

Abstract
Our aim was to study whether the aberrant amount or function of regulatory T cells is related to the development of type 1 diabetes (T1D) in children. We also set out to investigate the balance of different T cell subtype markers during the T1D autoimmune process. Treg cells were quantified with flow cytometric assay, and the suppression capacity was analysed with a carboxyfluorescein succinimidyl ester (CFSE)‐based T cell suppression assay in children in various phases of T1D disease process and in healthy autoantibody‐negative control children. The mRNA expression of different T cell subpopulation markers was analysed with real‐time qPCR method. The proportion and suppression capacity of regulatory T cells were similar in seroconverted children at an early stage of beta cell autoimmunity and also in children with T1D when compared to healthy and autoantibody‐negative children. Significant differences were observed in the mRNA expression of different T cell subpopulation markers in prediabetic children with multiple (≥2) autoantibodies and in children with newly diagnosed T1D when compared to the control children. In conclusion, there were no quantitative or functional differences in regulatory T cells between the case and control groups in any phase of the autoimmune process. Decreased mRNA expression levels of T cell subtype markers were observed in children with multiple islet autoantibodies and in those with newly diagnosed T1D, probably reflecting an exhaustion of the immune system after the strong immune activation during the autoimmune process or a generally aberrant immune response related to the progression of the disease.
Introduction
Type 1 diabetes (T1D) is a severe and chronic autoimmune disease which originates from the combination of genetic susceptibilities and environmental factors. Seroconversion to positivity for beta cell‐specific autoantibodies, such as IAA, GADA, IA‐2A and ZnT8A, strongly predicts progression to overt type 1 diabetes 1. However, it is generally accepted that autoreactive CD4+ and CD8+ T cells mediate the actual destruction of the insulin‐producing beta cells in the pancreatic islets 2.
According to the Th1/Th2 paradigm, Th1 cells are the most important players in many organ‐specific autoimmune diseases, while Th2 effector cells are perceived to have a protective role in autoimmunity 3. It has become clear that this Th1/Th2 paradigm is insufficient to explain all the immunopathology of different autoimmune diseases such as type 1 diabetes 4. The third member of effector T cells is the more recently found Th17 subset which secrete mainly pro‐inflammatory IL‐17 cytokine. Th17 cells play a role in the cell destruction at least in some of the autoimmune diseases, and it is possible that Th1 and Th17 cells are functioning at different phases of the autoimmune process 5, 6.
The most important immune regulators are CD4+, CD25+ Treg cells, which differentiate naturally in thymus (nTreg) or after induction in peripheral tissues (iTreg) 7. Treg cells suppress the proliferation and function of autoreactive Teff cells (self‐tolerance) and maintain the overall balance of the immune system. FOXP3 transcription factor is considered as a specific marker and the main regulator for the differentiation and function of Treg cells 8, 9. In humans, the mutation in the FOXP3 gene is known to cause the X‐linked syndrome (IPEX) characterized by immune dysregulation, polyendocrinopathy, enteropathy and X‐linked inheritance 10. The suppressive mechanisms of the Treg cells are still quite poorly defined, but it is likely that different cytokines (TGF‐β and IL‐10) and cell–cell contacts play a significant role in the suppression 11.
Multiple mechanisms may lead to the loss of self‐tolerance in autoimmunity 12. Defects in Treg number and/or function have been implicated in the development of different autoimmune diseases such as type 1 diabetes 13, 14, 15. However, there are also contrasting results 16, 17, 18. Some studies have shown increased Teff resistance to Treg suppression, which may lead to the defective regulation in type 1 diabetes 19, 20. Homeostatic cytokines affect markedly the balance between Teffs and Tregs and are necessary to maintain peripheral self‐tolerance 21, 22.
Most of the previous Treg analyses in type 1 diabetes have been performed after the clinical diagnosis of T1D. The aim of this study was to investigate whether there are differences in regulatory T cells or T cell subpopulation markers between children with type 1 diabetes‐associated autoimmunity, analysed from the early preclinical phase to long‐lasting type 1 diabetes and healthy controls.
Methods
Subjects
The study population includes children aged 0–15 years from various stages of type 1 diabetes autoimmunity, who were divided into four different case groups (Table 1). The first case group comprises 20 healthy children who had recently seroconverted to positivity for one beta cell‐specific autoantibody with the mean age of 0.5 years when first positive for islet autoantibodies (range 0.0–1.0 years). The second case group includes 22 children testing positive for multiple autoantibodies. These two case groups were recruited from the prospective Type 1 Diabetes Prediction and Prevention (DIPP) study at the Department of Paediatrics, Oulu University Hospital. The third case group consists of a total of 30 children with newly diagnosed type 1 diabetes, time from diagnosis ranging between 5–17 days at sample collection. The fourth case group includes 22 patients with long‐lasting type 1 diabetes (mean disease duration 6.9 years, range 2.8–13.1 years). Children in the latter two case groups were recruited from the Diabetes Clinic at the Department of Paediatrics, Oulu University Hospital. The control children selected from the DIPP study for each of the case groups were age‐, sex‐ and sampling date‐matched healthy and autoantibody‐negative children carrying HLA‐DQB1 risk‐associated genotypes (control groups 1–4). There were altogether 55 control children. Thirty controls were included in more than one control group. However, the controls were never used as a duplicate in the same control group (Table 1).
Table 1.
Demographic and laboratory data of the case and control groups
| Number of cases | Male (n) | Female (n) | Age at sampling/years (range) | Plasma glucose at sampling mmol/l (range) | HbA1c at sampling mmol/mol (range) | HbA1c at sampling % (range) | Average time since detected positive for one autoantibody/years (range) | Average time after T1D diagnosis/years (range) | |
|---|---|---|---|---|---|---|---|---|---|
| Case group 1 (recently seroconverted children) | 20 | 9 | 11 | 7.4 (0.6–15.4) | 5.1 (4.2–6.4) | 31.8 (27–38) | 5.1 (4.6–5.6) | 0.5 (0.0–1.0) | NA |
| Control group 1 | 20 | 9 | 11 | 7.7 (1.5–15.4) | 5.1 (4.3–6.6) | 35.4 (30–42) | 5.4 (4.9–6.0) | NA | NA |
| Case group 2 (children with multiple AAbs) | 22 | 9 | 13 | 9.7 (2.4–14.7) | 5.2 (4.5–6.0) | 36.6 (30–44) | 5.5 (4.9–6.2) | 5.8 (0.6–14.0) | NA |
| Control group 2 | 22 | 9 | 13 | 9.7 (2.8–15.4) | 4.9 (3.5–6.6) | 36.8 (30–42) | 5.5 (4.9–6.0) | NA | NA |
| Case group 3 (children with newly diagnosed type 1 diabetes) | 30 | 14 | 16 | 7.6 (0.8–15.7) | 11.0 (3.4–21.5) | 85.4 (51–149) | 10.0 (6.8–15.8) | NA | 0.02 (0.02–0.05) |
| Control group 3 | 30 | 14 | 16 | 7.8 (1.5–15.0) | 4.9 (4.2–6.2) | 35.5 (29–42) | 5.4 (4.8–6.0) | NA | NA |
| Case group 4 (children with long‐term type 1 diabetes) | 22 | 8 | 14 | 11.5 (7.6–15.0) | 10.3 (2.7–15.7) | 69.8 (47–97) | 8.5 (6.4–11.0) | NA | 6.9 (2.8–13.1) |
| Control group 4 | 22 | 8 | 14 | 11.6 (8.1–15.4) | 5.0 (3.8–6.6) | 34.8 (30–42) | 5.3 (4.9–6.0) | NA | NA |
NA = not applicable.
Quantitation of the regulatory T cells with the flow cytometric assay
Peripheral blood samples were collected in BD Vacutainer Cell Preparation Tubes (CPT). Peripheral blood mononuclear cells (PBMCs) were freshly isolated and frozen at −70 °C. For the flow cytometry analysis, stored PBMCs were thawed in +37 °C water bath. Next the cells were permeabilized and fixed with FOXP3 fix/perm kit (eBiosciences) according to the manufacturer's instructions. Non‐specific interactions between TCR and fluorochromes were blocked by incubation for 20 min with human IgG. Finally, the cells were stained with PerCP‐CD4, APC‐CD25 (BD Pharmingen, San Diego, CA) and Alexa Fluor 488‐FOXP3 (eBiosciences) antibodies and analysed with BD LSRFortessa. Representative plots of unlabelled and labelled samples from the FACS analysis and gating information are presented in Fig. 1A, B.
Figure 1.
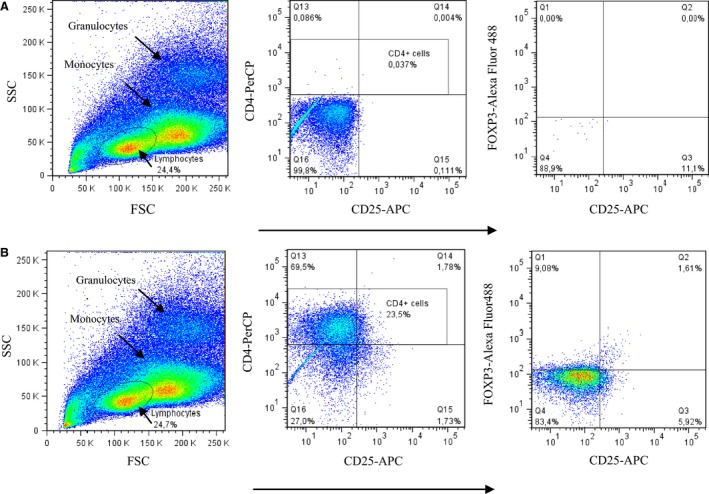
Representative plots and the gating information from the Treg FACS analysis. Lymphocytes were first gated from peripheral blood mononuclear cells (left). Second, the CD4+ cells were selected (middle). Third, FOXP3+ CD25+ cells were visualized and considered as regulatory T cells (gate Q2 on the right plot). A: An unlabelled sample represents a negative control. B: A sample after labelling the PBMCs with PerCP‐CD4, Alexa Fluor 488‐FOXP3 and APC‐CD25. The samples were measured with BD LSRFortessa and then analysed with FlowJo cell analysis software. FSC stands for forward scatter and SSC stands for side scatter in the flow cytometry analysis.
Suppression assay for regulatory T cells
CD4+ cells were first enriched from fresh peripheral blood samples collected in BD Vacutainer Cell Preparation Tubes (CPT). The enrichment of CD4+ cells was performed <2 h after venipuncture with Human CD4 + T Cell Enrichment Cocktail (Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer's instructions. Isolated CD4+ cells were cultured in RPMI‐1640 medium containing 1 mmol/l L‐glutamine, 5 mmol/l HEPES, 5% male human AB serum‐inactivated for 20 min at 45 °C (Lonza) and 1% penicillin/streptomycin overnight at 37 °C and 5% CO2. On the next day, the cells were suspended in 1xPBS–0.1% human AB serum buffer and stained with PerCP‐CD4, PE‐CD127 and APC‐CD25 (BD Pharmingen, San Diego, CA) antibodies. After the labelling, CD4+ cells were sorted for CD4+CD25+CD127−/low T cell (Treg) and CD4+CD25−CD127+ T cell (Teff) fractions with BD FACSAria II. Sorted Teff cells were labelled with 2 mmol/l carboxyfluorescein diacetate succinimidyl ester (CFSE) for 7 min at 37 °C and 5% CO2. After the labelling, the Teff cells were centrifuged and resuspended in the cell culture medium. CFSE was inactivated by 15‐min incubation at 37 °C and 5% CO2. For the suppression assay, Teff cells were cultured in U‐bottom 96‐well plates as a single culture and also with Treg cells as a co‐culture (Treg:Teff, 1:2 ratio) and stimulated with anti‐CD3/CD28 antigen‐coated beads (1:20 ratio). As a control, Teff and Treg cells were also co‐cultured without any stimulating agent. After 6 days, the cultured cells were stained with PE‐CD4 (BD Pharmingen, San Diego, CA) to analyse the proliferation of CFSE‐labelled Teff cells with BD LSRFortessa flow cytometer. The suppression percentage was calculated with the following formula: 100−[(% proliferation in the presence of Treg/% proliferation in the absence of Treg) × 100] (Fig. 2) 23.
Figure 2.
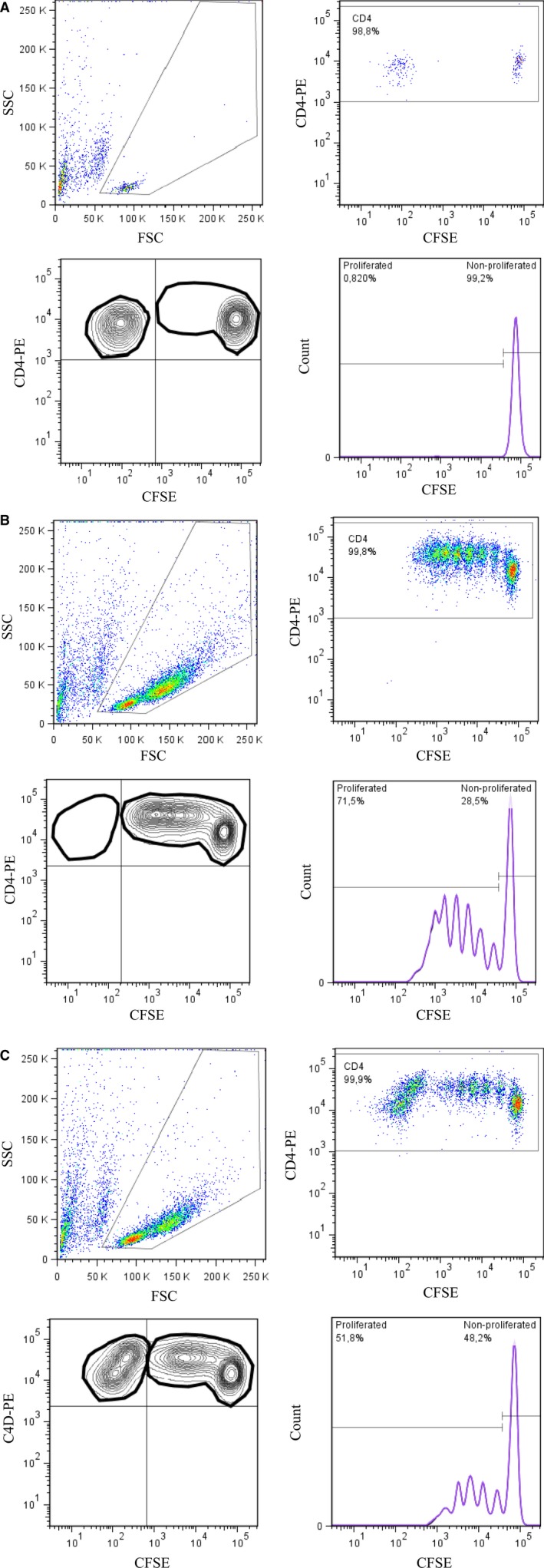
Representative plots and the gating information from the Treg suppression assay. After 6 days' stimulation with anti‐CD3/CD28 beads, viable lymphocytes were first gated (upper left). The second gate was created for the CD4+ cells (upper right). The third gate (lower left) represents proliferated Teff cells and the proliferation % was expressed by the histogram (lower right). The suppression % was calculated from stimulated Teff cells with or without Tregs, as, for example, 100−(51.8/71.5) × 100% = 27.6% (Fig. 2B and 2C). A: Representative figure from a cell co‐culture (Teff + Treg) with no stimulating agent. B: Single culture where Teff cells were stimulated with anti‐CD3/CD28 beads without suppressing Treg cells. C: In co‐culture, Treg and Teff cells were cultured and stimulated with anti‐CD3/CD28 beads in the same well.
RNA extraction and real‐time quantitative PCR (qPCR) analysis
Peripheral blood samples were collected into Paxgene RNA Tubes (Qiagen, Valencia, CA) and frozen at −70 °C according to the manufacturer's instructions. For analysing mRNA levels, the RNA was first isolated with the Blood RNA extraction kit (Applied Biosciences) and then converted to cDNA with the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real‐time qPCR was then carried out using the TaqMan® Gene Expression Assay Kit (Applied Biosystems). The housekeeping gene 18S RNA was used for normalization, and the real‐time qPCR was performed as a multiplex reaction. The relative mRNA gene expression of T cell markers (Th1 = T‐bet, Th2 = GATA3, Th17 = RORC, Treg=FOXP3 and CTLA‐4 as a co‐stimulatory factor that affects Treg cell function) was quantitated as a fold change against the common control sample by using the 2−∆∆CT method 24. A sample from the RT‐PCR performed without a reverse transcriptase enzyme was used as a negative control for detecting possible genomic DNA contaminations.
Beta cell‐specific autoantibodies
Autoantibodies against pancreatic beta cells were analysed from serum samples with immunofluorescence (islet cell autoantibodies, ICA) and specific radiobinding assays (IAA, GADA and IA‐2A) as previously described 25.
Statistical analysis
Differences in the mRNA gene expression levels, relative amount of Treg cells and percentage of suppression capacity between the case and control children were analysed with the nonparametric Mann–Whitney U‐test using the SPSS statistical program (version 22.0). A two‐tailed P value <0.05 was considered statistically significant.
Ethics
Informed consent has been obtained from all study subjects and/or their guardians, and the studies have been carried out in accordance with the principles of the Declaration of Helsinki as revised in 2008.
Results
Flow cytometric analysis of regulatory T cells
We quantitated the relative Treg number by analysing CD4+CD25+FOXP3+ cells from peripheral blood samples (Fig. 3). There were no statistically significant differences in the relative quantity of peripheral Tregs between the case and control groups at any stages of beta cell autoimmunity (case–control group 1: 1.52% versus 1.61% P = 0.256, case–control group 2: 1.66% versus 1.86% P = 0.372, case–control group 3: 1.56% versus 1.43% P = 0.980, case–control group 4: 2.14% versus 1.70% P = 0.218).
Figure 3.
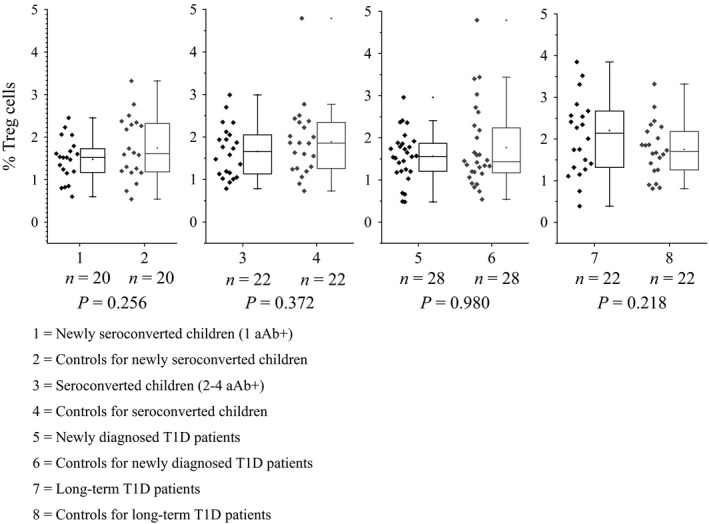
Proportion of lymphocytes expressing CD4+ CD25+ FOXP3+ (Treg) from all peripheral blood CD4+ cells in various phases of type 1 diabetes autoimmunity. Previously freshly isolated and frozen peripheral blood mononuclear cells (PBMCs) were thawed and stained with PerCP‐CD4, Alexa Fluor 488‐FOXP3 and APC‐CD25 fluorochromes and measured with BD FACSFortessa. Results were analysed with FlowJo software. Box and whisker plots represent the relative number of Treg cells (%), and every plot includes the median (solid line), interquartile range (IQR), range and outliers. The whiskers represent 1.5 interquartile ranges.
The T cell suppression assay
The function of the Treg cells was of the same magnitude between children with beta cell autoimmunity and their age‐ and sex‐matched autoantibody‐negative controls during preclinical type 1 diabetes (case–control group 1: 59.6% versus 52.7% P = 0.626; case–control group 2: 43.8 versus 50.6 P = 0.340) when autologous Tregs and Teffs were used in the assay protocol (Fig. 4). Neither were any significant differences observed when comparing the children with clinical type 1 diabetes to control children (case–control group 3: 53.7% versus 46.8% P = 0.820; case–control group 4: 38.9% versus 44.1% P = 0.694).
Figure 4.
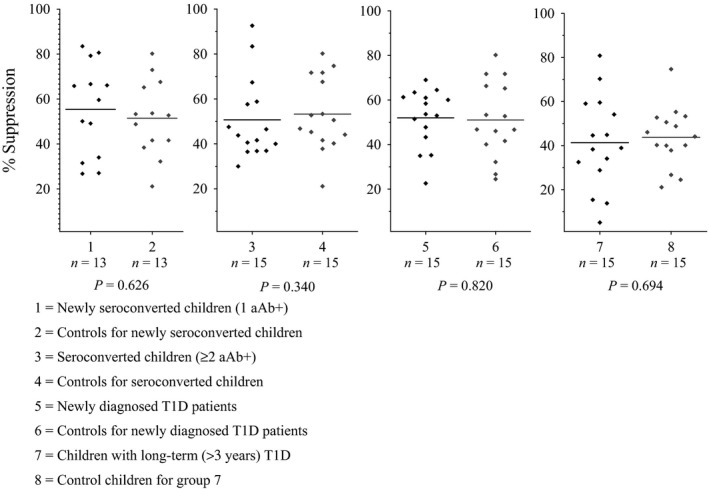
Suppression capacity (%) of autologous Treg cells during the T1D progression. Subjects from various stages of T1D autoimmunity (case groups 1, 3, 5 and 7) were compared with age‐ and sex‐matched control children who remained autoantibody negative (control groups 2, 4, 6 and 8). The suppression capacity was analysed with a carboxyfluorescein succinimidyl ester (CFSE)‐based T cell suppression assay. After 6 days in vitro stimulation with CD3/CD28 beads, the single (Teff) and co‐cultured (Teff + Treg) cells were labelled with PE‐CD4. The suppression capacity of Treg cells was measured with BD FACSFortessa, and the percentage of suppression was calculated as 100‐(Teff+Treg)/Teff × 100%. Mean values for each group are indicated by a horizontal line.
Quantitative real‐time qPCR analysis
The balance of the T cell‐mediated immune system was analysed by measuring the transcription factor mRNAs expressed specifically by different T cell subsets. An overall decrease in the mRNA expression of several transcription factors was observed in prediabetic children positive for multiple islet autoantibodies in comparison with autoantibody‐negative control children (RORC (Th17): 0.44 versus 0.53 P = 0.163; FOXP3 (Treg): 0.56 versus 0.62 P = 0.151; T‐bet (Th1): 0.34 versus 0.63 P < 0.001; GATA3 (Th2): 0.35 versus 0.46 P = 0.003; CTLA4 (Treg): 0.59 versus 0.68 P = 0.026) (Fig. 5B) and in children with newly diagnosed type 1 diabetes (RORC (Th17): 0.36 versus 0.52 P = 0.046; FOXP3 (Treg): 0.47 versus 0.61 P = 0.005; T‐bet (Th1): 0.24 versus 0.37 P < 0.001; GATA3 (Th2): 0.30 versus 0.50 P < 0.001; CTLA4 (Treg): 0.59 versus 0.64 P = 0.126) (Fig. 5C).
Figure 5.
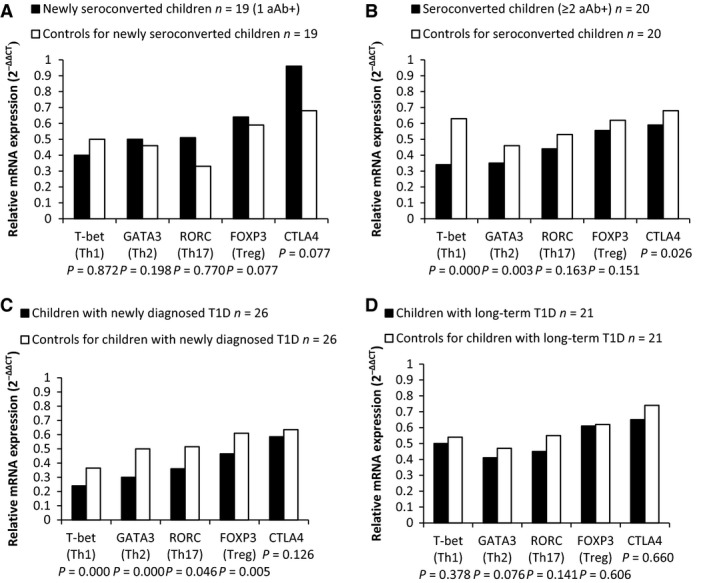
Relative mRNA expression levels, measured from total peripheral blood RNA, of different T cell subset markers in the various stages of type 1 diabetes. The relative mRNA expression analysis for T cell markers (Th1 = T‐bet, Th2 = GATA3, Th17 = RORC, Treg = FOXP3 and CTLA‐4) was performed with real‐time qPCR assay and quantitated as a fold change against the common control by using the 2−∆∆ CT method. A: recently seroconverted children, B: children positive for multiple autoantibodies, C: children with newly diagnosed T1D, D: children with long‐lasting T1D. Case groups are represented by black bars, and age‐ and sex‐matched control children for each group are shown in white bars.
There was a similar but non‐significant trend towards decreased expression of the analysed transcription factors among subjects with long‐standing type 1 diabetes (RORC (Th17): 0.45 versus 0.55 P = 0.141; FOXP3 (Treg): 0.61 versus 0.62 P = 0.606; T‐bet (Th1): 0.50 versus 0.54 P = 0.378; GATA3 (Th2): 0.41 versus 0.47 P = 0.076; CTLA4 (Treg): 0.65 versus 0.74 P = 0.660) (Fig. 5D).
No differences were seen in the early stage of beta cell autoimmunity when children with recent seroconversion to positivity for a single autoantibody were compared to autoantibody‐negative controls (RORC (Th17): 0.51 versus 0.33 P = 0.770; FOXP3 (Treg): 0.64 versus 0.59 P = 0.077; T‐bet (Th1): 0.40 versus 0.50 P = 0.872; GATA3 (Th2):0.50 versus 0.46 P = 0.198; CTLA4 (Treg):0.96 versus 0.68 P = 0.077) (Fig. 5A).
Discussion
A reduced number of regulatory T cells is one of the potential factors affecting the development of childhood type 1 diabetes. Previous studies have shown that there are, however, no alterations in the relative quantity of Treg cells in adult patients with clinical type 1 diabetes 14, 16, 17. We wanted to investigate whether the results are similar also in children (<15 years) with type 1 diabetes and to study the preclinical phase of type 1 diabetes before the clinical diagnosis. Our results are in line with previous studies. There was no difference in the relative quantity of Treg cells in children with newly diagnosed or long‐standing type 1 diabetes when compared to the non‐diabetic autoantibody‐negative control children. Furthermore, we did not observe any changes in the relative proportion of Tregs in children testing positive for beta cell‐specific autoantibodies.
There are conflicting results from studies analysing the functionality of regulatory T cells in type 1 diabetes 14, 17, 26, 27. We could not observe any differences in the suppression capacity of Treg cells between children with type 1 diabetes autoimmunity and autoantibody‐negative controls. It is noteworthy, however, that the in vitro suppression assay is very sensitive for changes in the protocol. Treg sorting and gating methods influence greatly the composition of the isolated Treg pool and hence the degree of suppression. Also, the strength and type of the stimulus used in the assay is critical and may affect the results remarkably 28, 29.
Decreased gene expression of several transcription factors specific for various T cell subpopulations was observed at the mRNA level in children with newly diagnosed type 1 diabetes as well as during the preclinical period in children testing positive for multiple islet autoantibodies when compared to healthy children. There was a similar but non‐significant trend among subjects with long‐lasting type 1 diabetes. No difference was seen at the early stage of beta cell autoimmunity when recently seroconverted children positive for a single autoantibody only were compared to autoantibody‐negative controls.
Decreased Th1 immune response and low cytokine secretion have been reported previously in patients with recently diagnosed type 1 diabetes 30. Other studies have also shown a general downregulation of genes involved in immune response pathways in patients with type 1 diabetes 31, 32. Rydén et al. have speculated that the reduced Th1‐associated immunity and decreased spontaneous cytokine secretion could be an effect of exhaustion of the immune system after the strong immune activation preceding the diagnosis of T1D. These observed abnormalities could be normalized in children with long‐standing T1D when the autoimmune process is coming to an end and the immune system is partly recovering 33. Our observed results of a decrease in expression of T cell‐specific transcription factors during the active T1D‐associated autoimmune process can be interpreted to support this hypothesis. Decrease seen in gene expression analysis, but not in Treg cell proportion, may reflect post‐transcriptional or translational regulation.
The strength of our study is a very comprehensive study population from the early stage of the development of beta cell‐specific autoimmunity to long‐lasting type 1 diabetes. Our study cohort consists of population‐based cases and age‐, sex‐ and sampling date‐matched controls. All samples were handled immediately after the sampling by the same personnel, and there was no shipping of the samples. Treg cell research is a challenging field due to the plastic and unstable nature of Treg cells. There are a complex set of regulatory T cell subtypes, and in certain conditions, Treg cells may lose their suppressive capacity and switch their phenotype to Teff cells 27, 28, 34. Also, the lack of specific markers for reliable characterization and sorting regulatory T cells may lead to the inconsistent study results. Most of the conventional Treg markers are also expressed in activated effector T cells 8. The main limitation in this study is using the peripheral blood T cells for in vitro assays and analysis. The local milieu in the pancreatic islets of Langerhans may differ from the systemic state. It is possible that there are certain inflammatory cytokines and antigen‐presenting cell types which influence Treg cell function and promote the development of the autoimmune process. Human post‐mortem samples from the pancreas have revealed that FOXP3+ cells are not commonly found in inflamed islets 35. Patients with type 1 diabetes have been observed to have functionally defective CD4+CD25+ Treg cells in their pancreatic lymph nodes, but not in peripheral blood 36.
Cytokines play a critical role in Treg differentiation and survival. Dysregulation of such inflammatory molecules could lead to the loss of function in Treg cells and imbalanced equilibrium between regulatory and effector T cells 8, 21. Previous studies have shown that impaired cytokine secretion is related to type 1 diabetes progression 33, 37, 38. In future, it would be interesting to analyse also in our study population whether there are any changes in cytokine expression in children with T1D autoimmunity.
In conclusion, we demonstrate that there are no alterations in the proportion or suppression capacity of Tregs in children with type 1 diabetes autoimmunity in the preclinical or clinical phase of type 1 diabetes. The gene expression of several specific transcription factors for different T cell subtypes was clearly declined in prediabetic children with multiple islet autoantibodies and also in children with newly diagnosed type 1 diabetes in comparison with controls. These results may be interpreted as reflecting an exhaustion of the immune system or reflecting a general abnormal immune reactivity during the actively progressing T1D‐associated autoimmunity.
Acknowledgment
The authors wish to thank all the DIPP families and personnel for the participation in the study. This study was supported by the Juvenile Diabetes Research Foundation (JDRF, grants 4‐1998‐274, 4‐1999‐731, 4‐2001‐435), Oulu University Hospital Research Funds, Alma, and K.A. Snellman Foundation, The Foundation for Pediatric Research in Finland, The Finnish Cultural Foundation/North Ostrobothnia Regional Fund, The Emil Aaltonen Foundation, Medical Research Center Oulu and The Diabetes Research Foundation in Finland.
References
- 1. Achenbach P, Warncke K, Reiter J, Naserke HE, Williams AJ, Bingley PJ et al Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes 2004;53 :384–92. [DOI] [PubMed] [Google Scholar]
- 2. Roep BO. T‐cell responses to autoantigens in IDDM. The search for the Holy Grail. Diabetes 1996;45 :1147–56. [DOI] [PubMed] [Google Scholar]
- 3. Bradley LM, Asensio VC, Schioetz LK, Harbertson J, Krahl T, Patstone G et al Islet‐specific Th1, but not Th2, cells secrete multiple chemokines and promote rapid induction of autoimmune diabetes. J Immunol 1999;162 :2511–20. [PubMed] [Google Scholar]
- 4. Bedoya SK, Lam B, Lau K Larkin J III. Th17 cells in immunity and autoimmunity. Clin Dev Immunol 2013;2013 :986789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li CR, Mueller EE, Bradley LM. Islet antigen‐specific Th17 cells can induce TNF‐alpha‐dependent autoimmune diabetes. J Immunol 2014;192 :1425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol 2007;19 :652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knip M, Siljander H. Autoimmune mechanisms in type 1 diabetes. Autoimmun Rev 2008;7 :550–7. [DOI] [PubMed] [Google Scholar]
- 8. Bin Dhuban K, Kornete MS, Mason E, Piccirillo CA. Functional dynamics of Foxp3(+) regulatory T cells in mice and humans. Immunol Rev 2014;259 :140–58. [DOI] [PubMed] [Google Scholar]
- 9. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S et al Crucial role of FOXP3 in the development and function of human CD25 + CD4 + regulatory T cells. Int Immunol 2004;16 :1643–56. [DOI] [PubMed] [Google Scholar]
- 10. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L et al The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001;27 :20–1. [DOI] [PubMed] [Google Scholar]
- 11. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med 2007;13 :108–16. [DOI] [PubMed] [Google Scholar]
- 12. Long SA, Buckner JH. CD4 + FOXP3 + T regulatory cells in human autoimmunity: more than a numbers game. J Immunol 2011;187 :2061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hughson A, Bromberg I, Johnson B, Quataert S, Jospe N, Fowell DJ. Uncoupling of proliferation and cytokines from suppression within the CD4 + CD25 + Foxp3 + T‐cell compartment in the 1st year of human type 1 diabetes. Diabetes 2011;60 :2125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T‐cells from patients with type 1 diabetes. Diabetes 2005;54 :92–9. [DOI] [PubMed] [Google Scholar]
- 15. Ryba‐Stanislawowska M, Rybarczyk‐Kapturska K, Mysliwiec M, Mysliwska J. Elevated levels of serum IL‐12 and IL‐18 are associated with lower frequencies of CD4(+)CD25 (high)FOXP3 (+) regulatory t cells in young patients with type 1 diabetes. Inflammation 2014;37 :1513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D et al No alterations in the frequency of FOXP3 + regulatory T‐cells in type 1 diabetes. Diabetes 2007;56 :604–12. [DOI] [PubMed] [Google Scholar]
- 17. Putnam AL, Vendrame F, Dotta F, Gottlieb PA. CD4 + CD25 high regulatory T cells in human autoimmune diabetes. J Autoimmun 2005;24 :55–62. [DOI] [PubMed] [Google Scholar]
- 18. Bacchetta R, Gambineri E, Roncarolo MG. Role of regulatory T cells and FOXP3 in human diseases. J Allergy Clin Immunol 2007;35: quiz236–7. [DOI] [PubMed] [Google Scholar]
- 19. Lawson JM, Tremble J, Dayan C, Beyan H, Leslie RD, Peakman M et al Increased resistance to CD4 + CD25hi regulatory T cell‐mediated suppression in patients with type 1 diabetes. Clin Exp Immunol 2008;154 :353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4 + FOXP3 + regulatory T cells. J Immunol 2008;181 :7350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupta S, Cerosaletti K, Long SA. Renegade homeostatic cytokine responses in T1D: drivers of regulatory/effector T cell imbalance. Clin Immunol 2014;151 :146–54. [DOI] [PubMed] [Google Scholar]
- 22. Ishigame H, Zenewicz LA, Sanjabi S, Licona‐Limon P, Nakayama M, Leonard WJ et al Excessive Th1 responses due to the absence of TGF‐beta signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc Natl Acad Sci U S A 2013;110 :6961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruitenberg JJ, Boyce C, Hingorani R, Putnam A, Ghanekar SA. Rapid assessment of in vitro expanded human regulatory T cell function. J Immunol Methods 2011;372 :95–106. [DOI] [PubMed] [Google Scholar]
- 24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25 :402–8. [DOI] [PubMed] [Google Scholar]
- 25. Siljander HT, Simell S, Hekkala A, Lahde J, Simell T, Vahasalo P et al Predictive characteristics of diabetes‐associated autoantibodies among children with HLA‐conferred disease susceptibility in the general population. Diabetes 2009;58 :2835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S et al Defects in IL‐2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T‐cells of type 1 diabetic subjects. Diabetes 2010;59 :407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA et al Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol 2011;186 :3918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol 2010;10 :849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McMurchy AN, Levings MK. Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol 2012;42 :27–34. [DOI] [PubMed] [Google Scholar]
- 30. Lohmann T, Laue S, Nietzschmann U, Kapellen TM, Lehmann I, Schroeder S et al Reduced expression of Th1‐associated chemokine receptors on peripheral blood lymphocytes at diagnosis of type 1 diabetes. Diabetes 2002;51 :2474–80. [DOI] [PubMed] [Google Scholar]
- 31. Orban T, Kis J, Szereday L, Engelmann P, Farkas K, Jalahej H et al Reduced CD4 + T‐cell‐specific gene expression in human type 1 diabetes mellitus. J Autoimmun 2007;28 :177–87. [DOI] [PubMed] [Google Scholar]
- 32. Elo LL, Mykkanen J, Nikula T, Jarvenpaa H, Simell S, Aittokallio T et al Early suppression of immune response pathways characterizes children with prediabetes in genome‐wide gene expression profiling. J Autoimmun 2010;35 :70–6. [DOI] [PubMed] [Google Scholar]
- 33. Ryden A, Ludvigsson J, Fredrikson M, Faresjo M. General immune dampening is associated with disturbed metabolism at diagnosis of type 1 diabetes. Pediatr Res 2014;75 :45–50. [DOI] [PubMed] [Google Scholar]
- 34. Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev 2014;259 :173–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009;155 :173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L et al Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes 2011;60 :2903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stechova K, Bohmova K, Vrabelova Z, Sepa A, Stadlerova G, Zacharovova K et al High T‐helper‐1 cytokines but low T‐helper‐3 cytokines, inflammatory cytokines and chemokines in children with high risk of developing type 1 diabetes. Diabetes Metab Res Rev 2007;23 :462–71. [DOI] [PubMed] [Google Scholar]
- 38. Foss NT, Foss‐Freitas MC, Ferreira MA, Cardili RN, Barbosa CM, Foss MC. Impaired cytokine production by peripheral blood mononuclear cells in type 1 diabetic patients. Diabetes Metab 2007;33 :439–43. [DOI] [PubMed] [Google Scholar]