

Abstract
Background:
Hyperoside, a flavonoid which is mainly found in Hypericum perforatum L., has many biological effects. One of the most important effects is to prevent the oxidative stress induced by reactive oxygen species. However, the molecular mechanisms underlying its effect are not fully understood. Oxidative stress is implicated in the occurrence of various physical diseases. A wide array of enzymatic antioxidant defense systems include NADH: quinone oxidoreductase 1, superoxide dismutase, and heme oxygenase-1 (HO-1). In the present study, the protective effects of hyperoside against hydrogen peroxide-induced oxidative stress in human lens epithelial cells, HLE-B3, were investigated in terms of HO-1 induction.
Methods:
The protein and mRNA expressions of HO-1 were examined by Western blotting and reverse transcriptase-PCR assays, respectively. To evaluate the ability of hyperoside to activate nuclear factor erythroid 2-related factor 2 (Nrf2), Western blotting and electrophoretic mobility shift assay were performed with nuclear extracts prepared from HLE-B3 cells treated with hyperoside. The activation of extracellular signal-regulated kinase (ERK), the upstream kinase of Nrf2 signaling, was monitored by Western blot analysis. The protective effect of hyperoside in HLE-B3 cells against hydrogen peroxide was performed by MTT assay.
Results:
Hyperoside increased both the mRNA and protein expression of HO-1 in a time- and dose-dependent manner. In addition, hyperoside elevated the level of of Nrf2 and its antioxidant response element-binding activity, which was modulated by upstream of ERK. Moreover, it activated ERK and restored cell viability which was decreased by hydrogen peroxide.
Conclusions:
Hyperoside is an effective compound to protect cells against oxidative stress via HO-1 induction.
Keywords: Hyperoside, Heme oxygenase-1, Oxidative stress, Antioxidants
INTRODUCTION
Flavonoids have many biological effects including inhibiting carcinogenesis, reducing the level of low-density lipoprotein, decreasing hypertension, and reducing the levels of reactive oxygen species (ROS).1–4 Hyperoside is a type of flavonoid and has many biological functions including the antidepressant activity, increasing the levels of serotonin, and a protective effect against oxidative stress.4,5 One of hyperoside’ effects is antioxidant activity, which can prevent cytotoxicity induced by ROS. However, the effects and molecular mechanisms underlying hyperoside-mediated antioxidant activity are not fully understood.
ROS include the superoxide anion (O2−), the hydroxyl radical (HO •), and hydrogen peroxide (H2O2). Oxidative stress has been strongly linked with the onset of various physical diseases.6,7 There exist enzymatic antioxidant defense systems including NADH: quinone oxidoreductase 1, superoxide dismutase, and heme oxygenase-1 (HO-1). HO-1 is a rate-limiting enzyme in heme degradation and also known as 32 kDa heat shock protein. In various model systems, HO-1 induction protects tissues from oxidative injuries, while abrogation of HO-1 induction accelerates cellular injuries.8
A number of intracellular signaling molecules are involved in regulating the induction of HO-1. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a major transcription factor of HO-1. In normal conditions, Nrf2 is located in the cytosol fraction and bound to the inhibitor protein Kelch-like ECH-associated protein-1 (Keap1). Upon signaling, Nrf2 separates from Keap1 and translocates into the nucleus. Nrf2 heterodimerizes with small Maf and binds to antioxidant response element (ARE) sequences, thereby transcriptionally activates HO-1.9–11
The present study investigated the protective activity of hyperoside against hydrogen peroxide-induced oxidative stress in HLE-B3 human lens epithelial cells, which is used for investigating human lens epithelial physiology and studying oxidative stress.12,13 The results suggest that hyperoside efficiently protects cells against oxidative stress by inducing HO-1 expression. Moreover, we also examined the molecular mechanism underlying HO-1 induction in hyperoside-treated cells, which involved activation of extracellular signal-regulated kinase (ERK) and the Nrf2-ARE signaling pathway.
MATERIALS AND METHODS
1. Materials
Hyperoside (quercetin-3-D-galactoside; Fig. 1) and U0126 were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Dulbecco’s modified Eagle medium (DMEM), FBS, and a penicillin/streptomycin/fungizone mixture were obtained from Gibco BRL (Grand Island, NY, USA). Anti-phospho ERK and anti-ERK antibodies were provided by Cell Signaling Technology (Beverly, MA, USA). Anti-Nrf2 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and anti-HO-1 antibody was supplied by Stressgen Biotechnologies (Victoria, BC, Canada).
Figure 1.
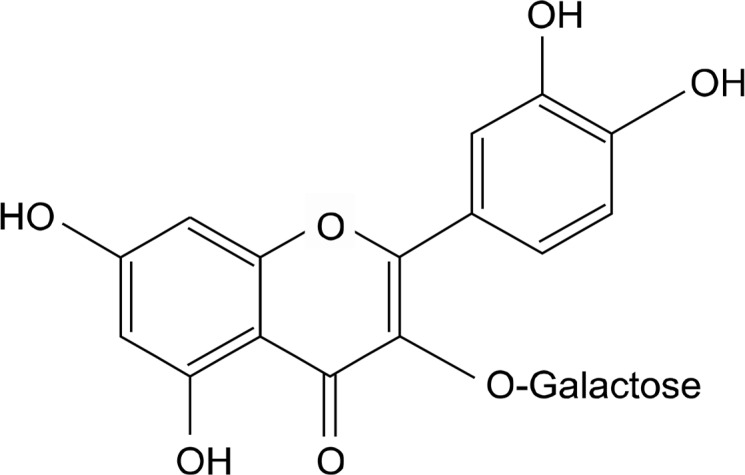
Chemical structure of hyperoside (quercetin-3-D-galactoside).
2. Cell culture
HLE-B3 cells were grown in DMEM supplemented with 20% FBS and penicillin/streptomycin in a humidified atmosphere containing 5% CO2 at 37°C.
3. Reverse transcriptase-polymerase chain reaction
Total RNA was isolated from cells using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Reverse transcriptase (RT)-PCR was performed following standard procedures. Amplification products were resolved by 1.2% agarose gel electrophoresis. Primers were purchased from Bionics (Seoul, Korea). PCR conditions were as follows: HO-1, 25 cycles of 95°C for 1 minute, 60°C for 1 minute, and 72°C for 2 minutes, and house-keeping gene glycera-ldehyde-3-phosphate dehydrogenase (GAPDH), 26 cycles of 94°C for 1 minute, 56°C for 2 minutes, and 72°C for 2 minutes. The primer pairs were as follows (forward and reverse, respectively): HO-1, 5′-CAG GCA GAG AAT GCT GAG TTC-3′ and 5′-GAT GTT GAG CAG GAA CGC AGT-3′, GAPDH, 5′-AAG GTC GGA GTC AAC GGA TTT-3′ and 5′-GCA GTG AGG GTC TCT CTC CCT-3′.
4. Western blot analysis
For immunoblot analysis, cells were washed two times in PBS, and proteins were extracted with lysis buffer (50 mM Tris-HCl, pH 8.0, 2 mM EDTA, and 1% Triton X-100) supplemented with a protease inhibitor cocktail tablet. After incubation on ice for 20 minutes, the lysates were centrifuged at 12,000 ×g for 15 minutes at 4°C. The protein concentration of supernatant was measured using bicinchoninic acid reagents (Pierce, Rockford, IL, USA). Protein samples (30–50 μg) were separated onto a 12% SDS PAGE gel and transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The blots were blocked for 1 hour at room temperature in fresh blocking buffer (0.1% Tween-20 prepared in PBS containing 5% non-fat dry milk). The blots were incubated with primary antibodies diluted with PBS containing 3% non-fat dry milk at 4°C and then further incubated with horseradish peroxidase-conjugated secondary antibodies diluted in PBS containing 3% non-fat dry milk for 1 hour at room temperature. The blots were incubated in enhanced chemiluminescence substrate solution (Amersham Pharmacia Biotech) for 1 minute according to the manufacturer’s instructions.
5. Preparation of nuclear proteins
After treatment with hyperoside, cells were washed with ice-cold PBS, scraped in 1 mL of PBS, and centrifuged at 12,000 ×g for 30 seconds at 4°C. Pellets were suspended in 200 μL of hypotonic buffer (10 mM HEPES, pH 7.8, 10 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol, 0.1 mM EDTA, and 0.1 mM phenylmethylsulfonyl fluoride) for 15 minutes on ice, and 12.5 μL of 10% Nonidet P-40 solution was added. Samples were incubated for 5 minutes and then centrifuged for 6 minutes at 12,000 ×g. The nuclei were washed once with 400 μL of PBS, suspended in 70 μL of buffer (50 mM HEPES, pH 7.8, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1 mM PMSF, and 10% glycerol) for 20 minutes on ice, and centrifuged for 6 minutes at 12,000 ×g. The supernatant containing nuclear proteins was collected and stored at −70°C after determination of the protein concentration.
6. Electrophoretic mobility shift assay
A synthetic double-stranded oligonucleotide containing the Nrf2-binding domain (ARE) was labeled with [γ-32P]ATP using T4 polynucleotide kinase and separated from unincorporated [γ-32P]ATP by gel filtration using a nick spin column (Pharmacia Biotech, Bjorkgatan, Sweden). Prior to addition of the 32P-labeled oligonucleotide (100,000 cpm), 10 μg of the nuclear extract was incubated on ice for 15 minutes in gel shift binding buffer (20% glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl, 50 mM Tris-HCl, pH 7.5, and 0.25 μg/mL poly [dI-dC]). DNA-protein complexes were resolved by 6% polyacrylamide gel electrophoresis at 200 V for 2 hours, followed by autoradiography.
7. Transient transfection and the reporter gene assay
Cells were plated at a confluency of 90% to 95% in a 6-well plate. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) with 1 μg of DNA (plasmids including ARE-luciferase gene, the GC-mut-Luc construct, and the control vector) per 2 μL of N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate. After 8 hours, the cells were treated with hyperoside for 12 hours and lysed using reporter lysis buffer. After mixing the cell extract with a luciferase substrate (Promega, Madison, WI, USA), luciferase activity was measured using a luminometer.
8. Immunofluorescence staining
Cells were washed with PBS, incubated for 1 hour in 4% formaldehyde, washed twice with PBS, and incubated with 0.2% Triton-X in PBS. Then, the cells were washed with PBS and blocked with 5% bovine serum albumin (BSA) solution for 2 hours at room temperature. Primary antibodies were diluted in PBS containing 3% BSA and incubated with samples for 1 hour at room temperature. Following three washes with 0.2% Triton-X in PBS, the cells were incubated with fluorescein isothiocyanate-conjugated secondary antibodies diluted in PBS containing 3% BSA for 1 hour at room temperature. After washing with PBS three times, the cells were treated with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector, Burlingame, CA, USA) and the image was assessed using fluorescence microscopy.
9. MTT assay
Cells were plated in 48-well plates, incubated for 12 hours after treatment with hyperoside (100 μM) and/or ZnPP (10 μM), and treated with hydrogen peroxide for 12 hours. MTT reagent was added and incubated at 37°C for 2 hours. After removing the media, samples were dissolved in 300 μL of dimethyl sulfoxide and were transferred to 96-well plates. Absorbance was read at 540 nm.
10. Statistical analysis
All values were demonstrated as mean ± SE. The results were subjected to ANOVA followed by the Tukey’s test to examine significant differences between values. P < 0.05 was considered statistically significant.
RESULTS
1. Increased heme oxygenase-1 mRNA and protein expression in hyperoside-treated cells
To investigate whether hyperoside can induce HO-1 gene expression in HLE-B3 cells, cells were treated with 100 μM hyperoside. RT-PCR analysis showed that expression of HO-1 mRNA was significantly increased in hyperoside-treated cells at 4 hours (Fig. 2A). In parallel with the elevated mRNA expression, the protein level of HO-1 was increased by hyperoside treatment in a time-dependent manner (Fig. 2B). In addition, hyperoside treatment induced both mRNA and protein expression of HO-1 in a concentration-dependent manner (Fig. 2C and 2D).
Figure 2.

Hyperoside upregulates mRNA and protein expression of heme oxygenase-1 (HO-1) in human lens epithelial cells. Cells were treated (A, B) with 100 μM hyperoside for various amounts of time or (C, D) with different concentrations of hyperoside for 4 and 12 hours to detect mRNA and protein expression of HO-1, respectively. (A, C) Reverse transcriptase-PCR and (B, D) Western blot analyses were conducted to measure the induced mRNA and protein expression. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and actin bands are shown to confirm the equal loading of RNA and protein, respectively.
2. Increases in the nuclear translocation and antioxidant response element-binding activity of nuclear factor erythroid2-related factor-2 in hyperoside-treated cells
Nrf2 is one of the transcription factors that modulate HO-1 expression. To evaluate the ability of hyperoside to stimulate Nrf2 activation, Western blotting and electrophoretic mobility shift assay were performed with nuclear extracts derived from hyperoside-treated cells. Nuclear Nrf2 expression was observed clearly at 3 hours after hyperoside treatment (Fig. 3A). ARE-binding activity was assessed by incubating nuclear extracts with radiolabelled oligonucleotides harboring the specific binding site of Nrf2. Nuclear extracts of hyperoside-treated cells showed increased ARE-binding at 3 hours (Fig. 3B).
Figure 3.

Hyperoside induces nuclear factor erythroid 2-related factor-2 (Nrf2) in human lens epithelial cells. (A) Nuclear extracts were prepared from cells after treatment with hyperoside for the indicated amount of time. Immunoblots of the nuclear lysates of hyperoside-treated cells were probed with an Nrf2-specific antibody. (B) Nuclear extracts prepared from hyperoside-treated cells for the indicated amount of time were subjected to the electrophoretic mobility shift assay to measure the antioxidant response element (ARE)-binding activity of Nrf2. (C) Cells were transfected with the control vector, the ARE-luciferase construct, and the GC-mut-Luc construct. After 8 hours, cells were treated with hyperoside for 12 hours, cell lysates were mixed with a luciferase substrate, and luciferase activity was measured using a luminometer. Results are expressed as the mean ± SE, *P < 0.05. (D) Cells were treated with hyperoside for 3 hours and immunocytochemistry was performed to investigate the nuclear translocation of Nrf2.
To determine whether hyperoside treatment induces Nrf2-dependent gene transcription, cells were transfected with the ARE-Luc vector (a construct containing the ARE-binding site ligated to the luciferase gene). When transfected cells were incubated with hyperoside for 12 hours, Nrf-ARE reporter gene activity increased by approximately 2.5 fold. In addition, transient transfection with GC-mut-Luc (a construct containing a mutant form of the GC dinucleotide at the 3′ end of the ARE core, which is essential for ARE function) rendered cells less responsive to hyperoside in terms of induction of ARE reporter gene activity (Fig. 3C). Immunocytochemistry also showed the nuclear translocation of Nrf2 at 3 hours after hyperoside treatment (Fig. 3D).
3. Activation of extracellular signal-regulated kinase and upstream kinase in nuclear factor E2-related factor-2-antioxidant response element signaling of hyperoside-treated cells
Activation of ERK was monitored by Western blot analysis using phospho-specific antibodies. The phosphor form of ERK (active ERK) was detected at 15 minutes in hyperoside-treated cells (Fig. 4A). ERK is considered to be critical for inducible expression of Nrf2; therefore, we examined whether pharmacologic inhibitor of ERK could suppress the activation of Nrf2 in hyperoside-treated cells. The hyperoside-induced protein expression were inhibited by U0126, a pharmacological inhibitor of MEK upstream of ERK (Fig. 4B). Also, the increased HO-1 level in hyperoside-treated cells was inhibited by U0126 (Fig. 4C).
Figure 4.

Hyperoside upregulates heme oxygenase-1 (HO-1) expression via nuclear factor erythroid 2-related factor-2 (Nrf2) and phosphorylation of extracellular signal-regulated kinase (ERK). (A) Cells were treated with hyperoside for various amounts of time. Western blot analysis was performed to measure the activation of ERK protein (B,C) Cells were pre-incubated with the inhibitor U0126 for 1 hour and then treated with hyperoside for 12 hours. The levels of (B) nuclear Nrf2 and (C) HO-1 were analyzed by Western blotting.
4. Protective effect of hyperoside via heme oxygenase-1 induction against oxidative stress
Cell viability was significantly suppressed in hydrogen peroxide-treated cells, but hyperoside treatment recovered the reduced cell viability (Fig. 5A). Because HO-1 is considered to be critical for removal of hydrogen peroxide, we examined whether selective inhibition of HO-1 could affect cell viability. When cells were co-treated with hyperoside and the HO-1 inhibitor ZnPP, cell viability was decreased compared to only hyperoside-treated cells (Fig. 5B).
Figure 5.

Hyperoside inhibits hydrogen peroxide-induced cell death. Cells were pretreated (A) with hyperoside (100 μM) or (B) hyperoside and ZnPP (10 μM) for 12 hours and then incubated with 100 μM hydrogen peroxide for 12 hours. Cell viability was measured by MTT assay as described in the Materials and Methods. Results are expressed at the mean ± SE, *P < 0.05.
DISCUSSION
The present study demonstrated that hyperoside had protective effects against oxidative stress induced by hydrogen peroxide in HLE-B3 cells. Hyperoside increased the viability of hydrogen peroxide-exposed cells (Fig. 5A). A low concentration of ROS may have benefits, such as intracellular messenger. By contrast, the inadequate removal of ROS and higher amounts of ROS may be harmful to organisms. Unbalanced ROS production has been postulated to play a pathological role in cancer, allergies, cardiological and vessel injuries, metabolic malfunctions, neuro-degenerative diseases, and ophthalmologic problems.6 Therefore, we investigated whether the protective effect of hyperoside is due to a reduction in ROS, which is associated with antioxidant enzymes, especially HO-1. HO is the rate-limiting enzyme and has three isoforms, namely, HO-1, HO-2, and HO-3. HO-1, known as heat shock protein 32, is highly inducible by some stimuli. HO degrades heme into biliverdin, carbon monoxide, and iron. Biliverdin is subsequently converted into bilirubin by biliverdin reductase. HO-1 is induced as a protective mechanism in response to various stimuli; therefore, targeted induction of this stress response enzyme may be considered to be an important therapeutic strategy for protection against oxidative tissue damage.9,12 Transfection of the human HO-1 gene into rabbit ocular tissues in vivo demonstrated that it could be a promising means for protecting against oxidative stress that contributes to the pathogenesis of cataracts, light-induced injury, age-related macular degeneration, and diabetic retinopathy.14
There is much evidence that food-derived antioxidants can reduce oxidative stress-induced damage by increasing the levels of cellular defense enzymes. For example, quercetin plays a role in inhibition of hydrogen peroxide-induced cataracts.15 Hyperoside is a galactose-conjugated form of quercetin. Quercetin-induced HO-1 production increases in a time- and dose- dependent fashion, and this regulation occurs at both the transcriptional and translational levels.16,17 However, some studies revealed that other glycosides of quercetin, such as rutin and quicitrin, do not induce HO-1.16,18 In the present study, the mRNA and protein expression of HO-1 increased in a time- and dose-dependent manner in hyperoside-treated cells (Fig. 2). A number of intracellular signaling molecules are involved in regulating the induction of HO-1, including activator protein-1, nuclear factor-κB, and Nrf2.19–22 Nrf2, a 66 kDa protein and a member of the Cap’n’Collar family of bZIP transcription factors, plays an important role in ARE-mediated expression of phase II detoxifying antioxidants.23 Nuclear translocation of Nrf2 and its ARE-binding activity were increased after 3 hours of hyperoside treatment (Fig. 3). ERK was phosphorylated maximally at 30 minutes in hyperoside-treated HLE-B3 cells (Fig. 4A). When cells were pretreated with a pharmacological inhibitor of ERK for 30 minutes, both Nrf2-ARE signaling and HO-1 induction were decreased (Fig. 4B and 4C). This study suggested that ERK and Nrf2-ARE signaling are upstream of HO-1. The HO-1 inhibitor, ZnPP, decreased cell viability recovered by hyperoside treatment in hydrogen peroxide-treated cells (Fig. 5B).
In summary, hyperoside increased expression of HO-1 and protected cells against hydrogen peroxide-induced cytotoxicity. Furthermore, hyperoside significantly induced ERK phosphorylation and Nrf2-ARE binding. This process may contribute to protect cell against oxidative stress-induced cell damage.
Acknowledgments
This work was supported by National Research Foundation of Korea (NRF) grant funded by Korea government (No. NRF-2015M2A2A7061657).
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Hu J, Huang Y, Xiong M, Luo S, Chen Y, Li Y. The effects of natural flavonoids on lipoxygenase-mediated oxidation of compounds with a benzene ring structure: a new possible mechanism of flavonoid anti-chemical carcinogenesis and other toxicities. Int J Toxicol. 2006;25:295–301. doi: 10.1080/10915810600746122. [DOI] [PubMed] [Google Scholar]
- 2.Naderi GA, Asgary S, Sarraf-Zadegan N, Shirvany H. Anti-oxidant effect of flavonoids on the susceptibility of LDL oxidation. Mol Cell Biochem. 2003;246:193–6. doi: 10.1023/A:1023483223842. [DOI] [PubMed] [Google Scholar]
- 3.Koçyildiz ZC, Birman H, Olgaç V, Akgün-Dar K, Melikoğlu G, Meriçli AH. Crataegus tanacetifolia leaf extract prevents L-NAME-induced hypertension in rats: a morphological study. Phytother Res. 2006;20:66–70. doi: 10.1002/ptr.1808. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z, Tao X, Zhang C, Lu Y, Wei D. Protective effects of hyperoside (quercetin-3-o-galactoside) to PC12 cells against cytotoxicity induced by hydrogen peroxide and tert-butyl hydroperoxide. Biomed Pharmacother. 2005;59:481–90. doi: 10.1016/j.biopha.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Zou Y, Lu Y, Wei D. Antioxidant activity of a flavonoid-rich extract of Hypericum perforatum L. in vitro. J Agric Food Chem. 2004;52:5032–9. doi: 10.1021/jf049571r. [DOI] [PubMed] [Google Scholar]
- 6.Matés JM, Sánchez-Jiménez F. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci. 1999;4:D339–45. doi: 10.2741/Mates. [DOI] [PubMed] [Google Scholar]
- 7.Orozco-Ibarra M, Chirino YI, Pedraza-Chaverrí J. Role of hemeox-ygenase-1 in the neurodegenerative disorders. Rev Neurol. 2006;43:556–62. [PubMed] [Google Scholar]
- 8.Takahashi T, Morita K, Akagi R, Sassa S. Heme oxygenase-1: a novel therapeutic target in oxidative tissue injuries. Curr Med Chem. 2004;11:1545–61. doi: 10.2174/0929867043365080. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, et al. Molecular cloning and functional characterization of a new Cap’n’ collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443–52. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 10.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–80. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 11.Owuor ED, Kong AN. Antioxidants and oxidants regulated signal transduction pathways. Biochem Pharmacol. 2002;64:765–70. doi: 10.1016/S0006-2952(02)01137-1. [DOI] [PubMed] [Google Scholar]
- 12.Andley UP, Rhim JS, Chylack LT, Jr, Fleming TP. Propagation and immortalization of human lens epithelial cells in culture. Invest Ophthalmol Vis Sci. 1994;35:3094–102. [PubMed] [Google Scholar]
- 13.Ni S, Yu Y, Zhang Y, Wu W, Lai K, Yao K. Study of oxidative stress in human lens epithelial cells exposed to 1.8 GHz radiofrequency fields. PLoS One. 2013;8:e72370. doi: 10.1371/journal.pone.0072370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham NG, da Silva JL, Lavrovsky Y, Stoltz RA, Kappas A, Dunn MW, et al. Adenovirus-mediated heme oxygenase-1 gene transfer into rabbit ocular tissues. Invest Ophthalmol Vis Sci. 1995;36:2202–10. [PubMed] [Google Scholar]
- 15.Kang DG, Moon MK, Sohn EJ, Lee DH, Lee HS. Effects of morin on blood pressure and metabolic changes in fructose-induced hypertensive rats. Biol Pharm Bull. 2004;27:1779–83. doi: 10.1248/bpb.27.1779. [DOI] [PubMed] [Google Scholar]
- 16.Chow JM, Shen SC, Huan SK, Lin HY, Chen YC. Quercetin, but not rutin and quercitrin, prevention of H2O2-induced apoptosis via anti-oxidant activity and heme oxygenase 1 gene expression in macrophages. Biochem Pharmacol. 2005;69:1839–51. doi: 10.1016/j.bcp.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Lin HC, Cheng TH, Chen YC, Juan SH. Mechanism of heme oxygenase-1 gene induction by quercetin in rat aortic smooth muscle cells. Pharmacology. 2004;71:107–12. doi: 10.1159/000076947. [DOI] [PubMed] [Google Scholar]
- 18.Chen TJ, Jeng JY, Lin CW, Wu CY, Chen YC. Quercetin inhibition of ROS-dependent and -independent apoptosis in rat glioma C6 cells. Toxicology. 2006;223:113–26. doi: 10.1016/j.tox.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Hartsfield CL, Alam J, Choi AM. Transcriptional regulation of the heme oxygenase 1 gene by pyrrolidine dithiocarbamate. FASEB J. 1998;12:1675–82. doi: 10.1096/fasebj.12.15.1675. [DOI] [PubMed] [Google Scholar]
- 20.Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J Biol Chem. 2002;277:17950–61. doi: 10.1074/jbc.M108317200. [DOI] [PubMed] [Google Scholar]
- 21.Farombi EO, Surh YJ. Heme oxygenase-1 as a potential therapeutic target for hepatoprotection. J Biochem Mol Biol. 2006;39:479–91. doi: 10.5483/BMBRep.2006.39.5.479. [DOI] [PubMed] [Google Scholar]
- 22.Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, et al. A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J Biol Chem. 2002;277:3456–63. doi: 10.1074/jbc.M110244200. [DOI] [PubMed] [Google Scholar]
- 23.Marini MG, Chan K, Casula L, Kan YW, Cao A, Moi P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J Biol Chem. 1997;272:16490–7. doi: 10.1074/jbc.272.26.16490. [DOI] [PubMed] [Google Scholar]