

Abstract
The addition of AlCl3 to four-coordinate boranes of the general formula (C–N-chelate)BCl2 results in halide abstraction and formation of three-coordinate borenium cations of the general formula [(C–N-chelate)BCl]+. The latter react with both arylstannanes and arylsilanes by boro-destannylation and -desilylation, respectively, to form arylated boranes. Catalytic quantities of AlCl3 were sufficient to effect high-yielding arylation of (C–N-chelate)BCl2. Boro-destannylation is more rapid than boro-desilylation and leads to double arylation at the boron center, whereas in reactions with arylsilanes either single or double arylation occurs dependent on the nucleophilicity of the arylsilane and on the electrophilicity of the borenium cation. The electrophilicity of the borenium cation derived from 2-phenylpyridine was greater than that of the benzothiadiazole analogues, enabling the boro-desilyation of less nucleophilic silanes and the direct electrophilic borylation of 2-methylthiophene.
Introduction
Four-coordinate boron compounds containing a chelating π-conjugated C/N donor and two exocyclic aromatic moieties, termed (C–N-chelate)BAr2 (e.g., 1-BAr2 right Scheme 1), have been extensively studied for application in optoelectronic devices.1,2 Changing the exocyclic aryl groups in 1-BAr2 significantly modulates the key optoelectronic properties including the frontier orbital energies and the photoluminescence quantum yield.2,3 Therefore, efficient and versatile routes to libraries of these compounds are important to optimize the materials properties and deliver improved device performance. A particularly attractive approach is the arylation of (C–N-chelate)BX2 (e.g., 1-BX2, X = Cl or Br) to form a wide range of (C–N-chelate)BAr2 compounds, as the starting compounds are readily accessed by electrophilic C–H borylation (Scheme 1).3,4
Scheme 1. Electrophilic C–H Borylation Followed by Previously Reported High-Yielding Arylation Methodologies.

Installation of aromatic moieties at three-coordinate boron species is generally achieved by reaction with either arylithium or aryl Grignard reagents.5 However, reaction of these reagents with Lewis base adducts of boranes often gives the desired product in poor yield.4 Instead functionalization of borane-Lewis adducts such as (2-phenylpyridyl)BBr2 (1-BBr2, Scheme 1) requires organozinc or organoaluminum reagents to achieve high-yielding transmetalation.3,4 Unfortunately these nucleophiles are highly sensitive to protic species (ROH), and the synthesis of organozinc reagents often results in mixtures containing ionic species (termed zincates) and coordinated etherate solvent, which can complicate transmetalation.6 Alternative nucleophiles are required that are readily synthesized, are well-defined, can be handled in air, and enable the boron-containing products to be easily isolated, preferably without column chromatography. Arylsilanes and arylstannanes meet these criteria; however, while three-coordinate boranes (e.g., ArBBr2) undergo transmetalation with arylsilanes and arylstannanes, four-coordinate boranes do not due to the Lewis acidity at boron being effectively quenched by the dative bond.4 We hypothesized that conversion of (C–N-chelate)BX2 into borenium cations,7 [(C–N-chelate)BX]+, using a halophilic Lewis acid (e.g., AlCl3) will enable transmetalation using arylstannanes and arylsilanes. The process is potentially catalytic in the halophile, as the byproduct from transmetalation will react as a functional equivalent of [R3Si]+ or [R3Sn]+, abstracting halide to generate further equivalents of borenium cations for subsequent transmetalation (Scheme 2). Related, albeit stoichiometric in halophile, approaches have been reported for activating chloro-boron subphthalocyanine and F2B-dipyrromethenes toward substitution of B–X with chalcogen-based nucleophiles.8 In contrast, the use of borenium cations in boro-desilylation has extremely limited precedence,9 while their use in boro-destannylation has not been reported to date to the best of our knowledge. Herein is reported catalytic (in AlCl3 activator) borenium cation mediated borylation as a simple method to functionalize (C–N-chelate)BCl2 species based on benzothiadiazole (BT) and pyridyl with aryl and heteroaryl groups.
Scheme 2. Borenium Cation Mediated Transmetalation.

Results and Discussion
Our initial attempts to access new 2-BAr2 compounds used an isolated organozinc reagent synthesized from ZnBr2 and p-tolylMgBr in THF, but this led to low yields of the desired arylated product. The low conversion was attributed to the “Zn(p-tolyl)2” formed under these conditions actually being the zincate [Mg(THF)4(μ-Br)2(Zn(p-tol)2)2]n.10 Due to the significant challenge presented in forming etherate-free arylzinc reagents,10 ArylSiMe3 and ArylSnBu3 nucleophiles were investigated for expanding the exocyclic boron substituents.
Mixing 2-BCl2 (readily formed from the unborylated precursor 2 (F8-BT-F8) and BCl3)3 with 2 equiv of PhSnBu3 in CH2Cl2 at room temperature led to no reaction until catalytic (ca. 5 mol %) AlCl3 was added to the reaction mixture. Compound 2-BCl2 then slowly transformed into diphenylated 2-BPh2 at 20 °C (Scheme 3). Heating of the reaction resulted in a more rapid reaction and good conversion to 2-BPh2 (89% isolated yield after 16 h at 60 °C in CH2Cl2 in a sealed tube). The addition of AlCl3 results in chloride abstraction from 2-BCl2 and borenium cation formation (indicated by downfield shifts in the 1H NMR spectrum and formation of [AlCl4]− in the 27Al NMR spectrum), consistent with previous studies on related compounds.3 The borenium cation [2-BCl]+ is then sufficiently electrophilic to boro-destannylate PhSnBu3. An alternative mechanism where AlCl3 and PhSnBu3 react to form Al-Ph species (which have been previously reported to transmetalate to four-coordinate boron halides)4 is precluded based on previous work where the combination of these reagents (in the absence of 2-BCl2) in haloalkane solvents (such as CH2Cl2) leads to solvent activation via C–Cl···AlCl3 interactions (Friedel–Crafts-type reactivity) and carbodestannylation to form R3C-Ph.11 Friedel–Crafts products are not observed in the reaction with 2-BCl2, which is attributed to AlCl3 reacting rapidly to form the borenium cation, thus disfavoring solvent activation. The ability to form 2-BPh2 in high conversion using catalytic AlCl3 confirmed that the electrophilic [Bu3Sn]+ (or a functional equivalent thereof) byproduct can react with further 2-BCl2, directly or via initial reaction with [AlCl4]−, to provide access to additional equivalents of borenium cations.
Scheme 3. AlCl3-Catalyzed Transmetalation from Arylstannanes to 2-BCl2, with Isolated Yields in Parentheses.

The installation on boron of heteroaryl substituents using 5-Bu3Sn-2-Me-thiophene, 3 (prepared by lithiation of 2-methylthiophene and quenching with Bu3SnCl) was also explored. Mixing 2-BCl2 with 2.2 equiv of 3 gave no reaction, but addition of catalytic AlCl3 (ca. 5 mol %) resulted in rapid arylation at 20 °C (complete within 10 min), and facile isolation simply by filtration through silica allowed 2-B(MeT)2 to be isolated in 67% yield. It is noteworthy that arylation using 3 is considerably more rapid at 20 °C than reactions with PhSnBu3, consistent with the enhanced nucleophilicity of the thienylstannane. Furthermore, the use of 3 indicates that transmetalation occurs via direct boro-destannylation, as the α C-Me in 3 precludes an alternative mechanism involving C–H borylation followed by proto-destannylation, as determined by Jäkle and co-workers for the borylation of a stannylated ferrocene.12
Borenium cation mediated transmetalation with organostannanes is effective for tetraarylation of [4-(BCl)2]2+. The diborenium cation [4-(BCl)2]2+ (Scheme 4) is produced by double borylative fusion of the unborylated precursor 4 (BT-F8-BT)3 as previously reported. With a slight excess of PhSnBu3 (4.2 equiv) [4-(BCl)2]2+ forms the previously characterized tetra-arylated product 4-(BPh2)2 as the major boron-containing complex after 72 h at 20 °C or 24 h at 60 °C (by multinuclear NMR spectroscopy) in 1,2-Cl2C6H4. Transmetalation with ZnPh2 to form 4-(BPh2)2 required prior conversion of [4-(BCl)2]2+ to neutral 4-(BCl2)2 by addition of NMe4Cl for acceptable conversion.4 In contrast, the boro-destannylation methodology requires the borenium for transmetalation; therefore it proceeds directly from [4-(BCl)2]2+.
Scheme 4. Tetraarylation of [4-(BCl)2]2+ with PhSnBu3.

The thiophene analogue of 2-BCl2, 5-BCl2 (Scheme 5), can be readily prepared from the unborylated precursor 5 as previously reported.4 Again while arylation with etherate-free diaryl zinc reagents proceeds with high fidelity, the addition of [Mg(THF)4(μ-Br)2(Zn(p-tol)2)2]n to 2-BCl2 led to an extremely low conversion to 5-B(p-tolyl)2 (isolated in only 13% yield). Analogous to the fluorene congener 2-BCl2, the addition of stoichiometric AlCl3 to 5-BCl2 resulted in halide abstraction and formation of the borenium cation [5-BCl][AlCl4] (based on the significant downfield chemical shift of aryl 1H NMR resonances and the observation of [AlCl4]− in the 27Al NMR spectrum), indicating the feasibility of borenium-mediated transmetalation with organostannanes. The addition of stannane 3 to 5-BCl2 again resulted in no reaction until addition of catalytic AlCl3 (ca. 5 mol %), at which point double arylation proceeded rapidly (complete within 10 min at 20 °C) to form 5-B(MeT)2. This product could be isolated by column chromatography in 51% yield (Scheme 5). It should be noted that both 2-B(MeT)2 and 5-B(MeT)2 undergo slow proto-deboronation of the exocyclic thienyl groups on standing in wet solvents but are stable in the solid state under ambient atmosphere for at least three months. An alternative synthesis of 5-B(MeT)2 by electrophilic C–H borylation was explored based on our previous success using PhBCl2 to form 5-B(Ph(Cl)) directly from 5.4 However, (5-(2-methylthiophene))2BCl ((MeT)2BCl) does not react with 5 (Scheme 5, right), presumably due to the reduced Lewis acidity at boron (relative to BCl3 and PhBCl2). Furthermore, (MeT)BCl213 also fails to borylate 5. Thus, C–H borylation using BCl3 followed by transmetalation is necessary to access this compound.
Scheme 5. Transmetalation to 5-BCl2 with Isolated Yield in Parentheses.

The boro-destannylation reaction was extended to 2-Bu3Sn-9,9-dioctylfluorene (6), synthesized by standard procedures. The reaction of 5-BCl2 with 2.2 equiv of 6 and catalytic AlCl3 (ca. 5 mol %) proceeded at room temperature, but required 18 h for formation of 5-(F8)2 in high conversion. The longer reaction time compared to transmetalation with 3 is attributable to the variation in arene nucleophilicity. Attempts to selectively form the monoarylated product by addition of 1 equiv of 6 to 5-BCl2 (with catalytic AlCl3) led to a mixture of 5-BCl2/5-BCl(F8) and 5-B(F8)2. 5-(F8)2 also can be synthesized from 5 in a two-step, one-pot reaction without the use of a glovebox in 88% yield. Compound 5-BCl2 is prepared by reaction of 5 with BCl3, followed by degassing (removing excess BCl3 and the HCl byproduct from C–H borylation) and subsequent addition of catalytic AlCl3 and 2.2 equiv of 6 (both weighed and handled under ambient atmosphere). The product, 5-(F8)2, is then simply isolated by filtering through silica.
The use of arylsilanes in place of arylstannanes is preferable from a toxicity perspective. However, reacting PhSiMe3 and 2-BCl2 with a range of AlCl3 loadings and reaction conditions (at 20 and 60 °C) consistently resulted in minimal transmetalation. It is well documented that silicon–boron exchange only proceeds with highly electrophilic boranes, in contrast with tin–boron exchange.14 This suggests that the borenium cation [2-BCl]+ is insufficiently electrophilic to effect boro-desilylation of PhSiMe3. A more nucleophilic silane, 2-Me-5-Me3Si-thiophene, 7, was therefore utilized. Compound 2-BCl2 was combined with an excess (2.2 equiv) of 7, resulting in no reaction. Addition of AlCl3 (ca. 5 mol %) to the reaction mixture initiated transmetalation, leading to only one transmetalation per boron, producing 2-BCl(MeT) (Scheme 6), even after long reaction times. As the borenium cation [2-B(MeT)]+ formed after the first transmetalation and subsequent halide abstraction contains a thienyl π donor, its Lewis acidity is presumably reduced relative to [2-BCl]+, disfavoring boro-desilylation of 7. Analogous trends have been previously observed when comparing the Lewis acidity of [PhBCl(amine)]+ and [Cl2B(amine)]+ borocations.15 Compound 2-BCl(MeT) then can be further arylated using other organometallic reagents; for example reaction with Zn(C6F5)2 gave the mixed arylated complex 2-B(MeT)(C6F5) (in 81% isolated yield). This provides a simple route to mixed arylated compounds, (C–N-chelate)BAr1(Ar2). It is notable that current routes to unsymmetrically substituted borane derivatives are challenging and require multiple steps and purifications. This is due to the formation of Ar1Ar2BX (for reaction with lithiated C–N-precursors), often leading to mixtures generally necessitating purification by fractional distillation.16
Scheme 6. Transmetalation Outcomes Using Varying Arylsilanes and 2-BCl2.

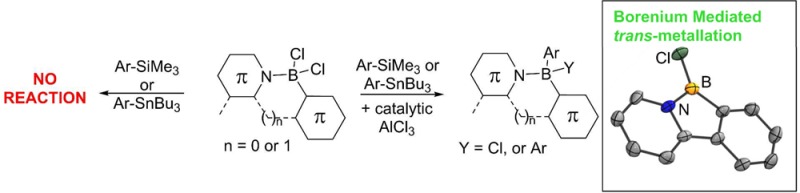
(C–N-chelate)BAr2 compounds based on 2-arylpyridyls and derivatives have been more extensively studied than the benzothiadiazole systems for a range of optoelectronic applications.2,6a,17 Therefore, the borenium cation mediated boro-destannylation/boro-desilylation reactions of these species were explored. 2-Phenylpyridine, 1, was readily borylated by a modification of a literature method4 using BCl3, 2,4,6-tri-tBu-pyridine (TBP), and AlCl3 to form 1-BCl2. Compound 1-BCl2 was stable to ambient conditions and could be readily isolated in air simply by sequential washing with H2O/MeOH and pentane. In contrast BT derivatives (e.g., 2-BCl2) are sensitive to water and column chromatography. The enhanced stability of 1-BCl2 is attributed to a stronger N→B dative bond in the pyridyl congener. The addition of an equivalent of AlCl3 to 1-BCl2 led to formation of the borenium salt [1-BCl][AlCl4], as indicated by a signal at +39.0 ppm in the 11B NMR spectrum and further confirmed by X-ray diffraction studies (crystallized by cooling a saturated CH2Cl2 solution to 4 °C, Figure 1).
Figure 1.
Structure of [1-BCl][AlCl4] showing the two closest [AlCl4]− anions in the extended structure. Thermal ellipsoids at the 50% probability level. Selected bond lengths and angles: B–N = 1.525(9); B–C1 = 1.522(9); B–Cl1 = 1.708(7); B–Cl2 = 3.223 Å; B–Cl3(A) = 3.737 Å; N–B–C1 = 105.0(5)°.
The solid-state structure of [1-BCl][AlCl4] reveals a planarized tricyclic structure and a trigonal planar environment at boron (∑ = 359.8°). Although two [AlCl4]− anions are proximal, the four Al–Cl (two participating in Al–Cl–B bridges and two not) distances are all identical (within 3σ), suggesting that these close contacts are principally due to electrostatic forces and packing effects. The ability of the borenium cation [1-BCl]+ to mediate boro-destannylation was investigated. Addition of 2.2 equiv of PhSnBu3 to 1-BCl2 resulted in no reaction until addition of ca. 5 mol % of AlCl3, which resulted in rapid boro-destannylation at 20 °C to form 1-BPh2. This compound has been previously synthesized by Murakami and co-workers via 1-BBr2 and AlPh3.4 The synthesis of 1-BPh2 in one pot in two steps from 2-phenylpyridine via electrophilic C–H borylation and subsequent AlCl3-catalyzed boro-destannylation can be performed without use of a glovebox in high conversion (72% isolated yield).
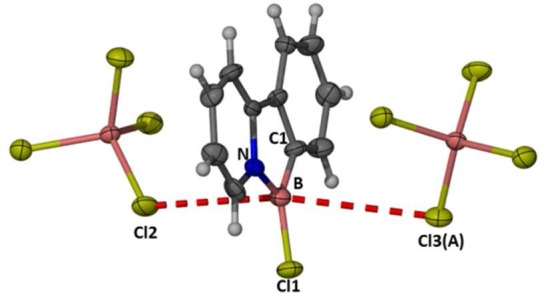
The rapid room-temperature double boro-destannylation observed on combination of 1-BCl2, catalytic AlCl3, and PhSnBu3 is in contrast to the BT congener 2-BCl2 (which requires heating to 60 °C). This suggests an enhanced electrophilicity of the boron center in [1-BY]+ (Y = Cl and Ph) relative to that in [2-BY]+. This was confirmed by the observation that addition of 2.2 equiv of PhSiMe3 to 1-BCl2 in the presence of catalytic (ca. 5 mol %) AlCl3 rapidly led to monoarylation (<10 min) and complete double arylation of boron within 10 h at 20 °C to form 1-BPh2. Thus, with 1-BCl2 double transmetalation is possible using the less toxic arylsilane reagent. This methodology can also be performed without the aid of a glovebox with no significant loss in yield, and the doubly arylated products can be isolated simply by filtration through a short plug of silica followed by drying in vacuo. The electronically deactivated silane (meta-Br-C6H4)SiMe3 was also a viable reagent for transmetalation to boron; however, at 20 °C this led only to a single arylation of 1-BCl2 (using ca. 5 mol % AlCl3), with no further arylation proceeding at 20 °C (Scheme 7). Double arylation of 1-BCl2 can be realized with (meta-Br-C6H4)SiMe3 by heating 1-BCl2/catalytic AlCl3 in 1,2-Cl2C6H4. The change in solvent is essential, as in this case heating a mixture of AlCl3, CH2Cl2, and an arylsilane for prolonged periods of time led to Friedel–Crafts alkyation reactions.11 Analogous conditions enabled the synthesis of the spiro complex 1-B(biphenyl) (Scheme 7, bottom) in good yield (82%) from the commercially available 9,9-dimethyl-9H-9-silafluorene. Spiro complexes such as 1-B(biphenyl) have been extensively explored as electron transport materials in electroluminescent devices.18 It is notable that attempts to make the analogous spiro compound from 2-BCl2 using catalytic AlCl3 failed with no reaction observed at 20 or 60 °C, again indicating the lower electrophilicity of the [2-BCl]+ borenium cation relative to [1-BCl]+.
| 1 |
Scheme 7. Boro-destannylation and Boro-desilylation Reactions Using 1-BCl2 Activated by AlCl3 (Isolated Yields in Parentheses).
Inset bottom right: structure of 1-B(biphenyl), thermal ellipsoids at 50% probability.
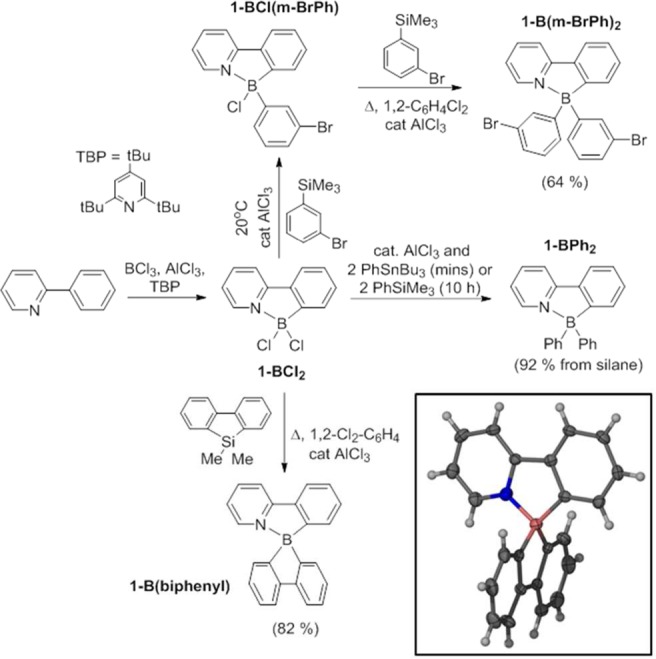
The greater reactivity of [1-BCl]+ relative to [2-BCl]+ suggested an enhanced electrophilicity at boron; to assess if this was due to the change in the aromatic moiety (i.e., thienyl/fluorenyl vs phenyl), calculations comparing [1-BCl]+ with the model BT analogue [8-BCl]+ were performed at the M06-2X/6311G(d,p) (PCM DCM) level (Figure 2). The optimized structure of [1-BCl]+ was in excellent agreement with the solid-state structure of [1-BCl][AlCl4]. Using a previously reported approach the hydride ion affinity (HIA, eq 1)15 relative to BEt3 was assessed and found to be 6.7 kcal mol–1 greater for [1-BCl]+ compared to [8-BCl]+. This indicates a greater Lewis acidity for the pyridyl congener toward soft nucleophiles (such as π systems) consistent with the relative reactivity observed. The nitrogen sites in BT are weakly basic relative to that in pyridyl; however examination of the calculated structure of [8-BCl]+ indicates a greater N→B π donation than in [1-BCl]+ (B–N in [8-BCl]+ = 1.474 Å; B–N in [1-BCl]+ = 1.514 Å). Furthermore, the N–S distances in [8-BCl]+ are different with a longer S–N bond involving the nitrogen bound to boron (N1–S = 1.69 vs N2–S = 1.59 Å, Scheme 8). Natural bond orbital analysis also indicates a significant positive charge on sulfur (+1.073e) and a greater negative charge on N1 in [8-BCl]+ (−0.757e for N1, −0.511e for N2) relative to that of the nitrogen in [1-BCl]+ (−0.588e). This indicates a significant contribution from a resonance form where sulfur is formally in the +4 oxidation state for [8-BCl]+ (Scheme 8, right). Presumably this effect combined with the preference for the five-membered boracycle to pyramidalize relative to the six-membered boracycle (N–B–C angles of 104.8° in [1-BCl]+ and 115.7° in [8-BCl]+) leads to the observed Lewis acidity and reactivity trend.
Figure 2.
Hydride ion affinity of [1-BCl]+ and [8-BCl]+ relative to BEt3 (at the M06-2X/6311G(d,p) (PCM DCM) level).
Scheme 8. Resonance Structures of [8-BCl]+.

The significant electrophilicity of [1-BCl]+ suggested it may be sufficiently reactive to directly borylate C–H bonds of activated arenes.7c,7d This would remove the requirement for preinstallation of R3E– groups on the desired aryl moiety. The addition of 1.1 equiv of 2-methylthiophene and TBP (to sequester the proton) to [1-BCl][AlCl4] (generated in situ) resulted in full consumption of [1-BCl][AlCl4], to form two new resonances in the 11B NMR spectrum. However, multinuclear NMR spectroscopy showed that ca. 0.6 equiv of 2-methylthiophene and 0.5 equiv of TBP and 1-BCl2 were present in the reaction mixture. The observations are consistent with the second new boron resonance being [1-B(MeT)]+. Minor variations in starting stoichiometry (between 1-BCl2 and AlCl3) led to the new 11B NMR resonance varying between 18 and 40 ppm. This is attributed to a fast exchange between differing quantities of [1-B(MeT)][AlCl4] and 1-B(MeT)Cl (Scheme 9). [1-B(MeT)]+ does not react with further 2-methylthiophene (presumably due to insufficient Lewis acidity) and is less chlorophilic than [1-BCl]+, resulting in the consumption of 0.5 equiv of the latter by rapid halide transfer from the expected initial product 1-B(MeT)Cl.19 The addition of a second equivalent of AlCl3 to this reaction mixture led to consumption of all 1-BCl2 and full conversion to [1-B(MeT)]+ (45 ppm in 11B NMR spectrum, Scheme 9). With only a single C–H borylation of 2-methyl thiophene possible using the 2-phenylpyridyl-chelated borenium cation double arylation at boron requires addition of an organometallic nucleophile, e.g., an arylsilane or arylstannane reagent. Alternatively, conversion of [1-B(MeT)]+ to form 1-BCl(MeT) is achieved by addition of a halide source to form 1-BCl(MeT).
Scheme 9. C–H Borylation of 2-Me-thiophene with [1-BCl]+.

Conclusions
The catalytic (in AlCl3) borenium cation mediated arylation of four-coordinate boron compounds using aryl stannanes and aryl silanes represents a simple route to (C–N-chelate)B(aryl)2 species, which are useful for optoelectronic applications. The methodology proceeds with a range of arylstannanes and arylsilanes without the requirement for a glovebox or isolation of the (C–N-chelate)BCl2. Single and double arylation of each boron center can be selected by appropriate choice of reagents, thus enabling facile access to unsymmetrically substituted four-coordinate boron compounds that are challenging to access via other methodologies.
Experimental Section
Unless otherwise stated, all manipulations were carried out using standard Schlenk techniques under argon or in an MBraun UniLab glovebox, under an atmosphere of argon (<0.1 ppm of O2/H2O). Unless otherwise indicated, solvents were distilled from appropriate drying agents: tetrahydrofuran (potassium); toluene (potassium); n-hexane (NaK); and dichloromethane (CaH2). Tetrahydrofuran and dichloromethane were stored over activated 3 Å molecular sieves, while toluene and n-hexane were stored over potassium mirrors. 2, 2-BCl2, 4, 5, 2-methyl-5-tributylstannylthiophene, trimethyl(5-methylthiophen-2-yl)silane, tributyl(9,9-dioctyl-9H-fluoren-2-yl)stannane, and [Mg(THF)4(μ-Br)2(Zn(p-tol)2)2]n were prepared according to previously published procedures.3,10 All other compounds were purchased from commercial sources and used as received. NMR spectra were recorded on Bruker AvanceIII-400 or Bruker Ascend-400 spectrometers. Chemical shifts are reported as dimensionless δ values and are referenced relative to residual protio-impurities in the NMR solvents for 1H and 13C{1H}, respectively, while 11B and 19F{1H} shifts are referenced relative to external BF3-etherate and hexafluorobenzene, respectively. Coupling constants J are given in hertz (Hz) as positive values regardless of their real individual signs. The multiplicities of the signals are indicated as “s”, “d”, “t”, “pent”, “sept”, or “m” for singlet, doublet, triplet, pentet, septet, or multiplet, respectively. Carbon atoms directly bonded to boron are not always observed in the 13C{1H} NMR spectra due to quadrupolar relaxation leading to considerable signal broadening. In a number of compounds individual carbon resonances are not observed for all inequivalent protons (particularly in the octyl chains) due to resonance coincidence. High-resolution mass spectra (HRMS) were recorded on a Waters QTOF mass spectrometer. Microanalysis was performed by Stephen Boyer at the London Metropolitan University microanalytical service. For the arylated compounds accurate combustion data were not obtainable with consistently low %C content observed. This is attributed to boron carbide formation and persisted even when V2O5 was used as an oxidant. For these compounds NMR spectra are included in the SI to support compound purity,
Synthesis of 2-B(MeT)2
BCl3, 1 M in DCM (0.3 mL, 0.3 mmol), was added to a solution of 2 (95 mg, 0.10 mmol) in DCM (3 mL), and the solution was stirred overnight under the dynamic flow of nitrogen. The solvent was then removed under reduced pressure. The resulting residue was dissolved in DCM (3 mL), and AlCl3 (1 mg) was added to the solution. 2-Methyl-5-tributylstannylthiophene (90 mg, 0.22 mmol) was added to the reaction mixture, which was then stirred overnight. The solvent was then removed under reduced pressure, and the purification was performed under ambient atmosphere using nonpurified solvents thereon. The residue was dissolved in hexane and was passed through (using hexane initially and then 10% DCM/90% hexane as eluent) a short plug of base-treated silica gel (pretreated with 5% NEt3/hexane), and only the purple-colored solution was retained. The solvent was removed to afford a purple residue. Yield: 78 mg, 67%.
1H NMR (400 MHz, CD2Cl2): δ = 8.50 (d, J = 7.7 Hz, 1 H), 8.15 (s, 1 H), 8.09 (d, J = 7.6 Hz, 1 H), 8.04–7.96 (m, 3 H), 7.93–7.87 (m, 1 H), 7.82 (d, J = 2.3 Hz, 1 H), 7.76–7.69 (m, 1 H), 7.48–7.27 (m, 6 H), 6.82 (d, J = 3.3 Hz, 2 H), 6.69 (d, J = 3.1 Hz, 2 H), 2.44 (s, 6 H), 2.20–2.01 (m, 8 H), 1.27–1.04 (m, 40 H), 0.89–0.67 (m, 20 H) ppm. 13C NMR (101 MHz, CD2Cl2): δ = 155.1 (br), 154.4, 152.0, 151.9, 150.8 (br), 150.1, 148.0, 142.5, 142.5, 142.2, 141.4, 140.9, 134.7, 133.2, 131.3, 131.2, 130.0, 128.7, 128.4, 128.2, 127.9, 127.5, 127.3, 126.3, 126.1, 125.2, 124.3, 123.6, 123.5, 120.9, 120.6, 120.5, 117.0, 55.8, 55.5, 41.1, 40.7, 32.4, 32.4, 30.6, 30.6, 29.8, 29.8, 29.8, 24.5, 24.4, 23.2, 15.6, 14.4. ppm. 11B NMR (128 MHz, CD2Cl2): δ = −2 (v br). HRMS (APCI): calcd for C74H94BN2S3+ (M + H) 1117.6667, found 1117.6664.
Synthesis of 2-BPh2
BCl3, 1 M in DCM (0.1 mL, 0.1 mmol), was added to a solution of 2 (50 mg, 0.055 mmol) in DCM (3 mL), and the solution was stirred overnight under the dynamic flow of nitrogen. The solvent was then removed under reduced pressure. The resulting residue was dissolved in DCM (3 mL), and AlCl3 (1 mg) was added to the solution. Tributylphenylstannane (40 mg, 0.121 mmol) was added to the solution, and the reaction mixture was stirred and heated overnight at 60 °C. The solvent was then removed under reduced pressure, and the purification was performed under ambient atmosphere using nonpurified solvents thereon. The residue was dissolved in hexane and was passed through (using hexane initially and then 10% DCM/90% hexane as eluent) a short plug of base-treated silica gel (pretreated with 5% NEt3/hexane), and only the purple-colored solution was retained. The solvent was removed to afford a purple residue. Yield: 53 mg, 89%. The spectra agree with that previously reported.3
Synthesis of 4-(BPh2)2
BCl3, 1 M solution in DCM (0.30 mL, 0.3 mmol), was added to a bright yellow solution of 4 (50 mg, 0.076 mmol) and 2,4,6-tritbutylpyridine (38 mg, 0.154 mmol) in DCM (3 mL). The solution rapidly changed color to a dark red. AlCl3 (20 mg, 0.15 mmol) was then added to the reaction mixture. After rotating for 16 h, an additional portion of AlCl3 (20 mg, 0.15 mmol) was added to the reaction mixture. The solution was rotated for a further 16 h, whereupon the solution turned dark green. The DCM was removed under reduced pressure, and the reaction mixture was dissolved in o-DCB (4 mL). Tributylphenylstannane (0.15 mL, 0.456 mmol) was added to the reaction mixture, which was then stirred at 20 °C for 48 h and heated at 40 °C for 16 h. NMe4Cl (50 mg, 0.456 mmol) was added to the reaction mixture, and after 1 h the solvent was removed under reduced pressure. The purification was performed under ambient atmosphere using nonpurified solvents thereon. The residue was purified via column chromatography on base-treated silica gel (5% NEt3/hexane) [eluent chloroform/hexane (2:8)] to afford a purple residue. Yield: 24 mg, 32%. The spectra agree with that previously reported.3
Synthesis of 5-B(MeT)2
BCl3, 1 M in DCM (0.2 mL, 0.20 mmol), was added to a solution of 5 (95 mg, 0.18 mmol) in DCM (3 mL), and the solution was stirred overnight under the dynamic flow of nitrogen. The solvent was then removed under reduced pressure. The resulting residue was dissolved in DCM (3 mL), and AlCl3 (1 mg) was added to the solution. 2-Methyl-5-tributylstannylthiophene (154 mg, 0.40 mmol) was added to the reaction mixture, which was then stirred overnight. The solvent was then removed under reduced pressure, and the purification was performed under ambient atmosphere using nonpurified solvents thereon. The residue was purified via column chromatography on base-treated silica gel (5% NEt3/hexane) [eluent DCM/hexane (1:9)] to afford a dark blue residue. Yield: 67 mg, 51%.
1H NMR (400 MHz, CD2Cl2): δ = 7.71 (d, J = 3.7 Hz, 1 H), 7.58 (d, J = 7.6 Hz, 1 H), 7.35 (d, J = 7.6 Hz, 1 H), 6.94–6.83 (m, 1 H), 6.82–6.77 (m, 1 H), 6.76 (d, J = 3.2 Hz, 2 H), 6.71–6.64 (m, 2 H), 2.87 (q, J = 7.1 Hz, 4 H), 2.45 (s, 6 H), 1.83–1.68 (m, 4 H), 1.51–1.22 (m, 20 H), 1.00–0.82 (m, 6 H). 13C NMR (101 MHz, CD2Cl2): δ = 158.7 (br), 153.8 (br), 151.9, 149.4, 148.8, 147.0, 142.3, 135.8, 131.6, 131.3, 130.9, 128.1, 128.1, 126.4, 126.1, 124.5, 124.4, 122.8, 32.5, 32.5, 32.2, 31.0, 30.8, 29.9, 29.9, 29.9, 29.8, 29.8, 23.3, 15.5, 14.5. 11B NMR (128 MHz, CD2Cl2): δ = −2 ppm (v br). HRMS (APCI): calcd for C35H44BN2S4+ (M – C5H5S) 631.2486, found 631.2477.
Synthesis of 5-B(F8)2
BCl3, 1 M in DCM (0.1 mL, 0.1 mmol), was added to a solution of 5 (30 mg, 0.057 mmol) in DCM (3 mL), and the solution was stirred overnight under the dynamic flow of nitrogen. The solvent was then removed under reduced pressure. The resulting residue was dissolved in DCM (3 mL), and AlCl3 (1 mg) was added to the solution. Tributyl(9,9-dioctyl-9H-fluoren-2-yl)stannane (85 mg, 0.125 mmol) was added to the reaction mixture, which was then stirred overnight. The solvent was then removed under reduced pressure, and the purification was performed under ambient atmosphere using nonpurified solvents thereon. The residue was dissolved in hexane and was passed through a short plug of base-treated silica gel (5% NEt3/hexane), and only the dark blue colored solution was retained. The solvent was removed to afford a purple residue. Yield: 66 mg, 88%.
1H NMR (400 MHz, CDCl3): δ = 7.88 (d, J = 3.7 Hz, 1 H), 7.84 (d, J = 7.6 Hz, 1 H), 7.66 (dd, J = 1.3, 6.2 Hz, 2 H), 7.61–7.50 (m, 5 H), 7.37–7.22 (m, 7 H), 7.04 (dd, J = 0.9, 7.6 Hz, 2 H), 6.86 (d, J = 3.7 Hz, 1 H), 2.89 (t, J = 7.6 Hz, 2 H), 2.81 (t, J = 7.7 Hz, 2 H), 2.07–1.85 (m, 8 H), 1.81–1.64 (m, 4 H), 1.42–0.98 (m, 60 H), 0.97–0.83 (m, 18 H), 0.68 (dt, J = 6.0, 13.8 Hz, 8 H). 13C NMR (101 MHz, CDCl3): δ = 162.0 (br), 153.3 (br), 151.9, 150.9, 149.9, 149.0, 147.9, 147.6, 141.7, 139.3, 135.5, 132.0, 130.9, 130.2, 128.2, 127.4, 126.5, 126.4, 125.5, 125.2, 123.9, 122.8, 121.6, 119.3, 118.9, 54.9, 40.5, 31.9, 31.9, 31.8, 31.6, 30.7, 30.3, 30.3, 29.6, 29.5, 29.5, 29.4, 29.4, 29.3, 29.3, 29.2, 24.1, 24.1, 22.7, 22.7, 14.2, 14.2. 11B NMR (128 MHz, CDCl3): no 11B NMR peak was observed at 20 °C. HRMS (APCI): calcd for C88H122BN2S3+ (M + H) 1313.8858, found 1313.8862.
Synthesis of 2-B(MeT)(C6F5)
AlCl3 (1 mg) was added to a solution of 2-BCl2 (50 mg, 0.5 mmol) and trimethyl(5-methylthiophen-2-yl)silane (20 μL, 0.1 mmol) in DCM (0.7 mL). After inverting for 14 h at room temperature NMR investigation showed only one arylation had occurred. The reaction mixture was then evaporated to dryness, and the residue was dissolved in DCM (0.7 mL). Zn(C6F5)2 (24 mg, 0.6 mmol) was added to the reaction mixture. After stirring for 3 h the reaction mixture was filtered through a plug of base-treated silica gel (5% NEt3/hexane). The reaction mixture was then purified via column chromatography on base-treated silica gel (5% NEt3/hexane) [eluent DCM/hexane (1:9)] to afford a dark purple residue. Yield: 48 mg, 81%.
1H NMR (400 MHz, CD2Cl2): δ = 8.51 (d, J = 7.7 Hz, 1 H), 8.19–8.03 (m, 3 H), 8.01 (s, 1 H), 7.98 (d, J = 7.9 Hz, 1 H), 7.90 (d, J = 7.8 Hz, 1 H), 7.85–7.77 (m, 1 H), 7.74 (dd, J = 3.4, 5.1 Hz, 1 H), 7.47–7.27 (m, 6 H), 6.78 (d, J = 3.2 Hz, 1 H), 6.70–6.64 (m, 1 H), 2.44 (s, 3 H), 2.20–1.94 (m, 8 H), 1.26–1.01 (m, 42 H), 0.86–0.58 (m, 20 H). 13C NMR (101 MHz, CD2Cl2): δ = 154.4, 152.0, 151.9, 150.7, 148.1, 142.9, 142.7, 142.5, 141.2, 140.9, 134.6, 133.4, 131.3, 130.9, 130.1, 128.5, 128.5, 128.2, 128.1, 127.5, 127.4, 126.6, 125.8, 125.5, 124.4, 123.6, 123.6, 120.8, 120.6, 120.5, 117.2, 55.9, 55.6, 41.1, 40.9, 40.8, 32.4, 32.4, 32.3, 30.6, 30.6, 30.6, 29.8, 29.8, 24.6, 24.5, 24.5, 23.2, 23.1, 15.6, 14.4, 14.4. 19F NMR (376 MHz, CD2Cl2): δ = −131.7 (dd, J = 9.0, 24.8 Hz, 2 F), −158.6 (t, J = 20.7 Hz, 1 F), −164.0 (m, 2 F). 11B NMR (128 MHz, CD2Cl2): δ ≈ −3 ppm. HRMS (APCI): calcd for C75H89BN2S2+ (M + H) 1187.6475, found 1187.6471.
Synthesis of 1-BCl2
BCl3, 1 M in DCM (4.0 mL, 4 mmol), 2,4,6-tri-tert-butylpyridine (0.8 g, 3.2 mmol), and 2-phenylpyridine (0.5 g, 3.2 mmol) were dissolved in DCM (40 mL). AlCl3 (0.854 mg, 6.4 mmol) was added to the reaction mixture, whereupon a color change from colorless to yellow was observed. After stirring for 4 h the reaction mixture was degassed under vacuum and NMe4Cl (0.351 g, 3.2 mmol) was added, whereupon the reaction mixture changed color from yellow to colorless. The reaction mixture was evaporated to dryness and washed with water (3 × 100 mL) and hexane (100 mL). The resulting white powder was dried under reduced pressure. Yield: 0.584 g, 77%.
1H NMR (400 MHz, CDCl3): δ = 8.82 (d, J = 5.8 Hz, 1 H), 8.18 (dt, J = 1.5, 7.8 Hz, 1 H), 7.98–7.91 (m, 1 H), 7.86 (d, J = 7.3 Hz, 1 H), 7.77 (d, J = 7.8 Hz, 1 H), 7.62–7.53 (m, 2 H), 7.47–7.39 (m, 1 H). Carbon NMR data are not reported due to the extremely low solubility of the product in a range of common organic solvents. 11B NMR (128 MHz, CDCl3): δ = 7 (v br) ppm. Anal. Calcd for C11H8BNCl2: C, 56.01; H, 3.43; N, 5.94. Found: C, 56.09; H, 3.32; N, 5.88.
Synthesis of [1-BCl][AlCl4]
AlCl3 (57 mg, 0.42 mmol) was added to a suspension of 1-BCl2 (100 mg, 0.42 mmol) in DCM (10 mL). This was stirred overnight, whereupon all the 1-BCl2 had dissolved and the reaction mixture had changed color from colorless to yellow. The reaction mixture was then concentrated to form a saturated solution (∼4 mL), which was then filtered via cannula, and the solution transferred to a 10 mL Young’s ampule. The sample was then held at 2 °C for 16 h, whereupon amber-colored crystals formed. The crystals were isolated via filtration. Yield: 93 mg, 60%.
1H NMR (400 MHz,CD2Cl2): δ = 8.76 (d, J = 5.6 Hz, 1 H), 8.62–8.50 (m, 1 H), 8.09 (d, J = 8.1 Hz, 1 H), 7.94 (d, J = 7.1 Hz, 1 H), 7.90–7.79 (m, 2 H), 7.75 (t, J = 8.1 Hz, 1 H), 7.69–7.59 (m, 1 H). 13C NMR (101 MHz, CDCl3): δ = 158.8, 153.0, 144.6, 140.4, 137.2, 135.8, 135.0, 127.0, 124.8, 121.3. 27Al (104 MHz, CD2Cl2): δ = 104 (br) ppm. 11B NMR (128 MHz, CD2Cl2): δ = 39 (v br). Anal. Calcd for C11H8BNAlCl5: C, 35.78; H, 2.18; N, 3.79. Found: C, 35.84; H, 2.32; N, 3.82
Synthesis of 1-BPh2 via Tributylphenylstannane
AlCl3 (2 mg) was added to a suspension of 1-BCl2 (31 mg, 0.13 mmol) and tributylphenylstannane (106 mg, 0.286 mmol) in DCM (4 mL). 1-BCl2 dissolved almost instantly upon the addition of AlCl3. The reaction mixture was stirred overnight, and the solution was passed through a short plug of silica gel. The purification was performed under ambient atmosphere using nonpurified solvents thereon. The reaction mixture was evaporated to dryness under reduced pressure, and the resulting white residue was washed with hexane to yield the desired product as a white crystalline solid. Yield: 30 mg, 72%.
Synthesis of 1-BPh2 via Trimethylphenylsilane
In a J.Young’s NMR tube AlCl3 (1 mg) was added to a suspension of 1-BCl2 (23 mg, 0.10 mmol) and trimethylphenylsilane (33 mg, 0.21 mmol) in DCM (0.7 mL). The reaction mixture was inverted for 10 h, whereupon NMR investigation showed the reaction had gone to completion. The purification was performed under ambient atmosphere using nonpurified solvents thereon The reaction mixture was then evaporated to dryness and dissolved in DCM (5 mL), and the solution was passed through a short plug of silica gel. The resulting solution was evaporated to dryness under reduced pressure to yield the desired product as a white crystalline solid. Yield: 29 mg, 92%.
The spectra agree with that previously reported.4
Synthesis of 1-B(m-BrPh)2
AlCl3 (15 mg, 0.11 mmol) was added to a suspension of 1-BCl2 (31 mg, 0.13 mmol) and trimethyl(3-bromophenyl)silane (55 μL, 0.29 mmol) in o-DCB (0.7 mL). The reaction mixture was heated at 60 °C for 16 h. NMe4Cl (15 mg, 0.14 mmol) was added to the reaction mixture, and the solvent was removed under reduced pressure. The resulting residue was purified via column chromatography on silica gel [eluent DCM/hexane (5:5)] to afford the desired product as a white crystalline solid. Yield: 38 mg, 62%.
1H NMR (400 MHz, CDCl3): δ = 8.43 (d, J = 5.7 Hz, 1 H), 8.13–8.00 (m, 2 H), 7.88 (d, J = 7.6 Hz, 1 H), 7.69 (d, J = 7.3 Hz, 1 H), 7.48 (t, J = 7.3 Hz, 1 H), 7.44–7.33 (m, 2 H), 7.33–7.27 (m, 2 H), 7.23 (s, 2 H), 7.16 (d, J = 7.5 Hz, 2 H), 7.09 (t, J = 7.6 Hz, 2 H). 13C NMR (101 MHz, CDCl3): δ = 161.3 (br), 158.4, 153.2 (br), 143.8, 141.1, 135.8, 135.2, 131.6, 130.6, 129.3, 128.9, 126.5, 122.6, 122.3, 121.9, 118.5. 11B NMR (128 MHz, CDCl3): δ = 3 (v br) ppm. HRMS (APCI): calcd for C23H17BBr2N+ (M + H) 477.9795, found 477.9796.
Synthesis of 1-B(biphenyl)
AlCl3 (2 mg) was added to a suspension of 1-BCl2 (30 mg, 0.13 mmol) and 9,9-dimethyl-9H-9-silafluorene (30 mg, 0.14 mmol) in o-DCB (0.7 mL). The reaction mixture was then heated overnight at 60 °C. The solvent was then removed under reduced pressure, and the purification was performed under ambient atmosphere using nonpurified solvents thereon. The resulting residue was dissolved in hexane and was filtered through a short plug of silica gel; hexane (100 mL) and then DCM (100 mL) were then passed through the silica gel, and the DCM fraction was collected and evaporated to dryness under reduced pressure to give the desired product as a white crystalline solid. Yield: 33 mg, 82%.
1H NMR (400 MHz, CDCl3): δ = 8.10–8.05 (m, 1 H), 8.02–7.93 (m, 2 H), 7.85 (td, J = 1.1, 5.6 Hz, 1 H), 7.83–7.75 (m, J = 7.6 Hz, 2 H), 7.44–7.36 (m, 3 H), 7.31 (dt, J = 1.2, 7.5 Hz, 2 H), 7.14 (ddd, J = 1.2, 5.8, 7.2 Hz, 1 H), 7.05 (dt, J = 1.0, 7.2 Hz, 2 H), 6.93–6.83 (m, J = 7.1 Hz, 2 H). 13C NMR (101 MHz, CDCl3): δ = 159.3 (br), 158.8, 154.1 (br), 150.8, 143.1, 140.5, 137.5, 131.0, 130.7, 130.1, 127.2, 126.4, 126.2, 121.8, 121.4, 119.3, 117.8. 11B NMR (128 MHz, CDCl3): δ = 2 (v br) ppm. HRMS (APCI): calcd for C23H17BN+ (M + H) 318.1447, found 318.1449.
Acknowledgments
The research leading to these results has received funding from Cambridge Display Technology (CDT/EPSRC Case Award to D.L.C.), the EPSRC (EP/K03099X/1), and the European Research Council (FP/2007-2013/ERC Grant Agreement 305868). M.J.I. acknowledges the Royal Society (for the award of a University Research Fellowship), and M.L.T. thanks InnovateUK for financial support of the Knowledge Centre for Material Chemistry. The authors would also like to acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London in carrying out this work. Dr. Martin Humphries at CDT is also thanked for useful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.5b00857.
The authors declare no competing financial interest.
Supplementary Material
References
- For reviews of four-coordinate boranes in optoelectronics see:; a Rao Y.-L.; Wang S. Inorg. Chem. 2011, 50, 12263–12274. 10.1021/ic200658v. [DOI] [PubMed] [Google Scholar]; b Frath D.; Massue J.; Ulrich G.; Ziessel R. Angew. Chem., Int. Ed. 2014, 53, 2290–2310. 10.1002/anie.201305554. [DOI] [PubMed] [Google Scholar]; c Li D.; Zhang H.; Wang Y. Chem. Soc. Rev. 2013, 42, 8416–8433. 10.1039/c3cs60170f. [DOI] [PubMed] [Google Scholar]
- For selected examples utilizing C–N-chelate substituents see:; a Amarne H.; Baik C.; Murphy S. K.; Wang S. Chem. - Eur. J. 2010, 16, 4750–4761. 10.1002/chem.200903582. [DOI] [PubMed] [Google Scholar]; b Matsumoto T.; Tanaka K.; Tanaka K.; Chujo Y. Dalton Trans. 2015, 44, 8697–8707. 10.1039/C5DT00718F. [DOI] [PubMed] [Google Scholar]; c Wakamiya A.; Taniguchi T.; Yamaguchi S. Angew. Chem., Int. Ed. 2006, 45, 3170–3173. 10.1002/anie.200504391. [DOI] [PubMed] [Google Scholar]; d Heiden Z. M.; Schedler M.; Stephan D. W. Inorg. Chem. 2011, 50, 1470–1479. 10.1021/ic102044z. [DOI] [PubMed] [Google Scholar]; e Rao Y.-L.; Kusamoto T.; Sakamoto R.; Nishihara H.; Wang S. Organometallics 2014, 33, 1787–1793. 10.1021/om500138f. [DOI] [Google Scholar]; f Smith A. C.; Ranade D. S.; Thorat S.; Maity A.; Kulkarni P. P.; Gonnade R. G.; Munshi P.; Patil N. T. Chem. Commun. 2015, 51, 16115. 10.1039/C5CC06351E. [DOI] [PubMed] [Google Scholar]; g Dou C.; Ding Z.; Zhang Z.; Xie Z.; Liu J.; Wang L. Angew. Chem., Int. Ed. 2015, 54, 3648–3652. 10.1002/anie.201411973. [DOI] [PubMed] [Google Scholar]
- Crossley D. L.; Cade I. A.; Clark E. R.; Escande A.; Humphries M. J.; King S. M.; Vitorica-Yrezabal I.; Ingleson M. J.; Turner M. L. Chem. Sci. 2015, 6, 5144–5151. 10.1039/C5SC01800E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N.; Moriya T.; Goya T.; Murakami M. J. Org. Chem. 2010, 75, 8709–8712. 10.1021/jo101920p. [DOI] [PubMed] [Google Scholar]
- Pelter A.; Smith K.; Brown H. C.. Borane Reagents; Academic Press: London, 1988. [Google Scholar]
- For select recent studies on the role of zincates in transmetalation see:; a Jin L.; Liu C.; Liu J.; Hu F.; Lan Y.; Batsanov A. S.; Howard J. A. K.; Marder T. B.; Lei A. J. Am. Chem. Soc. 2009, 131, 16656–16657. 10.1021/ja908198d. [DOI] [PubMed] [Google Scholar]; b McCann L. C.; Organ M. G. Angew. Chem., Int. Ed. 2014, 53, 4386–4389. 10.1002/anie.201400459. [DOI] [PubMed] [Google Scholar]; c McCann L. C.; Hunter H. N.; Clyburne J. A.; Organ C.; Organ M. G. Angew. Chem., Int. Ed. 2012, 51, 7024–7027. 10.1002/anie.201203547. [DOI] [PubMed] [Google Scholar]; d Hevia E.; Chua J. Z.; Garcia-Alvarez P.; Kennedy A. R.; McCall M. D. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5294–5299. 10.1073/pnas.0913307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on borenium cations see:; a Koelle P.; Noeth H. Chem. Rev. 1985, 85, 399–418. 10.1021/cr00069a004. [DOI] [Google Scholar]; b Piers W. E.; Bourke S. C.; Conroy K. D. Angew. Chem., Int. Ed. 2005, 44, 5016–5036. 10.1002/anie.200500402. [DOI] [PubMed] [Google Scholar]; c De Vries T. S.; Prokofjevs A.; Vedejs E. Chem. Rev. 2012, 112, 4246–4282. 10.1021/cr200133c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ingleson M. J. Top. Organomet. Chem. 2015, 49, 39. 10.1007/978-3-319-13054-5_2. [DOI] [Google Scholar]
- For select examples of Lewis acid activation for nucleophilic substitution in other four-coordinate boranes see:; a Morse G. E.; Bender T. P. Inorg. Chem. 2012, 51, 6460–6467. 10.1021/ic2016935. [DOI] [PubMed] [Google Scholar]; b Tahtaoui C.; Thomas C.; Rohmer F.; Klotz P.; Duportail G.; Mely Y.; Bonnet D.; Hibert M. J. Org. Chem. 2007, 72, 269–272. 10.1021/jo061567m. [DOI] [PubMed] [Google Scholar]; c Lundrigan T.; Cameron T. S.; Thompson A. Chem. Commun. 2014, 50, 7028–7031. 10.1039/c4cc02706j. [DOI] [PubMed] [Google Scholar]
- For borocations in boro-desilylation see:; a De Vries T. S.; Prokofjevs A.; Harvey J. N.; Vedejs E. J. Am. Chem. Soc. 2009, 131, 14679–14687. 10.1021/ja905369n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Del Grosso A.; Pritchard R. G.; Muryn C. A.; Ingleson M. J. Organometallics 2010, 29, 241–249. 10.1021/om900893g. [DOI] [Google Scholar]; c Reus C.; Weidlich S.; Bolte M.; Lerner H.-W.; Wagner M. J. Am. Chem. Soc. 2013, 135, 12892–12907. 10.1021/ja406766e. [DOI] [PubMed] [Google Scholar]
- Dunsford J. J.; Clark E. R.; Ingleson M. J. Angew. Chem., Int. Ed. 2015, 54, 5688–5692. 10.1002/anie.201411403. [DOI] [PubMed] [Google Scholar]
- a Eaborn C. J. Organomet. Chem. 1975, 100, 43–57. 10.1016/S0022-328X(00)88933-0. [DOI] [Google Scholar]; b Klaukien H.; Lehnig M.; Reiche T.; Reiss S.; Such P. J. Chem. Soc., Perkin Trans. 2 1995, 2115–2119. 10.1039/p29950002115. [DOI] [Google Scholar]
- Chen J.; Lalancette R. A.; Jäkle F. Organometallics 2013, 32, 5843–5851. 10.1021/om400426g. [DOI] [Google Scholar]
- For the synthesis of thienyl-BCl2 see:Del Grosso A.; Helm M. D.; Solomon S. A.; Caras-Quintero D.; Ingleson M. J. Chem. Commun. 2011, 47, 12459–12461. 10.1039/c1cc14226g. [DOI] [PubMed] [Google Scholar]
- For recent examples exemplifying the disparate reactivity of arylsilanes and arylstannanes toward boranes see:; a Li H.; Jäkle F. Angew. Chem., Int. Ed. 2009, 48, 2313–2316. 10.1002/anie.200805863. [DOI] [PubMed] [Google Scholar]; b Yin X.; Chen J.; Lalancette R. A.; Marder T. B.; Jäkle F. Angew. Chem., Int. Ed. 2014, 53, 9761–9765. 10.1002/anie.201403700. [DOI] [PubMed] [Google Scholar]
- Clark E. R.; Del Grosso A.; Ingleson M. J. Chem. - Eur. J. 2013, 19, 2462–2466. 10.1002/chem.201203318. [DOI] [PubMed] [Google Scholar]
- For an early example of the synthesis an unsymmetric Ar1Ar2BCl see:Paetzold P. I.; Habereder P. P.; Muellbauer R. J. Organomet. Chem. 1966, 7, 45–50. 10.1016/S0022-328X(00)90824-6. [DOI] [Google Scholar]
- a Rao Y. L.; Amarne H.; Zhao S. B.; McCormick T. M.; Martic S.; Sun Y.; Wang R. Y.; Wang S. J. Am. Chem. Soc. 2008, 130, 12898. 10.1021/ja8052046. [DOI] [PubMed] [Google Scholar]; b Ishida N.; Nakanishi Y.; Moriya T.; Murakami M. Chem. Lett. 2011, 40, 1047–1049. 10.1246/cl.2011.1047. [DOI] [Google Scholar]; c Wong H.-L.; Wong W.-T.; Yam V. W.-W. Org. Lett. 2012, 14, 1862–1865. 10.1021/ol3004595. [DOI] [PubMed] [Google Scholar]; d Niu L.; Yang H.; Wang R.; Fu H. Org. Lett. 2012, 14, 2618–2612. 10.1021/ol300950r. [DOI] [PubMed] [Google Scholar]; e Zhao Z.; Chang Z.; He B.; Chen B.; Deng C.; Lu P.; Qiu H.; Tang B. Z. Chem. - Eur. J. 2013, 19, 11512. 10.1002/chem.201301815. [DOI] [PubMed] [Google Scholar]
- A patent search in Sept 2015 revealed seven separate patents filed on the use of spiro materials related to 1-B(biphenyl) in electroluminescent devices, the earliest being:Hasegawa M.; Morii K.; Kuerya T.; Akatsuka I.; Jpn. Kokai Tokkyo Koho, 2013, JP2013053123.
- Cade I.; Ingleson M. J. Chem. - Eur. J. 2014, 20, 12874. 10.1002/chem.201403614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.