

Conspectus
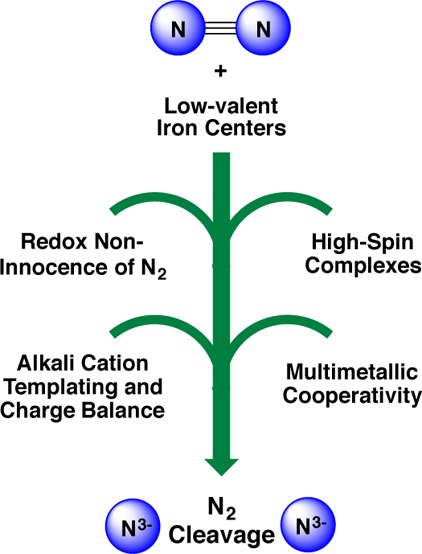
The iron–molybdenum cofactor of nitrogenase has unprecedented coordination chemistry, including a high-spin iron cluster called the iron-molybdenum cofactor (FeMoco). Thus, understanding the mechanism of nitrogenase challenges coordination chemists to understand the fundamental N2 chemistry of high-spin iron sites. This Account summarizes a series of studies in which we have synthesized a number of new compounds with multiple iron atoms, characterized them using crystallography and spectroscopy, and studied their reactions in detail. These studies show that formally iron(I) and iron(0) complexes with three- and four-coordinate metal atoms have the ability to weaken and break the triple bond of N2. These reactions occur at or below room temperature, indicating that they are kinetically facile. This in turn implies that iron sites in the FeMoco are chemically reasonable locations for N2 binding and reduction.
The careful evaluation of these compounds and their reaction pathways has taught important lessons about what characteristics make iron more effective for N2 activation. Cooperation of two iron atoms can lengthen and weaken the N–N bond, while three working together enables iron atoms to completely cleave the N–N bond to nitrides. Alkali metals (typically introduced into the reaction as part of the reducing agent) are thermodynamically useful because the alkali metal cations stabilize highly reduced complexes, pull electron density into the N2 unit, and make reduced nitride products more stable. Alkali metals can also play a kinetic role, because cation−π interactions with the supporting ligands can hold iron atoms near enough to one another to facilitate the cooperation of multiple iron atoms. Many of these principles may also be relevant to the iron-catalyzed Haber–Bosch process, at which collections of iron atoms (often promoted by the addition of alkali metals) break the N–N bond of N2.
The results of these studies teach more general lessons as well. They have demonstrated that N2 can be a redox-active ligand, accepting spin and electron density in complexes of N22–. They have shown the power of cooperation between multiple transition metals, and also between alkali metals and transition metals. Finally, alkali metal based cation−π interactions have the potential to be broadly useful for bringing metals close together with sufficient flexibility to allow multistep, multielectron reactions. At the same time, the positive charge on the alkali metal cation stabilizes charge buildup in intermediates.
Introduction: How Does the FEMOCO Work?
Nitrogenase enzymes provide the world’s largest input of fixed nitrogen, because they have the amazing ability to catalytically reduce dinitrogen to ammonia under ambient conditions of room temperature and 0.8 atm of N2. Though the activation barrier for this room temperature catalytic reaction is low compared to that for the high-temperature Haber–Bosch process, the biological reaction is driven thermodynamically by the hydrolysis of at least 16 mol equiv of ATP per molecule of N2.1 Kinetic studies on the molybdenum–iron enzyme, and the fact that the biological reductant transfers one electron at a time, suggest that there are least eight intermediates during the catalytic cycle, which strains the ability of bioinorganic chemists to establish the mechanism in detail.2 In addition, the structure of the iron–molybdenum cofactor, FeMoco (Figure 1), consists of eight metal ions, whose oxidation states are still the subject of debate.3 As a result, there are great opportunities and great challenges in understanding the fundamental chemistry that underlies biological dinitrogen reduction by nitrogenases.
Figure 1.

Structure of iron–molybdenum cofactor (FeMoco) of nitrogenase in its resting state.8
A number of studies over the last 15 years have led most chemists in the field to consider the most likely substrate binding site to be one or more of the central iron atoms, on the side facing His-195 and Val-70 of the α subunit. This idea was originally supported by the activity of enzyme variants derived from mutation of these residues as well as from ENDOR studies of trapped intermediates,4 and it has recently gained additional support in the crystal structure of a CO-inhibited state in which a sulfide on the same face is replaced by a bridging CO.5 Focus on the central part of the FeMoco is also bolstered by the identification of a carbide that bridges the six belt iron atoms. The observation of a carbide in an enzyme is unique to nitrogenase, and extremely surprising. However, the identification of the central atom as C, initially proposed on the basis of X-ray emission,6,7 crystallographic, and ESEEM measurements,8 has now been firmly established through specific 14C labeling of the carbide precursor and following the radiolabel through a series of biosynthetic precursors into the active protein.9 The presence of such an unusual structural feature, and the fact that there is cellular machinery devoted to installation of C into the FeMoco, implies that the carbide plays a crucial role in catalysis.
Figure 2 shows two hypothetical roles for the carbide during reduction of the cluster to the reactive, reduced form. One possibility (Figure 2a) proposes “hemilabile” bonding between the Fe and C atoms. In this hypothesis, flexing of the core to break or weaken a Fe–C bond would play a key role in N2 binding and further reduction steps. Peters and co-workers have established that Fe–C distances can vary widely in a series of N2-binding compounds capable of reducing N2 to NH3, which supports this idea.10 In a second possibility (Figure 2b), a Fe–S bond is cleaved and the Fe–C bonds remain intact. This possibility is supported by the observation of similar metal–metal distances (via EXAFS) in the FeMoco resting state and intermediates,11 by Fe–S cleavage in smaller Fe–S clusters upon protonation,12 and by the crystallographic observation of sulfide displacement from the FeMoco by CO.5 Each hypothetical mechanism gives a partially or completely open coordination site at which N2 could form a bond to an Fe atom.
Figure 2.

Two hypothetical activation modes in FeMoco: (a) Fe–C weakening or cleavage (indicated by a dashed line) and (b) Fe–S cleavage (indicated with one possible product after protonation).
The resulting Fe–N2 compounds are very unusual from the perspective of coordination chemistry.13 At the outset of our Fe–N2 research in 2000, there were no examples of Fe–N2 compounds in which the coordination number was less than five, no examples of Fe–N2 complexes with paramagnetic and/or high-spin metals, no Fe–N2 complexes in which the N–N bond was substantially weakened as judged by crystallography or vibrational spectroscopy, and no examples of Fe complexes that gave more than about 10% yield of the expected 2 NH4+ from reduction/protonation of N2. The lack of compounds of these types hindered efforts to determine which hypothetical intermediates are reasonable, and whether an iron-based mechanism was reasonable at all. We anticipated that new Fe–N2 complexes with Fe in environments more reminiscent of the enzyme could provide fundamental insights. This Account focuses on the evolution of my group’s research in this area. We also recommend reviews that cover a broader range of metals for N2 reduction.14−16
Low Coordination Numbers at Iron Enhance N–N Bond Weakening
Low-spin N2 complexes of iron(0) and iron(II) in the literature have relatively weak backbonding that leads to high N–N stretching frequencies and short N–N triple bonds. This generalization holds for both terminal and end-on/end-on bridging complexes of N2.13 However, when we prepared three-coordinate, formally diiron(I) complexes of N2, we discovered that they have much lower stretching frequencies (by more than 200 cm–1) and longer N–N bonds (by more than 0.07 Å).17 The low coordination number in these complexes is enforced by bulky β-diketiminate ligands (Figure 3).
Figure 3.

β-Diketiminate ligands stabilize low-coordinate iron complexes (X = Cl, Br), which react with N2 under reducing conditions to give products with weakened N2.
Studies on the electronic structure of the complex LMe,iPrFeNNFeLMe,iPr using ligand-field calculations and Mössbauer spectroscopy (in collaboration with Sebastian Stoian, Emile Bominaar, and Eckard Münck) suggested a resonance structure that has two iron(II) ions and a bridging N22– ligand.18 These two S = 2 Fe2+ sites can each couple antiferromagnetically with the S = 1 N22– group to give a total S = 3, consistent with Mössbauer and magnetic susceptibility data.19 We have also pursued density functional theory computations (BP86/TZVP) on this molecule in the septet (S = 3) ground state, which give excellent agreement (within 0.02 Å) with metrical parameters within the core and also excellent agreement with the Mössbauer parameters (exp, δ = 0.62 mm/s, |EQ| = 1.41 mm/s; calcd with TPSSh/TZVP at the BP86-optimized geometry, δ = 0.56 mm/s, |EQ| = 1.27 mm/s). These validated calculations show Mulliken spin populations of +3.2 on each iron atom and −0.6 on the N2 unit. Analysis of the ground state using broken-symmetry methods shows that two of the α/β pairs have an overlap (Sαβ) of only 89%, and the α spin is more localized on the iron atoms while the β spin is more localized on the N2 π* orbital (Figure 4). These “correlated pairs” are characteristic of redox noninnocent ligands, in agreement with the triplet N22– interpretation.
Figure 4.
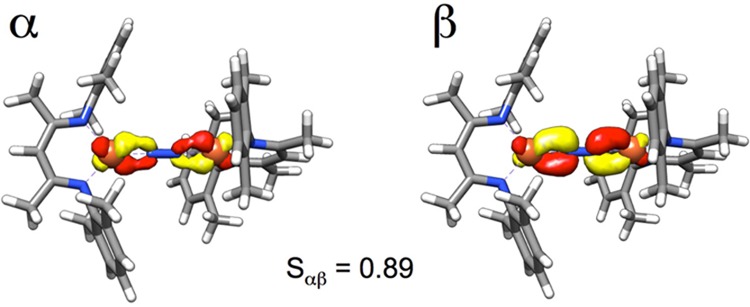
Key “correlated pair” of orbitals from broken-symmetry calculations on LMe,iPrFeNNFeLMe,iPr, showing that there is β spin more localized on the π* orbital of N2. (Sαβ is the value of the overlap integral between the α and β orbitals.) This is consistent with antiferromagnetic coupling of an N22– bridge to two Fe2+ ions.
Other iron species with lower coordination numbers are also capable of weakening N–N bonds, as shown in bridging diiron(I)–N2 complexes reported by Peters and Arnold and their respective co-workers.20,21 We have also shown the reduction of LMe,iPrFeNNFeLMe,iPr and its analogue LtBu,iPrFeNNFeLtBu,iPr by two electrons to yield complexes of the type M2[LFeNNFeL] (M = Na, K).17 Preliminary density functional theory calculations indicate greater spin population on N2 (−1.0), which is consistent with the lower observed stretching frequency (νNN = 1589–1625 cm–1). The greater extent of N−N activation is also indicated by the shift of the N2 σ* orbital energy in X-ray emission spectra.17c Therefore, though these complexes are formally diiron(0), it is likely that they are best viewed as Fe1+–N22––Fe1+.
Despite the negative charge on the N2 unit, reactions at N2 were not evident in the diketiminate-supported complexes. The most characteristic reaction was loss of N2, with transfer of electrons back to the iron, and coordination of an additional (typically π-acceptor) ligand or reaction with an oxidant.17b Binding of alkenes, alkynes, and phosphine gave iron(I) complexes in thermodynamically favorable reactions.22 Treatment of the N2 complexes with acids yielded no detectable ammonia. Unreported studies even used “tethered” acids: for example, since pyridine coordinates to LtBu,iPrFeNNFeLtBu,iPr, we hoped that 2-hydroxypyridine would suspend an acidic proton immediately above the N2 unit. However, this reaction yielded only the complex shown in Figure 5. Thus, the ability of the low-coordinate environment to “push” electron density into the π* orbitals of N2 did not translate into the desired reactions of coordinated N2.
Figure 5.

Reaction of LMe,iPrFeNNFeLMe,iPr with 2-hydroxypyridine leads not to protonation by the pendent acid, but gives N2 displacement instead. Reduction of 2 H+ to form H2 is assumed to account for the net oxidation from 2 Fe(I) to 2 Fe(II).
Cooperation Between Three Iron Atoms Enables N–N Cleavage to Nitrides
At this point, we hypothesized that the steric bulk of the ligands prevents N2 from reacting with acids and other potential electrophiles. Therefore, we decreased the size of the arene substituents from isopropyl to methyl in the new ligand LMe3 (Figure 6). Since our successful synthesis of LMe,iPrFeNNFeLMe,iPr in previous work used reduction of the iron(II) chloride complex [LMe,iPrFe(μ-Cl)]2 with 2 equiv of potassium graphite (KC8), we used the analogous reaction of [LMe3Fe(μ-Cl)]2 with 2 equiv of KC8. This reaction yielded a very different product, shown in Figure 6.23
Figure 6.

Smaller β-diketiminate, LMe3, gives a four-iron product where the N–N triple bond has been cleaved to form two nitrides.
This product has four iron atoms, two K+ ions, two Cl– ions, and two nitrides; isotope-labeling studies show that the nitrides come from N2. The complete reduction of N2 to two N3– ions requires six electrons, and this reduction half-reaction can be balanced by the net oxidation of four Fe1+ to two Fe2+ and two Fe3+, as indicated in Scheme 1b. These oxidation states agree with charge counting within the core, and with the results of Mössbauer spectra from collaborator Eckhard Bill.
Scheme 1. Series of Half-Reactions That Lead to the Bis-Nitride Product.

(a) Reduction of iron to the reactive iron(I) form and (b) cleavage of N2. Two molar equivalents of KCl are also lost in the second reaction; these are represented as “2 K+” in the overall redox reaction.
Though K+ is incorporated into the product, the complete reduction of N2 is not specific to potassium-containing reductants like KC8. Interestingly, RbC8 gives an analogous product, in which Rb+ ions occupy the K+ positions.24 Powdered sodium metal effects a similar transformation with full N–N cleavage, but the product has only one Na+ ion, and has lost one iron atom (presumably because Na+ is too small to bridge; [LMe3Fe(μ-Cl)]2 is observed as a byproduct that accounts for loss of this iron(II) ion).24 Mössbauer spectra of the three systems (Na, K, Rb) are similar, but the spectrum of the Na complex is missing a signal with parameters characteristic of tetrahedral high-spin iron(II), as expected for loss of the dangling Fe2+.
An important implication from these results is that N2 reduction is facilitated by cooperation between reduced iron species. Since three iron atoms are bound to the remnants of N2, our collaborators Travis Figg and Thomas Cundari explored potential pathways for three-iron N2 cleavage, using density functional calculations on simplified models having no diketiminate substituents (Ltrunc, Figure 7).25
Figure 7.

Computation of the energy barrier for the conversion from end-on/end-on/side-on (EES) geometry to the end-on/side-on/side-on (ESS) geometry using truncated ligands.25 (a) Computed barrier in the absence of potassium. (b) Computed barrier in the presence of potassium. These results indicate that the addition of an electron and a potassium ion stabilizes the N–N cleaved isomer substantially, favoring the rearrangement of the core.
These calculations showed that three diketiminate-bound iron(I) ions can interact with N2 in both end-on/end-on/side-on (EES) and end-on/side-on/side-on (ESS) binding modes that are connected by low activation barriers (one iron atom revolves partway around the core). The ESS binding mode can easily cleave the N–N bond without major geometric distortions, but the N–N cleaved product is not thermodynamically stable unless the negative charge on the two-coordinate nitride is stabilized by a positively charged ion like K+. N–N cleavage also requires an “extra” electron beyond those provided by these three iron(I) ions, which is provided either by the dangling iron atom or by the sacrificial iron in the Na reaction. These studies suggested a thermodynamically and kinetically reasonable pathway to N–N cleavage at a triiron site: initial formation of an LMe3FeNNFeLMe3 complex (analogous to those with bulkier ligands) is followed by coordination of a third iron(I) species in a side-on mode. The EES complex is only accessible with relatively small diketiminate supporting ligands for steric reasons, explaining why N–N cleavage was not observed with LtBu,iPr and LMe,iPr. After rotation to the ESS mode, reduction by a fourth iron(I) enables N–N cleavage to the final product. In this mechanism, a likely role for the alkali metal is to stabilize the N–N cleaved product: thus it is a fundamentally thermodynamic rather than kinetic explanation for the alkali metal effect.
Alkali Metals as Templating Elements for Multimetallic Reactions
It is also important to consider kinetic influences, in which alkali metals could facilitate the molecules achieving the correct transition state for the reaction. An important kinetic effect was suggested by our parallel reactions of [LMe3Fe(μ-Cl)]2 with cesium graphite (CsC8). Even though it should be an equally strong reductant as KC8 and RbC8, use of 2 equiv or 4 equiv of CsC8 per iron(II) dimer gave products with no N–N cleavage.24 A key product (Figure 8) had three end-on/end-on bridging N2 units and no nitrides. Subsequently, we learned that 4 equiv of KC8 or RbC8 gives analogous triangular products. (The N–N cleaved products described above are not intermediates along the way to the triangular products; the additional reductant apparently reacts with an intermediate prior to N–N cleavage.) This significant observation indicates that more reducing agent does not necessarily result in a greater degree of N–N reduction, and that thermodynamic driving force is not the main determinant of reaction outcome. Rather, we surmised that different alkali metal cations, which coexist with iron(I) species in the unobserved intermediates, can “steer” the iron ions into favorable conformations for different reactions.
Figure 8.
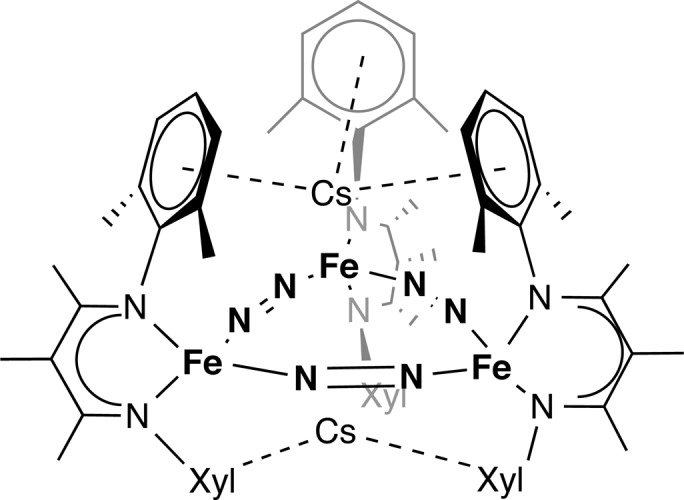
Product of reduction with 4 equiv of CsC8 is Cs2[LMe3Fe(μ-N2)]3, which has intact N–N bonds. This shows that more reducing conditions do not necessarily lead to N–N cleavage, and implies a kinetic influence on N2 activation.
The main way that alkali metal cations can favor different conformations is through cation−π interactions involving the supporting ligand, which we have observed in all anionic complexes of iron-diketiminate systems (except those where crown ethers were used to sequester these ions). In the formally diiron(0)–N2 complexes described above, the K+ and Na+ ions not only coordinate to the aromatic rings, but also to the bridging N2 ligand. We have also observed the first iron(I) sulfide complexes, which benefit from Na+ or K+ binding to the aromatic rings and to the bridging S2– ion.26 The ability to stabilize highly reduced species is thermodynamically facilitated by the presence of positive charge near the reduced core.
However, we have also gained evidence that cation-π interactions can influence the barriers for N2-binding reactions in a way that controls reactivity. For example, unusual iron(I) phenoxide complexes [KLtBu,iPrFeOPh]1,2 react with N2only when K+-arene interactions hold the compound together in a dimeric form, with a geometry that is well-adapted to give bridging N2.27 Computational studies supported the idea that initial Fe–N2 binding can happen equally well in both monometallic and bimetallic complexes, but the bimetallic complexes facilitate the bridging of N2 to give a more stable form. Thus, the bimetallic complexes, held together by cation−π interactions, are more active toward cooperative reactions.
Extending this concept, we hypothesize that in the N–N bond cleaving reaction, either larger cations (in the case of Cs+) or more cations (in the complexes from over-reduction) give a geometry that holds Fe atoms too far apart to achieve the necessary Fe3(N2) intermediate, whereas the correct size and number of Na+, K+, or Rb+ ions can steer the iron atoms into the correct relative orientation.24 This hypothesis is difficult to test, because the key intermediates have not been isolated, and because there are many possible orientations of cation−π interactions that would be difficult to evaluate computationally. However, this idea does suggest that the incorporation of alkali metal cations might be useful for a broad range of reactions in which multimetallic cooperativity is useful, for example multielectron redox reactions such as water oxidation/O2 reduction and CO2 reduction.
This approach complements the strategy of using covalently linked multinucleating ligands. For example, trimetallic complexes by Murray have been shown to bind N2 at a tricopper(I) species, and also to cleave N2 using a presumed triiron(I) intermediate.28,29 Limberg has also prepared dinucleating diketiminate ligands and their complexes.30 Multinucleating ligands from Betley have given multimetallic iron sites that reduce small molecules.31−33 We anticipate that the use of alkali metals will be an easy way to reversibly steer metal atoms into close proximity for multimetallic reactions, without the synthesis of complicated ligands.
Summary and Additional Perspectives on the Future
The fundamental work described above has led to important concepts, such as the use of low coordination numbers and a high-spin electronic configuration for N2 weakening, involvement of several metal ions for easier activation of strong bonds, and manipulation of geometries and charge buildup with alkali metal cations. In order for these coordination chemistry concepts to be applied to the challenges of the FeMoco mechanism, several barriers must be overcome.
One challenge is that very strong reducing agents must be replaced by less aggressive reductants. Our research shows that alkali metal cations give more positive redox potentials, which could be useful for enabling milder reduction. Note that the FeMoco in nitrogenase is surrounded by a number of positively charged amino acid residues, including a His and Arg near the active four-iron face, which might play an analogous role. Another potential route to reduced metal-N2 species without a strong reductant is by reductive elimination of hydrides as H2.34 We have demonstrated this method of N2 binding in both cobalt35 and iron36 complexes, and it has been proposed in the FeMoco based on H/D labeling and ENDOR studies.37 It may also be possible to reduce metal-N2 species at less negative potentials by coupling N2 reduction with N2 protonation. The study of metal-oxo intermediates has taught chemists that the ability to oxidize a substrate can be facilitated by increasing the basicity of the site that accepts a proton.38 Likewise, the ability to reduce a complex can in principle be eased by providing a proton simultaneously (proton-coupled electron transfer or PCET).39 PCET often has stringent geometric constraints, and geometric control is also necessary to make sure that protons and electrons go to N2 reduction rather than H2 formation.
Finally, the nature of the supporting ligands has not yet been evaluated in enough detail. The anionic nitrogen atoms in a diketiminate are weak-field π-donors like the biological sulfides, but the best supporting ligand would have S-based donors. Coordinatively unsaturated complexes rarely have thiolates or sulfides because of the tendency of sulfur to bridge between metals, forming clusters that fill any open coordination sites.40 However, the FeMoco belt iron atoms have only S and C coordination, and nothing is known about Fe–N2 binding in such an environment.
Overall, we hope that the perspectives above serve as encouragement for the study of more complexes with cooperative reactions, both between multiple transition metals and between transition metals and main-group metals, for the activation of strong bonds. The presence of clusters at some of the most potent bond-breaking and bond-forming enzymes (nitrogenase, photosystem II, methane monooxygenase, hydrogenase, CO dehydrogenase) is evidence that natural systems have benefitted from this strategy. Since there are so many permutations of metals and reactions, it is clear that the sky (with all of its N2) is the limit!
Acknowledgments
Research on N2 activation in the Holland lab has been supported by the National Institutes of Health (GM065313). Some of the computations described here were performed under the support of the Fulbright Foundation during a P.L.H. sabbatical at the Max Planck Institute for Chemical Energy Conversion, and we thank Frank Neese and his research group for their assistance. We thank our many collaborators on this project, including Eckhard Bill, Thomas Cundari, Serena DeBeer, Travis Figg, Brian Hoffman, Gudrun Lukat-Rodgers, Eckard Münck, Aaron Pierpont, Karl Pittard, Chris Pollock, Kenton Rodgers, and Sebastian Stoian. Crystallography has been crucial, and we thank Rene Lachicotte, Christine Flaschenriem, William Brennessel, and Brandon Mercado. Most of all, thanks go to the students who carried out this research, including Jeremy Smith, Javier Vela, Azwana Sadique, Ying Yu, Keying Ding, Thomas Dugan, Karen Chiang, Meghan Rodriguez, Cory MacLeod, and Kasia Grubel.
Biographies
Sean F. McWilliams received a B.S. in chemistry with highest honors from Temple University in 2013. He is currently pursuing graduate studies with Patrick L. Holland at Yale University, focusing on fundamental iron-dinitrogen chemistry relating to nitrogenase and the Haber–Bosch process.
Patrick L. Holland received an A.B. in chemistry with high honors from Princeton University, and a Ph.D. from the University of California at Berkeley working on organometallic chemistry with Richard Andersen and Robert Bergman. After postdoctoral work with William Tolman at the University of Minnesota, his independent research has addressed mimics of several iron-containing metalloenzymes, particularly nitrogenases. Prof. Holland was on the faculty at the University of Rochester from 2000 to 2013, and he is now Professor of Chemistry at Yale University.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Synthesis in Biological Inorganic Chemistry”.
References
- Burgess B. K.; Lowe D. J. Mechanism of Molybdenum Nitrogenase. Chem. Rev. 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- Hoffman B. M.; Dean D. R.; Seefeldt L. C. Climbing Nitrogenase: Toward a Mechanism of Enzymatic Nitrogen Fixation. Acc. Chem. Res. 2009, 42, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson R.; Lima F. A.; Spatzal T.; Weyhermuller T.; Glatzel P.; Bill E.; Einsle O.; Neese F.; DeBeer S. Identification of a spin-coupled Mo(III) in the nitrogenase iron-molybdenum cofactor. Chem. Sci. 2014, 5, 3096–3103. [Google Scholar]
- Dos Santos P. C.; Igarashi R. Y.; Lee H.-I.; Hoffman B. M.; Seefeldt L. C.; Dean D. R. Substrate interactions with the nitrogenase active site. Acc. Chem. Res. 2005, 38, 208–214. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Einsle O.; Howard J. B.; Rees D. C. Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Jaime M. U.; Dible B. R.; Chiang K. P.; Brennessel W. W.; Bergmann U.; Holland P. L.; DeBeer S. Identification of a Single Light Atom within a Multinuclear Metal Cluster Using Valence-to-Core X-ray Emission Spectroscopy. Inorg. Chem. 2011, 50, 10709–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Roemelt M.; Ettenhuber P.; Hu Y.; Ribbe M. W.; Neese F.; Bergmann U.; DeBeer S. X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L. A.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Lee C. C.; Ribbe M. W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337, 1672–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rittle J.; Peters J. C. Fe–N2/CO complexes that model a possible role for the interstitial C atom of FeMo-cofactor (FeMoco). Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Creutz S. E.; Peters J. C. Catalytic Reduction of N2 to NH3 by an Fe–N2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc. 2014, 136, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. I.; Burgess B. K.; Natoli C. R.; Filipponi A.; Gavini N.; Hedman B.; Di Cicco A.; Hodgson K. O. Direct evidence for the long-range Mo-Fe-Fe interaction and cyanide binding to the Mo in FeMo-cofactor. J. Am. Chem. Soc. 1994, 116, 2418–2423. [Google Scholar]
- Dance I.; Henderson R. A. Large structural changes upon protonation of Fe4S4 clusters: the consequences for reactivity. Dalton Trans. 2014, 43, 16213–16226. [DOI] [PubMed] [Google Scholar]
- a Hazari N. Homogeneous iron complexes for the conversion of dinitrogen into ammonia and hydrazine. Chem. Soc. Rev. 2010, 39, 4044–4056. [DOI] [PubMed] [Google Scholar]; b Crossland J. L.; Tyler D. R. Iron-dinitrogen coordination chemistry: Dinitrogen activation and reactivity. Coord. Chem. Rev. 2010, 254, 1883–1894. [Google Scholar]
- a MacKay B. A.; Fryzuk M. D. Dinitrogen Coordination Chemistry: On the Biomimetic Borderlands. Chem. Rev. 2004, 104, 385–401. [DOI] [PubMed] [Google Scholar]; b Gambarotta S.; Scott J. Multimetallic cooperative activation of N2. Angew. Chem., Int. Ed. 2004, 43, 5298–5308. [DOI] [PubMed] [Google Scholar]; c MacLeod K. C.; Holland P. L. Recent developments in the homogeneous reduction of dinitrogen by molybdenum and iron. Nat. Chem. 2013, 5, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köthe C.; Limberg C. Late Metal Scaffolds that Activate Both Dinitrogen and Reduced Dinitrogen Species NxHy. Z. Anorg. Allg. Chem. 2015, 641, 18–30. [Google Scholar]
- Khoenkhoen N.; de Bruin B.; Reek J. N. H.; Dzik W. I. Reactivity of Dinitrogen Bound to Mid and Late-Transition-Metal Centers. Eur. J. Inorg. Chem. 2015, 4, 567–598. [Google Scholar]
- a Smith J. M.; Lachicotte R. J.; Pittard K. A.; Cundari T. R.; Lukat-Rodgers G.; Rodgers K. R.; Holland P. L. Stepwise Reduction of Dinitrogen Bond Order by a Low-Coordinate Iron Complex. J. Am. Chem. Soc. 2001, 123, 9222–9223. [DOI] [PubMed] [Google Scholar]; b Smith J. M.; Sadique A. R.; Cundari T. R.; Rodgers K. R.; Lukat-Rodgers G.; Lachicotte R. J.; Flaschenriem C. J.; Vela J.; Holland P. L. Studies of Low-Coordinate Iron Dinitrogen Complexes. J. Am. Chem. Soc. 2006, 128, 756–769. [DOI] [PubMed] [Google Scholar]; c Pollock C. J.; Grubel K.; Holland P. L.; DeBeer S. Experimentally Quantifying Small-Molecule Bond Activation Using Valence-to-Core X-ray Emission Spectroscopy. J. Am. Chem. Soc. 2013, 135, 11803–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoian S. A.; Vela J.; Smith J. M.; Sadique A. R.; Holland P. L.; Münck E.; Bominaar E. L. Mössbauer and Computational Study of an N2-Bridged Diiron Diketiminate Complex. J. Am. Chem. Soc. 2006, 128, 10181–10192. [DOI] [PubMed] [Google Scholar]
- Holland P. L. Metal-dioxygen and metal-dinitrogen complexes: Where are the electrons?. Dalton Trans. 2010, 39, 5415–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Betley T. A.; Peters J. C. Dinitrogen Chemistry from Trigonally Coordinated Iron and Cobalt Platforms. J. Am. Chem. Soc. 2003, 125, 10782–10783. [DOI] [PubMed] [Google Scholar]; b Suess D. L. M.; Peters J. C. H–H and Si–H Bond Addition to Fe≡NNR2 Intermediates Derived from N2. J. Am. Chem. Soc. 2013, 135, 4938–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomitz W. A.; Arnold J. Synthesis and characterization of manganese and iron complexes supported by multidentate [N2P2] ligands. Dalton Trans. 2009, 10, 1714–1720. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Smith J. M.; Flaschenriem C. J.; Holland P. L. Binding Affinity of Alkynes and Alkenes to Low-Coordinate Iron. Inorg. Chem. 2006, 45, 5742–5751. [DOI] [PubMed] [Google Scholar]
- Rodriguez M. M.; Bill E.; Brennessel W. W.; Holland P. L. N2 Reduction and Hydrogenation to Ammonia by a Molecular Iron-Potassium Complex. Science 2011, 334, 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubel K.; Brennessel W. W.; Mercado B. Q.; Holland P. L. Alkali Metal Control over N–N Cleavage in Iron Complexes. J. Am. Chem. Soc. 2014, 136, 16807–16816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figg T. M.; Holland P. L.; Cundari T. R. Cooperativity Between Low-Valent Iron and Potassium Promoters in Dinitrogen Fixation. Inorg. Chem. 2012, 51, 7546–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. M.; Stubbert B. D.; Scarborough C. C.; Brennessel W. W.; Bill E.; Holland P. L. Isolation and Characterization of Stable Iron(I) Sulfide Complexes. Angew. Chem., Int. Ed. 2012, 51, 8247–8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang K. P.; Bellows S. M.; Brennessel W. W.; Holland P. L. Multimetallic cooperativity in activation of dinitrogen at iron-potassium sites. Chem. Sci. 2014, 5, 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray L. J.; Weare W. W.; Shearer J.; Mitchell A. D.; Abboud K. A. Isolation of a (Dinitrogen)Tricopper(I) Complex. J. Am. Chem. Soc. 2014, 136, 13502–13505. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Sloane F. T.; Blondin G.; Abboud K. A.; García-Serres R.; Murray L. J. Dinitrogen Activation Upon Reduction of a Triiron(II) Complex. Angew. Chem., Int. Ed. 2015, 54, 1499–1503. [DOI] [PubMed] [Google Scholar]
- For example:Haack P.; Limberg C.; Ray K.; Braun B.; Kuhlmann U.; Hildebrandt P.; Herwig C. Dinuclear Copper Complexes Based on Parallel β-Diiminato Binding Sites and their Reactions with O2: Evidence for a Cu–O–Cu Entity. Inorg. Chem. 2011, 50, 2133–2142. [DOI] [PubMed] [Google Scholar]
- Harris T. D.; Betley T. A. Multi-Site Reactivity: Reduction of Six Equivalents of Nitrite To Give an Fe6(NO)6 Cluster with a Dramatically Expanded Octahedral Core. J. Am. Chem. Soc. 2011, 133, 13852–13855. [DOI] [PubMed] [Google Scholar]
- Eames E. V.; Betley T. A. Site-Isolated Redox Reactivity in a Trinuclear Iron Complex. Inorg. Chem. 2012, 51, 10274–10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T. M.; Betley T. A. Testing the Polynuclear Hypothesis: Multielectron Reduction of Small Molecules by Triiron Reaction Sites. J. Am. Chem. Soc. 2013, 135, 12289–12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmann J.; Munha R. F.; Fryzuk M. D. The hydride route to the preparation of dinitrogen complexes. Chem. Commun. 2010, 46, 1013–1025. [DOI] [PubMed] [Google Scholar]
- Ding K.; Brennessel W. W.; Holland P. L. Three-Coordinate and Four-Coordinate Cobalt Hydride Complexes That React with Dinitrogen. J. Am. Chem. Soc. 2009, 131, 10804–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan T. R.; Holland P. L. New routes to low-coordinate iron hydride complexes: The binuclear oxidative addition of H2. J. Organomet. Chem. 2009, 694, 2825–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang Z.-Y.; Khadka N.; Lukoyanov D.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. On reversible H2 loss upon N2 binding to FeMo-cofactor of nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16327–16332. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. itrogenase: A Draft Mechanism. Acc. Chem. Res. 2013, 46, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Iron(IV) hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mayer J. M. Proton-coupled electron transfer: A reaction chemist’s view. Annu. Rev. Phys. Chem. 2004, 55, 363–390. [DOI] [PubMed] [Google Scholar]; b Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- a Bart S. C.; Lobkovsky E.; Bill E.; Wieghardt K.; Chirik P. J. Neutral-Ligand Complexes of Bis(imino)pyridine Iron: Synthesis, Structure, and Spectroscopy. Inorg. Chem. 2007, 46, 7055–7063. [DOI] [PubMed] [Google Scholar]; b Takaoka A.; Mankad N. P.; Peters J. C. Dinitrogen Complexes of Sulfur-Ligated Iron. J. Am. Chem. Soc. 2011, 133, 8440–8443. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Note added in proof: Peters recently described a thiolate-bridged diiron complex with bound N2: Creutz S. E.; Peters J. C. Diiron Bridged-Thiolate Complexes That Bind N2 at the FeIIFeII, FeIIFeI, and FeIFeI Redox States. J. Am. Chem. Soc. 2015, 137, 7310–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]