

Abstract
Objective
To determine the extent of respiratory chain abnormalities and investigate the contribution of mtDNA to the loss of respiratory chain complexes (CI–IV) in the substantia nigra (SN) of idiopathic Parkinson disease (IPD) patients at the single‐neuron level.
Methods
Multiple‐label immunofluorescence was applied to postmortem sections of 10 IPD patients and 10 controls to quantify the abundance of CI–IV subunits (NDUFB8 or NDUFA13, SDHA, UQCRC2, and COXI) and mitochondrial transcription factors (TFAM and TFB2M) relative to mitochondrial mass (porin and GRP75) in dopaminergic neurons. To assess the involvement of mtDNA in respiratory chain deficiency in IPD, SN neurons, isolated with laser‐capture microdissection, were assayed for mtDNA deletions, copy number, and presence of transcription/replication‐associated 7S DNA employing a triplex real‐time polymerase chain reaction (PCR) assay.
Results
Whereas mitochondrial mass was unchanged in single SN neurons from IPD patients, we observed a significant reduction in the abundances of CI and II subunits. At the single‐cell level, CI and II deficiencies were correlated in patients. The CI deficiency concomitantly occurred with low abundances of the mtDNA transcription factors TFAM and TFB2M, which also initiate transcription‐primed mtDNA replication. Consistent with this, real‐time PCR analysis revealed fewer transcription/replication‐associated mtDNA molecules and an overall reduction in mtDNA copy number in patients. This effect was more pronounced in single IPD neurons with severe CI deficiency.
Interpretation
Respiratory chain dysfunction in IPD neurons not only involves CI, but also extends to CII. These deficiencies are possibly a consequence of the interplay between nDNA and mtDNA‐encoded factors mechanistically connected via TFAM. ANN NEUROL 2016;79:366–378
Parkinson disease (PD) is a progressive movement disorder characterized by tremor, rigidity, bradykinesia, and postural instability that affects about 1% of those aged > 65 years.1 The pathological hallmarks of PD are selective loss of dopaminergic neurons and the presence of Lewy bodies in the substantia nigra (SN).2
Mitochondrial dysfunction has emerged as a potential mechanism in PD. Soon after the detrimental effects of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine on motor function were described and the inhibitory action of the toxin against respiratory chain complex I (CI) was unraveled,3 isolated CI deficiency was discovered in homogenates from postmortem SN samples of PD patients.4 The importance of this finding was emphasized when familial PD cases were found to harbor mutations in proteins involved in the removal of damaged mitochondria or the scavenging of reactive oxygen species that are predominantly generated by the electron transport chain.5
A study aiming to elucidate the molecular underpinnings of mitochondrial dysfunction in idiopathic PD (IPD) patients at the single‐cell level identified an accumulation of respiratory chain complex IV (CIV)‐deficient SN neurons with large somatic mtDNA deletions.6 Interestingly, the primary risk factor for PD development is ageing, which is itself correlated with the presence of CIV‐deficient neurons at similar levels to patients with IPD,6, 7 suggesting that age‐related damage accumulation could contribute to neuronal demise in IPD.6, 7
However, in the investigated IPD patients,6 CIV‐negative SN neurons constituted <3% of the total number of analyzed cells, a fraction well below the detection limit in traditional homogenate analysis. In comparison, the postmortem results from Schapira and colleagues4 imply an impairment of CI that is of sufficient magnitude to be detected in SN homogenates. Due to the lack of a robust histochemical method for the assessment of CI activity in single neurons, it has remained elusive whether mtDNA damage also underlies CI deficiency in IPD.
In this study, we aimed to determine the relative incidence of respiratory chain abnormalities and the molecular mechanisms underlying the loss of respiratory chain complexes in individual dopaminergic neurons from IPD patients. To establish the extent and relationship of these deficiencies in single neurons rather than SN homogenate, we employed quantitative quadruple immunofluorescence as an indicator of respiratory chain complex function at single‐cell resolution.8 In combination with laser‐capture microdissection (LCM) and real‐time polymerase chain reaction (PCR) analysis, this approach revealed altered levels of factors responsible for crosstalk between mitochondrial and nuclear compartments as a cause of respiratory chain dysfunction in IPD.
Subjects and Methods
Human Brain Tissue
Brain tissue was obtained from the Newcastle Brain Tissue Resource with ethical approval. Sections from paraffin‐embedded midbrain tissue of 10 IPD patients and 10 age‐matched controls (patients, mean ± standard error [SE]: 75.0 ± 1.8 years; controls, mean ± SE: 76.1 ± 4.1 years; p = 0.80) with comparable postmortem interval (PMI; patients [5 of them female], mean ± SE: 38.7 ± 4.5 hours; controls [4 of them female], mean ± SE: 45.8 ± 7.5 hours; p = 0.43) were used in this study. IPD patients had a mean (±SE) age at disease onset of 65.9 ± 1.2 years. Clinical and neuropathology information is summarized in the Table. Sections were cut at 5 μm using a microtome (Microm International, Walldorf, Germany) and mounted onto SuperFrostTM slides (Thermo Fisher Scientific, Waltham, MA).
Table 1.
Clinical and Neuropathology Features of IPD Patients
| Patient | Sex | Age at Onset, yr | Age at Death, yr | Clinical IPD | LB Pathology | Neuron Loss | Additional Features |
|---|---|---|---|---|---|---|---|
| IPD1 | M | 65 | 67 | Affected | Positive | +++ | Mild cognitive impairment, depression, anxiety |
| IPD2 | M | 66 | 77 | Affected | Positive | +++ | Dementia, visual hallucinations |
| IPD3 | M | 71 | 79 | Affected | Positive | +++ | Dementia, strokelike episodes, episodes of delirium |
| IPD4 | M | 66 | 81 | Affected | Positive | +++ | Cognitive impairment, visual hallucinations, confusion |
| IPD5 | M | 69 | 76 | Affected | Positive | +++ | Mild cognitive impairment, visual hallucinations, confusion |
| IPD6 | F | 60 | 65 | Affected | Positive | ++ | Mild cognitive impairment, depression, visual hallucinations, anxiety |
| IPD7 | F | 62 | 77 | Affected | Positive | ++ | Depression, anxiety, agitation |
| IPD8 | F | 62 | 71 | Affected | Positive | +++ | Mild cognitive impairment, psychosis, complex hallucinations (visual, auditory, olfactory, and tactile) |
| IPD9 | F | 66 | 81 | Affected | Positive | +++ | Dementia, impaired consciousness, visual and auditory hallucinations |
| IPD10 | F | 72 | 76 | Affected | positive | ++ | Confusion, dyskinesia |
| Average ± SE | 65.9 ± 1.2 | 75.0 ± 1.8 |
++ = moderate; + ++ = severe; F = female; IPD = idiopathic Parkinson disease; LB = Lewy body; M = male; SE = standard error.
For mtDNA analysis in single cells, sections were cut from frozen midbrain blocks on a cryostat (Bright Instruments, Huntingdon, UK) at a thickness of 15 μm. Sections were stored at −80 °C, thawed, and allowed to air dry for 60 minutes at room temperature (RT) before immunohistochemistry was performed.
Immunofluorescence Detection of Target Proteins in Formalin‐Fixed Brain Slices
We have previously established quantitative quadruple‐immunofluorescence as a sensitive tool to assess respiratory chain deficiencies at the single‐neuron level in postmortem tissue.8 In this study, this approach was applied to paraffin‐embedded midbrain sections using the following primary antibodies (all Abcam, Cambridge, MA) to target different subunits of respiratory chain complexes: CI, anti‐NDUFB8 (NADH dehydrogenase 1 beta subcomplex 8; ab110242) and anti‐NDUFA13 (NADH dehydrogenase 1 alpha subcomplex 13; ab110240); CII, anti‐SDHA (succinate dehydrogenase complex subunit A; ab14715); CIII, anti‐UQCRC2 (ubiquinol–cytochrome c reductase core protein II; ab14745); and CIV, anti‐COXI (cytochrome c oxidase subunit I; ab14705). The selected subunits are situated in functional domains correlating with the overall abundance of the respiratory chain complexes.8, 9, 10, 11, 12, 13 Additional mitochondrial antibodies included: anti‐porin (Abcam, ab14734), anti‐GRP75 (heat shock 70kDa protein 9 [mortalin]; Abcam, ab53098), anti‐TFAM (mitochondrial transcription factor A; Abcam, ab47517), and anti‐TFB2M (mitochondrial transcription factor B2; Sigma, St Louis, MO; HPA030265). To identify dopaminergic neurons, a rabbit anti–TH antibody (tyrosine hydroxylase; Sigma, T8700) was employed. Imaging using Axio Imager M1 (Zeiss, Oberkochen, Germany), and subsequent densitometric analysis of the acquired images was conducted as detailed in Grünewald et al.8 To rule out that the samples' PMIs negatively impacted on the abundance of the target proteins, we performed Spearman correlation analyses with PMI delay and mitochondrial protein levels as variables. This did not show a significant decline in protein levels as a result of higher PMI intervals.
Immunofluorescence Detection of Target Proteins in Frozen Brain Slices
In formalin‐fixed sections, the yield and quality of mtDNA are reduced to such an extent that molecular analyses, especially in single cells, are impeded. To simultaneously determine the cellular abundance of CI and the levels of mtDNA in single SN neurons from IPD patients and controls, we optimized our immunofluorescence protocol for use with frozen brain tissue. The previously employed anti‐porin and anti‐GRP75 antibodies showed nonspecific labeling in frozen midbrain sections and therefore could not be used. Furthermore, anti‐NDUFA13, which provided a stronger signal in frozen tissue, was preferred over anti‐NDUFB8 in this experiment. In view of the results from fixed tissue, we considered the mitochondrial mass unchanged in IPD patients and carried out immunofluorescence labeling to detect NDUFA13 only. Sections were dried for 60 minutes at RT, fixed in 4% (wt/vol) paraformaldehyde for 10 minutes, then washed in H2O for 10 minutes and in Tris‐buffered saline containing 3% Tween‐20 (TBST) for 5 minutes. Sections were blocked in TBST and 1% normal goat serum (NGS) for 1 hour at RT. Anti‐NDUFA13 was diluted at 1:100 in blocking solution, and slides were incubated overnight at 4 °C. Following the primary antibody incubation, sections were washed 3 times in TBST for 5 minutes. An anti‐mouse IgG2b secondary antibody (A‐21141; Life Technologies, Carlsbad, CA) was diluted at 1:100 in 1% NGS TBST and applied for 60 minutes at RT. Following 3 washes in TBST, sections were incubated in Sudan black solution for 10 minutes to quench autofluorescence. Finally, sections were washed in H2O for 10 minutes to remove Sudan black and air dried.
Mapping of SN Neurons for Isolation from Brain Slices by LCM
Following densitometric analysis of all neuromelanin‐positive SN neurons, neurons within a midbrain section were classified according to their absolute NDUFA13 levels. Ten neurons with high and 10 neurons with significantly (p < 0.0004) lower NDUFA13 levels but comparable neuromelanin content (as determined by densitometric analysis of the monochrome bright‐field image) were identified. These neurons were then mapped in a high‐resolution bright‐field image of the entire nigral region for each subject. The neurons of interest were located in the brain section, laser‐microdissected using a PALM MicroBeam (Zeiss), captured individually in 10 μl of lysis buffer, and incubated as previously described.14 This procedure was performed for 5 randomly selected IPD patients (compare in the Table: IPD2, IPD6, IPD7, IPD9, and IPD10).
Mitochondrial DNA Deletion, Copy Number, and Replication Analysis
A triplex real‐time PCR assay for simultaneous detection of 3 targets within the mitochondrial genome (ND1 [NADH dehydrogenase 1], ND4, and D‐loop) was performed.15 The quantification of ND1 and ND4 was conducted as previously published.16 For mtDNA copy number quantification, the ND1 concentration per unit area was calculated. To detect mtDNA major arc deletions, the ND1:ND4 was determined. The third target amplified in the triplex assay was located within an area of the noncoding region that is preserved in 99% of deleted mtDNA species reported to date.15, 17, 18 During transcription and replication, a linear DNA molecule, that is, 7S DNA, is incorporated in this particular region of the mitochondrial genome, resulting in a triple‐stranded structure denominated as “D‐loop.”17, 19 The D‐loop:ND1 ratio is therefore representative of the mtDNA replication status in a given cell, with a higher ratio representing more actively transcribed or replicating mtDNA molecules.20 Two microliters of cell lysates from individual SN neurons were used per real‐time PCR reaction, and all measurements were performed in triplicates. Standard curves were generated by serial dilution of a DNA plasmid containing a single copy of each mtDNA target.15
To explore a link between the mitochondrial genome and CI abundance at the single‐cell level, the mtDNA deletion levels, copy numbers, and replication efficiency were quantified in individually laser‐captured NDUFA13‐positive and NDUFA13‐negative patient SN neurons.
Moreover, to obtain a general picture of mtDNA integrity and copy number in patients and controls, pooled SN neurons from 10 controls and 9 IPD patients (frozen midbrain sections were unavailable for IPD5) were analyzed using the same real‐time PCR assay. Thirty randomly selected SN neurons containing comparable neuromelanin levels were isolated from each tissue section as described and captured in a single tube in 20 μl lysis buffer. The experiment was conducted on 3 separate occasions per subject.
Statistical Analysis
Densitometric analyses of individual SN neurons resulted in non‐normally distributed data in some cases. To account for this distribution, median values per person were calculated. In each run, the same control served as internal standard and its median abundance or median ratio of abundances was set to 100%. The differences between average patient and control group values in respect to protein abundances or mtDNA deletion levels, copy number, and mtDNA transcription/replication efficiency were assessed using unpaired 2‐sample t tests. To compare the ND1:area and D‐loop:ND1 ratios in individually laser‐captured CI‐deficient or CI‐normal neurons isolated from the same individual, paired 2‐sample t tests were employed. Finally, the Spearman rank correlation coefficient (r 2), associated p‐value, and frequency distributions were calculated to evaluate the relationship between different parameters in the study. For statistical analysis, Prism 6.0 software (GraphPad, San Diego, CA) was used.
Results
CI and CII Deficiency in Single Nigral Neurons from IPD Patients
IPD patient characteristics are presented in the Table. Cell size analysis, immunofluorescence detection, and densitometric quantification of mitochondrial protein levels were performed in individual SN neurons from 10 IPD patients and 10 age‐matched controls (Fig 1). For each investigated parameter, means per person were derived. Calculation of the mean cell area showed that patient neurons were significantly smaller than control neurons. By contrast, the assessment of the mitochondrial mass marker porin at the single‐neuron level indicated comparable mitochondrial densities in patients and controls.
Figure 1.
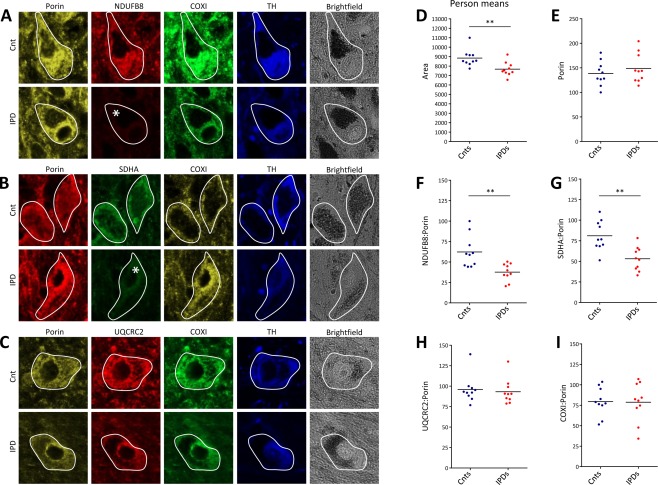
Abundances of subunits of respiratory chain complexes CI–IV and mitochondrial mass in dopaminergic substantia nigra (SN) neurons from idiopathic Parkinson disease (IPD) patients and controls (Cnt). (A–C) Epifluorescence microscopy after quadruple‐label immunofluorescence indicated reduced levels of the CI subunit NDUFB8 and the CII subunit SDHA in IPD patient neurons, whereas the CIII subunit UQCRC2, the CIV subunit COXI, and the mitochondrial mass marker porin were unchanged. The investigated neurons were tyrosine hydroxylase (TH) ‐positive and contained neuromelanin deposits indicative of their dopaminergic character. Deficiencies are marked by an asterisk. (D) Despite their significantly smaller size, (E) densitometric analysis of area‐normalized porin levels indicated unchanged mitochondrial mass in patient neurons. In agreement with the results from the visual inspection, the average (F) NDUFB8:porin and (G) SDHA:porin ratios in IPD patient neurons were significantly reduced. By contrast, (H) UQCRC2:porin and (I) COXI:porin ratios were comparable in control and patient SN neurons. Lines indicate mean group ratios. **p < 0.01. COXI = cytochrome c oxidase subunit I; NDUFB8 = NADH dehydrogenase 1 beta subcomplex 8; SDHA = succinate dehydrogenase complex subunit A; UQCRC2 = ubiquinol–cytochrome c reductase core protein II.
Next, we quantified the average neuronal protein abundance of the CI subunit NDUFB8 relative to mitochondrial mass (NDUFB8:porin ratio) in the patients and controls. In agreement with previous immunohistochemical studies,9, 21 we detected a significant reduction (60.5% of the control level) of NDUFB8 levels in the patient neurons (see Fig 1). Subsequently, we determined the abundance of the CII subunit SDHA. CII is the only complex of the electron transport chain entirely composed of nuclear‐encoded subunits. Quantification of SDHA:porin ratios in the SN neurons of all individuals showed a decrease in the amount of CII (65.5% of the control level) in patients. By contrast, assessment of the UQCRC2:porin ratio in individual neurons indicated no difference in the average CIII levels between patients and controls. Similarly, comparable CIV levels were found in neurons from patients and controls as quantified by the COXI:porin ratio.
Relationship of CI, CII, and CIV in Single IPD Patient Neurons
To correlate CI, CII, and CIV abundancies in single IPD patient and control neurons, we immunolabeled the CI subunit NDUFA13 along with SDHA (CII) and COXI (CIV). To simultaneously assess mitochondrial mass in neurons in this experimental setting, the anti‐porin antibody was replaced by an antibody against the outer mitochondrial membrane protein GRP75.22 Thus, antibody isotype mismatches between anti‐NDUFB8 and anti‐SDHA as well as between anti‐porin and anti‐NDUFA13 were circumvented. Results obtained using the anti‐GRP75 antibody confirmed that mitochondrial content is similar in patients and controls (Fig 2). Quantification of the mean ratios per person of NDUFA13:GRP75, SDHA:GRP75, and COXI:GRP75 also confirmed reduced neuronal CI and CII abundances as well as normal CIV levels in patients.
Figure 2.
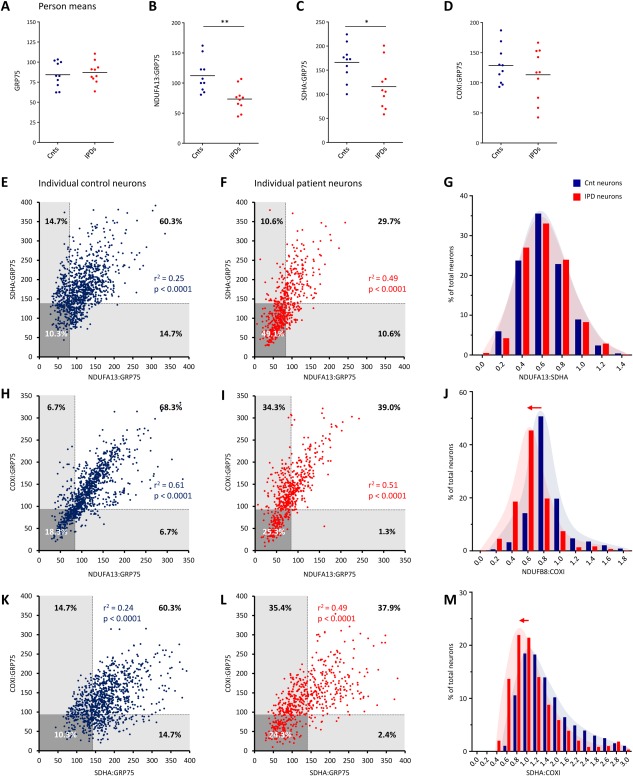
Dependencies between respiratory chain complexes CI, CII, and CIV in single control (Cnt) and idiopathic Parkinson disease (IPD) patient substantia nigra (SN) neurons. (A–D) Densitometric analysis of the mitochondrial mass marker GRP75, the CI subunit NDUFA13, the CII subunit SDHA, and the CIV subunit COXI in patient and control SN neurons. Calculation of mean neuronal protein abundances per person confirmed reduced levels of mitochondria‐normalized CI and CII in IPD patients. (E) Spearman correlation analysis of NDUFA13:GRP75 and SDHA:GRP75 ratios indicated a weak link between the protein levels of both complexes in individual control neurons. The lower 25th percentile of the control ranges for NDUFA13:GRP75 and SDHA:GRP75 were used to define thresholds of deficiency (broken lines and shaded areas). (F) In patient neurons, the association between NDUFA13:GRP75 and SDHA:GRP75 was increased. Applying the deficiency thresholds, 49.1% of cells lacked both NDUFA13 and SDHA. Equal proportions of cells (10.6%) showed either isolated NDUFA13 or SDHA deficiency. (G) This caused overlapping frequency distributions of NDUFA13:SDHA in patient and control cells. (H, I) Spearman correlation analysis of NDUFA13:GRP75 and COXI:GRP75 revealed coregulation of CI and CIV in individual control and patient neurons. About a quarter of patient cells (25.3%) were deficient for NDUFA13 and COXI. In 34.3% of all patient neurons, isolated NDUFA13 deficiency occurred, whereas isolated COXI deficiency was only present in 1.3% of cells. (J) An overall decrease in NDUFA13 resulted in a shift of the NDUFA13:COXI ratio in IPD patient neurons relative to the control population. (K) Spearman correlation analysis of the SDHA:GRP75 and COXI:GRP75 ratios indicated a weak dependency between CII and CIV in control cells. (L) In patient neurons, this coregulation increased as a result of the combined loss of CII and CIV in 24.3% of cells. A large proportion of patient SN neurons (35.4%) also showed isolated SDHA deficiency, (M) causing the frequency distribution of SDHA:COXI to shift toward lower SDHA levels. *p < 0.05, **p < 0.01. COXI = cytochrome c oxidase subunit I; GRP75 = heat shock 70kDa protein 9 (mortalin); NDUFA13 = NADH dehydrogenase 1 alpha subcomplex 13; r 2 = Spearman correlation coefficient; SDHA = succinate dehydrogenase complex subunit A. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Regarding the relationship of CI and CII, Spearman correlation analysis revealed that the variance of NDUFA13:GRP75 accounts for 25% of the variance of SDHA:GRP75 in single control neurons (r 2 = 0.25, p < 0.0001; see Fig 2). In patient neurons, this interdependency was increased to 49% (r 2 = 0.49, p < 0.0001). To explore the link between loss of CI and CII in IPD in more detail, we defined objective thresholds for mitochondria‐normalized NDUFA13 and SDHA. SN neurons that fell below the 25th percentile of the control range for NDUFA13:GRP75 or SDHA:GRP75 were considered to express low levels of the respective respiratory chain subunit and are denominated as “deficient” hereafter. Interestingly, 49.1% of all IPD patient SN neurons lacked both CI and CII. Patient cells lacking only CI (10.6%) or only CII (10.6%) were equally frequent, resulting in overlapping frequency distributions of NDUFA13:SDHA for patient and control neurons.
Because CIV deficiency has previously been implicated in IPD, we also wanted to explore the relationships between CIV and CI, as well as between CIV and CII at the single‐neuron level. In respect to CI and CIV, Spearman correlation analysis showed that 61% of the variance of NDUFA13:GRP75 was explained by the variance of COXI:GRP75 in single control neurons (r 2 = 0.61, p < 0.0001; see Fig 2). In patient neurons, this correlation was slightly reduced to 51% (r 2 = 0.51, p < 0.0001). Employing the above‐described strategy to establish deficiency cutoffs for NDUFA13:GRP75 and COXI:GRP75, 34.3% of patient neurons showed NDUFA13 deficiency without COXI involvement. About a quarter of the cells (25.3%) lacked both NDUFA13 and COXI. By contrast, neurons with isolated COXI deficiency were extremely rare (1.3%). In agreement with these observations, an overall shift of the NDUFB8:COXI ratio toward COXI was detected in IPD patient SN neurons. Further supporting a coregulation of CI and CIV, the average neuronal abundance of mitochondrial‐normalized NDUFB8 and COXI showed a similar rate of decline with age in controls (Fig 3).
Figure 3.
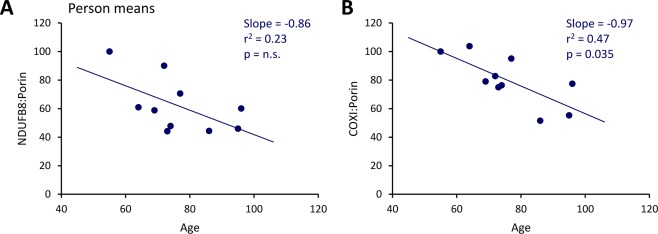
Correlation of respiratory chain complex abundances in substantia nigra neurons of controls with age. Plotting of the average neuronal NDUFB8:porin (A) and COXI:porin (B) against age at death revealed age‐associated declines. COXI = cytochrome c oxidase subunit I; NDUFB8 = NADH dehydrogenase 1 beta subcomplex 8; n.s. = not significant; r 2 = Spearman correlation coefficient. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Assessment of the relationship between CII and CIV (ie, SDHA:GRP75 and COXI:GRP75) indicated little correlation in control neurons (r 2 = 0.24, p < 0.0001; see Fig 2) but a markedly increased dependency in patient neurons (r 2 = 0.49, p < 0.0001), suggesting enhanced coregulation of CII and CIV in IPD patient neurons. The majority of deficient patient neurons lacked either SDHA and COXI (24.3%) or SDHA alone (35.4%). By contrast, isolated COXI deficiency was rare (2.4%), resulting in a shift of the SDHA:COXI ratio toward COXI.
CI Deficiency Coincides with Loss of TFAM and TFB2M
Next, we explored whether the loss of the partially mtDNA‐encoded CI is associated with impaired mtDNA transcription in IPD. The expression of mtDNA‐encoded subunits of the respiratory chain is controlled by TFAM.23, 24 TFAM protein abundance was quantified along with NDUFB8 and porin in the same cells. The average neuronal TFAM:porin ratio per person indicated significantly (p = 0.02) reduced protein levels of the transcription factor in IPD patients (64.0% of the control level) compared to controls (Fig 4A, B). In addition, the NDUFB8:porin and TFAM:porin ratios in single neurons were strongly correlated for both patients (r 2 = 0.74, p < 0.0001) and controls (r 2 = 0.72, p < 0.0001). The relative frequency of neurons with different NDUFB8:TFAM ratios was comparable in patients and controls, as indicated by overlapping frequency distributions (see Fig 4C).
Figure 4.
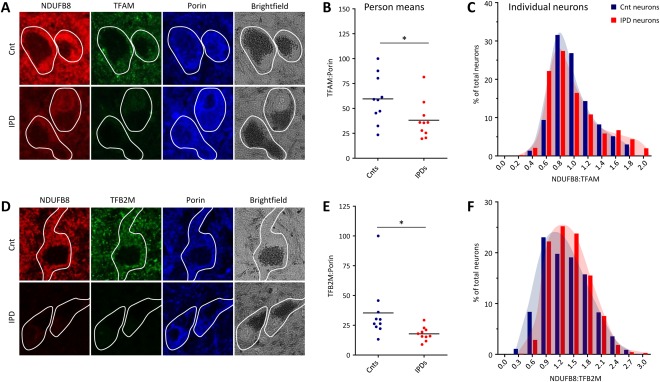
Abundance of mtDNA‐associated TFAM and TFB2M in patient and control (Cnt) substantia nigra neurons. (A) Epifluorescence microscopy after triple‐label immunofluorescence indicated joint loss of NDUFB8 and TFAM in idiopathic Parkinson disease (IPD) patients, whereas porin staining showed normal mitochondrial mass. (B) Densitometric analysis of investigated proteins confirmed significantly reduced TFAM:porin ratios in IPD patients. Lines represent mean group ratios. (C) In patients and controls, NDUFB8 and TFAM strongly correlated at the single‐neuron level, resulting in similar behaviors of the frequency histograms for NDUFB8:TFAM. (D) Triple‐label immunohistochemistry showed reduced abundances of NDUFB8 and TFB2M but normal porin levels in IPD neurons. (E) Densitometric analysis of average neuronal TFB2M and porin levels in patients and controls revealed a significant reduction of TFB2M:porin in the former. (F) The frequency distribution of the NDUFB8:TFB2M ratio in single neurons showed a slight shift toward lower TFB2M in patient cells. *p < 0.05. NDUFB8 = NADH dehydrogenase 1 beta subcomplex 8; TFAM = mitochondrial transcription factor A; TFB2M = mitochondrial transcription factor B2.
To further ascertain a possible link between mtDNA transcription and CI deficiency in IPD, the protein levels of another mitochondrial transcription factor, TFB2M,25 were assessed. As for TFAM, the average neuronal TFB2M:porin ratio per person was significantly (p = 0.04) reduced in IPD patients (50.3% of the control level) compared to controls (see Fig 4D, E). Furthermore, mitochondria‐normalized NDUFB8 and TFB2M were moderately and equally correlated in individual IPD patient (r 2 = 0.57, p < 0.0001) and control (r 2 = 0.54, p < 0.0001) SN neurons, resulting in similar frequency distributions of NDUFB8:TFB2M (see Fig 4F).
mtDNA Depletion in IPD Patient Neurons
In addition to their role as transcription factors, TFAM and TFB2M are involved in mitochondrial genome packaging26, 27 and replication.23, 28 Consequently, we sought to test whether loss of either factor is associated with mtDNA copy number and stability in SN neurons of IPD patients.
Real‐time PCR quantification of the mitochondrial copy number (ND1:area) in pooled SN neurons from controls and patients showed significantly (p = 0.026) fewer mtDNA copies in IPD patient cells (73.1% of the control level; Fig 5A). In addition, using our triplex real‐time PCR assay, we measured the levels of 7S DNA, which associates with the mitochondrial genome in the D‐loop region in a transcription/replication‐dependent manner. The D‐loop:ND1 ratio indicated significantly (p = 0.004) fewer 7S DNA molecules per mitochondrial genome in patient neurons (90.9% of the control level; see Fig 5B), linking mtDNA depletion to impaired mtDNA replication in IPD. Of note, the prevalence of mtDNA deletions in pooled neurons did not differ between patients and controls (data not shown).
Figure 5.
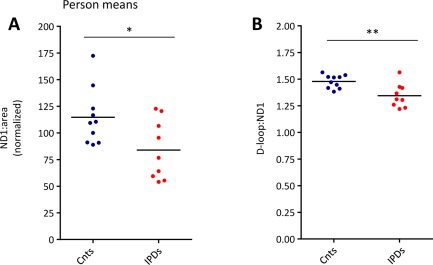
mtDNA analysis in pooled substantia nigra neurons isolated from control (Cnt) and idiopathic Parkinson disease (IPD) patient midbrain sections. Simultaneous real‐time polymerase chain reaction quantification of mtDNA fragments in ND1 and the D‐loop in 30 neurons per person and experiment from 10 controls and 9 IPD patients are shown. This analysis indicated significantly reduced (A) mtDNA copy numbers (ND1:area) and (B) D‐loop:ND1 ratios in IPD patients compared to controls. *p < 0.05, **p < 0.01. ND1 = NADH dehydrogenase 1. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
MtDNA Depletion Correlates with CI Deficiency
To explore whether the observed mtDNA depletion in IPD patient SN neurons correlates with CI deficiency at the single‐cell level, we isolated individual NDUFA13‐positive and NDUFA13‐negative neurons from 5 IPD patients. Real‐time PCR analysis revealed significantly (p = 0.041) lower mtDNA copy numbers (ND1:area) in NDUFA13‐deficient neurons (Fig 6). On average, NDUFA13‐negative patient neurons harbored 29.4% fewer mtDNA copies per cell area than NDUFA13‐positive patient neurons. Consistent with this result, quantification of mtDNA‐associated 7S DNA indicated significantly (p = 0.014) fewer mtDNA transcription/replication events in NDUFA13‐negative IPD patient neurons. By contrast, mtDNA major arc deletions did not correlate with CI abundance in individually laser‐captured patient neurons (data not shown).
Figure 6.
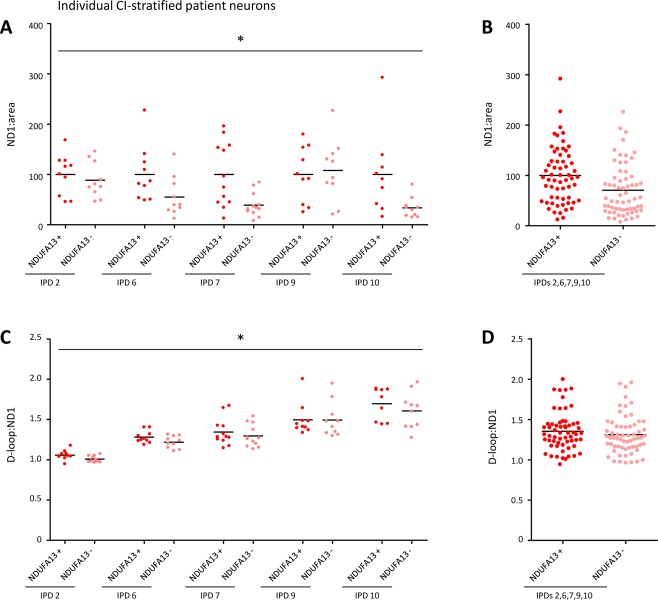
mtDNA real‐time polymerase chain reaction analysis in individually isolated patient substantia nigra neurons with high or low CI abundance. (A) Determination of the ND1:area ratio in single NDUFA13‐positive and NDUFA13‐negative neurons from 5 idiopathic Parkinson disease (IPD) patients showed significantly reduced mtDNA copy numbers in the latter cells. (B) On average, NDUFA13‐negative patient neurons contained 29.4% fewer mtDNA copies than NDUFA13‐positive patient neurons. (C, D) Impaired mtDNA transcription/replication was indicated by a significant reduction in the D‐loop:ND1 ratio in NDUFA13‐negative neurons of all patients. *p < 0.05. ND1 = NADH dehydrogenase 1; NDUFA13 = NADH dehydrogenase 1 alpha subcomplex 13. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Discussion
Applying quadruple immunofluorescence to postmortem SN sections, we performed a detailed single‐neuron level characterization of respiratory chain deficiency in IPD. This high‐resolution quantitative approach enabled us to evaluate the relationship between the mitochondria‐normalized abundances of respiratory chain CI–IV. First, we confirmed experimental results from the late 1980s indicating CI (nicotinamide adenine dinucleotide ubiquinone oxidoreductase) deficiency in SN homogenates in IPD.4 In our hands, this deficiency in individual IPD SN neurons corresponded to a loss of CI (NDUFB8 and NDUFA13) protein levels normalized for mitochondrial mass. We furthered our analysis by quantifying the abundance of a subunit of CII (SDHA), which represents the only respiratory chain complex entirely composed of nuclear‐encoded proteins. To our surprise, IPD patient SN neurons contained significantly lower levels of SDHA, suggesting an involvement of both nuclear and mitochondrial factors in IPD pathogenesis. By contrast, the average protein levels of subunits of respiratory chain CIII (UQCRC2) and CIV (COXI) were comparable in nigral neurons of IPD patients and aged controls. CI and CII deficiencies occurred in the absence of changes in neuronal mitochondrial content, thus implicating specific molecular mechanisms that result in qualitative mitochondrial respiratory chain deficiency.
In addition to the well‐established CI deficit, also CIV dysfunction has previously been implicated in the pathogenesis of IPD.6, 7 Benefiting from the high specificity and resolution of our single‐cell immunofluorescence approach, we studied the quantitative relationships between CI, CII, and CIV as an index of their coregulation in SN neurons. In agreement with the common dependency of CI and CIV assembly and function on the mitochondrial genome,29 NDUFA13 and COXI abundances were strongly correlated in single control and patient neurons. The majority of dysfunctional patient SN neurons demonstrated either isolated CI, or combined CI and CIV deficiencies, with few cells exhibiting selective loss of CIV. From this observation, we deduced that CIV deficiency in nigral neurons only occurs in the presence of CI deficiency in IPD. This molecular signature may arise from mtDNA alterations as discussed below.
CI and CII as well as CIV and CII were loosely linked in control neurons. However, in patient SN neurons, the associations between NDUFA13 and SDHA as well as between COXI and SDHA increased to the level of coregulation of NDUFA13 and COXI. The joint occurrence of CI, CII, and CIV deficiencies in IPD patient neurons (despite none of the subunits of CII being encoded by the mitochondrial genome) suggested that nuclear signaling pathways, such as the nuclear respiratory factor (NRF)1/2 pathway, are involved in the loss of respiratory chain complexes. The NRF1/2 pathway30, 31 not only controls the transcription of all CII subunits (and of some subunits of CI and CIV) but also directly regulates the mRNA expression of the mtDNA transcription factors TFAM24 and TFB2M.28
One reason why CII deficiency was not detected in a previous study on postmortem midbrain sections from the Newcastle PD cohort may be different methodology.6 Whereas our new image analysis method allows detection of subtle changes in protein levels within each tested neuron in a quantitative manner, previously applied visual classification schemes distinguished only between enzymatically completely SDH‐negative and ‐positive neurons. The same holds true for COXI deficiency in single neurons. Employing quantitative immunofluorescence, we detected subtle CIV deficits in SN neurons of aged controls that appear to even out the previously observed small shift toward CIV deficiency in the PD patients.6
To test the potential role of nuclear signaling in IPD further, we investigated the abundance of TFAM and TFB2M in nigral neurons from patients and controls. Both proteins correlated with CI levels at the single‐cell level and were reduced in patient tissue, implicating downregulated transcription of mtDNA genes as a potential factor in the development of CI deficiency in IPD. However, both TFAM and TFB2M are also required for transcription‐primed replication of the mitochondrial genome,24, 28 opening the possibility that mtDNA depletion contributes to respiratory chain deficiency in IPD.
To explore this hypothesis, we performed real‐time PCR analysis of the mitochondrial genome in pooled patient and control SN neurons. Patient neurons contained fewer mtDNA copies and less mtDNA replication/transcription‐associated 7S DNA than control cells. Further linking mtDNA depletion to loss of CI in IPD, individually isolated CI‐deficient patient neurons contained even fewer mtDNA copies and 7S molecules than their counterparts with normal CI levels. In keeping with these findings, increased exposure of the mitochondrial genome to TFAM has been reported to enhance 7S DNA association and mtDNA replication in an in organello DNA synthesis system.32 In line with our hypothesis stating that TFAM deficiency induces mtDNA depletion in IPD, conditional silencing of TFAM provokes PD‐like features such as respiratory chain dysfunction, degeneration of dopaminergic SN neurons, and progressive impairment of motor function in mice.33, 34 Remarkably, in addition to its effect as a CI inhibitor, the PD‐inducing reagent 1 methyl‐4‐phenylpyridinium also interferes with mtDNA replication by destabilization of the mitochondrial D‐loop.35, 36 Hence, our molecular results converge with these findings to indicate that toxin‐induced parkinsonism and the actual movement disorder may share a common molecular origin, namely disrupted mtDNA transcription and replication.
It is curious why CI and CII are seemingly more affected by impaired nuclear transcription activation and mtDNA depletion in IPD SN neurons than CIII and CIV. This discrepancy, however, was previously observed in a histopathology study on Alper syndrome, a primary mtDNA replication defect. In these individuals with a mutation in the mtDNA polymerase gamma (POLG) gene, mtDNA depletion resulted in isolated CI (but not CIV) deficiency.37 Preferential degradation of CI in Alper syndrome and of CI and CII in IPD may be due to lower total abundances of these complexes in respect to CIII and CIV. The stoichiometric ratios of the respiratory chain complexes have been measured as CI, 0.8–1.3; CII, 1.2–1.4; CIII, 3; and CIV, 6.0–7.5 in bovine heart mitochondria.38 Thus, CIV may exist in excess, whereas CI levels may be limiting. Moreover, the quantification of respiratory chain protein turnover rates in mice showed that the subunits of CII have the shortest average half‐life, followed by the components of CI and CIV, whereas the components of CIII have the longest half‐life.39 The high stability of the CIII subunits potentially explains why this complex behaves differently from CI and CII, despite relatively similar stoichiometric proportions. An alternative scenario underlying our immunohistochemical and real‐time PCR results may be that SN neurons lacking functional CIII and CIV (and harboring deletions) are more prone to degeneration and are thus undetectable in postmortem tissue. This would lead to a selection bias where CIII and CIV deficiencies would escape our analysis, yielding the current histopathological picture where CI and CII deficiencies predominate.
Whereas our study exclusively focused on respiratory chain deficiencies in dopaminergic neurons of the SN, it would be interesting to explore whether the described respiratory chain and mtDNA phenotypes extend to other areas of the brain known to be affected in the early and late phases of IPD. According to the staging scheme by Braak and colleagues,40 neuronal pathology in IPD evolves from the olfactory bulb and the dorsal motor nucleus via the locus ceruleus and the SN to the basal forebrain and amygdala, finally reaching the medial temporal lobe structures and the cerebral cortex.40, 41 Single‐cell analyses in these specific brain regions would help to further elucidate the contribution of mitochondrial dysfunction to disease progression.
In summary, our findings link respiratory chain deficiencies to both nDNA and mtDNA elements in nigral neurons of IPD patients. Possibly as a consequence of impaired NRF1/2 signaling, the transcription of nuclear‐encoded subunits of the respiratory chain complexes and of the mitochondrial transcription factors TFAM and TFB2M is compromised in IPD. Lack of TFAM packaging of the mitochondrial genome causes D‐loop destabilization and hampers mtDNA transcription and replication. In turn, a reduction in mtDNA copy number and transcripts potentiates respiratory chain dysfunction, which is primarily reflected in the loss of CI. By contrast, CIV deficiency appears only together with CI deficiency, possibly representing a later phase of respiratory chain impairment that may precipitate neuronal loss. Although mtDNA deletions have previously been associated with CIV deficiency in IPD, our results reveal that quantitative differences in mtDNA abundance and replication more specifically discriminate between CI‐normal and CI‐deficient SN neurons. Our detailed single‐cell analysis of respiratory chain proteins further expands the mitochondrial phenotype of IPD to include CII deficiency, which largely correlates with the loss of CI. The heretofore‐suspected predominant defect of CI is likely only the most pronounced sign of a manifold picture of respiratory chain complex deficiencies arising from impaired mitochondrial–nuclear signaling in IPD.
Author Contributions
Concept and study design: A.G., K.A.R., D.M.T. Data acquisition and analysis: A.G., P.D.H., M.P., C.M.M. Drafting the manuscript and figures: A.G., K.A.R., M.P., D.M.T.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
This work was funded by the Newcastle University Centre for Brain Ageing and Vitality (supported by the Biotechnology and Biological Sciences Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council, and Medical Research Council [MRC; G0700718]), the Wellcome Trust Centre for Mitochondrial Research (G906919), the MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK; G0800674), the UK National Institute for Health Research (NIHR) Biomedical Research Centre in Age and Age Related Diseases award to the Newcastle upon Tyne Hospitals National Health Service (NHS) Foundation Trust, and the UK NHS Specialist Commissioners “Rare Mitochondrial Disorders of Adults and Children” Service. Additional funding was received from the German Research Foundation (A.G.; GR 3731/2‐1 and GR 3731/2‐2) and the Luxembourg National Research Funds (A.G.; ATTRACT fellowship). Tissue for this study was provided by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK MRC (G0400074) and the Alzheimer's Society and Alzheimer's Research Trust through the Brains for Dementia Research Initiative, the NIHR Biomedical Research Centre Grant in Ageing and Health, and the NIHR Biomedical Research Unit in Lewy Body Dementia.
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
References
- 1. de Rijk MC, Tzourio C, Breteler MM, et al. Prevalence of parkinsonism and Parkinson's disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson's Disease. J Neurol Neurosurg Psychiatry 1997;62:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schapira AH. Mitochondrial pathology in Parkinson's disease. Mt Sinai J Med 2011;78:872–881. [DOI] [PubMed] [Google Scholar]
- 3. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine‐analog synthesis. Science 1983;219:979–980. [DOI] [PubMed] [Google Scholar]
- 4. Schapira AH, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989;1:1269. [DOI] [PubMed] [Google Scholar]
- 5. Kumar KR, Djarmati‐Westenberger A, Grunewald A. Genetics of Parkinson's disease. Semin Neurol 2011;31:433–440. [DOI] [PubMed] [Google Scholar]
- 6. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006;38:515–517. [DOI] [PubMed] [Google Scholar]
- 7. Kraytsberg Y, Kudryavtseva E, McKee AC, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 2006;38:518–520. [DOI] [PubMed] [Google Scholar]
- 8. Grünewald A, Lax NZ, Rocha MC, et al. Quantitative quadruple‐label immunofluorescence of mitochondrial and cytoplasmic proteins in single neurons from human midbrain tissue. J Neurosci Methods 2014;232:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reeve AK, Park TK, Jaros E, et al. Relationship between mitochondria and alpha‐synuclein: a study of single substantia nigra neurons. Arch Neurol 2012;69:385–393. [DOI] [PubMed] [Google Scholar]
- 10. Lax NZ, Grady J, Laude A, et al. Extensive respiratory chain defects in inhibitory interneurons in patients with mitochondrial disease. Neuropathol Appl Neurobiol. 2015 Mar 18. doi:10.1111/nan.12238. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reeve A, Meagher M, Lax N, et al. The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J Neurosci 2013;33:10790–10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hiniker A, Wong LJ, Berven S, et al. Axial mitochondrial myopathy in a patient with rapidly progressive adult‐onset scoliosis. Acta Neuropathol Commun 2014;2:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arthur CR, Morton SL, Dunham LD, et al. Parkinson's disease brain mitochondria have impaired respirasome assembly, age‐related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol Neurodegener 2009;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murphy JL, Blakely EL, Schaefer AM, et al. Resistance training in patients with single, large‐scale deletions of mitochondrial DNA. Brain 2008;131:2832–2840. [DOI] [PubMed] [Google Scholar]
- 15. Rygiel KA, Grady JP, Taylor RW, et al. Triplex real‐time PCR—an improved method to detect a wide spectrum of mitochondrial DNA deletions in single cells. Sci Rep 2015;5:9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krishnan KJ, Bender A, Taylor RW, Turnbull DM. A multiplexreal‐time PCR method to detect and quantify mitochondrial DNA deletions in individual cells. Anal Biochem 2007;370:127–129. [DOI] [PubMed] [Google Scholar]
- 17. Nicholls TJ, Minczuk M. In D‐loop: 40 years of mitochondrial 7S DNA. Exp Gerontol 2014;56:175–181. [DOI] [PubMed] [Google Scholar]
- 18. Damas J, Samuels DC, Carneiro J, et al. Mitochondrial DNA rearrangements in health and disease—a comprehensive study. Hum Mutat 2014;35:1–14. [DOI] [PubMed] [Google Scholar]
- 19. Clayton DA. Transcription and replication of mitochondrial DNA. Hum Reprod 2000;15(suppl 2):11–17. [DOI] [PubMed] [Google Scholar]
- 20. Brown TA, Clayton DA. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′‐dideoxycytidine. Nucleic Acids Res 2002;30:2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hattori N, Tanaka M, Ozawa T, Mizuno Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson's disease. Ann Neurol 1991;30:563–571. [DOI] [PubMed] [Google Scholar]
- 22. Rakovic A, Grünewald A, Seibler P, et al. Effect of endogenous mutant and wild‐type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum Mol Genet 2010;19:3124–3137. [DOI] [PubMed] [Google Scholar]
- 23. Campbell CT, Kolesar JE, Kaufman BA. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 2012;1819:921–929. [DOI] [PubMed] [Google Scholar]
- 24. Ekstrand MI, Falkenberg M, Rantanen A, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 2004;13:935–944. [DOI] [PubMed] [Google Scholar]
- 25. Ringel R, Sologub M, Morozov YI, et al. Structure of human mitochondrial RNA polymerase. Nature 2011;478:269–273. [DOI] [PubMed] [Google Scholar]
- 26. Hallberg BM, Larsson NG. TFAM forces mtDNA to make a U‐turn. Nat Struct Mol Biol 2011;18:1179–1181. [DOI] [PubMed] [Google Scholar]
- 27. Ngo HB, Lovely GA, Phillips R, Chan DC. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat Commun 2014;5:3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cotney J, Wang Z, Shadel GS. Relative abundance of the human mitochondrial transcription system and distinct roles for h‐mtTFB1 and h‐mtTFB2 in mitochondrial biogenesis and gene expression. Nucleic Acids Res 2007;35:4042–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bellance N, Lestienne P, Rossignol R. Mitochondria: from bioenergetics to the metabolic regulation of carcinogenesis. Front Biosci (Landmark Ed) 2009;14:4015–4034. [DOI] [PubMed] [Google Scholar]
- 30. Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF‐1 and NRF‐2) and PGC‐1 family coactivators. Mol Cell Biol 2005;25:1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta 2009;1793:1540–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gensler S, Weber K, Schmitt WE, et al. Mechanism of mammalian mitochondrial DNA replication: import of mitochondrial transcription factor A into isolated mitochondria stimulates 7S DNA synthesis. Nucleic Acids Res 2001;29:3657–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Galter D, Pernold K, Yoshitake T, et al. MitoPark mice mirror the slow progression of key symptoms and L‐DOPA response in Parkinson's disease. Genes Brain Behav 2010;9:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ekstrand MI, Terzioglu M, Galter D, et al. Progressive parkinsonism in mice with respiratory‐chain‐deficient dopamine neurons. Proc Natl Acad Sci U S A 2007;104:1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Umeda S, Muta T, Ohsato T, et al. The D‐loop structure of human mtDNA is destabilized directly by 1‐methyl‐4‐phenylpyridinium ion (MPP+), a parkinsonism‐causing toxin. Eur J Biochem 2000;267:200–206. [DOI] [PubMed] [Google Scholar]
- 36. Iwaasa M, Umeda S, Ohsato T, et al. 1‐Methyl‐4‐phenylpyridinium ion, a toxin that can cause parkinsonism, alters branched structures of DNA. J Neurochem 2002;82:30–37. [DOI] [PubMed] [Google Scholar]
- 37. Tzoulis C, Tran GT, Coxhead J, et al. Molecular pathogenesis of polymerase gamma‐related neurodegeneration. Ann Neurol 2014;76:66–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schagger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I‐V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem 2001;276:37861–37867. [DOI] [PubMed] [Google Scholar]
- 39. Karunadharma PP, Basisty N, Chiao YA, et al. Respiratory chain protein turnover rates in mice are highly heterogeneous but strikingly conserved across tissues, ages, and treatments. FASEB J 2015;29:3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Braak H, Ghebremedhin E, Rub U, et al. Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res 2004;318:121–134. [DOI] [PubMed] [Google Scholar]
- 41. Dickson DW. Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]