) in vitro (Casaccia-Bonnefil et al., 1996) or proNGF in vivo after spinal cord injury (Beattie et al., 2002). In immature oligodendrocytes, p75NTR can be upregulated in response to demyelination insults (Chang et al., 2000; Petratos et al., 2004), but its function in oligodendrocyte differentiation and maturation remains to be determined. Some in vitro studies indicate that p75NTR can partner with Trk receptors to facilitate basal forebrain oligodendrocytes maturation induced by NGF or neurotrophin-3 (NT-3) (Du et al., 2006), or form a complex with LINGO-1 to mediate inhibition of oligodendrocytes differentiation (Mi et al., 2004; Mi et al., 2005; Bourikas et al., 2010). These studies suggest that p75NTR can serve diverse functions in immature oligodendrocytes depending on the ligand present and the receptor with which p75NTR is paired. p75NTR and sortilin family members can interact directly and are both required to mediate pro-neurotrophins-induced cell death (Teng et al., 2005) or growth cone retraction in neurons (Deinhardt et al., 2011). Whether they have similar functions in oligodendrocytes development in HD needs further investigation.
Nonetheless, since p75NTR antagonism has been proposed as a therapeutic strategy (Brito et al., 2013), and shown to rescue striatal long-term potentiation deficit in a HD mouse model (Plotkin et al., 2014), it would also be important to examine whether downregulating p75NTR will facilitate the remyelination process and modulate disease progression in the late stage of this devastating disease.
Supplementary Material
Highlights.
Accurate quantitation of endogenously expressed proBDNF and mature BDNF in murine striatum
Age and region-dependent loss of mBDNF in the cortex and striatum of zQ175 HD mice
No proBDNF induction observed in the brain of aged zQ175 HD mice
p75NTR and sortilin induction associates with myelin defects in the striatum of aged zQ175 HD mice
Acknowledgments
This work was supported by The Cure Huntington’s Disease Initiative (CHDI) Foundation HD 23315 and National Institute of Health grants NS030687, NS064114 (B.L.H.); and HL96571, HL098351, DA08259 (T.A.M.). We thank all members from the B.L.H. laboratory for their assistance. We also express our appreciation to Drs. Francis S. Lee and Moses V. Chao for helpful discussions and for editing the manuscript.
Footnotes
Conflict of Interest:
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Anastasia A, Deinhardt K, Chao MV, Will NE, Irmady K, Lee FS, Hempstead BL, Bracken C. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nature communications. 2013;4:2490. doi: 10.1038/ncomms3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S. Myelin breakdown and iron changes in Huntington’s disease: pathogenesis and treatment implications. Neurochemical research. 2007;32:1655–1664. doi: 10.1007/s11064-007-9352-7. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemelmans AP, Horellou P, Pradier L, Brunet I, Colin P, Mallet J. Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington’s disease, as demonstrated by adenoviral gene transfer. Hum Gene Ther. 1999;10:2987–2997. doi: 10.1089/10430349950016393. [DOI] [PubMed] [Google Scholar]
- Bobrowska A, Paganetti P, Matthias P, Bates GP. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PloS one. 2011;6:e20696. doi: 10.1371/journal.pone.0020696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourikas D, Mir A, Walmsley AR. LINGO-1-mediated inhibition of oligodendrocyte differentiation does not require the leucine-rich repeats and is reversed by p75(NTR) antagonists. Molecular and cellular neurosciences. 2010;45:363–369. doi: 10.1016/j.mcn.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Brito V, Puigdellivol M, Giralt A, del Toro D, Alberch J, Gines S. Imbalance of p75(NTR)/TrkB protein expression in Huntington’s disease: implication for neuroprotective therapies. Cell death & disease. 2013;4:e595. doi: 10.1038/cddis.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nature reviews Neuroscience. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charalampopoulos I, Vicario A, Pediaditakis I, Gravanis A, Simi A, Ibanez CF. Genetic dissection of neurotrophin signaling through the p75 neurotrophin receptor. Cell reports. 2012;2:1563–1570. doi: 10.1016/j.celrep.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Conforti P, Mas Monteys A, Zuccato C, Buckley NJ, Davidson B, Cattaneo E. In vivo delivery of DN:REST improves transcriptional changes of REST-regulated genes in HD mice. Gene Ther. 2013;20:678–685. doi: 10.1038/gt.2012.84. [DOI] [PubMed] [Google Scholar]
- Crook ZR, Housman D. Huntington’s disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA, Neubert TA, Chao MV, Hempstead BL. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Science signaling. 2011;4:ra82. doi: 10.1126/scisignal.2002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Fischer TZ, Clinton-Luke P, Lercher LD, Dreyfus CF. Distinct effects of p75 in mediating actions of neurotrophins on basal forebrain oligodendrocytes. Molecular and cellular neurosciences. 2006;31:366–375. doi: 10.1016/j.mcn.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain research. 2000;866:257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. Journal of neurochemistry. 2008;105:369–379. doi: 10.1111/j.1471-4159.2007.05137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampa C, Montagna E, Dato C, Melone MA, Bernardi G, Fusco FR. Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PloS one. 2013;8:e64037. doi: 10.1371/journal.pone.0064037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, Wheeler VC, Persichetti F, MacDonald ME. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Human molecular genetics. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- HDCRG. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Lehtimaki K, Vartiainen N, Puolivali J, Hendricks SJ, Glaser JR, Bradaia A, Wadel K, Touller C, Kontkanen O, Yrjanheikki JM, Buisson B, Howland D, Beaumont V, Munoz-Sanjuan I, Park LC. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PloS one. 2012;7:e50717. doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Peng Q, Hou Z, Jiang M, Wang X, Langseth AJ, Tao M, Barker PB, Mori S, Bergles DE, Ross CA, Detloff PJ, Zhang J, Duan W. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Human molecular genetics. 2015 doi: 10.1093/hmg/ddv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nature neuroscience. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kells AP, Henry RA, Connor B. AAV-BDNF mediated attenuation of quinolinic acid-induced neuropathology and motor function impairment. Gene Ther. 2008;15:966–977. doi: 10.1038/gt.2008.23. [DOI] [PubMed] [Google Scholar]
- Kells AP, Fong DM, Dragunow M, During MJ, Young D, Connor B. AAV-mediated gene delivery of BDNF or GDNF is neuroprotective in a model of Huntington disease. Mol Ther. 2004;9:682–688. doi: 10.1016/j.ymthe.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Liot G, Zala D, Pla P, Mottet G, Piel M, Saudou F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:6298–6309. doi: 10.1523/JNEUROSCI.2033-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB. Knock-in mouse models of Huntington’s disease. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2005;2:465–470. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PloS one. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nature neuroscience. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nature neuroscience. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Milner TA, Waters EM, Robinson DC, Pierce JP. Degenerating processes identified by electron microscopic immunocytochemical methods. Methods in molecular biology. 2011;793:23–59. doi: 10.1007/978-1-61779-328-8_3. [DOI] [PubMed] [Google Scholar]
- Petratos S, Gonzales MF, Azari MF, Marriott M, Minichiello RA, Shipham KA, Profyris C, Nicolaou A, Boyle K, Cheema SS, Kilpatrick TJ. Expression of the low-affinity neurotrophin receptor, p75(NTR), is upregulated by oligodendroglial progenitors adjacent to the subventricular zone in response to demyelination. Glia. 2004;48:64–75. doi: 10.1002/glia.20056. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, Stavarache M, Kaplitt MG, Rosinski J, Chan CS, Surmeier DJ. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron. 2014;83:178–188. doi: 10.1016/j.neuron.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Seo H, Kim W, Isacson O. Compensatory changes in the ubiquitin-proteasome system, brain-derived neurotrophic factor and mitochondrial complex II/III in YAC72 and R6/2 transgenic mice partially model Huntington’s disease patients. Human molecular genetics. 2008;17:3144–3153. doi: 10.1093/hmg/ddn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulson I, Chase TN. Huntington’s disease. Annu Rev Med. 1975;26:419–436. doi: 10.1146/annurev.me.26.020175.002223. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M, Jing D, Massa SM, Longo FM. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:18712–18727. doi: 10.1523/JNEUROSCI.1310-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Rocha EM, McLean JR, Hayes MA, Izen SC, Isacson O, Hallett PJ. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Human molecular genetics. 2014;23:4510–4527. doi: 10.1093/hmg/ddu166. [DOI] [PubMed] [Google Scholar]
- Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A, Finkbeiner S, Glabe C, Marsh JL, Muchowski PJ, Thompson LM. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:11109–11119. doi: 10.1523/JNEUROSCI.0895-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Kooperberg C, Olson JM, Jones KR. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Sato Y, Watabe D, Okamoto Y, Nakata T, Kawarabayashi T, Oddo S, Laferla FM, Shoji M, Matsubara E. Sortilin is required for toxic action of Abeta oligomers (AbetaOs): extracellular AbetaOs trigger apoptosis, and intraneuronal AbetaOs impair degradation pathways. Life sciences. 2012;91:1177–1186. doi: 10.1016/j.lfs.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Kubo T, Matsuda K, Fujiwara T, Yano K, Winograd JM, Tohyama M, Hosokawa K. The neurotrophin receptor p75NTR in Schwann cells is implicated in remyelination and motor recovery after peripheral nerve injury. Glia. 2007;55:1199–1208. doi: 10.1002/glia.20533. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nature neuroscience. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nature neuroscience. 2005;8:1069–1077. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, Cattaneo E, Krainc D. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:9544–9553. doi: 10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. Neuronal release of proBDNF. Nature neuroscience. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Harte-Hargrove LC, Siao CJ, Marinic T, Clarke R, Ma Q, Jing D, Lafrancois JJ, Bath KG, Mark W, Ballon D, Lee FS, Scharfman HE, Hempstead BL. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell reports. 2014;7:796–806. doi: 10.1016/j.celrep.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain pathology. 2008;18:225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacological research : the official journal of the Italian Pharmacological Society. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nature genetics. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.