

Abstract
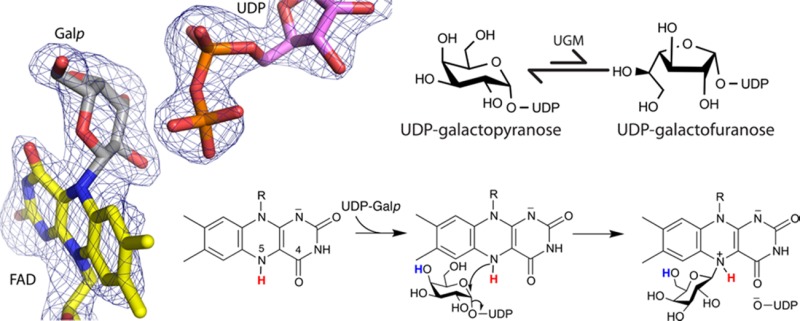
UDP-galactopyranose mutase (UGM) plays an essential role in galactofuranose biosynthesis in pathogens by catalyzing the conversion of UDP-galactopyranose to UDP-galactofuranose. Here we report the first crystal structure of a covalent intermediate in the UGM reaction. The 2.3 Å resolution structure reveals UDP bound in the active site and galactopyranose linked to the FAD through a covalent bond between the anomeric C of galactopyranose and N5 of the FAD. The structure confirms the role of the flavin as nucleophile and supports the hypothesis that the proton destined for O5 of galactofuranose is shuttled from N5 of the FAD via O4 of the FAD.
UDP-galactopyranose mutase (UGM) catalyzes the interconversion of UDP-galactopyranose (UDP-Galp) and UDP-galactofuranose (UDP-Galf) (Scheme 1). The latter sugar nucleotide is the donor substrate for enzymes that incorporate Galf into myriad biomolecules that form the host–pathogen barriers in bacteria, fungi, protozoan parasites, and nematodes.1 Because Galf has never been found in animals, the inhibition of Galf biosynthesis is a potential drug design strategy.
Scheme 1. Reaction Catalyzed by UGM.

Galf originates in the UGM reaction, which suggests that UGM is a good drug target. Indeed, UGM is an essential enzyme in Mycobacterium tuberculosis(2,3) and a virulence factor in eukaryotic pathogens, including the fungus Aspergillus fumigatus (causative agent of aspergillosis) and the trypanosomal parasite Leishmania spp.4,5 Also, Galf-containing glycoconjugates are involved in the mechanism of myocardial damage by Trypanosoma cruzi, the causative agent of Chagas disease.6 Furthermore, Galf has been identified in nematodes,7−9 suggesting that UGMs from Brugia malayi (causing elephantiasis) and Onchocerca volvulus (river blindness) are potential targets.
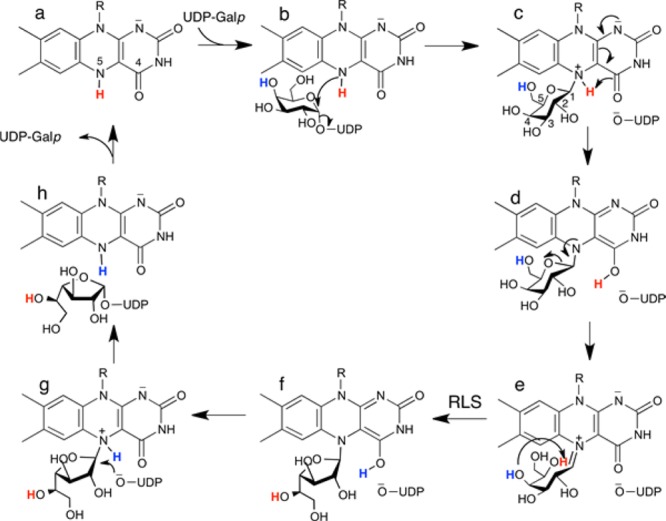
UGM also is important to basic science as the prototype of noncanonical flavoenzymes. Unlike traditional flavoenzymes, the redox state of the flavin in UGM is unchanged during the catalytic cycle. Rather, the FAD coenzyme in UGM functions as a nucleophile that attacks the anomeric C atom of the substrate (C1). This role requires that the flavin be in the reduced state for activity (Scheme 2a). The accepted mechanism begins with nucleophilic attack of the FAD N5 atom at the substrate C1 atom (Scheme 2b), generating a flavin–Galp intermediate with transient release of UDP (Scheme 2c). Subsequent proposed steps also involving flavin–sugar intermediates include proton transfers, opening of the Galp ring, and ring contraction to Galf (Scheme 2c–f). Finally, the sugar–UDP bond is re-formed to generate UDP-Galf (Scheme 2g,h).
Scheme 2. Mechanism of UGM.
Covalent flavin–sugar intermediates are distinguishing features of this unusual flavoenzyme mechanism, and much effort has been spent in seeking evidence of them. Landmark studies by Kiessling’s group using borohydride trapping and tritium labeling of the substrate revealed the first evidence of the flavin–iminium intermediate (Scheme 2e).10 Later studies by the same group confirmed the structural identity of this intermediate using nuclear magnetic resonance of the trapped species.11 Since then, additional indirect evidence implicating flavin–sugar intermediates in the UGM reaction has been obtained.8,12,13 However, despite more than a decade of research on the UGM reaction mechanism, direct structural evidence of a flavin–sugar adduct has remained elusive. Herein, we report the first crystal structure of a UGM having a covalent bond between the FAD and galactose.
The structure was determined using an active site mutant of A. fumigatus UGM (AfUGM) in which His63 is mutated to Ala (H63A). His63 is part of the conserved histidine loop, which has the sequence GGHVIF in AfUGM. All UGMs have Gly and His at positions 1 and 3 of the loop, respectively.14 As described previously, the conformation of the His loop of AfUGM depends on the redox state of the flavin.14 In the reduced (active) state, the carbonyl of Gly62 accepts a hydrogen bond from flavin N5, while the side chain of His63 forms a hydrogen bond with the 2′-OH of the FAD ribityl group. These protein–flavin interactions are thought be essential for maintaining the active conformation of UGM.14−17
Consistent with the universal conservation of the eponymous residue of the histidine loop, the catalytic properties of H63A are highly perturbed. H63A lacks catalytic activity. Although the flavin in the mutant enzyme can be reduced by sodium dithionite, reduced H63A is highly susceptible to oxidation by O2 compared to the wild-type (wt) enzyme (Figure S1). Furthermore, reduction of the flavin in H63A by NADPH is very slow. The efficiency of NADPH reduction (kred/KD) is 114 M–1 s–1, compared to 120000 M–1 s–1 for wt AfUGM (Figure S2). These results are consistent with our previous study showing that this mutation in T. cruzi UGM (H62A) decreased kcat for the mutase reaction by >300-fold.16
We serendipitously discovered that H63A could be used to capture a covalent FAD–Galp adduct in crystallo. Electron density maps from crystals of H63A that had been soaked simultaneously in sodium dithionite and UDP-Galp prior to being flash-cooled in liquid nitrogen surprisingly showed features consistent with covalent modification of the FAD at the N5 atom (Figure 1A). The soaking time and reagent concentrations were optimized to maximize the occupancy of the apparent intermediate, which required X-ray diffraction analysis of approximately 14 crystals. The structure reported here has a crystallographic resolution of 2.3 Å and was obtained from a crystal soaked for 2 h in 80 mM dithionite and 100 mM UDP-Galp prior to flash-cooling (Table S1).
Figure 1.
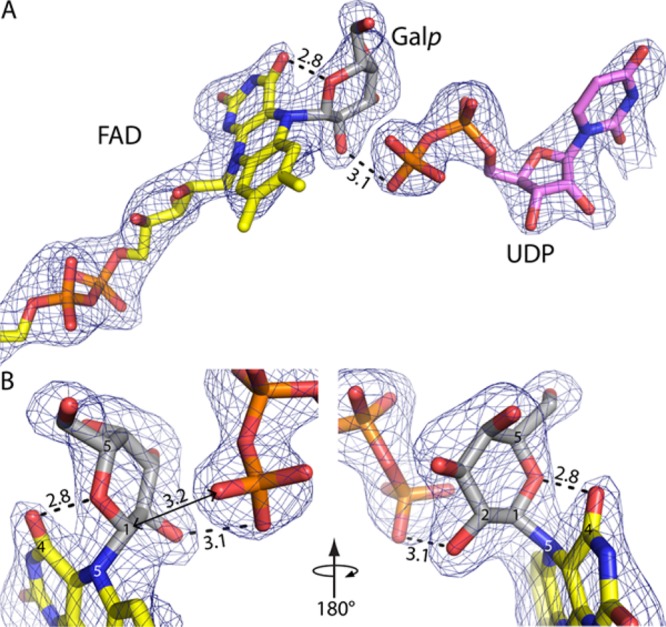
Electron density evidence of a covalent intermediate in the UGM reaction. The cage represents a simulated annealing Fo – Fc omit map contoured at 3σ. Prior to map calculation, the FAD, Galp, and UDP were deleted and simulated annealing refinement was performed. Distances are given in angstroms. (A) Overview of the ligands. FAD and Galp are colored yellow and gray, respectively. UDP is colored pink. (B) Close-up views of the covalent adduct. This figure and others were made with PyMOL.18
AfUGM crystallizes with a tetramer in the asymmetric unit, and the electron density in chain A provides the clearest depiction of a reaction intermediate. The map shows a strong feature indicating that the FAD is covalently modified at its N5 atom (Figure 1A). Also, strong electron density is present for UDP bound in the expected location (Figure 1A). The maps clearly indicated that the active site flaps are closed, which is expected when UDP is bound.14 The electron density for UDP is disconnected from that of the covalent modification when viewed at a level of 1.0σ for the refined 2Fo – Fc map or >2.5σ for the simulated annealing Fo – Fc omit map (Figure 1A), indicating the presence of two distinct ligands rather than an intact sugar nucleotide. These electron density features could be satisfactorily interpreted as Galp bound to FAD through a Galp C1–FAD N5 bond plus a detached UDP, which corresponds to intermediate c or d in Scheme 2.
The FAD–Galp intermediate was refined using geometrical restraints obtained from quantum mechanics/molecular mechanics (QM/MM) calculations (Supporting Information).19 The target Galp C1–FAD N5 bond distance was 1.547 Å. This bond refined to 1.6 Å, indicating that the crystallographic data are consistent with the QM/MM calculations. The average B factors of the FAD and Galp refined to 41.6 and 59.4 Å2, respectively, with fixed occupancies of 1.0 (Table S1). After refinement, the shortest distance between C1 of Galp and the O atoms of the UDP β-phosphate is 3.2 Å (Figure 1B), which is obviously outside of covalent bonding distance and consistent with rupture of the glycosidic bond during soaking.
Electron density in chain B was also modeled as intermediate c/d, but the density is not as strong as in chain A (Figure S3A). In chains C and D, the density suggests a possible mixture of covalent adducts without bound UDP (see Model Building Methods in the Supporting Information and Figure S3B,C). We thus focus the remaining discussion on the active site of chain A.
The trapped intermediate resembles the noncovalent complex of wt AfUGM with UDP-Galp (E–S complex), as one might expect for consecutive steps in a chemical mechanism. The conformation of UDP and its interactions with the enzyme are very similar to those of the E–S complex (Figure 2A). The Galp of the intermediate likewise has a pose and protein environment similar to those in the E–S complex (Figure 2A). The O2 and O3 hydroxyls of the covalently bound Galp form hydrogen bonds with Arg182. The Galp O3 and O4 hydroxyls contact the side chain of Phe66 (3.4 Å). The Galp O4 hydroxyl also forms a hydrogen bond with Asn207. The Galp O6 hydroxyl packs tightly between Trp315 (3.6 Å) and Arg327 (3.4 Å). All of these enzyme–sugar interactions are also seen in the E–S complex. The similarity of the H63A active structure to the genuine E–S complex implies that the species captured in crystallo is meaningful despite the use of a mutant enzyme.
Figure 2.
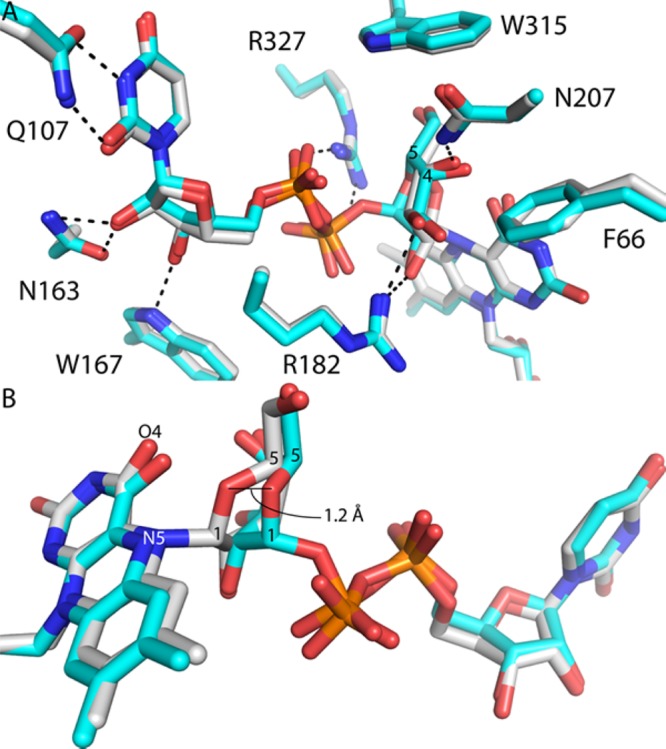
Comparison of the covalent intermediate in H63A (white) and the noncovalent E–S complex (cyan, Protein Data Bank entry 3UTH(15)). (A) Superposition of the two structures emphasizing the similarity of the UDP conformations and protein environment. The dashed lines indicate interaction distances of <3.1 Å. (B) Close-up showing how formation of the N5–C1 bond draws Galp O5 closer to flavin O4.
A difference between the trapped intermediate and the E–S complex is that formation of the FAD N5–Galp C1 bond draws O5 of Galp 1.2 Å closer to the pyrimidine ring of the FAD isoalloxazine (Figure 2B). In the covalent intermediate, Galp O5 is 2.8 Å from FAD O4 (Figure 1), compared to 3.3 Å in the E–S complex. The close approach of Galp O5 and FAD O4 in the intermediate is consistent with a proposal from QM/MM calculations20 that FAD O4 accepts a proton from FAD N5 and donates it to Galp O5, facilitating ring opening and formation of the iminium ion (Scheme 2c–e).
Electron density for the histidine loop of the intermediate (Figure 3A) suggests a conformation similar to that of the reduced wt enzyme, except for one important aspect (Figure 3B). In reduced wt AfUGM (ligand-free or complexed with UDP-Galp), the carbonyl of Gly62 accepts a hydrogen bond from flavin N5. Because N5 of the reduced FAD is an obligate hydrogen bond donor, the hydrogen bond with Gly62 is considered to be a key stabilizing interaction of the reduced enzyme.14−17 Indeed, this hydrogen bond is found in other structures of reduced UGMs. In the H63A–Galp adduct, however, Gly62 is rotated by ∼90° from the expected orientation so that it is not within hydrogen bonding distance of N5. This rotation appears to be necessary to avoid steric clash with Galp O5. The rotation may also reflect a change in the hydrogen bond capacity of FAD N5 in going from intermediate c to intermediate d in Scheme 2. In the latter state, N5 cannot donate a hydrogen bond, which could induce rotation of the Gly62 carbonyl, an obligate acceptor, away from N5. The orientation of Gly62 perhaps suggests that the trapped species is intermediate d rather than intermediate c, although it is impossible to distinguish between these species solely on the basis of the electron density at this resolution. Finally, it is also possible that mutation of His63 causes the atypical orientation of Gly62, and it is this structural perturbation that allowed us to capture the intermediate in crystallo.
Figure 3.
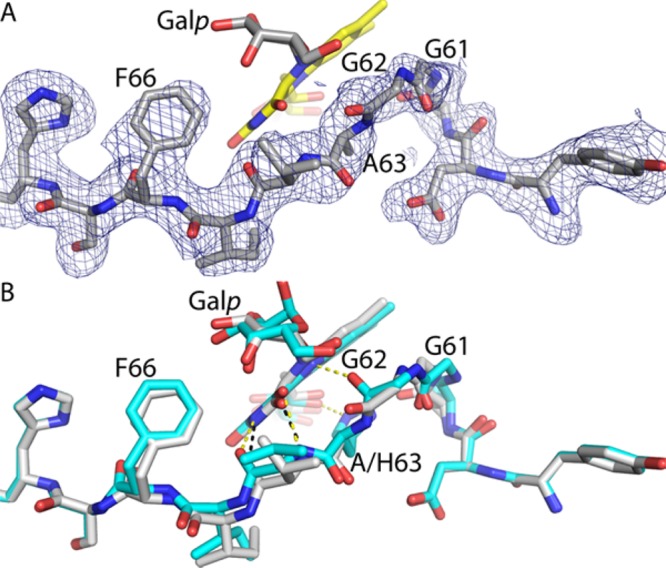
Conformation of the histidine loop. (A) Electron density for the histidine loop of H63A. The cage represents a simulated annealing Fo – Fc omit map (2.25σ). Prior to map calculation, residues 58–68 were deleted and simulated annealing refinement was performed. The FAD is colored yellow. (B) Comparison of the loops of H63A (white) and the E–S complex (cyan, Protein Data Bank entry 3UTH). Black and yellow dashes indicate hydrogen bonds in H63A and the E–S complex, respectively.
In conclusion, we have determined the first crystal structure of a covalent intermediate in the UGM reaction. To the best of our knowledge, it is the first structure of a substrate-derived covalent intermediate for any noncanonical flavoenzyme. The structure confirms the role of FAD N5 as a nucleophile and supports the hypothesis that the proton destined for O5 of Galf is transferred from FAD N5 via the FAD O4 carbonyl.
Acknowledgments
We thank Dr. Jay Nix for help with X-ray data collection and Dr. Isabel Da Fonseca for help with site-directed mutagenesis. Part of this research was performed at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00035.
Experimental methods, a table of X-ray data collection and refinement statistics, and supporting figures (PDF)
Accession Codes
Atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank as entry 5HHF.
Author Contributions
R.M.-C. and Y.D. contributed equally to this work.
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health via Grant R01GM094469 and National Science Foundation Grant CHE-1506206 (to P.S. and J.J.T.).
The authors declare no competing financial interest.
Supplementary Material
References
- Tefsen B.; Ram A. F.; van Die I.; Routier F. H. (2012) Glycobiology 22, 456–469. 10.1093/glycob/cwr144. [DOI] [PubMed] [Google Scholar]
- Pan F.; Jackson M.; Ma Y.; McNeil M. (2001) J. Bacteriol. 183, 3991–3998. 10.1128/JB.183.13.3991-3998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. (2003) Mol. Microbiol. 48, 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Schmalhorst P. S.; Krappmann S.; Vervecken W.; Rohde M.; Muller M.; Braus G. H.; Contreras R.; Braun A.; Bakker H.; Routier F. H. (2008) Eukaryotic Cell 7, 1268–1277. 10.1128/EC.00109-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczka B.; Lamerz A. C.; van Zandbergen G.; Wenzel A.; Gerardy-Schahn R.; Wiese M.; Routier F. H. (2007) J. Biol. Chem. 282, 10498–10505. 10.1074/jbc.M700023200. [DOI] [PubMed] [Google Scholar]
- Turner C. W.; Lima M. F.; Villalta F. (2002) Biochem. Biophys. Res. Commun. 290, 29–34. 10.1006/bbrc.2001.6189. [DOI] [PubMed] [Google Scholar]
- Beverley S. M.; Owens K. L.; Showalter M.; Griffith C. L.; Doering T. L.; Jones V. C.; McNeil M. R. (2005) Eukaryotic Cell 4, 1147–1154. 10.1128/EC.4.6.1147-1154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesener D. A.; May J. F.; Huffman E. M.; Kiessling L. L. (2013) Biochemistry 52, 4391–4398. 10.1021/bi400264d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli J. F.; Chaudhary K.; Canovas J.; Benner J. S.; Madinger C. L.; Kelly P.; Hodgkin J.; Carlow C. K. (2009) Dev. Biol. 335, 340–355. 10.1016/j.ydbio.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Soltero-Higgin M.; Carlson E. E.; Gruber T. D.; Kiessling L. L. (2004) Nat. Struct. Mol. Biol. 11, 539–543. 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]
- Gruber T. D.; Westler W. M.; Kiessling L. L.; Forest K. T. (2009) Biochemistry 48, 9171–9173. 10.1021/bi901437v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H. G.; Ruszczycky M. W.; Chang W. C.; Thibodeaux C. J.; Liu H. W. (2012) J. Biol. Chem. 287, 4602–4608. 10.1074/jbc.M111.312538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer M.; Valenciano A. L.; Kizjakina K.; Qi J.; Sobrado P. (2012) PLoS One 7, e32918. 10.1371/journal.pone.0032918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J. J.; Boechi L.; Andrew McCammon J.; Sobrado P. (2014) Arch. Biochem. Biophys. 544, 128–141. 10.1016/j.abb.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhatwalia R.; Singh H.; Oppenheimer M.; Karr D. B.; Nix J. C.; Sobrado P.; Tanner J. J. (2012) J. Biol. Chem. 287, 9041–9051. 10.1074/jbc.M111.327536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhatwalia R.; Singh H.; Oppenheimer M.; Sobrado P.; Tanner J. J. (2012) Biochemistry 51, 4968–4979. 10.1021/bi300498c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhatwalia R.; Singh H.; Solano L. M.; Oppenheimer M.; Robinson R. M.; Ellerbrock J. F.; Sobrado P.; Tanner J. J. (2012) J. Am. Chem. Soc. 134, 18132–18138. 10.1021/ja308188z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC, Portland, OR. [Google Scholar]
- Pierdominici-Sottile G.; Cossio Perez R.; Galindo J. F.; Palma J. (2014) PLoS One 9, e109559. 10.1371/journal.pone.0109559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Gauld J. W. (2012) J. Phys. Chem. B 116, 14040–14050. 10.1021/jp310952c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.