

Abstract
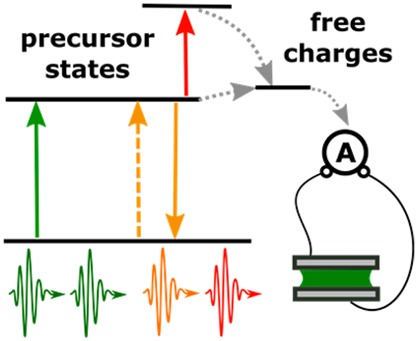
While ultrafast spectroscopy with photocurrent detection was almost unknown before 2012, in the last 3 years, a number of research groups from different fields have independently developed ultrafast electric probe approaches and reported promising pilot studies. Here, we discuss these recent advances and provide our perspective on how photocurrent detection successfully overcomes many limitations of all-optical methods, which makes it a technique of choice when device photophysics is concerned. We also highlight compelling existing problems and research questions and suggest ways for further development, outlining the potential breakthroughs to be expected in the near future using photocurrent ultrafast optical probes.
Ultrafast transient absorption (TA) methods and their multidimensional extensions are currently well-developed approaches to study electronic and structural dynamics of optoelectronic nanosystems like organic, organic–inorganic hybrid or quantum dot photovoltaic devices, biological light conversion systems, or electrochemical cells. Some successful examples include elucidation of charge separation and transport pathways in organic materials,1−6 observation of structural7,8 and vibrational9 dynamics, as well as elucidation of correlation of multiple excited states and coherent phenomena involved.10,11 The essence of the approach comes down to using a sequence of two or more subpicosecond optical pulses. First, pump pulse(s) bring the system into the excited state, leading to the modulation of the system’s optical properties. These photoinduced changes affect the probe pulse(s) intensity as they pass through the sample. Therefore, the detection of the outcoupled probe field as a function of delay between pump and probe pulses provides an opportunity to resolve the dynamics of the optoelectronic system in time.
Despite the long history of development and a broad choice of particular technique modifications, most varieties of TA methods suffer from a range of limitations when applied to optoelectronic systems and devices:
-
(i)
At low excitation pulse energies, the photoinduced modulation in optical properties can be small and insufficient to provide a detectable variation in optical density. To tackle this problem, high pump intensities are usually used to boost the signal, which typically scales linearly with the excitation intensity. However, this may cause photodegradation. It was also proven that high photon fluxes are unsuitable for some optoelectronic materials and devices that should be studied at “working condition” illumination.12,13 Finally, multistep absorption to higher-lying states at moderate fluence may display apparent linear intensity behavior but lead to unexpected population products.14
-
(ii)
The observed variations of optical properties are broad and featureless and can be similar for different photoexcited states, complicating the interpretation of the results. Also, the optical cross sections of some transition can be much weaker compared to the others, making the signatures of the most important processes (for example, charge dynamics that determine device performance) hidden behind other trivial but strong spectroscopic responses.15
-
(iii)
The detection scheme in any TA technique requires outcoupling of probe light. This makes these methods of limited use for samples with high optical density in the probe region and for certain types of devices. In addition, highly scattered samples are inaccessible for one-color TA-type experiments unless for sophisticated techniques like phase cycling16 or double modulation.17
-
(iv)
Due to the all-optical detection nature, the spatial resolution of TA methods is limited by the size of the probe beam. Therefore, even in optical microscopy configurations, the typical dimension of resolved features cannot reach below a few hundred nanometers, which may be insufficient for nanomaterial characterization.
TA spectroscopy, therefore, is an effective and well-developed tool to address charge dynamics in model systems like solutions or homogeneous films. However, all of the restrictions mentioned above make this method of limited use for addressing time-resolved dynamics in optoelectronic devices and functional systems at working conditions.
In this Perspective, we review recent developments in device-based ultrafast spectroscopies with photocurrent (PC) detection (Figure 1). In this method, a sequence of short optical pulses first brings the system to the excited state and then modulates the excited-state dynamics, while the effect of modulation is observed through the device performance. Though this approach was not widely used before 2012, a number of research groups from different fields have independently developed electric probe schemes and reported promising pilot studies in the last 3 years. Here, we show that the ultrafast spectroscopy with PC detection successfully overcomes the limitations of all-optical methods mentioned above and that it becomes a technique of choice when (nano)devices are concerned. Our paper also reviews existing problems on the way to further development and outlines the potential breakthroughs to be achieved in the near future.
Figure 1.
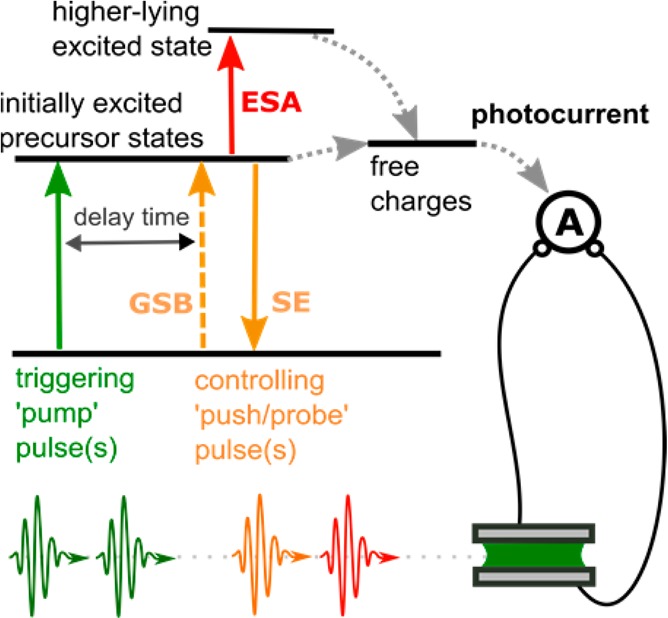
Concept of the ultrafast experiment with PC detection. The pump pulses bring the system to the excited state, and the push pulses modulate the excited-state dynamics. The detection is done by observing the effect of the push on the device performance.
Toward Ultrafast PC Spectroscopy. A promising alternative to all-optical methods in application to optoelectronic devices can be hybrid spectroscopic methods with electrical detection. The simplest and most widely used example of this approach is PC excitation spectroscopy. In this technique, an actual optoelectronic device is illuminated by a continuous-wave (CW), tunable, monochromatic light source, and the produced PC is measured as a function of photon energy. Alternatively, a broad-band interferometric realization of this technique can be used.18 PC spectroscopy is widely used to study the nature of photoexcitations in semiconductors. From an application point of view, it is an essential technique to unravel the charge generation process for the optimization of solar cells based on novel materials.19 It is particularly useful for systems where photocarriers are not produced directly by interband transitions but are mediated by precursor states such as excitons in quantum-confined or molecular semiconductors. PC detection directly probes those states via their excitation spectral line shapes.18 Being purely absorption-sensitive, PC spectroscopy easily eliminates scattering and reflection artifacts, which gives it an outstanding dynamic range, sufficient for the identification of very low concentrations of defect states and intermolecular interactions. The main limitation of the conventional PC spectroscopy is that it is a steady-state technique incapable of addressing and resolving in time the dynamics of the excited states.
To our knowledge, the first successful attempt to resolve excited-state electronic dynamics in molecular systems using PC detection was reported in 1981 by Lukin and co-workers.20 They used (Figure 2a) a combination of two laser sources to identify an intermediate state responsible for the PC generation after anthracene ionization in an electrochemical device. Though Lukin et al. used two CW light sources, the lifetime of the intermediate state formed after the illumination with a 347 nm laser (trapped electron in solution) was sufficiently long to allow its sequential re-excitation by a 694 nm source, leading to a very substantial (40 times) increase in PC. The application area for the developed two-color CW PC spectroscopy was limited to the systems with a very long lived intermediate excited states, and this method did not attract broad interest despite its clear potential.
Figure 2.

Early two-color PC spectroscopy studies. (a) The setup for two-color PC spectroscopy on electrochemical devices. Reproduced from ref (20) with permission, Elsevier, 2015. Note the elegant stability-control solution with a reference cell. (b) Comparison between TA, two-pulse photoluminescence, and 2PPC measurements from ref (22) with permission, APS, 2015.
To elucidate the dynamics of intermediate states with shorter lifetimes, a pulsed modification of the technique was developed.21 For simplicity, we will from now on address this technique as the two-pulse PC (2PPC) method. In a 2PPC experiment, an optoelectronic device is illuminated by a sequence of two so-called pump and push laser pulses interacting with the active material in the device. The result of these interactions is detected by observing the variations in the current flow through the device as a function of time delay between the pump and push and their spectra. 2PPC combines the sensitivity and device relevance of electronic methods with the excitation selectivity and ultrafast time resolution of optical techniques.
Later, Frankevich et al.23 exploited advances in solid-state ultrafast lasers and applied ultrafast 2PPC techniques to investigate charge dynamics in organic nanodevices. This pioneering study featured subpicosecond time resolution and discovered the existence of precursor states for the photocarrier formation in organic semiconductors. The development has triggered substantial interest and inspired more detailed work from the Feldman group.22 These studies were the first to compare different ultrafast techniques including TA, 2PPC, and two-pulse photoluminescence (Figure 2b). As seen from the figure, even if all of above measurements are performed on the same device with identical pulses, the observed kinetics are similar for TA and photoluminescence as those reflect purely exciton dynamics but are different for the 2PPC response. This reveales that 2PPC is sensitive to a subensemble of excited states particularly critical for efficient device performance.20 Despite the initial success, the visible push realizations of the 2PPC method used by Frankevich et al. and Muller et al. have not acquired wide recognition and application. We believe the main reason behind this was that visible push pulses were used in these techniques, which brings several issues for the interpretation of data. Primarily, the high (1–2 eV) photon energy of visible push pulses is sufficient to generate charge carriers in organic semiconductors through both sub- and above-gap states, which leads to high background currents. Furthermore, such strong visible push pulses strongly perturb the excited-state dynamics. Both factors complicated the interpretation of data and limited the use of the technique.
2PPC Spectroscopy with an IR Push Pulse. The limitation of using high-energy push photons in 2PPC methods was overcome with an IR push pulse that is not resonant with the absorption of the system in the ground state.24 Gentle and targeted IR re-excitation allowed application of this technique to study charge separation and charge trapping dynamics in organic and hybrid optoelectronic systems. Initially, IR 2PPC was applied to identify loss channels in organic photovoltaic cells by switching optically the electronic states of the molecules.24−26 The 2PPC kinetics presented in Figure 3a reflect the probability to enhance the dissociation of a bound interfacial charge-transfer state using the energy from an additional IR photon.24 This study found that for a range of organic photovoltaic systems, charge-transfer state formation is the efficiency-limiting step. It also demonstrated that the dissociation of bound carriers requires a transition from a local polaronic level to a delocalized “band” state, which can be induced by a ∼0.5 eV photon. The opportunities that this new method provided have been applied to different material systems, including multicomponent organic photocells,27 oxide-based hybrid optoelectronic devices,28,29 and colloidal quantum dot p–i–n diodes.30
Figure 3.
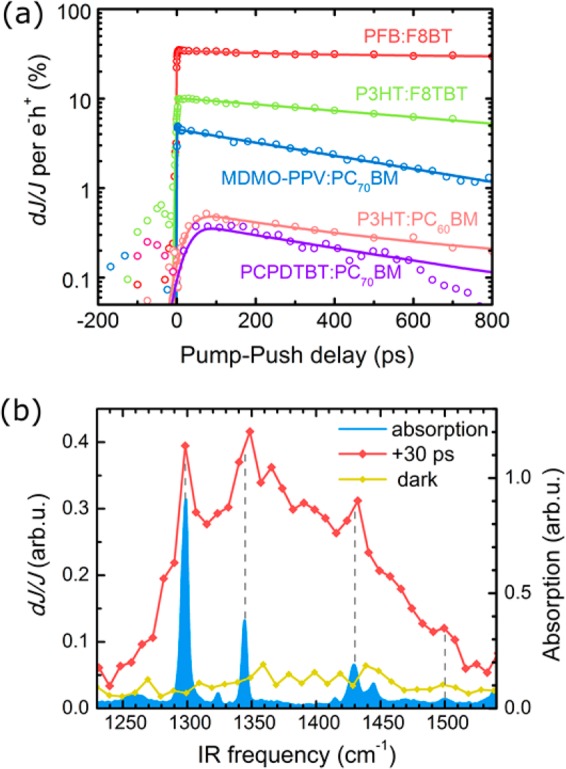
(a) 2PPC transients reflecting the recombination dynamics of bound electron–hole states in a range of organic photovoltaic devices, adapted from ref (24) with permission, AAAS, 2015. (b) Frequency-resolved effect of IR irradiation on the photoconductivity of the pentacene molecular crystal. Vibrational (narrow) and electronic (broad) response are observed. A 30 ps time delay was set between the visible and IR pulses. Adapted from ref (31) with permission, Nature Publishing Group, 2015.
The application of IR push pulses potentially provides an opportunity to access vibrational modes and their coupling to electronic dynamics. Using the 2PPC approach, it has recently been demonstrated experimentally that the performance of an organic optoelectronic system can be controlled by selectively exciting vibrational modes of the molecules involved in charge transport using a pentacene/C60 photoresistor as a model system.31 When addressing vibronic phenomena, a selective excitation of different vibrational modes is required. This can be achieved using narrow-band IR pulses32,33 at the expense of time resolution or by applying an ultrafast interferometry approach,34,35 which allows for precise control over the time/frequency domain structure of the IR optical pulses. The latter approach was used in the 2PPC study, where an interferometric sequence of two ultrafast mid-IR laser pulses created a coherent superposition of molecular vibrational motions inside of the active layer of a device and then vibrational excitation was correlated with the device performance. Importantly, this interferometric approach combines sufficient <10 cm–1 frequency resolution to identify vibrational modes with a 100 fs time resolution despite using the broad-band IR push spectrum. In fact, a spectral bandwidth of at least 300 cm–1 is essential for keeping the ultrafast (sub-100 fs) time resolution needed for observation of nuclear and electronic dynamics.
When the PC signal is resolved as a function of the push photon energy, two different types of responses can be identified (Figure 3b). One is a broad response, which has a weak dependence on the IR photon energy. This component is associated with a low-energy polaronic transition,4 and the effect of IR excitation is probably similar to one observed for charge-transfer states in organic photovoltaic devices.24 At the same time, a narrow-band response is observed only at certain push photon frequencies, for example, ∼1300 or ∼1445 cm–1. These frequencies correspond to the vibrational modes of the studied organic semiconductor (pentacene). This indicates that the observed increase in PC originates from the coupling between vibrational and electronic degrees of freedom, the so-called vibronic phenomena. Both the experiment and theoretical calculations demonstrate that different nonequilibrium geometries and atomic motions have dissimilar effects on the charge dynamics. For example, the vibrations along the long axis of pentacene molecules (e.g., ∼1445 cm–1 mode) lead to a larger increase in the charge-hopping rate than the molecular motions along the short axis (e.g., 1300 cm–1 mode). Such mode-selective “vibrational control” of charge dynamics opens a number of new opportunities, including utilization of vibronic phenomena for ultrafast switching of organic devices and identification of charge-transport mechanisms and pathways in (bio)molecular junctions.
2DPC Spectroscopy. 2D electronic spectroscopies are the extensions of visible-region TA methods that permit identification of homogeneous and inhomogeneous spectral line shape contributions and can reveal microscopic interchromophore couplings. In a 2D spectroscopic measurement, the response of a system driven by multiple electromagnetic fields is recorded in a multidimensional frequency or time space. This involves exciting the sample with a sequence of phased ultrafast laser pulses, typically three or four depending on the adopted geometry, and picking up the nonlinear signal with the appropriate phase relationship. In the context of photocarrier generation dynamics, the potential to identify interstate couplings is key to investigate energy- and charge-transfer phenomena that eventually lead to photoconversion. Because of this, coherent optical spectroscopy has seen intensive development during the past decade, shedding key insights on a wide variety of phenomena, such as energy- and electron-transfer dynamics in photosyntetic systems,36−38 in addressing local conformations of nucleotides in DNA constructs,39 in excited-state energy transfer of exciton-coupled molecular dimers in biological membranes,40,41 in coherent response of the optically created excitations in semiconductors and semiconductor nanostructures,42−44 in probing excitons in molecular dimers and aggregates,45−49 and finally multipolariton correlations in inorganic–semiconductor optical microcavities,50 just to give a few examples of note.
Experimentally, 2D spectroscopy is most commonly implemented using a noncollinear geometry of the excitation beams.46,51−53 Within this approach, three noncollinear laser pulses interact with the sample, inducing a third-order polarization, which radiates in a phase-matched direction, with the amplitude and phase of that radiation detected by means of spectral interferometry with a fourth replica pulse (heterodyne detection).54
As PC detection does not require outcoupling and spatially separating light pulses, a different experimental scheme has been recently implemented, based on four collinear femtosecond pulses and acousto-optic phase modulation.55 The first pulse sets the system in a coherent nonequilibrium superposition of states. The second pulse converts this superposition to a population in the excited state, which keeps the phase of the superposition state. The third pulse generates again a coherent superposition of states, which the fourth pulse converts to an observable fourth-order population. The signal being measured might, then, be considered as proportional to this excited-state fourth-order population. This collinear experimental approach was developed by Marcus and co-workers to investigate the nonlinear optical response of fluorescent systems like atomic Rb vapor.55 Recently, the technique was extended by Nardin et al. to detect PC instead of the luminescence in GaAs quantum wells produced for 2D electron–gas studies,56 followed by Karki et al. to address carrier multiplication processes in PbS solar cells.57 This modification makes the technique particularly valuable for the investigation of charge carrier generation dynamics in photocells because it offers direct access to the physical quantity of interest, that is, PC, at the operating regime of the devices.
One advantage of the collinear approach with PC detection comes from the employment of acousto-optic modulation to achieve the phase-locking condition of the four excitation pulses, with a pulse sequence depicted in Figure 4a. The phase modulation scheme determines a PC signal evolving in time at specific frequencies, thus allowing one to isolate the fourth-order population contribution to the overall signal by phase-sensitive detection. Each measurement, obtained by scanning the time variables t21 and t43 (see Figure 4a for a definition of these interpulse delay times), simultaneously produces four maps: the in-phase and the in-quadrature ones for the rephasing and nonrephasing frequencies.58 The maps so obtained in the time domain are, finally, converted in the energy domain by Fourier-transforming the time variables t21 and t43 (see Figure 4a for a definition of these interpulse delay times) and recorded as a function of the population time, t32. It is worth noting that, in contrast to the partitioning of resonant dispersive and absorptive features between the real and imaginary parts of the linear optical response, the rephasing and nonrephasing spectra contain mixed absorptive and dispersive contributions.
Figure 4.
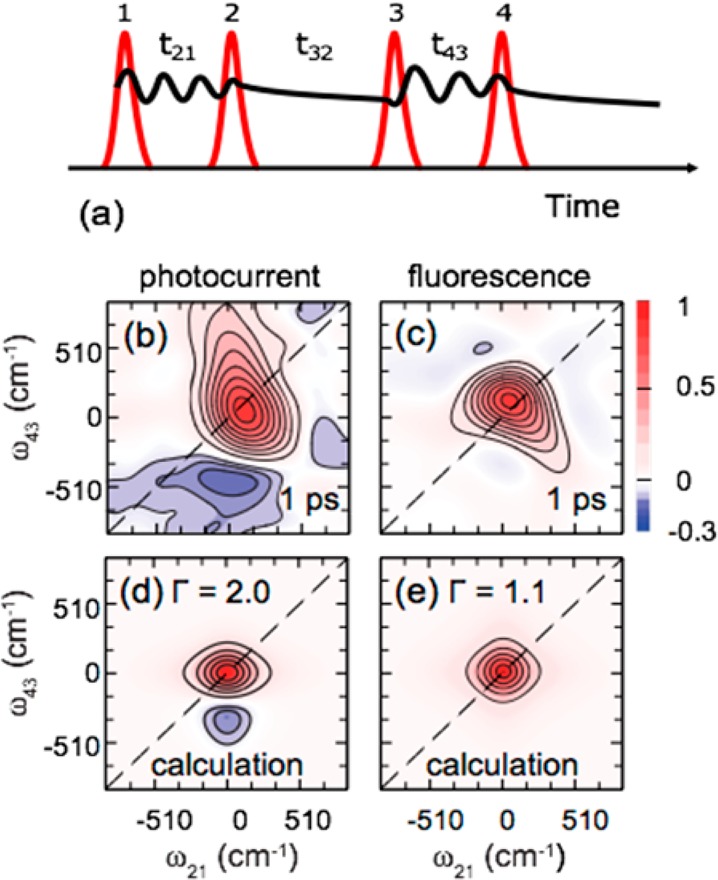
(a) Schematic of the pulse sequence used in a 2DPC experiment. (b–e) Reproduced from ref (57) with permission, Nature Publishing Group, 2015. The real part of the 2D total correlation spectra (that is, the sum of the rephasing and nonrephasing signals) as measured by (b) PC on a PbS colloidal quantum dot photocell and (c) by photoluminescence on a PbS colloidal suspension. The qualitative forms of the 2D line shape in each case reproduced from calculations are reported in (d) and (e).
The work by Karki et al., which addressed carrier multiplication processes in solar cells based on colloidal semiconductor quantum dots,57 demonstrated a subpicosecond evolution of the line shape of the 2DPC-detected spectra from absorptive to dispersive character, interpreted as a time-dependent shift of the resonant transition energies during exciton multiplication. The specific sensitivity accessible through directly probing the PC is illustrated in Figure 4, which compares the components of 2D total correlation spectra as measured by both PC and fluorescence detection.57 The PC data were acquired on the photocell, whereas the photoluminescence spectra were measured on a colloidal suspension sample under the same excitation conditions. As is evident, the two probes provide remarkably different spectra. In particular, the PC-detected spectrum shows a dispersive line shape, whereas the fluorescence detected one exhibits primarily absorptive features. It was speculated57 that the observed responses are different because the multiple excitons in quantum dots do not contribute efficiently to luminescence due to their rapid nonradiative Auger recombination. On the other hand, in the photocell, the electron–hole pairs in the multiple exciton states separate within a few picoseconds, thus contributing to a significant PC quantum yield. The results reported by Karki et al. provide an eloquent example of how different final observables, in that case, photoluminescence and PC, provide insights on the dynamics through the different excited-state pathways in optoelectronic materials and devices.
Another example of implementing PC detection in 2D coherent techniques probes a photovoltaic device based on the benchmark polymer–fullerene blend PCDTBT:PCBM.59Figure 5a displays J–V curves measured under solar illumination and under pulsed femtosecond excitation. In particular, the latter curve was acquired under the same excitation conditions as the 2D maps shown in Figure 5b–e. The comparison reported in Figure 5a illustrates yet another relevant advantage of the multidimensional PC-detected spectroscopy over TA methods; this PC-detected variant of 2D spectroscopy probes the photocarrier generation process in photovoltaic devices under actual working conditions.
Figure 5.
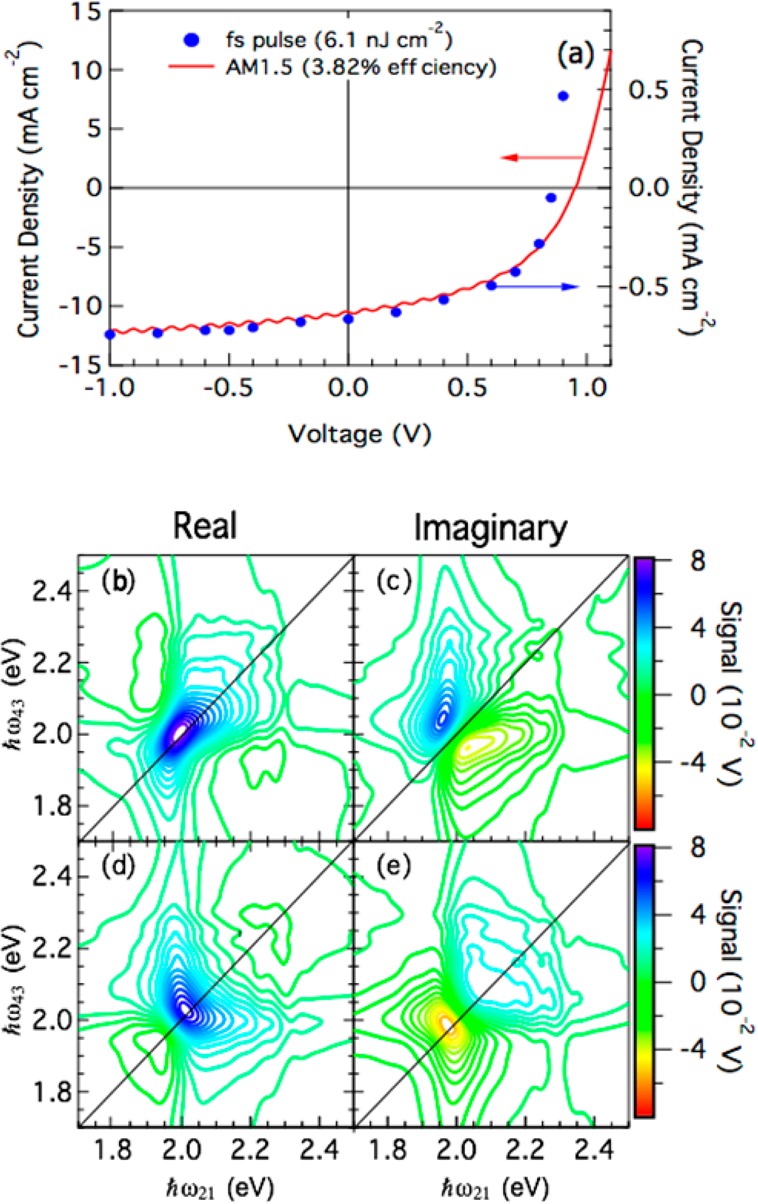
(a) J–V curve on an organic photovoltaic device under pulsed femtosecond excitation and under illumination with a solar simulator. 2DPC spectra on the same device: (b,c) rephasing signal, (d,e) nonrephasing signal. Reproduced from ref (59).
Figure 5 shows the PC-detected 2D real and imaginary spectra on this photocell; rephasing (Figure 5b,c) and nonrephasing spectra (Figure 5d,e) were acquired simultaneously using a two-channel lock-in amplifier. It highlights a powerful feature of the application of PC detection in the context of 2D coherence spectroscopies, which measure correlation spectra related to the full complex nonlinear permittivity function, composed of absorptive and dispersive contributions, in the PC excitation spectrum. This is an important aspect to evaluate the evolution of charged photoexcitations in excitonic solar cells; the details of the photocarrier production dynamics are contained in the complex permittivity function, as underlined eloquently by the work of Karki et al.57 These data represent, to the best of our knowledge, the first attempt to measure 2DPC spectra on an organic-based photocell. Beyond the field of photovoltaic devices based on novel materials, 2DPC might also be relevant in the study of single nanostructures where PCs have already been reported.60,61 Moreover, this technique could be particularly helpful in the investigation of polariton many-body correlations in inorganic quantum well Fabry–Pérot microcavities, where electrodes can be applied and PC easily detected.
Ultrafast PC Spectroscopy down to the Single-Molecule Level. An important advantage of electrical methods is their outstanding sensitivity and capacity to probe local phenomena. One well-established example of electrical spatially sensitive methods is scanning tunneling microscopy (STM; see Figure 6), which uses current detection to observe objects as small as single atoms or organic molecules. Such sensitivity is hardly achievable by all-optical methods due to the complexities with overcoming the optical diffraction limit, relatively low absorption cross section, and photobleaching. At the same time, single molecules can be stable enough to conduct well-detectable currents for long periods of time, providing large statistics and high accuracy of measured values. Unfortunately, conventional STM-type techniques are not suitable for observing fast (submicrosecond) dynamics of nanosystems due to the limitations of electrical circuits. The accessible time window is usually defined by the time constants of current injection and the response time of electric amplifiers.62,63 To resolve this limitation, ultrafast optical pulses can be used to modulate the current flow. There have already been a number of successful developments along this path. Femtosecond lasers were used to gate the STM tip,64 to switch the transmission lines,65 or to modulate the tunneling current by the tip and/or sample electronic excitation,66 which allowed, in some exceptional cases, one to reach a few-picoseconds dynamics regime. Unfortunately, the outlined approaches have not yet become a robust solution due to the complexity of the experimental approach.
Figure 6.
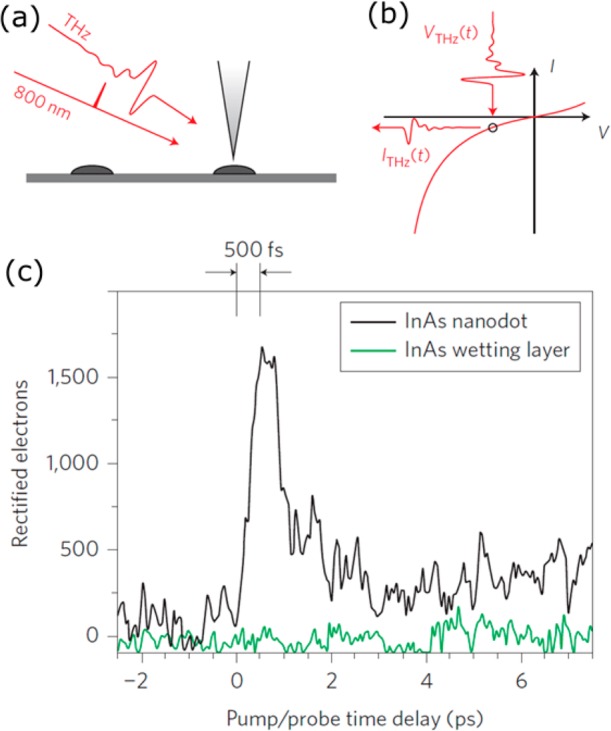
(a,b) The concept of a THz ultrafast tunneling microscope. (c) A representative time-resolved tunneling transient provide by such a microscope, which features subpicosecond time and 2 nm spatial resolution. Reproduced from ref (67) with permission, Nature Publishing Group, 2015.
An interesting development for ultrafast STM was recently proposed by Cocker and co-workers using THz spectroscopy instrumentation.67 In their approach, the STM junction bias was modulated by irradiating the scanning probe tip with subpicosecond terahertz pulses. This modulation introduced strong tunneling current pulses in a STM only for the period of THz pulse transiting the tip area, therefore gating the nanojunction between the tip and the sample in different moments of time. When combined with an additional, optical “dynamics trigger” pulse, the terahertz STM provided simultaneous <500 fs time resolution and nanometer (2 nm) imaging resolution under ambient laboratory conditions, which was sufficient, for example, to image ultrafast carrier capture into a single InAs quantum dot. We note that, despite the very different optical range and current generation mechanism, the concept of THz STM is quite similar to the 2PPC/2DPC described above. All of the methods first use visible pump pulse(s) to create the excited state of interest, and then, the THz or vis/IR “push” samples the dynamics of the excited states after a controllable delay time. The practical difference comes from the fact that the “reference” current is generated by visible light in 2PPC and by THz light in THz STM, which implies that different modulation schemes for lock-in detection should be employed.
The idea of using tunneling current is not limited to THz modulation and the STM approach. There is ongoing progress in applying electric detection and multipulse techniques to study different types of molecular junctions, down to the single-molecule level,68 or any other type of system that is conductive on the nanoscale. The above-mentioned studies illustrate the strength of ultrafast spectroscopy with PC detection when individual nanodimension systems or devices are concerned. This approach provides a unique opportunity to combine the outstanding sensitivity of electrical measurements with unprecedented time resolution of ultrafast optical techniques.
Information Provided by PC versus Optical Detection. On the basis of the existing studies, it is possible to make a few general conclusions about the type of responses that can be observed in ultrafast PC detection spectroscopies (Figure 1). First, only the processes affecting charge generation can be observed using PC detection. This allows neglect of many “background” responses, such as coherent artifacts, scattering, and photochromic effects, narrowing down the observable to the dynamics of interest. Second, the stimulated emission (SE) and ground-state bleach (GSB) responses are very observable with ultrafast PC methods. Particularly, SE signals can be interpreted in a similar way as they are in TA data, just by inverting the sign of the response.20 Indeed, whereas the all-optical methods register the increase of transmitted light, the PC methods should register the drop in the device output as fewer excited states are formed in the sample. Third, the origins of photoinduced absorption (PIA) responses are quite different in TA and PC. As the lifetime of higher-lying excited states in most optoelectronic materials is very short, the effect of the system dynamics due to the additional absorbed photon is determined by the thermalization pathway to the first excited state. If after the push pulse the system repopulates back the same state that it was excited from, no changes will be observed in the long-term charge dynamics and in the PC output. However, if the push pulse leads to a different or modified excited state, the charge dynamics are likely to be affected, and the device performance will vary. Both negative and positive PIA responses have been observed in ultrafast PC experiments. While the former are usually indicative of releasing a bound charge carrier, the latter are a sign of enhanced bimolecular processes like exciton–exciton annihilation or recombination.24 From this perspective, the positive PIA signals provide the most valuable information for the development of the optoelectronic devices as they help to identify bottleneck states in the excited-state dynamics. Positive PC signals present direct evidence that the device performance can be improved if additional energy is given to the system at the certain moments of time, which can serve as an efficient feedback parameter for device optimization.
Conclusions and Perspective for the Future. PC detection schemes in ultrafast optical probes have a distinct advantage in terms of sensitivity compared to optical probe techniques. Namely, when photocarriers are generated efficiently, this detection scheme has the potential to detect every photoexcitation event. This is certainly the case in semiconductor nanostructures56 but is also the case in molecular systems that are designed for applications in photodetectors and solar cells. PC detection opens the door for implementations that are simply not possible using optical detection schemes, such as single-molecule nonlinear optical probes under low-intensity illumination conditions representative of solar light. For example, we envision the development of single-molecule multidimensional spectroscopy that exploits the extraordinarily sensitivity of scanning probe techniques.
The opportunities presented by PC detection schemes will clearly facilitate the investigation of photosynthetic processes in natural systems with detail that goes beyond what is achievable with all-optical nonlinear probes such as four-wave mixing implementations of multidimensional spectroscopies. This is because a key product of the photosynthetic reaction—photocharge—can be isolated and interrogated. We highlight recent breakthroughs in processing of native pigment proteins in device architectures that probe PC. For example, Friese et al. have recently exploited the high quantum efficiency of photosynthetic pigment proteins by incorporating them directly in bioelectronic devices.69 These consist of bacterial reaction centers and light-harvesting-1 complexes, self-assembled on an electrochemically roughened silver electrode. Due to enhanced light absorption facilitated by moderate plasmonic effects from the rough surface, this biohybrid nanostructured architecture displays a peak PC of >165 μA cm–2, solely derived from the photosynthetic material, under 1 sun illumination, and up to 3-fold higher currents under more intense illumination.69 We anticipate that nonincremental progress in the understanding of natural light-harvesting mechanisms in diverse native protein environments will be achieved as a result of the opportunities presented by ultrafast PC probes.
Finally, the opportunity to examine the evolution of the complex permittivity function via PC probes in multidimensional excitation techniques promises breakthroughs in the understanding of photocarrier generation in state-of-the-art thin-film solar cells, such as emerging perovskite technologies. Measurement of this optical response over ultrafast time scales provides a key window into the materials physics among the most relevant for solar energy conversion: how are charges that can do work generated by solar light? Our perspective is that PC-detected optical probes will play a central role in unraveling these underlying physics.
Acknowledgments
A.A.B. is currently a Royal Society University Research Fellow. A.A.B. also acknowledges a VENI grant from the NWO. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 639750). C.S. acknowledges funding from the Natural Science and Engineering Research Council of Canada, the Fonds de recherche du Québec–nature et technologies, the Canada Research Chair in Organic Semiconductor Materials, and the Université de Montréal Research Chair. C.S. and E.V. acknowledge collaboration with Sachetan Tuladhar, Michelle Vezie, Sheridan Few, Jenny Nelson, Hao Li, and Eric Bittner. Finally, C.S. and E.V. acknowledge essential discussions with Andy Marcus and Julia Widom for the implementation of the two-dimensional spectroscopy apparatus.
Biographies
Artem A. Bakulin received his M.Sc. degree from Lomonosov Moscow State University and Ph.D. from the University of Groningen. He is currently a Royal Society University Research Fellow at the University of Cambridge, focusing on ultrafast spectroscopy of nanodevices and vibronic phenomena in organic materials.
Carlos Silva received a Ph.D. from the the University of Minnesota in 1998 and was a Postdoctoral Associate and then EPSRC Advanced Research Fellow in the Cavendish Laboratory, University of Cambridge. In 2005, he began his tenure at the Université de Montréal, where he held the Canada Research Chair in Organic Semiconductor Materials from 2005 to 2010. He currently holds a University Research Chair. His group focuses on optical and electronic properties of complex semiconductor materials.
Eleonora Vella received her Ph.D. form the University of Palermo and is currently a postdoctoral fellow in Prof. Silva’s group at Université de Montréal. Her work focuses on ultrafast and multidimensional coherent spectroscopy of devices and microcavities based on organic materials.
The authors declare no competing financial interest.
Notes
§C.S.: Visiting Professor, Experimental Solid State Physics, Department of Physics, Imperial College London, South Kensington Campus, London SW7 2AZ, United Kingdom.
References
- van Hal P. A.; Janssen R. A. J.; Lanzani G.; Cerullo G.; Zavelani-Rossi M.; De Silvestri S. Full Temporal Resolution of the Two-Step Photoinduced Energy-Electron Transfer in a Fullerene-Oligothiophene-Fullerene Triad Using Sub-10 Fs Pump-Probe Spectroscopy. Chem. Phys. Lett. 2001, 345, 33–38. 10.1016/S0009-2614(01)00874-0. [DOI] [Google Scholar]
- Scheblykin I. G.; Yartsev A.; Pullerits T.; Gulbinas V.; Sundstrom V. Excited State and Charge Photogeneration Dynamics in Conjugated Polymers. J. Phys. Chem. B 2007, 111, 6303–6321. 10.1021/jp068864f. [DOI] [PubMed] [Google Scholar]
- Gelinas S.; Pare-Labrosse O.; Brosseau C. N.; Albert-Seifried S.; McNeill C. R.; Kirov K. R.; Howard I. A.; Leonelli R.; Friend R. H.; Silva C. The Binding Energy of Charge-Transfer Excitons Localized at Polymeric Semiconductor Heterojunctions. J. Phys. Chem. C 2011, 115, 7114–7119. 10.1021/jp200466y. [DOI] [Google Scholar]
- Sheng C.-X.; Tong M.; Singh S.; Vardeny Z. V. Experimental Determination of the Charge/Neutral Branching Ratio in the Photoexcitation of P-Conjugated Polymers by Broadband Ultrafast Spectroscopy. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 75, 085206. 10.1103/PhysRevB.75.085206. [DOI] [Google Scholar]
- Bridewell V. L.; Alam R.; Karwacki C. J.; Kamat P. V. Cdse/Cds Nanorod Photocatalysts: Tuning the Interfacial Charge Transfer Process through Shell Length. Chem. Mater. 2015, 27, 5064–5071. 10.1021/acs.chemmater.5b01689. [DOI] [Google Scholar]
- Aly S. M.; Ahmed G. H.; Shaheen B. S.; Sun J.; Mohammed O. F. Molecular-Structure Control of Ultrafast Electron Injection at Cationic Porphyrin–Cdte Quantum Dot Interfaces. J. Phys. Chem. Lett. 2015, 6, 791–795. 10.1021/acs.jpclett.5b00235. [DOI] [PubMed] [Google Scholar]
- Backus E. H. G.; Bloem R.; Donaldson P. M.; Ihalainen J. A.; Pfister R.; Paoli B.; Caflisch A.; Hamm P. 2d-Ir Study of a Photoswitchable Isotope-Labeled A-Helix. J. Phys. Chem. B 2010, 114, 3735–3740. 10.1021/jp911849n. [DOI] [PubMed] [Google Scholar]
- Middleton C. T.; Marek P.; Cao P.; Chiu C.-c.; Singh S.; Woys A. M.; de Pablo J. J.; Raleigh D. P.; Zanni M. T. Two-Dimensional Infrared Spectroscopy Reveals the Complex Behaviour of an Amyloid Fibril Inhibitor. Nat. Chem. 2012, 4, 355–360. 10.1038/nchem.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse N.; Ashihara S.; Nibbering E. T. J.; Elsaesser T. Ultrafast Vibrational Relaxation of O–H Bending and Librational Excitations in Liquid H2o. Chem. Phys. Lett. 2005, 404, 389–393. 10.1016/j.cplett.2005.02.007. [DOI] [Google Scholar]
- Ostroumov E. E.; Mulvaney R. M.; Cogdell R. J.; Scholes G. D. Broadband 2d Electronic Spectroscopy Reveals a Carotenoid Dark State in Purple Bacteria. Science 2013, 340, 52–56. 10.1126/science.1230106. [DOI] [PubMed] [Google Scholar]
- Romero E.; Augulis R.; Novoderezhkin V. I.; Ferretti M.; Thieme J.; Zigmantas D.; van Grondelle R. Quantum Coherence in Photosynthesis for Efficient Solar-Energy Conversion. Nat. Phys. 2014, 10, 676–682. 10.1038/nphys3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piris J.; Dykstra T. E.; Bakulin A. A.; Loosdrecht P. H. M. v.; Knulst W.; Trinh M. T.; Schins J. M.; Siebbeles L. D. A. Photogeneration and Ultrafast Dynamics of Excitons and Charges in P3ht/Pcbm Blends. J. Phys. Chem. C 2009, 113, 14500–14506. 10.1021/jp904229q. [DOI] [Google Scholar]
- Marsh R. A.; Hodgkiss J. M.; Friend R. H. Direct Measurement of Electric Field-Assisted Charge Separation in Polymer:Fullerene Photovoltaic Diodes. Adv. Mater. 2010, 22, 3672–3676. 10.1002/adma.201001010. [DOI] [PubMed] [Google Scholar]
- Silva C.; Russell D. M.; Dhoot A. S.; Herz L. M.; Daniel C.; Greenham N. C.; Arias A. C.; Setayesh S.; Müllen K.; Friend R. H. Exciton and Polaron Dynamics in a Step-Ladder Polymeric Semiconductor: The Influence of Interchain Order. J. Phys.: Condens. Matter 2002, 14, 9803. 10.1088/0953-8984/14/42/302. [DOI] [Google Scholar]
- Rao A.; Wilson M. W. B.; Albert-Seifried S.; Di Pietro R.; Friend R. H. Photophysics of Pentacene Thin Films: The Role of Exciton Fission and Heating Effects. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 195411. 10.1103/PhysRevB.84.195411. [DOI] [Google Scholar]
- Bloem R.; Garrett-Roe S.; Strzalka H.; Hamm P.; Donaldson P. Enhancing Signal Detection and Completely Eliminating Scattering Usingquasi-Phase-Cycling in 2d Ir Experiments. Opt. Express 2010, 18, 27067–27078. 10.1364/OE.18.027067. [DOI] [PubMed] [Google Scholar]
- Augulis R.; Zigmantas D. Two-Dimensional Electronic Spectroscopy with Double Modulation Lock-in Detection: Enhancement of Sensitivity and Noise Resistance. Opt. Express 2011, 19, 13126–13133. 10.1364/OE.19.013126. [DOI] [PubMed] [Google Scholar]
- Vandewal K.; Gadisa A.; Oosterbaan W. D.; Bertho S.; Banishoeib F.; Van Severen I.; Lutsen L.; Cleij T. J.; Vanderzande D.; Manca J. V. The Relation between Open-Circuit Voltage and the Onset of Photocurrent Generation by Charge-Transfer Absorption in Polymer: Fullerene Bulk Heterojunction Solar Cells. Adv. Funct. Mater. 2008, 18, 2064–2070. 10.1002/adfm.200800056. [DOI] [Google Scholar]
- Congreve D. N.; Lee J.; Thompson N. J.; Hontz E.; Yost S. R.; Reusswig P. D.; Bahlke M. E.; Reineke S.; Van Voorhis T.; Baldo M. A. External Quantum Efficiency above 100% in a Singlet-Exciton-Fission–Based Organic Photovoltaic Cell. Science 2013, 340, 334–337. 10.1126/science.1232994. [DOI] [PubMed] [Google Scholar]
- Lukin L. V.; Tolmachev A. V.; Yakovlev B. S. The Photoexcitation of Trapped Electrons Produced in the Photoionization of Anthracene in Liquid N-Hexane. Chem. Phys. Lett. 1981, 81, 595–598. 10.1016/0009-2614(81)80471-X. [DOI] [Google Scholar]
- Brazgun F. F.; Nadtochenko V. A.; Rubtsov I. V.; Lukin L. V. Dynamics of Geminate Charge Separation in Liquid Methylcyclohexane Studied by the Photoassisted Ion Pair Separation Technique. Chem. Phys. 1996, 211, 469–488. 10.1016/0301-0104(96)00149-8. [DOI] [Google Scholar]
- Muller J. G.; Lemmer U.; Feldmann J.; Scherf U. Precursor States for Charge Carrier Generation in Conjugated Polymers Probed by Ultrafast Spectroscopy. Phys. Rev. Lett. 2002, 88, 147401. 10.1103/PhysRevLett.88.147401. [DOI] [PubMed] [Google Scholar]
- Frankevich E.; Ishii H.; Hamanaka Y.; Yokoyama T.; Fuji A.; Li S.; Yoshino K.; Nakamura A.; Seki K. Formation of Polaron Pairs and Time-Resolved Photogeneration of Free Charge Carriers in P-Conjugated Polymers. Phys. Rev. B: Condens. Matter Mater. Phys. 2000, 62, 2505–2515. 10.1103/PhysRevB.62.2505. [DOI] [Google Scholar]
- Bakulin A. A.; Rao A.; Pavelyev V. G.; van Loosdrecht P. H. M.; Pshenichnikov M. S.; Niedzialek D.; Cornil J.; Beljonne D.; Friend R. H. The Role of Driving Energy and Delocalized States for Charge Separation in Organic Semiconductors. Science 2012, 335, 1340–1344. 10.1126/science.1217745. [DOI] [PubMed] [Google Scholar]
- Dimitrov S. D.; Bakulin A. A.; Nielsen C. B.; Schroeder B. C.; Du J.; Bronstein H.; McCulloch I.; Friend R. H.; Durrant J. R. On the Energetic Dependence of Charge Separation in Low-Band-Gap Polymer/Fullerene Blends. J. Am. Chem. Soc. 2012, 134, 18189–18192. 10.1021/ja308177d. [DOI] [PubMed] [Google Scholar]
- Savoie B. M.; Rao A.; Bakulin A. A.; Gélinas S.; Movaghar B.; Friend R. H.; Marks T. J.; Ratner M. A. Unequal Partnership: Asymmetric Roles of Polymeric Donor and Fullerene Acceptor in Generating Free Charge. J. Am. Chem. Soc. 2014, 136, 2876–2884. 10.1021/ja411859m. [DOI] [PubMed] [Google Scholar]
- Mangold H.; Bakulin A. A.; Howard I. A.; Kastner C.; Egbe D. A. M.; Hoppe H.; Laquai F. Control of Charge Generation and Recombination in Ternary Polymer/Polymer:Fullerene Photovoltaic Blends Using Amorphous and Semi-Crystalline Copolymers as Donors. Phys. Chem. Chem. Phys. 2014, 16, 20329–20337. 10.1039/C4CP01883D. [DOI] [PubMed] [Google Scholar]
- Vaynzof Y.; Bakulin A. A.; Gélinas S.; Friend R. H. Direct Observation of Photoinduced Bound Charge-Pair States at an Organic-Inorganic Semiconductor Interface. Phys. Rev. Lett. 2012, 108, 246605. 10.1103/PhysRevLett.108.246605. [DOI] [PubMed] [Google Scholar]
- Pachoumi O.; Bakulin A. A.; Sadhanala A.; Sirringhaus H.; Friend R. H.; Vaynzof Y. Improved Performance of Zno/Polymer Hybrid Photovoltaic Devices by Combining Metal Oxide Doping and Interfacial Modification. J. Phys. Chem. C 2014, 118, 18945–18950. 10.1021/jp506266f. [DOI] [Google Scholar]
- Bakulin A. A.; Neutzner S.; Bakker H. J.; Ottaviani L.; Barakel D.; Chen Z. Charge Trapping Dynamics in Pbs Colloidal Quantum Dot Photovoltaic Devices. ACS Nano 2013, 7, 8771–8779. 10.1021/nn403190s. [DOI] [PubMed] [Google Scholar]
- Bakulin A. A.; Lovrincic R.; Yu X.; Selig O.; Bakker H. J.; Rezus Y. L. A.; Nayak P. K.; Fonari A.; Coropceanu V.; Bredas J.-L.; et al. Mode-Selective Vibrational Modulation of Charge Transport in Organic Electronic Devices. Nat. Commun. 2015, 6, 7880. 10.1038/ncomms8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.; Lawrence C. M.; Xiao D.; Kireev V. V.; Skourtis S. S.; Sessler J. L.; Beratan D. N.; Rubtsov I. V. Modulating Unimolecular Charge Transfer by Exciting Bridge Vibrations. J. Am. Chem. Soc. 2009, 131, 18060–18062. 10.1021/ja907041t. [DOI] [PubMed] [Google Scholar]
- Delor M.; Scattergood P. A.; Sazanovich I. V.; Parker A. W.; Greetham G. M.; Meijer A. J. H. M.; Towrie M.; Weinstein J. A. Toward Control of Electron Transfer in Donor-Acceptor Molecules by Bond-Specific Infrared Excitation. Science 2014, 346, 1492–1495. 10.1126/science.1259995. [DOI] [PubMed] [Google Scholar]
- Helbing J.; Hamm P. Compact Implementation of Fourier Transform Two-Dimensional Ir Spectroscopy without Phase Ambiguity. J. Opt. Soc. Am. B 2011, 28, 171–178. 10.1364/JOSAB.28.000171. [DOI] [Google Scholar]
- Skoff D. R.; Laaser J. E.; Mukherjee S. S.; Middleton C. T.; Zanni M. T. Simplified and Economical 2d Ir Spectrometer Design Using a Dual Acousto-Optic Modulator. Chem. Phys. 2013, 422, 8–15. 10.1016/j.chemphys.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read E. L.; Engel G. S.; Calhoun T. R.; Mančal T.; Ahn T. K.; Blankenship R. E.; Fleming G. R. Cross-Peak-Specific Two-Dimensional Electronic Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14203–14208. 10.1073/pnas.0701201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womick J. M.; Moran A. M. Nature of Excited States and Relaxation Mechanisms in C-Phycocyanin. J. Phys. Chem. B 2009, 113, 15771–15782. 10.1021/jp908093x. [DOI] [PubMed] [Google Scholar]
- Panitchayangkoon G.; Hayes D.; Fransted K. A.; Caram J. R.; Harel E.; Wen J.; Blankenship R. E.; Engel G. S. Long-Lived Quantum Coherence in Photosynthetic Complexes at Physiological Temperature. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 12766–12770. 10.1073/pnas.1005484107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom J. R.; Johnson N. P.; von Hippel P. H.; Marcus A. H. Solution Conformation of 2-Aminopurine Dinucleotide Determined by Ultraviolet Two-Dimensional Fluorescence Spectroscopy. New J. Phys. 2013, 15, 025028. 10.1088/1367-2630/15/2/025028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott G. A.; Perdomo-Ortiz A.; Utterback J. K.; Widom J. R.; Aspuru-Guzik A.; Marcus A. H. Conformation of Self-Assembled Porphyrin Dimers in Liposome Vesicles by Phase-Modulation 2d Fluorescence Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16521–16526. 10.1073/pnas.1017308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo-Ortiz A.; Widom J. R.; Lott G. A.; Aspuru-Guzik A.; Marcus A. H. Conformation and Electronic Population Transfer in Membrane-Supported Self-Assembled Porphyrin Dimers by 2d Fluorescence Spectroscopy. J. Phys. Chem. B 2012, 116, 10757–10770. 10.1021/jp305916x. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhang T.; Borca C. N.; Cundiff S. T. Many-Body Interactions in Semiconductors Probed by Optical Two-Dimensional Fourier Transform Spectroscopy. Phys. Rev. Lett. 2006, 96, 057406. 10.1103/PhysRevLett.96.057406. [DOI] [PubMed] [Google Scholar]
- Stone K. W.; Gundogdu K.; Turner D. B.; Li X.; Cundiff S. T.; Nelson K. A. Two-Quantum 2d Ft Electronic Spectroscopy of Biexcitons in Gaas Quantum Wells. Science 2009, 324, 1169–1173. 10.1126/science.1170274. [DOI] [PubMed] [Google Scholar]
- Turner D. B.; Wen P.; Arias D. H.; Nelson K. A.; Li H.; Moody G.; Siemens M. E.; Cundiff S. T. Persistent Exciton-Type Many-Body Interactions in Gaas Quantum Wells Measured Using Two-Dimensional Optical Spectroscopy. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 85, 201303. 10.1103/PhysRevB.85.201303. [DOI] [Google Scholar]
- Moran A. M.; Maddox J. B.; Hong J. W.; Kim J.; Nome R. A.; Bazan G. C.; Mukamel S.; Scherer N. F. Optical Coherence and Theoretical Study of the Excitation Dynamics of a Highly Symmetric Cyclophane-Linked Oligophenylenevinylene Dimer. J. Chem. Phys. 2006, 124, 194904. 10.1063/1.2196041. [DOI] [PubMed] [Google Scholar]
- Stiopkin I.; Brixner T.; Yang M.; Fleming G. R. Heterogeneous Exciton Dynamics Revealed by Two-Dimensional Optical Spectroscopy. J. Phys. Chem. B 2006, 110, 20032–20037. 10.1021/jp062882f. [DOI] [PubMed] [Google Scholar]
- Womick J. M.; Miller S. A.; Moran A. M. Correlated Exciton Fluctuations in Cylindrical Molecular Aggregates. J. Phys. Chem. B 2009, 113, 6630–6639. 10.1021/jp810291d. [DOI] [PubMed] [Google Scholar]
- Milota F.; Prokhorenko V. I.; Mancal T.; von Berlepsch H.; Bixner O.; Kauffmann H. F.; Hauer J. Vibronic and Vibrational Coherences in Two-Dimensional Electronic Spectra of Supramolecular J-Aggregates. J. Phys. Chem. A 2013, 117, 6007–6014. 10.1021/jp3119605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin A.; Johnson P. J. M.; Tempelaar R.; Murphy R. S.; Knoester J.; Jansen T. L. C.; Miller R. J. D. Two-Dimensional Spectroscopy of a Molecular Dimer Unveils the Effects of Vibronic Coupling on Exciton Coherences. Nat. Chem. 2014, 6, 196–201. 10.1038/nchem.1834. [DOI] [PubMed] [Google Scholar]
- Wen P.; Christmann G.; Baumberg J. J.; Nelson K. A. Influence of Multi-Exciton Correlations on Nonlinear Polariton Dynamics in Semiconductor Microcavities. New J. Phys. 2013, 15, 025005. 10.1088/1367-2630/15/2/025005. [DOI] [Google Scholar]
- Hybl J. D.; Albrecht Ferro A.; Jonas D. M. Two-Dimensional Fourier Transform Electronic Spectroscopy. J. Chem. Phys. 2001, 115, 6606–6622. 10.1063/1.1398579. [DOI] [PubMed] [Google Scholar]
- Bristow A. D.; Karaiskaj D.; Dai X.; Zhang T.; Carlsson C.; Hagen K. R.; Jimenez R.; Cundiff S. T. A Versatile Ultrastable Platform for Optical Multidimensional Fourier-Transform Spectroscopy. Rev. Sci. Instrum. 2009, 80, 073108. 10.1063/1.3184103. [DOI] [PubMed] [Google Scholar]
- Turner D. B.; Stone K. W.; Gundogdu K.; Nelson K. A. Invited Article: The Coherent Optical Laser Beam Recombination Technique (Colbert) Spectrometer: Coherent Multidimensional Spectroscopy Made Easier. Rev. Sci. Instrum. 2011, 82, 081301. 10.1063/1.3624752. [DOI] [PubMed] [Google Scholar]
- de Boeij W. P.; Pshenichnikov M. S.; Wiersma D. A. Heterodyne-Detected Stimulated Photon Echo: Applications to Optical Dynamics in Solution. Chem. Phys. 1998, 233, 287–309. 10.1016/S0301-0104(98)00084-6. [DOI] [Google Scholar]
- Tekavec P. F.; Lott G. A.; Marcus A. H. Fluorescence-Detected Two-Dimensional Electronic Coherence Spectroscopy by Acousto-Optic Phase Modulation. J. Chem. Phys. 2007, 127, 214307. 10.1063/1.2800560. [DOI] [PubMed] [Google Scholar]
- Nardin G.; Autry T. M.; Silverman K. L.; Cundiff S. T. Multidimensional Coherent Photocurrent Spectroscopy of a Semiconductor Nanostructure. Opt. Express 2013, 21, 28617–28627. 10.1364/OE.21.028617. [DOI] [PubMed] [Google Scholar]
- Karki K. J.; Widom J. R.; Seibt J.; Moody I.; Lonergan M. C.; Pullerits T.; Marcus A. H. Coherent Two-Dimensional Photocurrent Spectroscopy in a Pbs Quantum Dot Photocell. Nat. Commun. 2014, 5, 5869. 10.1038/ncomms6869. [DOI] [PubMed] [Google Scholar]
- Mukamel S.Principles of Nonlinear Optical Spectroscopy; Oxford University Press: New York, 1999. [Google Scholar]
- Vella E.; Grégoire P.; Li H.; Tuladhar S. M.; Vezie M.; Few S.; Nelson J.; Bittner E. R.; Silva C.. Two-Dimensional Coherent Photocurrent Excitation Spectroscopy in a Polymer Solar Cell. arXiv preprint arXiv:1506.07837, 2015.
- Zrenner A.; Beham E.; Stufler S.; Findeis F.; Bichler M.; Abstreiter G. Coherent Properties of a Two-Level System Based on a Quantum-Dot Photodiode. Nature 2002, 418, 612–614. 10.1038/nature00912. [DOI] [PubMed] [Google Scholar]
- Bao W.; Melli M.; Caselli N.; Riboli F.; Wiersma D. S.; Staffaroni M.; Choo H.; Ogletree D. F.; Aloni S.; Bokor J.; et al. Mapping Local Charge Recombination Heterogeneity by Multidimensional Nanospectroscopic Imaging. Science 2012, 338, 1317–1321. 10.1126/science.1227977. [DOI] [PubMed] [Google Scholar]
- Kemiktarak U.; Ndukum T.; Schwab K. C.; Ekinci K. L. Radio-Frequency Scanning Tunnelling Microscopy. Nature 2007, 450, 85–88. 10.1038/nature06238. [DOI] [PubMed] [Google Scholar]
- Loth S.; Etzkorn M.; Lutz C. P.; Eigler D. M.; Heinrich A. J. Measurement of Fast Electron Spin Relaxation Times with Atomic Resolution. Science 2010, 329, 1628–1630. 10.1126/science.1191688. [DOI] [PubMed] [Google Scholar]
- Yarotski D. A.; Averitt R. D.; Negre N.; Crooker S. A.; Taylor A. J.; Donati G. P.; Stintz A.; Lester L. F.; Malloy K. J. Ultrafast Carrier-Relaxation Dynamics in Self-Assembled Inas/Gaas Quantum Dots. J. Opt. Soc. Am. B 2002, 19, 1480–1484. 10.1364/JOSAB.19.001480. [DOI] [Google Scholar]
- Khusnatdinov N. N.; Nagle T. J.; Nunes G. Ultrafast Scanning Tunneling Microscopy with 1 Nm Resolution. Appl. Phys. Lett. 2000, 77, 4434–4436. 10.1063/1.1336817. [DOI] [Google Scholar]
- Terada Y.; Yoshida S.; Takeuchi O.; Shigekawa H. Real-Space Imaging of Transient Carrier Dynamics by Nanoscale Pump-Probe Microscopy. Nat. Photonics 2010, 4, 869–874. 10.1038/nphoton.2010.235. [DOI] [Google Scholar]
- Cocker T. L.; Jelic V.; Gupta M.; Molesky S. J.; Burgess J. A. J.; Reyes G. D. L.; Titova L. V.; Tsui Y. Y.; Freeman M. R.; Hegmann F. A. An Ultrafast Terahertz Scanning Tunnelling Microscope. Nat. Photonics 2013, 7, 620–625. 10.1038/nphoton.2013.151. [DOI] [Google Scholar]
- Selzer Y.; Peskin U. Transient Dynamics in Molecular Junctions: Picosecond Resolution from Dc Measurements by a Laser Pulse Pair Sequence Excitation. J. Phys. Chem. C 2013, 117, 22369–22376. 10.1021/jp403005q. [DOI] [Google Scholar]
- Kamran M.; Friebe V. M.; Delgado J. D.; Aartsma T. J.; Frese R. N.; Jones M. R. Demonstration of Asymmetric Electron Conduction in Pseudosymmetrical Photosynthetic Reaction Centre Proteins in an Electrical Circuit. Nat. Commun. 2015, 6, 6530. 10.1038/ncomms7530. [DOI] [PMC free article] [PubMed] [Google Scholar]