

Abstract
Antibodies are widely used biomarkers for the diagnosis of many diseases. Assays based on solid-phase immobilization of antigens comprise the majority of clinical platforms for antibody detection, but can be undermined by antigen denaturation and epitope masking. These technological hurdles are especially troublesome in detecting antibodies that bind nonlinear or conformational epitopes, such as anti-insulin antibodies in type 1 diabetes patients and anti-thyroglobulin antibodies associated with thyroid cancers. Radioimmunoassay remains the gold standard for these challenging antibody biomarkers, but the limited multiplexability and reliance on hazardous radioactive reagents have prevented their use outside specialized testing facilities. Here we present an ultrasensitive solution-phase method for detecting antibodies, termed antibody detection by agglutination-PCR (ADAP). Antibodies bind to and agglutinate synthetic antigen–DNA conjugates, enabling ligation of the DNA strands and subsequent quantification by qPCR. ADAP detects zepto- to attomoles of antibodies in 2 μL of sample with a dynamic range spanning 5–6 orders of magnitude. Using ADAP, we detected anti-thyroglobulin autoantibodies from human patient plasma with a 1000-fold increased sensitivity over an FDA-approved radioimmunoassay. Finally, we demonstrate the multiplexability of ADAP by simultaneously detecting multiple antibodies in one experiment. ADAP’s combination of simplicity, sensitivity, broad dynamic range, multiplexability, and use of standard PCR protocols creates new opportunities for the discovery and detection of antibody biomarkers.
Short abstract
We have developed a qPCR assay to ultrasensitively detect antibodies using antigen−DNA conjugates. The multivalency and agglutination propensity of antibodies drive a proximity effect to facilitate generation of DNA amplicons upon ligation.
Introduction
Circulating antibodies represent one of the most prevalent classes of biomarkers for human disorders including infectious,1 autoimmune,2 neurological,3 and oncological4,5 diseases. Detection of low-abundance antibodies using highly sensitive assays improves patient outcomes significantly by enabling early diagnosis and therapeutic intervention.4−6 However, the physical deformation of antigen upon immobilization on solid supports impedes the detection of many disease-specific antibodies by enzyme-linked immunosorbent assays (ELISAs), protein microarrays, lateral flow assays, or immuno-PCR.7−16 Furthermore, the unpredictable orientation of surface-deposited antigen can conceal important epitopes for antibody binding.17
Solution-phase approaches to antibody detection offer significant advantages. The solution-phase radioimmunoassay (RIA) is the current gold standard detection method for antibodies that exclusively bind intact antigen,7 such as anti-insulin autoantibodies used for the early detection of type 1 diabetes.9,10 RIAs are more sensitive than ELISAs but use hazardous radioactive reagents and demand laborious washing and centrifugation steps. Additionally, the limited multiplexing capacity of RIA hinders its application to the discovery of new antibody biomarkers. Consequently, current methods do not meet the need for an assay that preserves the native conformation of antigens and enables sensitive, multiplexed detection of their cognate antibodies. Such a method would greatly improve diagnostic strategies for diseases with conformation-sensitive antibody biomarkers and accelerate the discovery of underexplored biomarkers in various human pathologies.
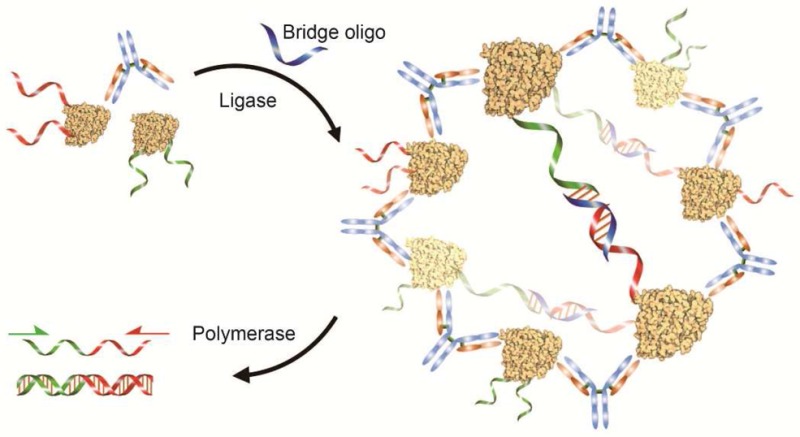
We report the development of a new assay, antibody detection by agglutination-PCR (ADAP), for the robust and rapid detection of antibodies in a solution-phase format (Figure 1). We took inspiration from two distinct assay formats: (1) the classic latex agglutination assay,18 where serum antibodies cluster antigen-latex particles into optically detectable complexes, and (2) proximity ligation assays in which protein–protein complexes are detected by PCR amplification.19−22 ADAP harnesses the agglutination power of antibodies to aggregate antigen–DNA conjugates and thereby drive ligation of oligonucleotides, in turn producing an amplifiable PCR amplicon (Figure 1). The ligation event converts the PCR-incompetent half-amplicons on each antigen–DNA conjugate into a new and distinct PCR reporter.19 Notably, this solution-phase step preserves the antigen’s native conformation and eliminates the need for washing and centrifugation protocols to remove unbound secondary reporters.19 These features significantly improved sensitivity over existing techniques while only requiring slight modifications to a standard PCR protocol.
Figure 1.

Schematic representation of antibody detection by agglutination-PCR (ADAP). (a) The sample containing the target antibody analyte is incubated with a pair of antigen–DNA conjugates. Each conjugate bears an oligonucleotide sequence comprising either the 5′-(red) or 3′-(green) half of a full amplicon. (b) Next, antibodies within the sample agglutinate the antigen–DNA conjugates and position them for ligation upon the addition of a bridging oligonucleotide (blue) and DNA ligase. (c) The newly generated amplicon (red/green) is exponentially amplified with primers that bind their respective sites (red and green arrows) and quantified by real-time qPCR. The immune complex of antibodies and antigen–DNA conjugates shown here represents the proposed mechanism for detecting polyclonal antibodies with relatively large antigens at high concentrations. For monoclonal and anti-small molecule antibody detection, as well as when antibody concentration is significantly lower than that of antigen–DNA conjugates, the complex likely consists of a single antibody bound to two antigen–DNA conjugates (Figure S5).
Results
Synthesis of Antigen–DNA Conjugates
Central to a sensitive ADAP assay is the creation of antigen–DNA conjugates. For protein antigens, we synthesized these components by lysine-to-thiol cross-linking using sulfosuccinimidyl 4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (sulfo-SMCC) and thiolated oligonucleotides.20 Briefly, maleimides were installed on lysines of purified antigen by reaction with sulfo-SMCC in PBS (Figure S1). Thiolated oligonucleotides were activated by dithiothreitol (DTT)-mediated reduction. Both antigen and oligonucleotides were desalted, pooled, and allowed to react overnight. Unreacted reagents were removed by extensive purification with size-exclusion spin columns. Antigen-DNA conjugation ratios were determined by UV–vis spectroscopy and by SDS-PAGE analysis. Typically, a 1:2 antigen-to-DNA conjugation ratio yielded the optimal signal in ADAP assays. We found that greater degrees of antigen–DNA conjugation can mask epitopes for antibody binding and thus lead to reduced assay sensitivity (Figure S11).
For small molecule antigens, N-hydroxysuccinimidyl (NHS) ester-activated derivatives were incubated with amine-modified oligonucleotides in a one-step conjugation (Figure S5a). The resulting small molecule–DNA conjugates were characterized by high-resolution mass spectrometry. In contrast to protein-based antigens, small molecules contain far fewer antibody epitopes due to their size. It is thus critical to design conjugation sites that still preserve the accessibility of epitopes to antibodies. For the dinitrophenol (DNP)–DNA conjugate (Figure 4a and Figure S5), we used the same conjugation site that was used to generate the immunogen for the antibody we tested (a DNP–BSA conjugate in which DNP was linked to lysine side chains).23,24
Figure 4.
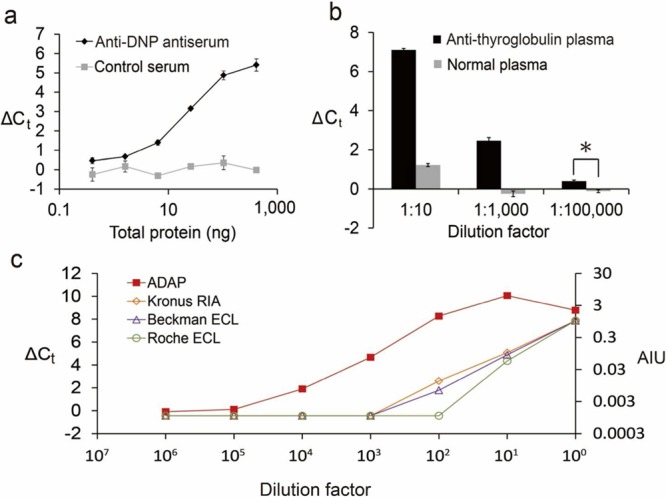
Detection of antibodies in mouse serum or human patient plasma and comparison with commercial diagnostics. (a) Detection of anti-dinitrophenol (DNP) from rabbit antiserum. Antiserum was serially diluted into PBS and analyzed by ADAP. A dilution series of antigen-naïve serum was analyzed as a negative control. (b) Detection of conformation sensitive antithyroglobulin from patient plasma. Antithyroglobulin-positive patient plasma was diluted into PBS as indicated and analyzed by ADAP. Antithyroglobulin-negative plasma from healthy subjects was analyzed as a negative control. (*p < 0.01) (c) Identical samples of antithyroglobulin-positive human plasma were analyzed by ADAP, an FDA-approved radioimmunoassay (Kronus RIA) and two electrochemiluminescent assays (Beckman and Roche ECL).
ADAP Workflow
In a typical ADAP experiment (Figure 1), pairs of antigen–DNA conjugates are diluted in buffer. One antigen–DNA conjugate bears the 5′ half of a PCR amplicon, while the other conjugate bears the 3′ half that is 5′ phosphorylated to enable ligation. The pooled conjugates are added to 2 μL of antibody-containing analyte and incubated for 30 min to allow binding. Next, DNA ligase and a bridging oligonucleotide are added and incubated for 15 min. Following selective hydrolysis of the bridging oligonucleotide, the ligation mixture is preamplified, and the resulting products are analyzed by qPCR. As high Ct values of qPCR are associated with low assay reproducibility,25 we included a preamplification step in the ADAP protocol to ensure high reproducibility and low intra-assay (<1%) and interassay (<3%) variations.42
Assay Validation and Specificity/Sensitivity Analysis
As a first target, we synthesized insulin–DNA conjugates to detect anti-insulin antibodies. Insulin autoantibodies are an important early biomarker of type 1 diabetes,9 but the development of a standard immunoassay is thought to be frustrated by the denaturation of insulin on solid supports.10,26 Currently, RIA is the principal technology for detecting insulin autoantibodies.10,26 A solution-phase PCR assay would reduce the amount of time needed for the test and remove the requirement of radioactive reagents.
We serially diluted affinity-purified anti-insulin antibodies into various biological matrices and analyzed them by ADAP. We observed a dose-dependent response across 5 orders of magnitude with very similar results obtained in different biological diluents (Figure 2a). The detection limit in serum was found to be 170 zeptomoles of antibody in a 2 μL sample (Table S1). We performed a head-to-head comparison with a direct ELISA and found an 865-fold improvement in limit of detection (Figure 2b and Table S1). The specificity of ADAP was determined by assaying samples containing isotype control antibodies, which yielded no detectable signal (Figure 2c). Similarly, no detectable signal was observed when the assay was performed with irrelevant antigen–DNA conjugates (Figure S12). These results demonstrate that ADAP detects target antibodies with superior sensitivity and specificity over traditional methods while using much smaller sample volumes.
Figure 2.

Sensitivity and specificity of ADAP. (a) Detection of serially diluted purified anti-insulin antibodies in phosphate-buffered saline (PBS) or bovine serum. The x-axis displays the moles of antibody in a 2 μL sample. The y-axis is ΔCt calculated by the difference of Ct value between the sample and a blank. (b) Head-to-head comparison with an ELISA for the detection of anti-insulin antibody. The right y-axis represents arbitrary intensity units (AIU) from the ELISA. (c) The specificity of ADAP was investigated by analysis of serially diluted isotype IgG in serum. No detectable signal was observed. Error bars represent standard deviation from triplicate samples, but for many data points are too small to be visualized.
To show that ADAP scales over a broad range of antigen molecular weights, we assayed antigen–antibody pairs for biotin (∼0.24 kDa), GFP (26 kDa), and mouse IgG (150 kDa). For all three pairs, ADAP consistently detected low attomoles of antibody (Figure 3, Table S1 and Figures S2–S4).
Figure 3.

ADAP detects zeptomoles to attomoles of antibodies that bind antigens across a wide molecular weight distribution. The limits of ADAP detection for antibiotin, anti-insulin, anti-GFP, and antimouse IgG antibodies (antigen molecular weights of 0.24, 5.8, 27, and 150 kDa respectively) was determined by analyzing antibodies added into PBS or bovine serum. Error bars represent the standard deviation from triplicate samples.
Detection of Serum-Derived Antibodies against Small Molecules
Antibodies against small molecules can mediate allergic responses to drugs, particularly those capable of covalently modifying host proteins.24,27 However, the detection of small molecule binding antibodies by solid-phase immunoassay is challenging. Small molecules do not adsorb readily to plastics used in common immunoassays and therefore require specialized surfaces to produce an appropriate substrate.28 We were curious whether anti-small molecule antibodies could be detected by ADAP given the limited ability of such a conjugate to form large aggregates (Figure S5b). As a model system, we synthesized DNP–DNA conjugates and incubated them with rabbit antisera from animals inoculated with DNP–BSA. Significantly, agglutination was detected with as little as 0.74 ng of total antiserum protein (Figure 4a and Figure S5 and Table S1). This experiment demonstrates that ADAP can sensitively detect natively produced antibodies from whole serum and has the potential to monitor allergic responses to small molecules.
ADAP Is 1000-fold More Sensitive than Clinically Used Techniques
Next, we used ADAP to detect antibodies directly from patient samples. Thyroglobulin autoantibodies mediate and are diagnostic of autoimmune thyroiditis.29 They can also be a critical biomarker for monitoring thyroid cancer recurrence following therapeutic thyroidectomy.12 A widely applicable, sensitive, and accurate detection assay for anti-thyroglobulin autoantibodies could prevent misdiagnosis of cancer recurrence and unnecessary treament for healthy patients.12 Currently, radioimmunoassays remain the gold standard for detecting this autoantibody.12 However, only specialized laboratories retain the full capacity to perform this test, as stringent regulations for use and disposal of radioactive reagents limit widespread adoption. We analyzed anti-thyroglobulin-positive patient plasma by ADAP with thyroglobulin–DNA conjugates using healthy human plasma as a negative control. A robust ADAP signal was observed from the anti-thyroglobulin-positive samples (2 μL) down to 105-fold dilution with nearly no background from healthy controls (Figure 4b). Identical samples were assayed using three FDA-approved clinical laboratory assays: the Kronus/RSR radioimmunoassay and two electrochemiluminescence assays (Beckman Coulter and Roche). Impressively, ADAP detected anti-thyroglobulin antibodies with a detection limit 3–4 orders of magnitude lower than these standard assays (Figure 4c).
Circumventing Interference from Anti-DNA Autoantibodies
One potential confounding issue for ADAP is the interference from endogenous anti-DNA autoantibodies. These antibodies might agglutinate antigen–DNA conjugates in an antigen-agnostic manner and result in false positives. Patients suffering from autoimmune disorders such as systemic lupus erythematosus (SLE) often produce anti-DNA antibodies in high titer.30 They are also generally present in small quantities in about 10% of healthy adults.31 We obtained patient plasma that was independently verified to harbor anti-DNA antibodies, as well as normal plasma with much lower levels of anti-DNA antibodies as a negative control. We used GFP–DNA conjugates as a control antigen to observe the extent of interference from anti-DNA autoantibodies, since there should be no naturally occurring anti-GFP antibodies in human plasma.
As expected, we observed strong signal from anti-DNA-positive patient plasma and weak yet robust signal from normal plasma (Figure 5a), demonstrating that these antibodies can interfere with ADAP analysis. Interestingly, after adding in anti-GFP antibodies, identical dose–response curves were observed for both anti-DNA-positive patient plasma and normal plasma (Figure S6). This observation is consistent with the notion that high affinity anti-GFP antibodies dominate the agglutination event and ADAP signal, regardless of the presence of anti-DNA antibodies.
Figure 5.

Circumventing interference from anti-DNA autoantibodies by competition with free DNA. (a) Investigation of interference from anti-DNA autoantibodies. GFP–DNA conjugates were used to analyze anti-DNA-positive patient plasma and healthy normal plasma. Patient samples were grouped into those containing anti-single-stranded DNA antibodies (ssDNA) and those with anti-dsDNA antibodies (dsDNA). Interference was observed at dilution factors of 1 and 10 for all sample types (b) Competitor DNA was titrated into undiluted patient and normal plasma. The addition of competitor DNA eliminated background signal from interfering antibodies. (c) The experiment in (a) was repeated but with the addition of 100 μM competitor DNA which eliminated interference. (d) Purified GFP antibodies were added to anti-DNA positive and normal plasma. Detection of GFP antibodies was performed in the presence of 100 μM competitor DNA in all samples to confirm that it did not disrupt ADAP performance.
In an abundance of caution, we sought a general solution to circumvent potential interference from anti-DNA autoantibodies. To this end, we titrated in free DNA as a competitor to “protect” the antigen–DNA conjugates from counterfeit aggregation (Figure 5b). At 100 μM of the competitor DNA, we no longer observed spurious signal from anti-DNA antibodies (Figure 5b,c). To validate that competitor DNA does not otherwise complicate ADAP performance, both anti-GFP antibodies and competitor DNA were added to anti-DNA positive plasma and normal plasma (Figure 5d). ADAP analysis of these samples showed the expected dose response with no interference from anti-DNA antibodies. The limit of detection of anti-GFP antibodies in human plasma was similar to that in buffer (48 and 27 attomoles, respectively). Together, these results demonstrate that the addition of competitor DNA allows us to circumvent interference in human plasma samples.
Multiplexed Detection of Antibodies by DNA Barcoding
Multiplexed detection of several antibodies can be accomplished by use of orthogonal DNA sequence pairs to barcode distinct antigens. Diseases such as type 1 diabetes have multiple autoantibody biomarkers (anti-insulin, anti-IA-2, anti-GAD, anti-ZnT8).9 Several clinical protocols use antibody panels to establish a diagnosis. A barcoded assay could help by detecting antibodies in a single test.
We generated a set of orthogonal antigen–DNA conjugates either with biotin (Sequence Set 1; Table S2) or mouse IgG (Sequence Set 2; Table S2) as the antigen. The amplicons were designed such that Set 1 primers did not amplify the Set 2 amplicon and vice versa. The two sets of antigen–DNA conjugates were pooled and incubated with anti-biotin antibodies, anti-IgG antibodies, or both and then analyzed by ADAP. The sample incubated with the anti-biotin antibodies showed signal only when analyzed with Set 1 primers, while the sample incubated with the anti-mouse IgG antibodies showed signal only with the Set 2 primers. The mixed sample containing both antibodies generated signal when analyzed with both sets of primers (Figure 6a and Figure S7). Importantly, there was no detectable cross-talk in this multiplexed experiment.
Figure 6.

ADAP can be multiplexed. (a) Detection of two orthogonal antibodies in one ADAP experiment. Biotin–DNA and mouse IgG–DNA conjugates bearing either DNA sequence 1 or 2 (Table S2), respectively, were incubated with either anti-biotin antibody only, antimouse IgG antibody only, or both antibodies together, and then analyzed by ADAP. (b) Multiplexed detection of anti-antigen antibody and total antibody levels by ADAP and proximity ligation assay (PLA), respectively. Biotin–DNA conjugates and anti-IgG–DNA conjugates were incubated with samples containing constant total IgG but varied fractions of anti-biotin antibodies. These samples were analyzed by ADAP and PLA. Error bars represent the standard deviation from triplicate samples, but for many data points are too small to be visualized.
Additionally, typical antibody tests do not take into account total immunoglobulin concentration. This can lead to false negatives for patients with immunoglobulin deficiency, which is a common problem in Celiac disease.32 We envisioned that simultaneous detection of antigen-binding capacity and total immunoglobulin content could differentiate false negatives from abnormally low immunoglobulin levels.
To multiplex the detection of total IgG and antigen-binding ability, we generated anti-IgG antibody–DNA conjugates from a single batch of anti-IgG polyclonal antibodies. The batch was split into two pools, and each was modified with either the upstream or downstream fragment of the Set 2 PCR amplicon. As in proximity ligation,19−22 the two halves of the amplicon are brought close together when the polyclonal antibody–DNA conjugates bind nearby epitopes, allowing for ligation and subsequent detection by PCR. Goat anti-biotin antibodies were diluted into total goat IgG such that the total amount of IgG remained constant, but the anti-biotin fraction varied. ADAP analysis with the Set 2 primers showed no change in signal, corresponding to the constant concentration of IgG in every sample, whereas signal generated from the Set 1 primers increased as the fraction of anti-biotin antibodies increased (Figure 6b and Figures S8–9). This shows that detection of the total antibody levels can be multiplexed with detection of antigen-specific antibodies.
Effect of Antibody Valency and Clonality on ADAP Performance
Finally, we wished to investigate the impact of antibody valency and clonality on the performance of ADAP. Serum antibodies are multivalent and polyclonal to allow optimal agglutination of pathogens for effective neutralization and clearance.33 However, the limited agglutination power of Fab fragment and monoclonal antibodies33 might preclude them from ADAP-based detection.
We incubated mouse IgG–DNA conjugates either with bivalent anti-mouse IgG or the corresponding monovalent Fab fragment and analyzed them by ADAP. As expected, robust signal was detected for the anti-mouse IgG sample and no signal from the Fab sample (Figure 7a). Next, we incubated either polyclonal or monoclonal anti-GFP antibodies with GFP–DNA conjugates. Interestingly, both antibodies displayed similar limits of detection (Figure 7b), but with very different dynamic ranges (about 6 or 4 orders of magnitude for polyclonal or monoclonal antibody, respectively). We hypothesized that this difference was due to the saturation of binding sites when the concentration of the antigen–DNA conjugates matches that of the antibody analytes (the “hook effect”).34 While the monoclonal antibody shows classic hook behavior when the antibody concentration (1.3 nM) is close to the antigen–DNA conjugate concentration (0.5 nM), the polyclonal antibody hook effect is delayed until a much higher antibody concentration (64 nM). We attribute this delayed hook effect to the availability of multiple antigen binding sites with polyclonal antibody. Polyclonal antibodies enjoy a higher effective epitope concentration and thus a wider dynamic range. These results demonstrate ADAP is well-suited for the detection of both poly- and monoclonal antibodies.
Figure 7.

Effects of valency and clonality of the target antibody on ADAP performance. (a) Mouse IgG–DNA conjugates were incubated either with polyclonal antimouse IgG antibodies, monovalent Fab fragment antimouse IgG antibodies, or a control Fab fragment that recognizes unrelated antigens, and analyzed by ADAP. (b) GFP–DNA conjugates are incubated either with polyclonal or monoclonal anti-GFP antibodies and analyzed by ADAP. Error bars represent the standard deviation from triplicate samples, but for many data points are too small to be visualized.
Discussion
Of all the protein types one might want to detect in a clinical setting, antibodies are by far the most numerous.1−6 They are used as biomarkers of autoimmune diseases, cancers, infectious diseases, neurological disorders, and vaccine efficacy.1−6 Despite the high value of antibodies as clinical diagnostic targets, conventional assays for their detection such as solid-phase ELISAs or RIAs have significant deficiencies. The ADAP technology uniquely addresses these limitations, being operationally simple, ultrasensitive, and multiplexable, as well as applicable to antibodies that recognize conformation-dependent epitopes.
Previous applications of proximity ligation share the common format of using DNA-conjugated antibodies to detect an analyte of interest.19−22 ADAP inverts this scenario, using a DNA-conjugated antigen for detection of antibodies. The assay exploits the intrinsic multivalency of antibodies to drive the proximity effect. The impressive dynamic range of ADAP appears related to the ability of antibodies to aggregate their antigens, as suggested by the superior performance of poly- versus monoclonal antibodies (Figure 7b). This mechanism is unique to antibody detection and to ADAP.
The advantages of the ADAP platform for antibody detection are considerable. As a solution phase assay, ADAP circumvents the protein denaturation and epitope masking problems of surface-immobilized-antigen based formats. While solution-phase assays such as the radioimmunoassay exist, they are difficult to perform, are slow, and have limited capacity for multiplexing. ADAP is 3 orders of magnitude more sensitive than clinically used assays, creating new possibilities for the early detection and treatment of disease. As a no-wash assay, ADAP eliminates the tedious optimization of washing and centrifugation steps that is necessary to minimize the loss of low-affinity antibodies. It does not require isolation of unique monoclonal antibodies as required for certain types of ELISAs. Since the ADAP does not rely on animal antibodies as capture reagents, it obviates interference from patient heterophilic antibodies.35
The reduction in sample consumption and multiplexing capability lessen the demand for patient serum to promote patient compliance in applications requiring repeated monitoring. Significantly, ADAP is readily deployable in many clinical settings, as it uses only conventional PCR equipment and reagents, which are standard devices in diagnostic laboratories. The custom reagents (antigen–DNA conjugates, ligation enzymes, and a bridging oligonucleotide) are used in ultralow quantities. For example, 100 μg of a 60 kDa antigen-DNA conjugate is sufficient to perform approximately ∼1.7 million assays.
Infectious diseases such as HIV increasingly rely on combined antibody and antigen tests to improve confidence in diagnosis.36 Combined with traditional proximity ligation19−22 and PCR tests, ADAP opens the possibility of performing all three types of tests (genome-derived nucleic acids, antigens, and antibodies) in a unified platform. ADAP could also be easily adapted to any number of novel point-of-care PCR platforms to provide highly sensitive solution phase antibody tests in low resource settings. Because of these favorable attributes, its operational simplicity, and the leveraging of existing technology, we predict that ADAP will provide a useful analytical platform for a multitude of clinical and research applications.
Methods
Synthesis of Antigen–DNA Conjugates
Insulin–DNA conjugate was synthesized by resuspending recombinant insulin (Sigma-Aldrich) to make a 1 mg/mL solution in reaction buffer (55 mM sodium phosphate, 150 mM sodium chloride, 20 mM EDTA, pH 7.2). One μL of a 4 mM solution of sulfo-SMCC (Pierce Biotechnologies) in anhydrous DMSO was added to 10 μL of the protein solution and incubated at RT for 2 h. Thiolated-DNA (IDT) was resuspended to 100 μM in reaction buffer. Three microliters of the 100 μM thiolated-DNA stock was then added to 50 μL of reaction buffer. To this solution, 4 μL of a 100 mM solution of DTT (Life Technologies) was added to reduce the oxidized thiolated-DNA. The solution was then incubated at 37 °C for 1 h. 7k MWCO gel microspin columns (Life Technologies) were equilibrated with reaction buffer. The reduced oligonucleotides were desalted by the equilibrated microspin columns twice. Unreacted sulfo-SMCC was removed from the insulin solution by a 3k MWCO centrifugal filter column (EMD Millipore) to a final volume of 60 μL. The thiolated-DNA and insulin solutions were then mixed, reacted overnight at 4 °C, and then purified by 10k MWCO filter column. Conjugate concentrations were determined by BCA assay (Life Technologies). Conjugation efficiencies were analyzed by SDS-PAGE and silver staining as described previously.37 A representative silver-stain is shown in Figure S1. DNA-to-antigen ratios of the conjugates were estimated by UV–vis absorption. Antigen–DNA conjugates were stored at 4 °C for short-term usage or aliquoted for long-term storage at −80 °C. GFP-, mouse-IgG-, and thyroglobulin–DNA conjugates were synthesized similarly with slight modifications. Briefly, unreacted SMCC was filtered by 7k MWCO gel microspin columns. Conjugates were purified from unconjugated DNA by centrifugal filter columns (GFP, 30k MWCO column; mouse IgG, 100k MWCO column; thyroglobulin, 100k MWCO column).
Finally, biotin–DNA conjugates was purchased from IDT. DNP–DNA conjugates were synthesized as follows.Twenty-five milligrams DNP-NHS ester (Life Technologies) was dissolved in anhydrous DMSO to make a 50 mM solution. 5′ or 3′ amine functionalized DNA (IDT) was resuspended in ddH2O to make a 1 mM solution. 40 μL of the 1 mM DNA solution was added to 300 μL of PBS with 50 mM NaHCO3. 80 μL of the NHS ester solution was added over 2 days at RT under constant rotational mixing. Modified DNA was then precipitated by adding 2.5 vol of ethanol and 0.1 vol of 10 M ammonium acetate and then incubated for 4 h. Precipitated DNA was pelleted by centrifugation for 15 min at 4 °C, followed by a gentle wash in ice cold 70% ethanol-H2O. The pellet was then resuspended in 100 μL of ddH2O and then purified again by precipitation as before to ensure complete removal of unreacted small molecules. After the second precipitation, the pellet was diluted in ddH2O to make a 100 μM stock solution, which was stored at −20 °C until used. Synthesis was confirmed by high resolution LC-MS.
Antibody Detection by Agglutination-PCR (ADAP)
One fmole of paired antigen–DNA conjugates was resuspended in 2 μL of incubation buffer C (2% BSA, 0.2% Triton X-100, 8 mM EDTA in PBS). Two microliters of analyte was added to the conjugates and then incubated at 37 °C for 30 min. 116 μL of ligation mix (20 mM Tris, 50 mM KCl, 20 mM MgCl2, 20 mM DTT, 25 μM NAD, 0.025 U/μL ligase, 100 nM bridge oligo, 0.01% BSA, pH = 7.5) was added, and then incubated at 30 °C for 15 min. Ten microliters uracil-excision mix (0.025 U/μL Epicenter Bio) was added and incubated for 15 min at 30 °C. Twenty-five microliters of the solution was added to 25 μL of 2x PCR Master Mix (Qiagen) with 10 nM primers and then amplified by PCR (95 °C for 10 min, 95 °C for 15 s, 60 °C for 30 s 12 cycles). The reaction was then diluted 1:20 in ddH2O. 8.5 μL of the diluted PCR samples were added to 10 μL of 2x qPCR Master Mix (Life Technologies) with 1.5 μL of primers (final concentration 690 nM). qPCR was performed on either a Bio-Rad CFX96 or a Bio-Rad iQ5 real-time PCR detection system.
The ADAP assays for affinity purified anti-insulin (Abcam), anti-biotin (Abcam), anti-GFP (Vector Laboratories), and anti-mouse IgG antibodies (Pierce Biotechnologies) were carried out as described above with the following modifications. For dilution in buffer, 2 μL of antigen–DNA conjugates was mixed with 2 μL of serial diluted antibodies (concentration range: 102–10–4 μg/mL) in buffer C or buffer only (blank). For dilution in fetal bovine serum (Sigma-Aldrich), antibodies were spiked in fetal bovine serum to obtain 2 wt %/wt antibodies solution, which was then serial diluted in buffer C (concentration range: 102–10–4 μg/mL) for ADAP assay. Isotype antibodies (Santa Cruz Biotech) subjected to the same preparation were analyzed side-by-side as negative controls.
ADAP Detection Assay for Anti-DNP Antibodies from Antiserum
The ADAP PCR detection assay for anti-DNP antiserum (Abcam) was carried out as described above with the following modifications. Anti-DNP antiserum was obtained from rabbit inoculated with DNP-conjugated carrier proteins without further purification. Two microliters of DNP–DNA conjugates was mixed with 2 μL of serial diluted anti-DNP antiserum (concentration range: 0.4–0.0002 mg/mL) in buffer C for ADAP detection.
ADAP Detection Assay for Anti-Thyroglobulin Patient Plasma
The ADAP detection assay for anti-thyroglobulin positive patient plasma (ImmunoVision) was carried out as described above with the following modifications. Two microliters of thyroglobulin–DNA conjugates were mixed with 2 μL of serially diluted patient plasma (dilution factor: 100–106) in buffer C for ADAP detection.
Multiplexed ADAP for Anti-Biotin and Anti-Mouse IgG Antibodies
Three sets of ADAP experiments were carried out to investigate the orthogonality of anti-biotin and anti-mouse IgG antibody detection. The multiplex ADAP assay for anti-biotin and anti-mouse IgG antibody was carried out as described above with the following modifications. 1 μL of biotin–DNA conjugates (sequence 1 as in Table S2) and 1 μL mouse-IgG–DNA conjugates (sequence 2 as in Table S2) are mixed with 2 μL of serial diluted either (1) anti-biotin antibodies (concentration range: 102–10–4 μg/mL) in buffer C or buffer only (blank) (2) anti-mouse antibodies (concentration range: 102–10–4 μg/mL) in buffer C or buffer only (blank) (3) both anti-biotin and anti-mouse antibodies (concentration range: 102–10–4 μg/mL) in buffer C or buffer only (blank). The antigen and antibody mixtures were processed and analyzed as described above.
Multiplexed ADAP and PLA Detection for Anti-Biotin Antibodies and Total IgG
ADAP and PLA19−22 (proximity ligation assay) were used in conjunction to quantify the specific antibodies and total antibodies amounts in a given sample. The multiplex ADAP detection assay for anti-biotin and total IgG was carried out as described above with the following modifications. 1 μL biotin-DNA conjugates (sequence 1) and 1 μL anti-goat-IgG–DNA conjugates (sequence 2) are mixed with 2 μL of serially diluted either (1) goat anti-biotin antibodies (concentration range: 102–10–4 μg/mL) in buffer C or buffer only (blank) (2) goat IgG (concentration range: 102–10–4 μg/mL) in buffer C or buffer only (blank) (3) both anti-biotin and goat IgG in buffer C, where total IgG is fixed at 0.7 μg/mL and anti-biotin antibodies fraction varied from 100%–0% or buffer only (blank). The antigen and antibody mixtures were processed and analyzed as described above.
Direct ELISA Detection of Anti-Insulin Antibodies
Recombinant human insulin (Sigma) was resuspended to 1 mg/mL in PBS. 75 μL of the insulin solution was added to wells of an ELISA plate (Santa Cruz Biotech). The plate was covered with a plastic membrane, and the insulin was allowed to adsorb to the plate overnight at 4 °C. Excess supernatant was decanted, and the wells were blocked with 5% BSA in PBS overnight at 4 °C. Anti-insulin antibodies were diluted into PBS and allowed to bind at RT for 1 h. The supernatant was decanted and the wells were washed 4× with PBS. Secondary antibody-HRP probes (Santa Cruz Biotech) were diluted 1:5000 in 5% BSA in PBS and added to the wells and allowed to incubate at RT for 1 h. The supernatant was decanted and then washed 4× in PBS. 50 μL of TMB substrate solution as added and allowed to develop for 15 min and then quenched by addition of 50 μL of 2 M H2SO4. Absorbance was read at 450 nm in a plate reader.
Circumventing Interference from Anti-DNA Antibodies
Anti-DNA antibodies positive patient
plasmas were purchased from
ImmunoVision. ADAP detection of anti-DNA plasma was carried out as
described above with slight modifications. For detection of anti-GFP
antibodies, anti-GFP antibodies are spiked into anti-DNA and normal
plasma. A sample of 2 μL serial diluted anti-GFP solution is
incubated with 2 μL solution containing GFP–DNA conjugates
and with or without 100 μM competition DNA. The competition
DNA is purchased from IDT with the sequence below:
Radioimmunoassay and Electrochemiluminescent Assays for Anti-Thyroglobulin Autoantibodies
Tg-RIAs (Kronus), the Beckman Access TgAb (Beckman Coulter) and Roche Elecsys TgAb (Roche Diagnostics) were performed per the manufactures’ instructions at University of Southern California. These assays are standardized against WHO reference serum 65/93. The assays were performed as previously described.38
Data Analysis
Three replicate ADAP measurements were carried out for each antibody sample in buffer C in addition to a blank. The replicates were measured by taking aliquots from the same dilution series and the same preparation of ligation, excision and preamplification steps but placing them in three different wells for qPCR analysis. A representative real-time qPCR measurement plot taken from an ADAP assay for the serial dilution of an anti-biotin antibody is shown in Figure S10. A single threshold fluorescence value was automatically chosen by Bio-Rad software. For each curve, the PCR cycle number with the fluorescence value corresponding to the chosen threshold value was defined as the cycle threshold (Ct) value. ΔCt is defined as the Ct value of the blank minus the Ct value of the samples.39 The value of ΔCt is proportional to the initial amplicon concentration. This amplicon concentration is then also proportional to the amount of target antibodies present in the initial dilution series. A volume of 2 μL from each serial dilution series was taken for ADAP measurement. Thus, the number of antibody molecules in each measurement is (2 × 10–6 L) × antibody concentration (M) × Avogadro’s number. A nonlinear four parameter logistic fit40 for an antibody dilution series is determined using custom software. The limit of detection for the ADAP assay is defined as the average ΔCt value of the buffer C only blank plus 3 standard deviations of the blank.41 The value of each limit of detection is calculated relative to the corresponding blank.
Intra-Assay and Interassay Variation for ADAP
The intra-assay variation for ADAP was determined by repeating ADAP measurements in triplicate for anti-GFP antibodies six times on the same plate. The intra-assay variation is defined as standard deviation of the triplicate divided by mean of the triplicate42 and is consistently <1%. The interassay variation for ADAP is evaluated by measuring anti-GFP antibody concentrations in triplicate on five different plates on different days. The interassay variation defined by standard deviation of the concentrations from five different plates divided by the mean of concentrations from five different plates42 and is <3%. Both the intra-assay and interassay variation of ADAP are far below the accepted biomedical assay variation values, which are 10% and 15% respectively.42 ADAP’s superior intra-assay and interassay performance is likely a result of having fewer overall handling steps, no wash steps, and no centrifugation steps. The extensive washing and centrifuging requirements for other assays might compromise their precision and reproducibility.
Acknowledgments
We graciously acknowledge M. C. Y. Chang and M. C. Hammond for technical assistance. We thank the UC Berkeley QB3/Chemistry mass spectrometry facility for help with characterization of small molecule–DNA conjugates. We also thank B. Belardi, C. M. Woo, M. Boyce, and P. J. Utz for fruitful discussions and D. Fox, A. E. Herr, and S. W. Lee for critical review of the manuscript. This work was funded by NIH Grant R37 GM058867 and R21 DK108781 to C.R.B.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00340.
Additional experimental methods and figures, DNA sequences, and synthetic schemes (PDF)
Author Contributions
# C.T. and P.V.R. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Bai T.; Zhou J.; Shu Y. Serologic study for influenza A (H7N9) among high-risk groups in China. N. Engl. J. Med. 2013, 368, 2339–2340. 10.1056/NEJMc1305865. [DOI] [PubMed] [Google Scholar]
- Meroni P. L.; Biggioggero M.; Pierangeli S. S.; Sheldon J.; Zegers I.; Borghi M. O. Standardization of autoantibody testing: a paradigm for serology in rheumatic diseases. Nat. Rev. Rheumatol. 2013, 10, 35–43. 10.1038/nrrheum.2013.180. [DOI] [PubMed] [Google Scholar]
- Reddy M. M.; Wilson R.; Wilson J.; Connell S.; Gocke A.; Hynan L.; German D.; Kodadek T. Identification of candidate IgG biomarkers for Alzheimer’s disease via combinatorial library screening. Cell 2011, 144, 132–142. 10.1016/j.cell.2010.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. S.; Sibani S.; Wallstrom G.; Qiu J.; Mendoza E. A.; Raphael J.; Hainsworth E.; Montor W. R.; Wong J.; Park J. G.; Lokko N.; Logvinenko T.; Ramachandran N.; Godwin A. K.; Marks J.; Engstrom P.; Labaer J. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J. Proteome. Res. 2011, 10, 85–95. 10.1021/pr100686b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford T. J.; Wang X.; Chinnaiyan A. M. Cancer immunomics: using autoantibody signatures in the early detection of prostate cancer. Urol. Oncol. 2006, 24, 237–242. 10.1016/j.urolonc.2005.11.033. [DOI] [PubMed] [Google Scholar]
- Kudo-Tanaka E.; Ohshima S.; Ishii M.; Mima T.; Matsushita M.; Azuma N.; Harada Y.; Katada Y.; Ikeue H.; Umeshita-Sasai M.; Miyatake K.; Saeki Y. Autoantibodies to cyclic citrullinated peptide 2 (CCP2) are superior to other potential diagnostic biomarkers for predicting rheumatoid arthritis in early undifferentiated arthritis. Clin. Rheumatol. 2007, 26, 1627–1633. 10.1007/s10067-007-0558-5. [DOI] [PubMed] [Google Scholar]
- Liu E.; Eisenbarth G. S. Accepting clocks that tell time poorly: fluid-phase versus standard ELISA autoantibody assays. Clin. Immunol. 2007, 125, 120–126. 10.1016/j.clim.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J. E.; Ni L.; Brown W. R.; Joshi K. S.; Chang J.; Rosenberg B.; Voss E. W. Jr. The immunochemistry of sandwich ELISAs—VI. Greater than 90% of monoclonal and 75% of polyclonal anti-fluorescyl capture antibodies (CAbs) are denatured by passive adsorption. Mol. Immunol. 1993, 30, 1165–1175. 10.1016/0161-5890(93)90135-X. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Kumar R. B.; Dai H.; Feldman B. J. A plasmonic chip for biomarker discovery and diagnosis of type 1 diabetes. Nat. Med. 2014, 20, 948–953. 10.1038/nm.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum C. J.; Palmer J. P.; Kuglin B.; Kolb H. Insulin autoantibodies measured by radioimmunoassay methodology are more related to insulin-dependent diabetes mellitus than those measured by enzyme-linked immunosorbent assay: results of the fourth international workshop on the standardization of insulin autoantibody measurement. J. Clin. Endocrinol. Metab. 1992, 74, 1040–1044. 10.1210/jcem.74.5.1569152. [DOI] [PubMed] [Google Scholar]
- Gentile F.; Conte M.; Formisano S. Thyroglobulin as an autoantigen: what can we learn about immunopathogenicity from the correlation of antigenic properties with protein structure. Immunology 2004, 112, 13–25. 10.1111/j.1365-2567.2004.01861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer C. A. Clinical utility of thyroglobulin antibody (TgAb) measurements for patients with differentiated thyroid cancers (DTC). J. Clin. Endocrinol. Metab. 2011, 96, 3615–3627. 10.1210/jc.2011-1740. [DOI] [PubMed] [Google Scholar]
- Mahler M.; Fritzler M. J. Epitope specificity and significance in systemic autoimmune diseases. Ann. N. Y. Acad. Sci. 2010, 1183, 267–287. 10.1111/j.1749-6632.2009.05127.x. [DOI] [PubMed] [Google Scholar]
- Robinson W. H.; DiGennaro C.; Hueber W.; Haab B. B.; Kamachi M.; Dean E. J.; Fournel S.; Fong D.; Genovese M. C.; de Vegvar H. E.; Skriner K.; Hirschberg D. L.; Morris R. I.; Muller S.; Pruijn G. J.; van Venrooij W. J.; Smolen J. S.; Brown P. O.; Steinman L.; Utz P. J. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 2002, 8, 295–301. 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- Simon-Vecsei Z.; Király R.; Bagossi P.; Tóth B.; Dahlbom I.; Caja S.; Csosz É.; Lindfors K.; Sblattero D.; Nemes É.; Mäki M.; Fésüs L.; Korponay-Szabó I. R. A single conformational transglutaminase 2 epitope contributed by three domains is critical for celiac antibody binding and effects. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 431–436. 10.1073/pnas.1107811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W.; Shi Y.; Baekkeskov S. Autoreactive epitopes defined by diabetes- associated human monoclonal antibodies are localized in the middle and C-terminal domains of the smaller form of glutamate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2832–2836. 10.1073/pnas.90.7.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson G. M.; Matsuura E.; Victoria E. J.; Cockerill K. A.; Linnik M. D. The orientation of β2GPI on the plate is important for the binding of anti- 2βGPI autoantibodies by ELISA. J. Autoimmun. 2002, 18, 289–297. 10.1006/jaut.2002.0590. [DOI] [PubMed] [Google Scholar]
- Adler S. P.; McVoy M.; Biro V. G.; Britt W. J.; Hider P.; Marshall D. Detection of cytomegalovirus antibody with latex agglutination. J. Clin. Microbiol. 1988, 26, 2116–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S.; Gullberg M.; Jarvius J.; Olsson C.; Pietras K.; Gústafsdóttir S. M.; Östman A.; Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Söderberg O.; Gullberg M.; Jarvius M.; Ridderstråle K.; Leuchowius K. J.; Jarvius J.; Wester K.; Hydbring P.; Bahram F.; Larsson L. G.; Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Fredriksson S.; Dixon W.; Ji H.; Koong A. C.; Mindrinos M.; Davis R. W. Multiplexed protein detection by proximity ligation for cancer biomarker validation. Nat. Methods 2007, 4, 327–329. 10.1038/nmeth1020. [DOI] [PubMed] [Google Scholar]
- Darmanis S.; Nong R. Y.; Vänelid J.; Siegbahn A.; Ericsson O.; Fredriksson S.; Bäcklin C.; Gut M.; Heath S.; Gut I. G.; Wallentin L.; Gustafsson M. G.; Kamali-Moghaddam M.; Landegren U. ProteinSeq: high-performance proteomic analyses by proximity ligation and next generation sequencing. PLoS One 2011, 6, e25583. 10.1371/journal.pone.0025583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.; Zhong P.; Qu L.; Wang C.; Yuan Y. Preparation and identification of anti-2, 4-dinitrophenyl monoclonal antibodies. J. Immunol. Methods 2006, 313, 20–28. 10.1016/j.jim.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Chipinda I.; Hettick J. M.; Siegel P. D. Haptenation: chemical reactivity and protein binding. J. Allergy 2011, 2011, 839682. 10.1155/2011/839682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling D.; Salvaterra P. M. Robust RT-qPCR data normalization: validation and selection of internal reference genes during post-experimental data analysis. PLoS One 2011, 6, e17762. 10.1371/journal.pone.0017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Miao D.; Scrimgeour L.; Johnson K.; Rewers M.; Eisenbarth G. S. Distinguishing persistent insulin autoantibodies with differential risk. Diabetes 2012, 61, 179–186. 10.2337/db11-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano A.; Gaeta F.; Valluzzi R. L.; Caruso C.; Rumi G.; Bousquet P. J. IgE-mediated hypersensitivity to cephalosporins: cross-reactivity and tolerability of penicillins, monobactams, and carbapenems. J. Allergy Clin. Immunol. 2010, 126, 994–999. 10.1016/j.jaci.2010.06.052. [DOI] [PubMed] [Google Scholar]
- Kaur J.; Boro R. C.; Wangoo N.; Singh K. R.; Suri C. R. Direct hapten coated immunoassay format for the detection of atrazine and 2, 4-dichlorophenoxyacetic acid herbicides. Anal. Chim. Acta 2008, 607, 92–99. 10.1016/j.aca.2007.11.017. [DOI] [PubMed] [Google Scholar]
- Stassi G.; De Maria R. Autoimmune thyroid disease: new models of cell death in autoimmunity. Nat. Rev. Immunol. 2002, 2, 195–204. 10.1038/nri750. [DOI] [PubMed] [Google Scholar]
- Riboldi P.; Gerosa M.; Moroni G.; Radice A.; Allegri F.; Sinico A.; Tincani A.; Meroni P. L. Anti-DNA antibodies: a diagnostic and prognostic tool for systemic lupus erythematosus?. Autoimmunity 2005, 38, 39–45. 10.1080/08916930400022616. [DOI] [PubMed] [Google Scholar]
- Ruffatti A.; Calligaro A.; Del Ross T.; Bertoli M. T.; Doria A.; Rossi L.; Todesco S. Anti-double-stranded DNA antibodies in the healthy elderly: prevalence and characteristics. J. Clin. Immunol. 1990, 10, 300–303. 10.1007/BF00917474. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Jarzabek-Chorzelska M.; Sulej J.; Karnewska K.; Farrell T.; Jablonska S. Celiac disease and immunoglobulin A deficiency: how effective are the serological methods of diagnosis?. Clin. Diag. Lab Immunol. 2002, 9, 1295–1300. 10.1128/CDLI.9.6.1295-1300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche A. M.; Richard A. L.; Rahkola J. T.; Janoff E. N.; Weiser J. N. Antibody blocks acquisition of bacterial colonization through agglutination. Mucosal Immunol. 2015, 8, 176–185. 10.1038/mi.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando S. A.; Wilson G. S. Studies of the ‘hook’ effect in the one-step sandwich immunoassay. J. Immunol. Methods 1992, 151, 47–66. 10.1016/0022-1759(92)90104-2. [DOI] [PubMed] [Google Scholar]
- Kricka L. Human anti-animal antibody interferences in immunological assays. J. Clin. Chem. 1999, 45, 942–956. [PubMed] [Google Scholar]
- Faraoni S.; Rocchetti A.; Gotta F.; Ruggiero T.; Orofino G.; Bonora S.; Ghisetti V. Evaluation of a rapid antigen and antibody combination test in acute HIV infection. J. Clin. Virol. 2013, 57, 84–87. 10.1016/j.jcv.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Ji Y. T.; Qu C. Q.; Cao B. Y. An optimal method of DNA silver staining in polyacrylamide gels. Electrophoresis 2007, 28, 1173–1175. 10.1002/elps.200600557. [DOI] [PubMed] [Google Scholar]
- Netzel B. C.; Grebe S. K.; Carranza Leon B. G.; Castro M. R.; Clark P. M.; Hoofnagle A. N.; Spencer C. A.; Turcu A. F.; Algeciras-Schimnich A. Thyroglobulin (Tg) testing revisited: Tg Assays, TgAb assays, and correlation of results with clinical outcomes. J. Clin. Endocrinol. Metab. 2015, 100, 1074–1083. 10.1210/jc.2015-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer C.; Adler M.; Wacker R. Detecting antigens by quantitative immuno-PCR. Nat. Protoc. 2007, 2, 1918–1930. 10.1038/nprot.2007.267. [DOI] [PubMed] [Google Scholar]
- Findlay J. W. A.; Dillard R. F. Appropriate calibration curve fitting in ligand binding assays. AAPS J. 2007, 9, 260–267. 10.1208/aapsj0902029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster D. A.; Pry T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29, S49–S52. [PMC free article] [PubMed] [Google Scholar]
- Bastarache J. A.; Koyama T.; Wickersham N. E.; Mitchell D. B.; Mernaugh R. L.; Ware L. B. Accuracy and reproducibility of a multiplex immunoassay platform: a validation study. J. Immunol. Methods 2011, 367, 33–39. 10.1016/j.jim.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.