

Abstract
A new TEMPO–Co tandem redox system with TEMPO and Co(bpy)3 2+/3+ has been investigated for the use in dye‐sensitized solar cells (DSSCs). A large open‐circuit voltage (V OC) increase, from 862 mV to 965 mV, was observed in the tandem redox system, while the short‐circuit current density (J SC) was maintained. The conversion efficiency was observed to increase from 7.1 % for cells containing the single Co(bpy)3 2+/3+ redox couple, to 8.4 % for cells containing the TEMPO–Co tandem redox system. The reason for the increase in V OC and overall efficiency is ascribed to the involvement of partial regeneration of the sensitizing dye molecules by TEMPO. This assumption can be verified through the observed much faster regeneration dynamics exhibited in the presence of the tandem system. Using the tandem redox system, the faster recombination problem of the single TEMPO redox couple is resolved and the mass‐transport of the metal‐complex‐based electrolyte is also improved. This TEMPO–Co tandem system is so far the most effienct tandem redox electrolyte reported not involving iodine. The current results show a promising future for tandem system as replacements for single redox systems in electrolytes for DSSCs.
Keywords: electrolyte, photoelectrochemistry, redox chemistry, solar cell, tandem‐redox
Since Grätzel and O’Regan significantly improved the efficiency of dye‐sensitized solar cells (DSCs) in 1991,1 the I−/I3 − system has remained as the most efficient redox system in DSSC electrolytes for nearly two decades.2 In 2010, the Hagfeldt and Sun groups combined an electrolyte based on metal–organic cobalt complexes with bulky organic dyes.3 This new strategy has attracted significant attention back to Co‐complex‐based redox systems. Recently, a new record DSSC efficiency, 13 %, was achieved using this kind of redox system.4 2,2,6,6‐tetramethyl‐1‐piperidinyloxy (TEMPO) is a relatively stable organic radical molecule, which is widely used as antioxidant and light‐stabilizer.5 In 2008, TEMPO was introduced into DSSCs by the Grätzel group, and a 5.4 % efficiency was reported. The low efficiency was caused by the fast recombination of TEMPO+, resulting in a low current density.6 A detailed study was recently carried out by the Hagfeldt group.7 A series of TEMPO derivatives were also investigated, but the simple TEMPO substance is considered the best among the derivatives examined.8 In 2014, Chu et al. linked TEMPO to N‐butyl imidazole iodide with a secondary amide group, forming a new type of ionic liquid. With this ionic liquid as redox couple, 8.1 % efficiency was achieved. The authors consider this redox couple to behave like a TEMPO–iodide tandem redox system.9
The tandem redox system in electrolytes for DSSCs has been demonstrated as potential alternatives since 2006, but generally such systems offer very low conversion efficiencies.10 However, very recently, it has been noticed that DSSCs based on tandem redox systems can generate considerably high conversion efficiencies, although nearly all of the studied systems contain the notorious iodine‐based redox system.11 Iodine is a reactive substance and may cause electrolyte leakage and electrode corrosion in solar cell modules. Here, an iodine‐free tandem redox system, the TEMPO–Co system, is investigated as a potential solution to the efficiency and corrosion problems associated with iodine‐based electrolyte systems.
Three new electrolytes containing TEMPO added to a standard Co‐based electrolyte were prepared. C0 denotes the standard Co‐complex‐based, single redox electrolyte, which consists of 0.22 m [CoII(bpy)3](B(CN)4)2, 0.05 m [CoIII(bpy)3](B(CN)4)3, 0.10 m lithium bis (trifluoromethanesulfonyl) imide (LiTFSI), and 0.20 m 4‐tert‐butylpyridine (TBP) in acetonitrile as solvent. The three tandem redox electrolytes, TC1, TC2 and TC3, contain 0.10 m, 0.20 m and 0.40 m, TEMPO in addition to the contents of the C0 electrolyte, respectively. A TEMPO‐based electrolyte T, which contains 0.22 m TEMPO, 0.02 m Nitrosonium tetrafluoroborate (NOBF4), 0.10 m LiTFSI, and 0.20 m TBP in acetonitrile, was also prepared as a reference electrolyte.
The photovoltaic properties of the DSSCs assembled containing the different electrolytes are shown in Table 1. The sensitizer LEG4 (Figure 1) adsorbed to a 5+5 μm double layer TiO2 film was used in the DSSC devices. A blocking layer, which is thicker than that used in common DSSCs, was applied in order to retard the recombination reaction between TEMPO+ and the dye/TiO2 interface (the details of fabrication can be found in the Supporting information). Under a 1‐sun AM 1.5 G illumination, higher open‐circuit voltages (V OC), short‐circuit current densities (J SC), and solar conversion efficiencies were observed for DSSCs containing the TEMPO–Co tandem systems, as compared to cells containing the electrolytes based on single redox‐systems. The electrolytes containing the higher TEMPO concentrations rendered cells with even higher V OC. Devices containing the tandem redox electrolytes with 0.10 m and 0.20 m TEMPO showed an increase in the V OC of about 60 mV and 100 mV, respectively. The reason may be ascribed to a larger contribution of TEMPO to the regeneration reaction of the oxidized dye in the solar cell system. Higher TEMPO concentrations did not render an increase in V OC. The reason might be that at 0.20 m concentration TEMPO already dominates the regeneration process. The tandem systems offer a solution to the problem of low J SC in the TEMPO‐only systems,7 since the presence of Co2+ cause an efficient reduction in TEMPO+ concentration by fast exchange at the photoelectrode. The higher J SC values observed in the presence of TEMPO may be caused by the faster dye regeneration by TEMPO, as compared to that of cobalt complex (more details to support these hypotheses can be found in the latter section of this paper). At higher TEMPO concentrations, the J SC starts to decrease. This is most likely caused by the increase of charge‐transfer resistance at the counter electrode (more details can be found in the section of this paper dealing with electrochemical impedance spectroscopy). The small increase in FF (fill factor) may be ascribed to the more efficient mass transport in tandem redox electrolytes. DSSCs containing a pure TEMPO‐based electrolyte showed lower V OC and J SC. This is most likely caused by the fast recombination between TEMPO+ and the photoelectrode, which was also observed in terms of a short electron lifetime in the photoelectrode substrate.6a DSSCs containing the electrolyte TC2 displayed the highest conversion efficiency of 8.4 % (V OC=963 mV, J SC=13.4 mA cm−2 and FF=65.0 %), which qualifies the system as the record efficiency for DSSCs with iodine‐free, tandem‐based redox electrolytes.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 1.
Molecular structures of the TEMPO0/+, the trisbipyridine cobalt complex and the sensitizing dye LEG4.
The photovoltaic properties of the DSSCs at different light densities were also recorded. The results are shown in Table 2, and the corresponding J–V curves are shown in Figure 2. At lower light densities, the DSSCs containing the tandem‐based redox systems also showed better V OC and efficiencies than those obtained from devices containing the single redox systems. However, at the lowest light intensity, 0.1 sun, devices based on the tandem redox electrolytes showed lower efficiencies than that at 1 sun intensity. The reason may be traced to a faster recombination reaction between TEMPO+ and injected electrons via the dye/TiO2 interface.
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 2.
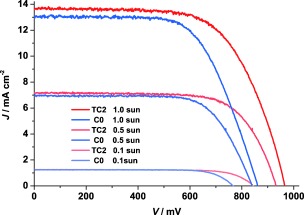
Current density versus applied potential under different light densities for the DSSC devices containing the single‐cobalt‐based redox electrolyte C0 and the tandem‐redox‐based electrolyte TC2.
The incident photon‐to‐current conversion efficiencies (IPCEs) of the solar cells based on the electrolytes C0 and TC2 are shown in Figure 3. Lower IPCE values of TC2 were found in the range 350 to 450 nm. This is caused by the light absorption of TEMPO in the electrolyte in this wavelength region. However, from 450 to 700 nm, DSSCs containing the electrolyte TC2 show higher IPCE. This can be explained by the more efficient dye regeneration and electrolyte mass transport in this electrolyte. These explanations will be discussed later in this work in combination with results from other experimental studies.
Figure 3.
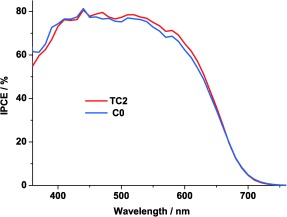
IPCE spectra of DSSC devices based on the electrolytes C0 and TC2.
Results from electrochemical impedance spectroscopy (EIS) of DSSCs containing the single and tandem redox systems are shown in Figure 4. All devices show similar recombination resistance at the TiO2/dye/electrolyte interface (20 Ω for C0, and 17 Ω for TC2 and TC3). The similar recombination resistances noted for the two types of electrolyte show that the fast recombination associated with TEMPO+ is successfully inhibited. For devices containing the electrolytes C0 and TC2, the resistance at the counter electrode are quite similar; about 11 Ω. With higher concentrations of TEMPO in the electrolyte, the resistance at the counter electrode increases slightly from 11 to 14 Ω. This effect may contribute to the lower current density in DSSCs based on electrolytes with higher TEMPO concentrations. At higher TEMPO concentration, there is a significant probability that also higher concentrations of TEMPO+ exist in the electrolyte. At high TEMPO+ concentrations, the charge exchange (see Figure 6) within the electrolyte is not efficient, and the TEMPO+ is likely to be also present at the counter electrode in higher concentrations. The charge transfer to TEMPO+ at the counter electrode is not as efficient as for Co3+ and may thus contribute to the lower overall current density. A discussion on the mechanism of regeneration can be found in the latter part of this paper, and results from the EIS studies can be found in Supporting Information.
Figure 4.
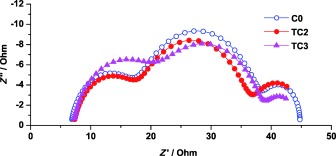
Nyquist plots of the EIS of the DSSC devices based on the C0, TC2, and TC3 electrolytes. The cells were studied at open‐circuit voltage bias under 1 sun illumination. C0 0.22 m [CoII(bpy)3](B(CN)4)2, [CoIII(bpy)3](B(CN)4)3, 0.10 m LiTFSI, and 0.20 m TBP in acetonitrile; TC2 and TC3 contains 0.20 m and 0.40 m TEMPO in C0, respectivly
Figure 6.
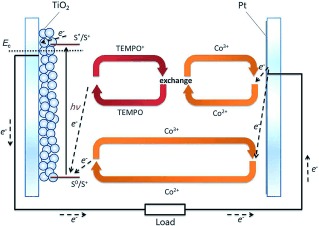
Schematic description of the expected electron transfer processes in the TEMPO–Co tandem redox electrolyte.
Transient absorption spectroscopy (TAS) was performed in order to study the regeneration processes between the sensitizing dye LEG4 and the redox systems of the electrolytes TC2, C0, T0 as well as a redox‐inert electrolyte (0.10 m LiTFSI and 0.20 m TBP in acetonitrile as solvent). The transient absorption kinetics of LEG4‐sensitized TiO2 electrodes in the presence of the different electrolytes are shown in Figure 5. The transient optical signal was observed at 750 nm after the laser pulse excitation at 560 nm, caused by the absorption of the oxidized dye LEG4+. From the absorbance signal of an inert electrolyte, a half‐time (t 1/2) of 57 μs for the recombination reaction was observed. The strongly accelerated decay of the signal in the presence of the redox‐active electrolytes suggests a rapid regeneration/reduction of the dye by the electrolyte redox mediators. The t 1/2 for regeneration of the dye molecules in the presence of the electrolyte TC2 is as short as 211 ns, which suggests a much faster reaction rate than in the presence of an electrolyte based on the single Co‐complex redox system displaying a half‐time of 690 ns. This indicates that the TEMPO–Co tandem redox electrolyte offer much faster regeneration of the oxidized dye molecules than those containing the single Co‐based redox system. The reason for the faster overall regeneration is the faster reaction rate between TEMPO and the LEG4+ cation than that between Co(bpy)3 2+ and LEG4+. For the single TEMPO electrolyte T0, the half‐time t 1/2 of regeneration is 176 ns, which is only slightly shorter than that with the TEMPO–Co tandem redox electrolyte TC2. From one aspect, this shows that the TEMPO component dominates the process of regeneration, but also that Co2+ still partially takes place in the regeneration reaction.
Figure 5.
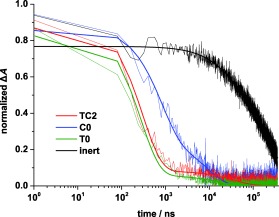
Transient absorption kinetics of LEG4‐sensitized TiO2 electrodes employing different electrolytes. Inert 0.10 m LiTFSI and 0.20 m TBP in acetonitrile solvent; C0 0.22 m [CoII(bpy)3](B(CN)4)2, [CoIII(bpy)3](B(CN)4)3, 0.10 m LiTFSI and 0.20 m TBP in acetonitrile; TC2 contains 0.20 m TEMPO added to C0; T0 contains 0.20 m TEMPO, 0.10 m LiTFSI and 0.20 m TBP in acetonitrile.
On the basis of the experimental results, a feasible mechanism can be suggested to model the electron‐transfer processes in the DSSCs containing the tandem redox electrolytes. After optical excitation of the sensitizing dye, electrons are injected into the TiO2 conduction band. In this process, the dye turns into a mono‐ionized state. The TEMPO/TEMPO+ redox potential is 0.18 V more positive than that of Co(bpy)3 2+/3+ under standard conditions.12 The corresponding potential difference as estimated from the Nernst equation invoking the different redox species concentrations in the TEMPO–Co tandem redox electrolyte is 0.22 V at room temperature. With 0.10 m TEMPO in electrolyte, the V OC of DSSC devices increases from 0.86 V to 0.92 V; and with 0.20 m and 0.40 m TEMPO, the V OC further increases to 0.96 V. The V OC increase is less than the potential difference between TEMPO/TEMPO+ and Co(bpy)3 2+/3+. This suggests that the oxidized dye cations react with both the Co2+ and TEMPO species in parallel, forming Co3+ and TEMPO+; with higher concentrations of TEMPO species in the electrolyte, the oxidized dye cations have a higher probability to react with TEMPO. Also, the addition of TEMPO leads to a faster regeneration of the oxidized dye molecules, and thus the unwanted reductive recombination reaction to the oxidized dye molecules by injected electrons is reduced under operating conditions with a higher open‐circuit voltage as a result. At 0.20 m concentration of TEMPO the increase in V OC is about 100 mV, which is lower than the energy gap between Co(bpy)3 2+/3+ and TEMPO/TEMPO+. Therefore, there is a high possibility that, in the presence of electrolyte TC2, the oxidized dye cations are still only partially regenerated by TEMPO; this can also be concluded from the TAS data. The faster regeneration reaction rate between the dye cations and TC2 than with C0 is also suggested from the TAS spectra. This is another indication that the TEMPO species play an important role in the regeneration process. It is expected that Co3+ is the main component to become reduced at the counter electrode, a hypothesis that gains some support from the observed highly similar charge‐transfer resistances in devices containing the two electrolytes TC2 and C0, as extracted from the EIS spectra. Just like for other tandem redox electrolytes,11e it is expected that there is an electron exchange between TEMPO+ and Co2+ in the liquid space between the working electrode and the counter electrode, regenerating TEMPO and Co3+, and closing the regenerative redox system cycle. The electron exchange decreases the concentration of TEMPO+ in the electrolyte, which successfully resolves the recombination problem7 observed for the single TEMPO redox system. Altogether, the processes can be formalized in the form of two cycles of redox exchange reactions in the electrolyte running in parallel. One is the single‐component Co2+/Co3+ redox cycle, and the other is the synergistic TEMPO/TEMPO+‐Co2+/Co3+ cycle. The whole process is schematically visualized in Figure 6.
In conclusion, a new TEMPO–Co tandem redox system with TEMPO0/+ (although the presence of TEMPO+ is very low) and Co(bpy)3 2+/3+ was investigated in DSSCs. A large increase in V OC, from 862 mV to 965 mV, was observed in the new tandem redox system, while the J SC was essentially unchanged. The conversion efficiency was observed to increase from 7.1 % in devices based on the single Co(bpy)3 2+/3+ redox system to 8.4 % for the DSSCs containing the TEMPO–Co tandem redox system. EIS and transient absorption spectroscopy were employed to study the possible mechanisms that lead to the observed photovoltaic improvements. The reason for the increase in V OC and overall efficiency can be ascribed to the partial regeneration of the sensitizing dye molecules by TEMPO. This assumption can be verified through the observed much faster regeneration dynamics exhibited in the presence of the tandem system, and the large increase in V OC and its dependence on the TEMPO concentration in the electrolyte. In contrast to the single redox system, TEMPO firstly reacts with the oxidized dye forming TEMPO+, and then the TEMPO+ may undergo an electron exchange with Co2+, re‐generating TEMPO and Co3+. Co3+ is presumed to be the main component reduced at the counter electrode rendering Co2+. The concentration of TEMPO+ in the tandem redox electrolyte will be reduced through the electron exchange, and the recombination was greatly inhibited compared with the single TEMPO redox system. Therefore, the low current density problem of the single TEMPO system is resolved. A current density that is even higher than that observed for devices based on the single Co redox system was achieved, benefiting from the faster regeneration reaction and the better mass‐transport properties. The new TEMPO–Co tandem system also successfully avoids the corrosive effects of iodine in the electrolyte; at the same time, it shows a much higher V OC and a conversion efficiency, outperforming the single redox electrolyte systems based on Co. This TEMPO–Co tandem system represents the most efficient tandem redox electrolyte so far that does not involve iodine . The current results show a promising future for tandem systems in replacing single redox systems in electrolytes for DSSCs.
Experimental Section
Device fabrication was done as follows. Fluorine‐doped tin oxide (FTO) glass substrates (Pilkington, TEC15) were cleaned (in the order of detergent/water solution, and ethanol) using an ultrasonic bath. Then, a solution of 10 mm titanium(IV) isopropoxide (Ti[OCH(CH3)2]4) in ethanol was spin‐coated on the glass at 2000 rpm for 20 s in air. The substrate was put in a water vapor environment for 30 min for a hydrolysis process to form TiO2. Then the substrate was heated to 450 °C for 30 min. The substrate was treated again, with 40 mm aqueous TiCl4 solution at 70 °C for 30 min to generate a good blocking layer on the substrate. A layer mesoporous TiO2 (DSL 18NR‐T, Dyesol) that was 5 μm thick and 18 nm in size and a layer of TiO2 (WER2‐O, Dyesol) that was 5 μm thick and 150–250 nm in size were screen‐printed on to the pre‐treated FTO glass surface. The double‐layer TiO2 electrodes (area: 5×5 mm) were heated in air at 480 °C for 30 min. The sintered films were treated with 40 mm aqueous TiCl4 solution again at 70 °C for 30 min, and heated to 450 °C for 15 min. The films were then immersed into a 2×10−4 m dye 3‐(6‐(4‐(bis(2′,4′‐dibutoxy‐[1,1′‐biphenyl]‐4‐yl)amino)phenyl)‐4,4‐dihexyl‐4H‐cyclopenta[1,2‐b:5,4‐b′]dithiophen‐2‐yl)‐2‐cyanoacrylic acid (LEG4) solution in ethanol and maintained in the dark for 18 h. The resulting sensitized TiO2 electrodes were then rinsed with ethanol and dried. Hermetically sealed cells were fabricated by assembling the dye‐loaded film as the working electrode and a platinized counter electrode (TEC7) separated with a hot‐melt Surlyn 1702 film (25 μm, Dupont).
Acknowledgements
This work is supported by the Swedish Research Council, the Swedish Energy Agency and the Knut & Alice Wallenberg Foundation.
References
- 1. O′Regan B., Grätzel M., Nature 1991, 353, 737–740. [Google Scholar]
- 2.
- 2a. Nazeeruddin M. K., De Angelis F., Fantacci S., Selloni A., Viscardi G., Liska P., Ito S., Takeru B., Grätzel M., J. Am. Chem. Soc. 2005, 127, 16835–16847; [DOI] [PubMed] [Google Scholar]
- 2b. Chiba Y., Islam A., Watanabe Y., Komiya R., Koide N., Han L., Jpn. J. Appl. Phys. 2006, 45, L638; [Google Scholar]
- 2c. Gao F., Wang Y., Shi D., Zhang J., Wang M., Jing X., Humphry‐Baker R., Wang P., Zakeeruddin S. M., Grätzel M., J. Am. Chem. Soc. 2008, 130, 10720–10728; [DOI] [PubMed] [Google Scholar]
- 2d. Chen C.‐Y., Wang M., Li J.‐Y., Pootrakulchote N., Alibabaei L., Ngoc‐le C.‐h., Decoppet J.‐D., Tsai J.‐H., Grätzel C., Wu C.‐G., Zakeeruddin S. M., Grätzel M., ACS Nano 2009, 3, 3103–3109; [DOI] [PubMed] [Google Scholar]
- 2e. Zeng W., Cao Y., Bai Y., Wang Y., Shi Y., Zhang M., Wang F., Pan C., Wang P., Chem. Mater. 2010, 22, 1915–1925. [Google Scholar]
- 3. Feldt S. M., Gibson E. A., Gabrielsson E., Sun L., Boschloo G., Hagfeldt A., J. Am. Chem. Soc. 2010, 132, 16714–16724. [DOI] [PubMed] [Google Scholar]
- 4. Mathew S., Yella A., Gao P., Humphry‐Baker R., CurchodBasile F. E., Ashari‐Astani N., Tavernelli I., Rothlisberger U., Nazeeruddin K., Grätzel M., Nat. Chem. 2014, 6, 242–247. [DOI] [PubMed] [Google Scholar]
- 5. Nishide H., Suga T., Electrochem. Soc. Interface 2005, 14, 32–36. [Google Scholar]
- 6.
- 6a. Zhang Z., Chen P., Murakami T. N., Zakeeruddin S. M., Grätzel M., Adv. Funct. Mater. 2008, 18, 341–346; [Google Scholar]
- 6b.Z. Zhang, EPFL 2008.
- 7.W. Yang, N. Vlachopoulos, A. Hagfeldt, G. Boschloo, Comparison between a triphenylamine dye and an indoline dye in dye‐sensitized solar cells with TEMPO‐based redox couple, unpublished results.
- 8. Kato F., Hayashi N., Murakami T., Okumura C., Oyaizu K., Nishide H., Chem. Lett. 2010, 39, 464–465. [Google Scholar]
- 9. Chu T.‐C., Lin R. Y.‐Y., Lee C.‐P., Hsu C.‐Y., Shih P.‐C., Lin R., Li S.‐R., Sun S.‐S., Lin J. T., Vittal R., Ho K.‐C., ChemSusChem 2014, 7, 146–153. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Cazzanti S., Caramori S., Argazzi R., Elliott C. M., Bignozzi C. A., J. Am. Chem. Soc. 2006, 128, 9996–9997; [DOI] [PubMed] [Google Scholar]
- 10b. Caramori S., Husson J., Beley M., Bignozzi C. A., Argazzi R., Gros P. C., Chem. Eur. J. 2010, 16, 2611–2618. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Cong J., Yang X., Hao Y., Kloo L., Sun L., RSC Adv. 2012, 2, 3625–3629; [Google Scholar]
- 11b. Liu J., Yang X., Cong J., Kloo L., Sun L., Phys. Chem. Chem. Phys. 2012, 14, 11592–11595; [DOI] [PubMed] [Google Scholar]
- 11c. Cheng M., Yang X., Zhang F., Zhao J., Sun L., Angew. Chem. Int. Ed. 2012, 51, 9896–9899; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10034–10037; [Google Scholar]
- 11d. Zhang W., Qiu L., Chen X., Yan F., Electrochim. Acta 2014, 117, 48–54; [Google Scholar]
- 11e. Cong J., Hao Y., Sun L., Kloo L., Adv. Energy Mater. 2014, 4, 1301273. [Google Scholar]
- 12. Cong J., Yang X., Kloo L., Sun L., Energy Environ. Sci. 2012, 5, 9180–9194.. [Google Scholar]