

Abstract
Purpose
Recent studies suggest that the neural retinal response to light is compromised in diabetes. Electroretinogram studies suggest that the dim light retinal rod pathway is especially susceptible to diabetic damage. The purpose of this study was to determine whether diabetes alters rod pathway signaling.
Methods
Diabetes was induced in C57BL/6J mice by three intraperitoneal injections of streptozotocin (STZ; 75 mg/kg), and confirmed by blood glucose levels > 200 mg/dL. Six weeks after the first injection, whole-cell voltage clamp recordings of spontaneous and light-evoked inhibitory postsynaptic currents from rod bipolar cells were made in dark-adapted retinal slices. Light-evoked excitatory currents from rod bipolar and AII amacrine cells, and spontaneous excitatory currents from AII amacrine cells were also measured. Receptor inputs were pharmacologically isolated. Immunohistochemistry was performed on whole mounted retinas.
Results
Rod bipolar cells had reduced light-evoked inhibitory input from amacrine cells but no change in excitatory input from rod photoreceptors. Reduced light-evoked inhibition, mediated by both GABAA and GABAC receptors, increased rod bipolar cell output onto AII amacrine cells. Spontaneous release of GABA onto rod bipolar cells was increased, which may limit GABA availability for light-evoked release. These physiological changes occurred in the absence of retinal cell loss or changes in GABAA receptor expression levels.
Conclusions
Our results indicate that early diabetes causes deficits in the rod pathway leading to decreased light-evoked rod bipolar cell inhibition and increased rod pathway output that provide a basis for the development of early diabetic visual deficits.
Keywords: diabetes, GABA, bipolar cells, amacrine cells, inhibition
Diabetic retinopathy is the leading cause of adult onset blindness in the United States.1 The most notable characteristic of diabetic retinopathy is the progressive decline of vascular function, which ultimately causes retinal damage and blindness.2 Additionally, there is mounting evidence from diabetic patients and animal models that show deficits in visual function such as contrast sensitivity, acuity, night vision, and retinal neuronal signaling.2–4 These deficits are likely due to direct changes in retinal signaling, as changes in the global retinal measurement electroretinogram (ERG) are also reported early in diabetes.2–6 Retinal signaling begins with light detection by photoreceptors that is transduced to bipolar, amacrine, and ganglion cells using glutamate.7 Retinal output is modulated by GABA or glycine release from inhibitory amacrine cells onto bipolar and ganglion cells.7 Light sensed by rod photoreceptors triggers rod bipolar cell excitation that is reflected in the b-wave of ERGs. Some studies have reported reduced b-wave amplitudes in diabetes8,9 suggesting reduced photoreceptor function, while others have shown the b-wave to be unaffected.3,10 Electroretinogram oscillatory potentials, which reflect signaling between bipolar and amacrine cells, are consistently altered in diabetes.3,4,6,8,10 However, the ERG technique is used to measure a population response to light and cannot discriminate responses of individual neurons or determine if signaling has changed due to loss of neurons.
We used the streptozotocin (STZ) mouse model of type 1 diabetes to determine what properties of retinal signaling were altered in early (6 weeks) diabetes. We recorded the response of single retinal neurons to light. We focused on neurons that comprise the rod pathway because ERG studies have suggested that they may be more susceptible to hyperglycemic damage.3,11 A recent study also found dysfunctional synaptic transmission between neurons in the rod pathway but did not determine whether the response to light, the natural stimulus of the retina, was altered.12 We found that diabetes decreases light-evoked GABAergic signaling from amacrine cells to rod bipolar cells, which increases the output of the rod pathway onto downstream neurons independent of rod photoreceptor dysfunction or loss of retinal neurons.
Research Design and Methods
Animals
Animal protocols conformed with the ARVO Statement for the Use of Animals in Ophthalmic and Visual Research and were approved by the University of Arizona Institutional Animal Care and Use Committee. Experiments used C57BL/6J male mice (Jackson Laboratories, Bar Harbor, ME, USA) that were housed in the University of Arizona animal facility and given the National Institutes of Health-31 rodent diet food and water ad libitum. Five-week-old mice were fasted for 4 hours and injected intraperitoneally with either STZ (Sigma-Aldrich Corp., St. Louis, MO, USA; 75 mg/kg body weight) dissolved in 0.01 M pH 4.5 citrate buffer or citrate buffer vehicle for three consecutive days.13 Body weight and urine glucose were monitored weekly. Six weeks after the first injection, mice were fasted for 4 hours and blood glucose was measured (OneTouch UltraMini; LifeScan, Milpitas, CA, USA). Streptozotocin-injected animals with blood glucose ≤ 200 mg/dL were eliminated from the study. Fasting blood glucose was 361 ± 9.5 mg/dL (n = 50 mice) for STZ-treated mice and 137 ± 5 mg/dL (n = 36 mice; P < 0.001 unpaired Student's t-test) for control mice. Body weights of diabetic and control mice were 21.6 ± 0.3 and 24.5 ± 0.4 g (P < 0.001), respectively.
Solutions and Drugs
Extracellular solution (bubbled with 95%/5% O2/CO2) contained (in mM): 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 20 glucose, 26 NaHCO3, 2 CaCl2. Extracellular solution for spontaneous GABAC receptor (R) recordings contained (in mM): 120 NaCl, 15 KCl, 1 MgCl2, 1.25 NaH2PO4, 5 glucose, 26 NaHCO3, 2 CaCl2. The recording pipette intracellular solution contained (in mM): 120 CsOH, 120 gluconic acid, 1 MgCl2, 10 HEPES, 10 EGTA, 10 TEA-Cl, 10 phosphocreatine-Na2, 4 Mg-ATP, 0.5 Na-GTP, 50 μM Alexa Fluor-488 (Invitrogen, Carlsbad, CA, USA) adjusted to pH 7.2 with CsOH. Strychnine (500 nM–1 μM), SR95531 (20 μM), TPMPA (1,2,5,6-Tetrahydropyridin-4-yl)methylphosphinic acid hydrate, 50 μM) were used to block glycine, GABAA, and GABAC receptors, respectively. Tetrodotoxin (TTX; 500 nm) and CdCl2 (100 μM) were used to block voltage-gated Na+ and Ca2+ channels. All solutions were applied (∼1 mL/minute) via gravity-driven superfusion system (Cell Microcontrols, Norfolk, VA, USA). Chemicals were purchased from Sigma-Aldrich Corp.
Preparation and Recordings
Six weeks after injections, retinal slices were prepared from mice dark-adapted overnight. Infrared illumination was used for dissections to preserve the light sensitivity.14 Briefly, eyes were enucleated from mice killed using carbon dioxide, corneas and lenses removed, eyecups incubated in extracellular solution with hyaluronidase (800 units/mL) for 20 minutes, and retinas removed. The retina was trimmed, mounted onto filter paper, and sliced into 250-μm slices. Whole-cell voltage-clamp recordings in dark-adapted retinal slices were made under infrared illumination at 32°C.14 Light-evoked inhibitory postsynaptic currents (L-IPSCs) and spontaneous (s)IPSCs were recorded at 0 mV (reversal potential for nonselective cation channels). Light-evoked excitatory postsynaptic currents (L-EPSCs), spontaneous (s)EPSCs, and miniature (m)EPSCs were recorded at −60 mV (reversal potential for Cl−). Borosilicate glass electrodes (World Precision Instruments, Sarasota, FL, USA) had resistances of 5 to 7 MΩ and the series resistance during recordings was typically 10 to 20 MΩ. Liquid junction potentials of 20 mV were corrected prior to recording. Responses were filtered at 5 kHz on an Axopatch 200B amplifier and digitized at 10 kHz using a Digidata 1440A A/D board and Clampex software (Molecular Devices, Sunnyvale, CA, USA). Alexa fluorescence was imaged at the end of each recording using an Intensilight fluorescence lamp and Digitalsight camera controlled by Elements software (Nikon Instruments, Tokyo, Japan) to confirm rod bipolar cell15 and AII amacrine cell16 morphology. Full-field light stimuli were generated by a light emitting diode (LED, λpeak = 525 nm) projected through the microscope camera port onto the retina. The light stimuli used were calibrated as photons/μm2/s and converted to rhodopsin isomerizations per second using a collecting area of 0.5 μm2.17 Light intensity and duration (30 ms) were controlled by varying the current through the LED.
Electrophysiology Analysis and Statistics
Light-evoked inhibitory postsynaptic currents and L-EPSCs for each condition were averaged, and the peak amplitude and charge transfer (Q - over the time of the response for each cell) were measured using Clampfit (Molecular Devices). Because the data were not normally distributed, log two-way repeated measures ANOVAs were used to compare L-IPSCs and L-EPSCs between conditions across light intensities, using the Student-Newman-Keuls (SNK) for pairwise comparisons. P-values reflect the main treatment effect of STZ unless otherwise indicated. Mini-Analysis software (Synaptosoft, Fort Lee, NJ, USA) was used to analyze the peak amplitude, tdecay, interevent interval and frequency of sIPSCs, sEPSCs, and mEPSCs.18–20 The threshold was set from 2 to 5 pA depending on the baseline noise, and events were subjected to visual confirmation. Events separated by an interval ≥ 5 ms were fit with a single exponential to measure the decay time constant. Values were compared using the Kolmogorov–Smirnov test (K–S). Average GABAA and GABAC receptor sIPSCs from rod bipolar cells and AII amacrine cell mEPSCs were generated using Mini-Analysis software. Differences were considered significant when P < 0.05.
The estimated amount of transmitter release mediating rod bipolar cell GABAA and GABAC receptor-mediated L-IPSCs, and AII amacrine cell L-EPSCs were calculated using custom Matlab software. Release functions were calculated by deconvolution analysis21 using the relationship20:
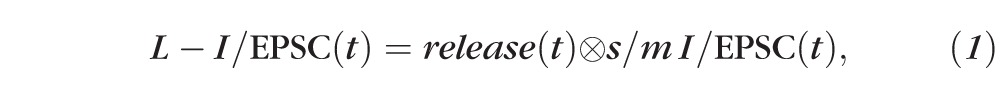 |
such that
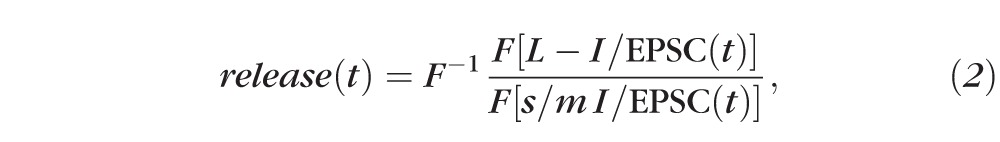 |
Data are reported as mean ± standard error of the mean (SEM) unless otherwise indicated.
Immunohistochemistry, Imaging, and Analysis
Whole-mounted retinas were fixed in 4% paraformaldehyde for 15 (GABAA α1 subunit and protein kinase C [PKC]) or 30 minutes (PKC and TO-PRO-3) at room temperature, and rinsed with 0.1 M phosphate-buffered saline (PBS; pH 7.4). The tissue was incubated overnight in blocking solution (5% Donkey serum and 0.5% Triton X-100 in 0.1 M PBS) then incubated with primary antibodies for 3 (GABAA α1 subunit and PKC) or 5 days (PKC) at 4°C. Anti-PKC (mouse monoclonal antibody, 1:1000; Sigma-Aldrich Corp.) primary antibody labelled rod bipolar cells. Anti-GABAA α1 subunit antibody (guinea pig polyclonal, 1:5000, provided by J.M. Fritschy) labelled GABAA receptors. Retinas were incubated overnight with secondary antibodies in PBS, then with TO-PRO-3 (Life Technologies, Carlsbad, CA, USA) nuclear stain (retinas labeled for only PKC), washed with PBS, and mounted in Vectashield (Vector Labs, Burlingame, CA, USA). Secondary antibodies utilized were anti-mouse Alexa 488 (1:1000; Invitrogen), anti-guinea pig Alexa 568 (1:1000; Invitrogen) or anti-mouse Alexa 649 (1:500; Jackson ImmunoResearch, West Grove, PA, USA).
To determine the numbers of cells per each layer, retinas were imaged using a Zeiss (Oberkochen, Germany) LSM 510 Meta confocal microscope with a 40× 1.2 NA objective. Stacked images (1-μm thick) were acquired from four regions 500 μm from the optic nerve head (142.9 × 142.9 μm2). Cells were counted using the ImageJ Cell Counter plug-in (http://rsbweb.nih.gov/ij/plugins/cell-counter.html; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). A different marker was used to mark unique cells in each vertical (Z-axis) layer to prevent double counting. Only cells with nuclei more than 50% within the borders of the region of interest were counted. The cell numbers were averaged among the four locations in each retina, compared between groups using an unpaired Student's t-test, and considered significant if P < 0.05.
To analyze GABAA receptor labeling, images were acquired on a FV1000 Olympus (Center Valley, PA, USA) laser scanning confocal microscope with a 60× 1.35 NA objective at a voxel size of 0.051, 0.051, 0.3 μm. Images were processed with MetaMorph (Molecular Devices) and Amira (FEI Visualization Sciences Group, Hillsboro, OR, USA) software programs. To isolate rod bipolar cell terminals, the PKC signal was masked in 3D using the Labelfield function in Amira. The mask was multiplied with the receptor channel using the Arithmetic function in Amira to exclude receptor signal outside the mask and isolate the receptor signal exclusively within the PKC labeled rod bipolar cell terminals. A constant threshold was applied on the receptor channel to exclude any background nonclustered signal and determine the receptor pixels within the mask. The volume of detected receptor pixels was expressed as a percentage of the total volume occupied by the pixels within the mask and compared between groups using an unpaired Student's t-test.22–24
Results
Early Diabetes Decreases Amacrine Cell Signaling to Rod Bipolar Cells
To determine if early diabetes (6 weeks post-STZ) reduces rod bipolar cell activation by rod photoreceptors, we recorded L-EPSCs from rod bipolar cells after 30 ms light stimuli of varying light intensities that cover both rod and cone activation (Fig. 1A).25 There were no significant differences in either the peak amplitudes (P = 0.2; Fig. 1B) or Q (P = 0.8; Fig. 1C) of the L-EPSCs. These results show that 6 weeks of diabetes does not reduce light-evoked activation of rod bipolar cells by rod photoreceptors and agree with ERG studies that show no difference in the b-wave.
Figure 1.

Light-evoked inhibition to rod bipolar cells is reduced after 6 weeks of diabetes. (A) Representative traces of L-EPSCs averaged from two responses at the maximum light intensity recorded from rod bipolar cells in response to a 30-ms light stimulus onto rod photoreceptors (R, inset) are shown. Light-evoked excitatory postsynaptic currents are not different between control (n = 7 cells from 5 mice) and diabetic (n = 8 cells from 7 mice) rod bipolar cells. (B, C) The peak amplitude (B) and magnitude (Q, C) of L-EPSCs at increasing intensities are similar between both groups (P = 0.2, 2-way ANOVA). (D) Representative traces of L-IPSCs averaged from two responses at the maximum light intensity recorded from rod bipolar cells in response to a 30-ms light stimulus (inset) are shown. (E, F) The light-evoked inhibitory postsynaptic currents peak amplitude (E) and Q (F) of diabetic (n = 18 cells from 12 mice) treated rod bipolar cells are reduced compared to control cells (n = 16 cells from 12 mice) at multiple intensities. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant; black bar = light stimulus.
To determine if amacrine cell-mediated inhibition to rod bipolar cells is changed in early diabetes, we recorded L-IPSCs from rod bipolar cells (Fig. 1D). Rod bipolar cell L-IPSCs from diabetic mice had lower peak amplitudes (P = 0.002; Fig. 1E) and Q's (P = 0.02; Fig. 1F) compared to control. Pairwise comparisons determined that peak amplitudes were reduced at dim and bright intensities (P < 0.05, SNK post hoc), and Q was reduced at a dim intensity (P = 0.03). Thus, light-evoked inhibitory input from amacrine cells to rod bipolar cells is reduced after 6 weeks of diabetes, independent of excitatory input from rod photoreceptors, suggesting an imbalance in excitatory and inhibitory inputs.
Early Diabetes Decreases Light-Evoked GABAergic Inhibition to Rod Bipolar Cells
Inhibition to rod bipolar cells is composed of glycinergic inputs onto glycine receptors and GABAergic inputs onto GABAA and GABAC receptors.26 However, proportionally GABAergic inputs contribute most of the inhibition to rod bipolar cells.19 Changes in GABA production and GABA receptor properties have been reported in diabetes,10,27,28 which may underlie our measured inhibition decrease. Therefore, we investigated whether light-evoked inhibition mediated by both GABA receptor types are decreased in early diabetes by recording L-IPSCs mediated by isolated GABAA (strychnine + TPMPA) or GABAC receptors (strychnine + SR95531; Figs. 2A, 2D). Diabetes reduced both the peak amplitudes and Q of GABAA receptor input to rod bipolar cells (peak: P = 0.05; Q: P = 0.003; Figs. 2B, 2C). Pairwise comparisons showed that the average peak amplitude was reduced at dim intensities (P = 0.05; Fig. 2B). The average Q was reduced at dim and bright intensities (P ≤ 0.01; Fig. 2C). The peak amplitudes (Figs. 2D, 2E) of GABAC receptor L-IPSCs were also significantly reduced (P = 0.006) at multiple intensities. Q was reduced overall, but pairwise comparisons did not determine a significant decrease at any one intensity (P = 0.02; Fig. 2F). Thus, reduced light-evoked rod bipolar cell inhibition after 6 weeks of diabetes may reflect reduced GABAergic inhibitory input from amacrine cells.
Figure 2.

GABAergic inhibition from amacrine cells to rod bipolar cells is reduced in early diabetes. (A) GABAA receptor L-IPSCs are reduced in diabetic rod bipolar cells as shown by representative traces averaged from two responses at the maximum light intensity. (B, C) The peak amplitudes and Q of GABAA receptor L-IPSCs are decreased at increasing intensities in diabetic rod bipolar cells compared to control (n = 7 cells from 6 mice for both groups). (D) Light-evoked inhibitory postsynaptic currents mediated by GABAC receptors are reduced in diabetic rod bipolar cells as shown by representative traces averaged from two responses at the maximum light intensity. (E, F) GABAC receptor L-IPSCs peak amplitudes (E) and Q (F) are decreased at multiple intensities in diabetic rod bipolar cells compared to control (n = 5 cells from 3 mice for both groups). *P < 0.05, **P < 0.01; black bar = light stimulus.
Early Diabetes Increases Spontaneous GABAergic Rod Bipolar Cell Activity
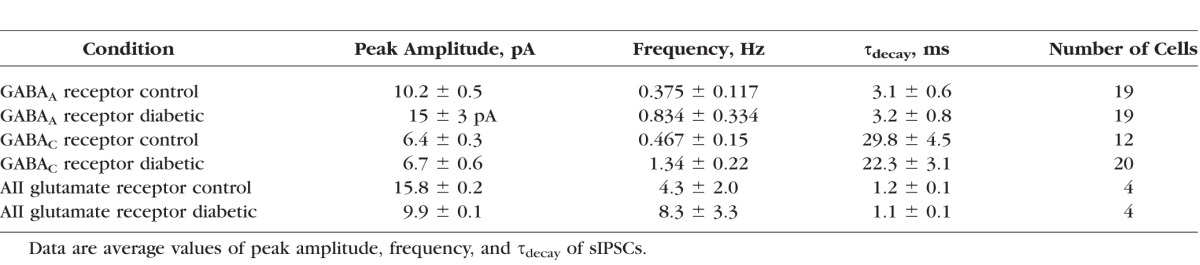
Reduced light-evoked GABAergic signaling may be explained by changes in presynaptic amacrine cell function, or changes in the postsynaptic GABAA and GABAC receptors on rod bipolar cell terminals. To distinguish between these possibilities, we measured spontaneous (s)IPSCs mediated by GABAA and GABAC receptors in the absence of a stimulus to determine if receptor and transmitter release properties, reflected by differences in sIPSC peak amplitude and τdecay values versus frequency, are altered18–20 (Fig. 3). GABAA receptor sIPSC peak amplitudes and frequencies were increased in diabetic mice (Figs. 3B, 3C; Table), with no difference in the τdecay (P = 0.8). In contrast, although the frequency of GABAC receptor sIPSCs increased (Figs. 3D, 3F; Table), the peak amplitude was unchanged (Fig. 3E) and there was also no difference in the τdecay (P = 0.2). These results suggest that spontaneous GABA release from amacrine cells and GABAA receptor cluster size or activity on rod bipolar cell terminals are increased after 6 weeks of diabetes.
Figure 3.

Spontaneous GABAergic input to rod bipolar cells from amacrine cells is increased after 6 weeks of diabetes. (A) Representative traces of spontaneous GABAA receptor activity recorded from rod bipolar cells are shown. There were fewer GABAA receptor sIPSCs detected in control (black trace) compared to diabetic (gray trace) rod bipolar cells. (B) Distribution of GABAA receptor sIPSCs normalized to the number of events shows increased peak amplitudes in diabetic (n = 19 cells from 12 mice, 3382 events) rod bipolar cells compared to control (n = 19 cells from 11 mice, 1768 events; P < 0.0001 K-S test). (C) Distribution of GABAA receptor sIPSCs normalized to the number of events shows decreased interevent intervals (IEI) in diabetic rod bipolar cells (P < 0.0001). (D) Representative traces of spontaneous GABAC receptor activity recorded from rod bipolar cells are shown. More GABAC receptor sIPSCs were detected in diabetic (gray trace) rod bipolar cells than in control (black trace). (E) The peak amplitudes of GABAC receptor-mediated sIPSCs are similar (P = 0.7). (F) Distribution of GABACR sIPSCs normalized to the number of events shows decreased IEI in diabetic (n = 12 cells from 7 mice, 9355 events) rod bipolar cells compared to control (n = 20 cells from 12 mice, 2167 events; P < 0.0001). Black and gray filled circles represent the median values for control and diabetic, respectively.
Table.
Values for sIPSC Properties in Control and Diabetic Rod Bipolar Cells, and mEPSC Properties in Control and Diabetic AII Amacrine Cells
Early Diabetes Increases Rod Bipolar Cell Output to AII Amacrine Cells
The decrease in rod bipolar cell inhibition in diabetic mice predicts an increase in rod bipolar cell output onto downstream targets. Unlike other retinal bipolar cell types that contact ganglion cells, the excitatory output of rod bipolar cells goes to AII amacrine cells.29,30 To determine if the reduction in light-evoked inhibition to rod bipolar cells causes an increase in light-evoked rod bipolar cell activation of AII amacrine cells in diabetes, we recorded L-EPSCs from AII amacrine cells (Fig. 4A). L-EPSC peak amplitudes of AII amacrine cells from diabetic mice increased compared to control (Fig. 4B), and pairwise analysis found increases at dim intensities (P ≤ 0.05). In contrast, there was no difference in the Q between control and diabetic mice (P = 0.2; Fig. 4C).
Figure 4.

Excitatory input from rod bipolar cells to downstream AII amacrine cells is increased in early diabetes. (A) Representative traces of L-EPSCs averaged from two responses at the 4750 Rh*/rod/sec light intensity recorded from AII amacrine cells after a 30-ms light stimulus (inset) are increased in diabetes. (B) The peak amplitudes are increased at multiple intensities in diabetic (n = 9 cells from 8 mice) AII amacrine cells compared to control (n = 7 cells from 5 mice, P = 0.05, 2-way ANOVA). (C) The magnitude (Q) of the L-EPSCS are similar (P = 0.2). (D, E) Spontaneous excitatory input from rod bipolar cells recorded from AII amacrine cells is increased in diabetic (gray trace) cells compared to control (black trace) determine by measuring mEPSCs (D) and sEPSCs (E). (F, G) Distribution of sEPSCs normalized to the number of events shows increased peak amplitudes (F) and reduced interevent intervals (G) in diabetic (n = 9 cells from 8 mice, 9859 events) cells compared to control (n = 7 cells from 5 mice, 6140 events; P < 0.0001 K-S test). (H) Distribution of mEPSCs show increased peak amplitudes in diabetic (n = 4 cells, 3995 events) cells compared to control (n = 4 cells, 2049 events; P < 0.0001 K-S test). Black and gray filled circles represent the median values for control and diabetic, respectively. *P < 0.05, n.s., not significant; black bar = light stimulus.
Our data showing that spontaneous GABAergic inhibition to the rod bipolar cell increases in diabetes predict reduced spontaneous excitatory output onto AII amacrine cells in diabetic animals. Surprisingly, we found that the peak amplitude (Fig. 4F, control: 25 ± 0.3 pA; diabetic: 33 ± 0.2 pA) and the frequency (Fig. 4G, control: 88 ± 15 Hz, diabetic: 111 ± 16 Hz) of sEPSCs of AII amacrine cells were increased in diabetic mice. These results could be explained by changes in either presynaptic glutamate release from rod bipolar cells or the postsynaptic AII amacrine cell receptors that mediate the EPSCs. Since spontaneous and evoked glutamate release at the rod bipolar-AII amacrine cell synapse occurs by multivesicular release, or the simultaneous fusion of multiple vesicles,31 it is possible that increased spontaneous GABAergic inhibition does not affect spontaneous release from rod bipolar cells because of a high amount of multivesicular release.
To determine if increased multivesicular release underlies the diabetes-induced increase in AII amacrine cell sEPSC amplitude, we recorded miniature (m) EPSCs evoked by the release of single vesicles. mEPSCs were isolated from dark-adapted AII amacrine cells by recording in the presence of TTX and Cd2+ to block action potentials and synaptic input, and limit multivesicular release. If increased spontaneous excitatory activity is due to multivesicular release, we would expect the peak amplitude of diabetic mEPSCs to be similar to control mEPSCs. We found that the average AII amacrine cell mEPSC peak amplitude was reduced in diabetic cells (Figs. 4D, 4H; Table) compared to control. There was no significant difference in the average frequency (P = 0.3) or τdecay (P = 0.4). These results suggest that increased AII amacrine cell spontaneous activity may be due to augmented multivesicular release from the rod bipolar cell in diabetes and that there may even be a small decrease in glutamate receptor expression on AII amacrine cells in diabetes.
Diabetes Leads to Decreased GABA Release From Amacrine Cells to Rod Bipolar Cells and Increased Rod Bipolar Cell Glutamate Release
Our results of decreased light-evoked and increased spontaneous inhibitory input to rod bipolar cells in diabetes suggest that increased spontaneous release of GABA may lead to decreased L-IPSCs by reducing the amount of GABA available to be released after a stimulus. To determine whether there was a decrease in light-evoked GABA release, we used deconvolution analysis,20,21 which assumes that the release of individual vesicles that evoke sIPSCs sum linearly to evoke a L-IPSC. Therefore, this analysis reflects the minimum amount of quantal release required to evoke a response. Diabetes reduced light-evoked GABA release onto both GABAA (P < 0.001) and GABAC (P < 0.01) receptors (Fig. 5). The peak of GABA release onto GABAA receptors, as well as the amount of GABA released over time (Q, vesicles*ms, P < 0.001, data not shown), were reduced in diabetic cells. Pairwise comparisons showed that the peak of release (Fig. 5B) was reduced at each light intensity. Similarly, the peak (Fig. 5E) and total amount (P < 0.05) of release onto GABAC receptors were reduced. We conclude that reduced rod bipolar cell light-evoked GABAergic inhibition is due to decreased GABA release from amacrine cells onto rod bipolar cell terminals.
Figure 5.

Amacrine cell GABA release is reduced and rod bipolar cell glutamate release is increased in early diabetes. (A, D) Representative traces of the time course of GABA release onto rod bipolar cell GABAA (A) and GABAC (D) receptors estimated from deconvolution analysis of GABA receptor-mediated sIPSCs and L-IPSCs are shown. (B, E) The peak of GABAergic vesicle release is decreased at multiple intensities in diabetic rod bipolar cells compared to control (GABAA: n = 7 cells from 6 mice for both groups; GABAC: n = 5 cells from 3 mice for both groups). (C, F) Representative traces (C) of the time course of glutamate release shows that release is increased (F) from rod bipolar cells to AII amacrine cells in diabetes. Deconvolution analysis of glutamate receptor mEPSCs and L-EPSCs shows that the peak of glutamate vesicle release is higher in diabetic (n = 9 cells from 8 mice) cells to control (n = 7 cells from 5 mice, P < 0.05, 2-way ANOVA). Representative traces are deconvolved from L-IPSCs recorded at the maximum intensity (GABA) or L-EPSCs recorded at the 4750 Rh*/rod/sec light intensity (glutamate). *P < 0.05, **P < 0.01, ***P < 0.001. q/ms, quanta/millisecond.
Our analysis of AII amacrine cell L-EPSCs and mEPSCs (Fig. 4) suggest that diabetes may cause an increase in glutamate release from rod bipolar cells. To determine if the increased L-EPSC peak amplitude we measured was due to increased glutamate release from rod bipolar cells, we used deconvolution analysis to estimate the amount of glutamate release at each light-intensity (Figs. 5C, 5F). Due to the high rate of multivesicular release at the rod bipolar-AII amacrine cell synapse, we used an averaged mEPSC in place of an averaged sEPSC to deconvolve with the L-EPSC and determine the estimated glutamate release. The peak (Fig. 5F) of light-evoked glutamatergic vesicle release was increased in diabetic mice (P < 0.05). Pairwise comparisons showed significant increases at dim intensities (P < 0.05). Thus, rod bipolar cell glutamate release is increased in diabetes as a result of reduced rod bipolar cell GABAergic inhibition.
Early Diabetes Does Not Cause Cell or Receptor Loss
Although the above results suggest a change in the physiological function of rod bipolar and amacrine cells, they do not exclude the possibility of changes in retinal morphology. To test whether there was loss of retinal neurons after 6 weeks of diabetes, we used TO-PRO-3 to label nuclei in whole-mounted retinas (Fig. 6A). We found no difference between control and diabetic mice in the numbers of cells in the outer nuclear layer (P = 0.9 unpaired Student's t-test), inner nuclear layer (P = 0.5), or ganglion cell layer (P = 0.5; Fig. 6B). It is possible that the loss of retinal neurons is limited to specific types of cells, as previously reported,32 since the loss of only one cell-type might not be detected in our analysis of total retinal neurons. Since ERGs are population measurements, loss of rod bipolar cells could explain decreases in rod pathway signaling. We labeled rod bipolar cells with an antibody against PKC,33 (Fig. 6A) and found similar numbers of rod bipolar cells in control and diabetic mice (P = 0.4; Fig. 6B).
Figure 6.

No loss of retinal neurons is detected in early diabetes. (A) Representative images of whole mounted retinas stained with PKCα (green) to label rod bipolar cells (RBCs) or the nuclei stain TO-PRO-3 (red) from control (top panels) and diabetic (bottom panels) mice are shown. The outer nuclear layer (ONL) contains photoreceptors. The inner nuclear layer (INL) contains horizontal, bipolar, and amacrine cells. The ganglion cell layer (GCL) contains ganglion cells and displaced amacrine cells. (B) Quantification of the number of cells per layer is shown. There is no significant difference in the average number of cells per layer between control and diabetic treated retinas (n = 7 mice for both groups). n.s., not significant. Scale bars: 25 μm.
Analysis of the spontaneous GABAA receptor input to rod bipolar cells suggested an increase in either rod bipolar cell GABAA receptor clusters or activity (Fig. 3B). Therefore, we labelled the α1 subunit of GABAA receptors on rod bipolar cells34 (Fig. 7). Since GABAA α1 receptor expression is abundant and not limited to rod bipolar cell terminals, GABAA α1 receptor labeling in locations other than rod bipolar cell terminals was excluded digitally (see Research Design and Methods).22–24 The volume of rod bipolar cell terminals occupied by GABAA α1 receptors was not different between control and diabetic mice (Fig. 7B, P = 0.3). Thus, decreased amacrine cell-mediated rod bipolar cell inhibition in early diabetes does not reflect changes in the amount of GABAA receptors that occupy the rod bipolar cell terminals.
Figure 7.

GABAA receptor expression on rod bipolar cell terminals is not changed in early diabetes. (A) GABAA α1 receptors (cyan) colabeled with PKC positive rod bipolar cell terminals (red) in control and diabetic retinas. Maximum intensity projection of a confocal image stack representing a thickness of 9 μm. A 3D mask was created for the PKC immunolabeled terminals to isolate the GABAA α1 signal exclusively within the rod bipolar cell terminals. After application of a threshold to eliminate background fluorescence, GABAA α1 receptor pixels could be detected (Mask panels, white). (B) There is no difference in the volume of the detected GABAA α1 pixels expressed as % occupancy of the PKC labeled rod bipolar terminals (RBCs) in control and diabetic retinas (n = 4 mice for both groups). P = 0.3 Student's t-test; n.s., not significant.
Discussion
We found that light-evoked amacrine cell-mediated inhibition to rod bipolar cells is reduced after 6 weeks of diabetes without changes in excitatory drive from rod photoreceptors or large scale loss of retinal neurons. Our results indicate that compromised GABAergic inhibition from amacrine cells due to reduced GABA release underlies the decreased inhibition that leads to increased light-evoked excitatory output from the rod pathway. These results are in agreement with a recent study that showed signaling at the rod bipolar-AII amacrine cell synapse is compromised; however, light-evoked responses of rod bipolar and amacrine cells were not measured.12 Here we have shown that the processing of a light signal by the inner retinal circuitry is compromised in early diabetes by directly measuring light-evoked inhibition from amacrine cells to rod bipolar cells. Dysfunctional retinal signaling in the rod pathway was detected after just 6 weeks of diabetes. This imbalance in retinal excitatory and inhibitory signaling likely contributes to visual deficits reported in diabetes.
Although it is not currently possible to measure the response of individual neurons or determine what mechanisms have changed in vivo, our results do support previous results from diabetic humans and animal models. Our results showing reduced inhibition are consistent with a commonly reported consequence of diabetes, reduced amplitudes, and increased latency of oscillatory potentials that reflect amacrine cell activity measured by ERGs.2,3,6,9,10,35 Differences in the oscillatory potentials in diabetes correlate directly between humans and animal models.5 Our results showing no deficits in photoreceptor inputs to rod bipolar cells also agree with ERG studies that show no differences in the in vivo ERG a- or b-waves in early diabetes,3,10 although there remains some controversy.8,9,11
Another advantage of recording from neural retinal tissue directly is that we can differentiate loss of neurons from dysfunctional neural activity as the cause of visual changes. Previous reports have shown loss of retinal neurons in humans with diabetes36,37 and in animal models as early as 4 weeks after the onset of experimental diabetes,32,38 which could explain the observed physiological changes. For example, loss of retinal neurons leads to changes in retinal neuronal signaling in the rd1 mouse model of retinitis pigmentosa.39,40 Given that we did not observe loss of cells in any retinal layer or rod bipolar cells from diabetic mice (Fig. 6), we conclude that there is no change in gross retinal morphology in early diabetes. Because we could only make recordings from live neurons, our results distinguish between changes due to dysfunctional neural signaling and changes due to cell loss, although it is still possible that subpopulations of neurons could be lost. Thus, unlike in the rd1 mouse, our results show changes in retinal neuronal signaling without significant loss of retinal neurons.
Although we did not observe changes in neuronal morphology, we cannot rule out the possibility that changes in the blood retinal barrier contribute to retinal neuronal dysfunction.41 In diabetes, retinal blood vessels exhibit deficits in light-induced dilation suggesting that neurovascular coupling is compromised.42 Also, ERG deficits are predictive of vascular growth.43 The connection between neuronal signaling and retinal vasculature is not well understood, but this neurovascular unit is likely disrupted in diabetes.
We found that diabetes reduced GABAergic light-evoked inhibition to rod bipolar cells (Fig. 2), which may be a common characteristic of neuronal diabetic damage as diabetes also decreases GABAergic inhibition in other neural systems,44 but the mechanisms for this dysfunction are not known. Reduced expression of hippocampal GABAA receptor mRNA occurs in diabetic rats.45 However, our results suggest that GABAA receptor expression is not decreased at 6 weeks of diabetes since we see an increase in GABAA receptor spontaneous activity (Fig. 3) and no change in GABAA α1 receptor expression on rod bipolar cell terminals (Fig. 7). Altered subunit expression and increased open time of GABAC receptors was reported in isolated rod bipolar cells from diabetic rats,46 but changes in synaptic or physiological properties of the rod bipolar cells were not investigated. Our results of increases in GABAC sIPSC frequency without a change in timing or peak amplitude suggest that the properties of synaptic GABAC receptors on rod bipolar cell terminals are not changed in early diabetes. Thus, changes to GABAergic input to rod bipolar cells are likely occurring presynaptically in amacrine cells. In agreement with this, we found that reduced light-evoked GABAergic inhibition was due to decreased GABA release from amacrine cells, but the mechanism is unclear. A recent study in diabetic mice found that A17 amacrine cells, which form reciprocal connections with rod bipolar cells and comprise ∼50% of rod bipolar cell inhibition,14,47,48 have reduced Ca2+-permeability when activated by exogenous glutamate.12 If A17 amacrine cell Ca2+ influx in response to a light stimulus is reduced, then GABA release from A17 amacrine cells onto rod bipolar cells would be limited, as blocking the accumulation of Ca2+ in amacrine cells attenuates release under nondiseased conditions.49 Additionally, a previous study showed a downregulation of synaptic proteins in early diabetes that would also limit transmitter release.50
Since loss of inhibition to rod bipolar cells increases the output of the rod pathway,20 we predicted that the reduced light-evoked inhibition in diabetes would increase the output of the rod pathway onto AII amacrine cells, the output neurons of the rod pathway. As there is a strong relationship between a single rod bipolar cell depolarization and AII amacrine cell excitation, and AII amacrine cells receive inputs from multiple rod bipolar cells, even a small increase in depolarization of the rod bipolar cell membrane potential due to loss of inhibition could result in a large change in AII excitatory currents.51 Our results show that disinhibition of the rod bipolar cell in diabetes increased the glutamate release (Fig. 5F) and consequently increased L-EPSC peak amplitude (Fig. 4B), but not the charge transfer of AII amacrine cells (Fig. 4C). A recent study found that the transient (peak) and sustained (charge transfer) components of AII amacrine cell responses have distinct functions with the transient component encoding contrast and the sustained component encoding luminance.52 Although the properties that underlie these distinct AII amacrine cell responses remain unclear, the peak and transient components may be governed by different mechanisms. It is possible that diabetes affects the mechanisms that control the peak of the response more than those that mediate the charge transfer.
We expected that the increased spontaneous GABAergic inhibitory inputs to rod bipolar cells (Fig. 3) might decrease spontaneous excitatory activity in AII amacrine cells, but actually observed an increase. Consistent with this, a recent study showed increased spontaneous excitatory activity of AII amacrine cells in diabetic rats, thought to be due to reduced tonic GABAC receptor input to rod bipolar cells.12 However, they did not measure spontaneous inhibition to rod bipolar cells directly, and experiments were performed in light-adapted conditions that significantly decrease spontaneous inhibition.14 We hypothesized that increased spontaneous excitatory activity of AII amacrine cells reflected increased multivesicular release of glutamate from rod bipolar cells. Our results showing decreased mEPSC peak amplitude in AII amacrine cells from diabetic mice support this hypothesis and suggest that the high rate of rod bipolar cell release is able to overcome increased spontaneous inhibition. This may be due to the properties inherent to rod bipolar cell ribbon synapses, which are designed to support graded potential changes with sustained multivesicular release,53 unlike amacrine cell synapses, which are more similar to conventional synapses. Thus, amacrine cells are likely to be more sensitive to vesicle depletion by increased spontaneous release than rod bipolar cells. Our present study is the first to investigate light-evoked responses of rod bipolar cells in diabetes that may be regulated differently than spontaneous activity. Increased output in diabetes is not limited to the rod pathway, as a different study has shown that ON ganglion cells, which likely receive ON cone bipolar cell inputs, have increased spontaneous bursting activity due to loss of inhibition.54
How does an imbalance between excitatory-inhibitory signaling translate into deficits in visual function? Amacrine cell inhibition significantly contributes to center-surround organization that refines spatial and temporal tuning in the inner retina.55–57 In the rod pathway, the AII amacrine cell center-surround is governed by excitatory rod bipolar cell input (center) and amacrine cell inhibition to rod bipolar cells (surround).57 Blocking GABAergic inhibition reduces the sensitivity of ganglion cells to stimuli size58 and may reduce contrast sensitivity. Thus, our results indicate that in the early stages of diabetes in a model system, the retinal circuitry has a reduced capacity to refine spatial and temporal tuning prior to sending it to the brain. This may underlie the deficits in visual acuity and contrast sensitivity detected in diabetic patients and animal models.
Acknowledgments
The authors thank Ronald Lynch, Ralph Fregosi, and Richard Levine of the University of Arizona, and Nii Addy of Yale University for helpful discussion and comments on the manuscript.
Supported by the Juvenile Diabetes Research Foundation, Innovative Grant Award 5-2013-163 (EDE) and Postdoctoral Fellowship 3-PDF-2014-105-A-N (JMM); the National Institutes of Health (NIH; R01-EY026027, EDE) and Cardiovascular Training Grant 2T32HL7249-36A1 appointment (JMM); a New Investigator Award from the Alcon Research Institute (EDE); and a grant from the International Retinal Research Foundation (EDE). MH was supported by NIH Grant EY10699 to R. Wong.
Disclosure: J.M. Moore-Dotson, None; J.J. Beckman, None; R.E. Mazade, None; M. Hoon, None; A.S. Bernstein, None; M.J. Romero-Aleshire, None; H.L. Brooks, None; E.D. Eggers, None
References
- 1. Klein R,, Klein BEK. Vision disorders in diabetes. Diabetes in America 2nd edition: National Diabetes Data Group Diabetes in America. Bethesda, MD: National Institutes of Health; 1995: 293–337.
- 2. Jackson GR,, Barber AJ. Visual dysfunction associated with diabetic retinopathy. Curr Diab Rep. 2010; 10: 380–384. [DOI] [PubMed] [Google Scholar]
- 3. Aung MH,, Kim MK,, Olson DE,, Thule PM,, Pardue MT. Early visual deficits in streptozotocin-induced diabetic long evans rats. Invest Ophthalmol Vis Sci. 2013; 54: 1370–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawasaki K,, Yonemura K,, Yokogawa Y,, Saito N,, Kawakita S. Correlation between ERG oscillatory potential and psychophysical contrast sensitivity in diabetes. Doc Ophthalmol. 1986; 64: 209–215. [DOI] [PubMed] [Google Scholar]
- 5. Pardue MT,, Barnes CS,, Kim MK,, et al. Rodent hyperglycemia-induced inner retinal deficits are mirrored in human diabetes. Trans Vis Sci Tech. 2014; 3: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juen S,, Kieselbach GF. Electrophysiological changes in juvenile diabetics without retinopathy. Arch Ophthalmol. 1990; 108: 372–375. [DOI] [PubMed] [Google Scholar]
- 7. Masland Richard H. The neuronal organization of the retina. Neuron. 2012; 76: 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shinoda K,, Rejdak R,, Schuettauf F,, et al. Early electroretinographic features of streptozotocin-induced diabetic retinopathy. Clin Exp Ophthalmol. 2007; 35: 847–854. [DOI] [PubMed] [Google Scholar]
- 9. Aung MH,, Park HN,, Han MK,, et al. Dopamine deficiency contributes to early visual dysfunction in a rodent model of type 1 diabetes. J Neurosci. 2014; 34: 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramsey DJ,, Ripps H,, Qian H. An electrophysiological study of retinal function in the diabetic female rat. Invest Ophthalmol Vis Sci. 2006; 47: 5116–5124. [DOI] [PubMed] [Google Scholar]
- 11. Holopigian K,, Seiple W,, Lorenzo M,, Carr R. A comparison of photopic and scotopic electroretinographic changes in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 1992; 33: 2773–2780. [PubMed] [Google Scholar]
- 12. Castilho A,, Ambrosio AF,, Hartveit E,, Veruki ML. Disruption of a neural microcircuit in the rod pathway of the Mammalian retina by diabetes mellitus. J Neurosci. 2015; 35: 5422–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keck M,, Romero-Aleshire MJ,, Cai Q,, Hoyer PB,, Brooks HL. Hormonal status affects the progression of STZ-induced diabetes and diabetic renal damage in the VCD mouse model of menopause. Am J Physiol Renal Physiol. 2007; 293: F193 –F. [DOI] [PubMed] [Google Scholar]
- 14. Eggers ED,, Mazade RE,, Klein JS. Inhibition to retinal rod bipolar cells is regulated by light levels. J Neurophysiol. 2013; 110: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghosh KK,, Bujan S,, Haverkamp S,, Feigenspan A,, Wassle H. Types of bipolar cells in the mouse retina. J Comp Neurol. 2004; 469: 70–82. [DOI] [PubMed] [Google Scholar]
- 16. Veruki ML,, Oltedal L,, Hartveit E. Electrical coupling and passive membrane properties of AII amacrine cells. J Neurophysiol. 2010; 103: 1456–1466. [DOI] [PubMed] [Google Scholar]
- 17. Field GD,, Rieke F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002; 34: 773–785. [DOI] [PubMed] [Google Scholar]
- 18. Eggers ED,, McCall MA,, Lukasiewicz PD. Presynaptic inhibition differentially shapes transmission in distinct circuits in the mouse retina. J Physiol. 2007; 582: 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eggers ED,, Lukasiewicz PD. GABA(A), GABA(C) and glycine receptor-mediated inhibition differentially affects light-evoked signalling from mouse retinal rod bipolar cells. J Physiol. 2006; 572: 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eggers ED,, Lukasiewicz PD. Receptor and transmitter release properties set the time course of retinal inhibition. J Neurosci. 2006; 26: 9413–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diamond JS,, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995; 15: 1097–1107. [DOI] [PubMed] [Google Scholar]
- 22. Schubert T,, Hoon M,, Euler T,, Lukasiewicz PD,, Wong RO. Developmental regulation and activity-dependent maintenance of GABAergic presynaptic inhibition onto rod bipolar cell axonal terminals. Neuron. 2013; 78: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Newkirk GS,, Hoon M,, Wong RO,, Detwiler PB. Inhibitory inputs tune the light response properties of dopaminergic amacrine cells in mouse retina. J Neurophysiol. 2013; 110: 536–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grimes WN,, Hoon M,, Briggman KL,, Wong RO,, Rieke F. Cross-synaptic synchrony and transmission of signal and noise across the mouse retina. Elife. 2014; 3: e03892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang JS,, Kefalov VJ. An alternative pathway mediates the mouse and human cone visual cycle. Curr Biol. 2009; 19: 1665–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masland RH. The tasks of amacrine cells. Vis Neurosci. 2012; 29: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishikawa A,, Ishiguro S,, Tamai M. Accumulation of gamma-aminobutyric acid in diabetic rat retinal Müller cells evidenced by electron microscopic immunocytochemistry. Curr Eye Res. 1996; 15: 958–964. [DOI] [PubMed] [Google Scholar]
- 28. Ambati J,, Chalam KV,, Chawla DK,, et al. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997; 115: 1161–1166. [DOI] [PubMed] [Google Scholar]
- 29. Strettoi E,, Raviola E,, Dacheux RF. Synaptic connections of the narrow-field, bistratified rod amacrine cell (AII) in the rabbit retina. J Comp Neurol. 1992; 325: 152–168. [DOI] [PubMed] [Google Scholar]
- 30. Chun MH,, Han SH,, Chung JW,, Wassle H. Electron microscopic analysis of the rod pathway of the rat retina. J Comp Neurol. 1993; 332: 421–432. [DOI] [PubMed] [Google Scholar]
- 31. Singer JH,, Lassova L,, Vardi N,, Diamond JS. Coordinated multivesicular release at a mammalian ribbon synapse. Nature Neurosci. 2004; 7: 826–833. [DOI] [PubMed] [Google Scholar]
- 32. Gastinger MJ,, Singh RS,, Barber AJ. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci. 2006; 47: 3143–3150. [DOI] [PubMed] [Google Scholar]
- 33. Greferath U,, Grunert U,, Wässle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990; 301: 433–442. [DOI] [PubMed] [Google Scholar]
- 34. Fletcher EL,, Koulen P,, Wässle H. GABAA and GABAC receptors on mammalian rod bipolar cells. J Comp Neurol. 1998; 396: 351–365. [DOI] [PubMed] [Google Scholar]
- 35. Ishikawa A,, Ishiguro S,, Tamai M. Changes in GABA metabolism in streptozotocin-induced diabetic rat retinas. Curr Eye Res. 1996; 15: 63–71. [DOI] [PubMed] [Google Scholar]
- 36. Van Dijk HW,, Kok PH,, Garvin M,, et al. Selective loss of inner retinal layer thickness in type 1 diabetic patients with minimal diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009; 50: 3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Valverde AM,, Miranda S,, Garcia-Ramirez M,, Gonzalez-Rodriguez A,, Hernandez C,, Simo R. Proapoptotic and survival signaling in the neuroretina at early stages of diabetic retinopathy. Mol Vis. 2013; 19: 47–53. [PMC free article] [PubMed] [Google Scholar]
- 38. Barber AJ,, Lieth E,, Khin SA,, Antonetti DA,, Buchanan AG,, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998; 102: 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marc RE,, Jones BW,, Watt CB,, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003; 22: 607–655. [DOI] [PubMed] [Google Scholar]
- 40. Stasheff SF. Emergence of sustained spontaneous hyperactivity and temporary preservation of OFF responses in ganglion cells of the retinal degeneration (rd1) mouse. J Neurophysiol. 2008; 99: 1408–1421. [DOI] [PubMed] [Google Scholar]
- 41. Antonetti DA,, Barber AJ,, Khin S,, Lieth E,, Tarbell JM,, Gardner TW. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes. 1998; 47: 1953–1959. [DOI] [PubMed] [Google Scholar]
- 42. Mishra A,, Newman EA. Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia. 2010; 58: 1996–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harrison WW,, Bearse MA,, Jr,, Ng JS,, et al. Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Invest Ophthalmol Vis Sci. 2011; 52: 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hassan Z,, Sattar MZ,, Suhaimi FW,, et al. Blunted endogenous GABA-mediated inhibition in the hypothalamic paraventricular nucleus of rats with streptozotocin-induced diabetes. Acta Neurologica Belgica. 2013; 113: 319–325. [DOI] [PubMed] [Google Scholar]
- 45. Sherin A,, Anu J,, Peeyush KT,, et al. Cholinergic and GABAergic receptor functional deficit in the hippocampus of insulin-induced hypoglycemic and streptozotocin-induced diabetic rats. Neuroscience. 2012; 202: 69–76. [DOI] [PubMed] [Google Scholar]
- 46. Ramsey DJ,, Ripps H,, Qian H. Streptozotocin-induced diabetes modulates GABA receptor activity of rat retinal neurons. Exp Eye Res. 2007; 85: 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chavez AE,, Singer JH,, Diamond JS. Fast neurotransmitter release triggered by Ca influx through AMPA-type glutamate receptors. Nature. 2006; 443: 705–708. [DOI] [PubMed] [Google Scholar]
- 48. Hartveit E. Reciprocal synaptic interactions between rod bipolar cells and amacrine cells in the rat retina. J Neurophysiol. 1999; 81: 2923–2936. [DOI] [PubMed] [Google Scholar]
- 49. Moore-Dotson JM,, Klein JS,, Mazade RE,, Eggers ED. Different types of retinal inhibition have distinct neurotransmitter release properties. J Neurophysiol. 2015; 113:jn 00447 02014. [DOI] [PMC free article] [PubMed]
- 50. VanGuilder HD,, Brucklacher RM,, Patel K,, Ellis RW,, Freeman WM,, Barber AJ. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur J Neurosci. 2008; 28: 1–11. [DOI] [PubMed] [Google Scholar]
- 51. Singer JH,, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003; 23: 10923–10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oesch NW,, Diamond JS. Ribbon synapses compute temporal contrast and encode luminance in retinal rod bipolar cells. Nature Neurosci. 2011; 14: 1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singer JH. Multivesicular release and saturation of glutamatergic signalling at retinal ribbon synapses. J Physiol. 2007; 580: 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu J,, Wang L,, Weng SJ,, Yang XL,, Zhang DQ,, Zhong YM. Hyperactivity of ON-type retinal ganglion cells in streptozotocin-induced diabetic mice. PLoS One. 2013; 8: e76049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cook PB,, McReynolds JS. Lateral inhibition in the inner retina is important for spatial tuning of ganglion cells. Nature Neurosci. 1998; 1: 714–719. [DOI] [PubMed] [Google Scholar]
- 56. Bloomfield SA,, Xin D. Surround inhibition of mammalian AII amacrine cells is generated in the proximal retina. J Physiol. 2000; 523( pt 3): 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Volgyi B,, Xin D,, Bloomfield SA. Feedback inhibition in the inner plexiform layer underlies the surround-mediated responses of AII amacrine cells in the mammalian retina. J Physiol. 2002; 539: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Protti DA,, Di Marco S,, Huang JY,, Vonhoff CR,, Nguyen V,, Solomon SG. Inner retinal inhibition shapes the receptive field of retinal ganglion cells in primate. J Physiol. 2014; 592: 49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]