

Abstract
The virion proteins of Pseudoalteromonas phage φRIO-1 were identified and quantitated by mass spectrometry and gel densitometry. Bioinformatic methods customized to deal with extreme divergence defined a φRIO-1 tail structure homology group of phages, which was further related to T7 tail and internal virion proteins (IVPs). Similarly, homologs of tubular tail components and internal virion proteins were identified in essentially all completely sequenced podoviruses other than those in the subfamily Picovirinae. The podoviruses were subdivided into several tail structure homology groups, in addition to the RIO-1 and T7 groups. Molecular phylogeny indicated that these groups all arose about the same ancient time as the φRIO-1/T7 split. Hence, the T7-like infection mechanism involving the IVPs was an ancestral property of most podoviruses. The IVPs were found to variably host both tail lysozyme domains and domains destined for the cytoplasm, including the N4 virion RNA polymerase embedded within an IVP-D homolog.
Keywords: Bacteriophage, Virion structure, Podoviruses
Introduction
Pseudoalteromonas phage φRIO-1 was isolated from the East Sea off the coast of South Korea and characterized as to genomic sequence and to have a podoviral morphology (Hardies et al., 2013). Its genome exhibits a large scale mosaicism. It shares a novel operon of genes involved in metabolizing γ-glutamyl amide linkages or unusual peptide bonds with a small number of other podoviruses typified by Pseudomonas phage LUZ24 (Ceyssens et al., 2008). However, the replicative functions, although generally related to other podoviruses, are not closely related to the LUZ24-like phages, and a small module apparently horizontally derived from φKMV-like phages (Lavigne et al., 2006) was noted. Structure and morphogenesis genes of φRIO-1 that could be identified, encoding large terminase, major capsid protein, portal (connector), and tubular tail B, were in a separate arm of the genome from the early and replicative genes.
Sequence similarities throughout the φRIO-1 structure and morphogenesis operon were noted to a collection of other podoviruses including Pseudomonas phage PA11 (Kwan et al., 2006), Salinivibrio phage CW02 (Shen et al., 2012), Roseophage SIO1 (Rohwer et al., 2000), and Vibrio parahaemolyticus phage VpV262 (Hardies et al., 2003). Of these, SIO1, VpV262, and CW02 have been described as members of a T7 supergroup. Rohwer et al. (2000) emphasized the distant relationship of the replicative functions of SIO1 to T7 to define a T7 supergroup. Hardies et al. (2003) emphasized an ancestral relationship in structure and morphogenesis proteins among SIO1, VpV262, and T7, however noting that VpV262 did not have T7-related replicative functions. It is now recognized that VpV262 has replicative functions closer to those of the φKMV-like podoviruses than to T7 (Hardies et al., 2013). Shen et al. (2012) applied the T7 supergroup terminology in describing similarity in the head structure at the level of cryoelectron microscopy (cryoEM) between CW02 and T7, but did not resolve the tail structures. CryoEM examination of φRIO-1 (Steven AC, personal communication) indicates a structural resemblance of φRIO-1 in the tail to a range of characterized podoviruses including T7 (Cuervo et al., 2013), Prochlorococcus (cyano) phage P-SSP7 (Liu et al., 2010), and enterobacteria phages K1E and K1-5 (Leiman et al., 2007), epsilon15 (Jiang et al., 2006; Chang et al., 2010) and P22 (Chang et al., 2006; Lander et al., 2009; Tang et al., 2011).
The concept of an overall T7 supergroup appears unable to accommodate the confusion caused by horizontal exchanges and mosaicism. However, we were interested in whether homology in the ensembles of proteins making up the tail structures could tie together some subset of the podoviruses. Our concept is similar to the “core genes” approach that Comeau et al. (2007) applied to T4-related phages, except that the members of an ensemble are defined by knowledge of which proteins interact to perform a function with gene synteny utilized when present, but not mandated. One such ensemble is the external tail structure, formed in T7 by tubular tail proteins A and B. Tubular tail A (also called the gatekeeper protein) forms the attachment for the side fibers (also called tail spikes) and is thought to mediate the initiation of infection through sensing the deflection of the side fibers upon cell wall binding. Tubular tail A was proposed to have structural homology between T7 and P22 (Cuervo et al., 2013) and also between podoviruses and siphoviruses (Olia et al., 2011). Tubular tail B (also called the nozzle) was recently found to be detectable in a wide range of podoviruses by a simple PSI-BLAST search (Hardies et al., 2013). In T7 there are 6 side fibers each composed of trimers of a single polypeptide; but the side fiber arrangement is expected to be intensively variable, and mosaic due to its content of the cell adhesin. A second tail ensemble consists of the internal virion proteins (IVPs), which in T7 extends upon infection to form a transient tail tube penetrating through the cell wall to the cellular membrane (Kemp et al., 2005; Hu et al., 2013, Guo et al., 2013). The IVP operon in T7 includes one small (IVP-B) and two very large (IVP-C and -D) proteins, plus an associated nonstructural IVP assembly protein known as IVP-A. The two ensembles might be considered separately or as a joint tail structure ensemble, depending on whether they descend coordinately or reassort independently in a given range of phages. Within the φRIO-1 genome there are candidates for genes encoding a similar set of proteins based on size alone, but sequence similarity has not been detectable by standard methods.
To complete the description of the structural proteins of φRIO-1 and related phages, we used several strategies to focus the study on structural proteins only and deal with extreme divergence between distant homologs. First, the full complement of φRIO-1 structural proteins was identified and quantitated using mass spectrometry, SDS-PAGE, and gel densitometry. This allowed a comprehensive approach to the structural proteome while ignoring mosaicism affecting other functions. It also allowed restricting candidates for divergent T7 homologs to only those proteins with appropriate virion copy numbers. Secondly, that collection of phages found to be cladistically related to φRIO-1 in the more conserved proteins was explored as a reduced database in which to search for homologs of the faster diverging proteins together with a positionally biased search approach (Hardies et al., 2003) to triage marginal similarities. This set of phages is designated as the φRIO-1 tail structure homology group. The φRIO-1 structural protein alignments were converted to hidden Markov models (HMMs) suitable for sensitive HMM–HMM comparisons to the structural protein families of T7 and other podoviral groups.
It became apparent both through the initial HMM–HMM comparisons, and the review of apparent structural homology found by cryoEM in the podoviruses P22 (Olia et al., 2011), and N4 (Choi et al., 2008), that there are additional podoviral tail structural homology groups distantly related to φRIO-1 and T7. It further became apparent that adding these other groups strengthened the φRIO-1/T7 comparisons. Hence the plan of the study evolved from ascertainment of φRIO-1/T7 homology to ascertainment of homology across several structural homology groups, which in the aggregate are referred to as the transient tail homology group. Besides φRIO-1, Autographivirinae (T7), P22, N4, and epsilon15 groups, this includes an additional structurally uncharacterized group for which we arbitrarily chose Pseudomonas phages F116 (Byrne and Kropinski, 2005) and H66 (GenBank: KC262634) to act as prototypes. The only major division of Podoviridae that does not appear to have tail structures related to T7 is Picovirinae, which has tail structures related to φ29.
Results
Proteomics
φRIO-1 virion proteins were separated by SDS-PAGE and analyzed by mass spectrometry as described in methods. The number of spectra assigned for suspected structural proteins ranged from 1884 for the major capsid protein down to 32 for gp44. There were three spectra observed that corresponded to a clearly nonstructural protein (gp8), and one peptide attributable to the scaffold protein (gp49), which is expected to be mostly removed from the virion during maturation. There was a relatively clear distinction in the total number of spectra observed between virion proteins found in a stoichiometric proportion, and nonstructural proteins found in trace amounts. Hence, the results provide a complete census of φRIO-1 structural proteins. Sequence coverage of the φRIO-1 structural proteins is given in Table 1. There was no clear indication of proteolytic processing, consistent with the lack of any identifiable protease encoded in the φRIO-1 genome.
Table 1.
φRIO-1 virion proteins detected by mass spectrometry.
| kDaa | % cov. | Copies from gel densityb | Proposed function | Expected copiesc | SC/MW | Terminal residues unsampled (N,C) | |
|---|---|---|---|---|---|---|---|
| gp37 | 174.9 | 65 | 4±2 | IVP-D | 4 | 4.3 | 0,0 |
| gp55 | 73.2 | 86 | 12±6 | Side fiber | 18 | 11.2 | 6,0 |
| gp36 | 63.8 | 70 | 6±3 | IVP-C | 8 | 7.8 | 0,0 |
| gp51 | 63.6 | 68 | 9±4 | Portal | 12 | 6.2 | 0,0 |
| gp40 | 44.4 | 78 | 7±3 | Tube B (C ter) | 6 | 3.6 | 7,5 |
| gp42 | 41.4 | 63 | 23±15d | 4.0 | 38,5 | ||
| gp44 | 35.0 | 59 | n.d.d | Tube B (N ter) | 6 | 0.9 | 5,0 |
| gp48 | 35.2 | 97 | 415e | Major capsid | 415 | 53.5 | 0,0 |
| gp46 | 28.8 | 49 | 15±7 | Tube A | 12 | 2.5 | 21,4 |
| gp47 | 17.6 | 86 | 275±140f | 14.6 | 0,0 | ||
| gp43 | 19.1 | 75 | n.d.f | 5.6 | 1,4 | ||
| gp38 | 14.8 | 79 | <17g | IVP-B | 12 | 9.4 | 43,0 |
Molecular weight adjusted for loss of N terminal methionine confirmed by semitryptic fragment observed for gp37, gp36, gp51, and gp43. Presence of N-terminal methionine was confirmed for gp48 and gp47.
Number of polypeptides per virion with estimated 95% confidence interval.
From the precedent in T7 (Molineux, 2006; Guo et al., 2013).
Gp42 and gp44 comigrate; the sum is given.
Peak area of gp48 was assumed to correspond to 415 copies for calibration purposes.
Gp47 and gp43 comigrate; the sum is given.
There was no Coomassie-staining peak observed in the gel profile within the slice exhibiting gp38 tryptic fragments. The total area above our best estimate of the baseline across the slice was used to project the indicated maximum copy number.
Each structural protein was clearly assignable to a particular gel slice according to the greatest abundance of its spectrum counts, and this further allowed identification of each structural protein with a peak in the Coomassie-stained SDS PAGE profile (Fig. 1) with the exceptions that gp44 and gp42 comigrated, gp43 and gp47 comigrated, and gp38 appeared to have diffused broadly within its slice. The relative abundance of each virion protein derived from integration of the Coomassie profile is shown in Table 1 (copy number) with a confidence interval as described in methods.
Fig. 1.
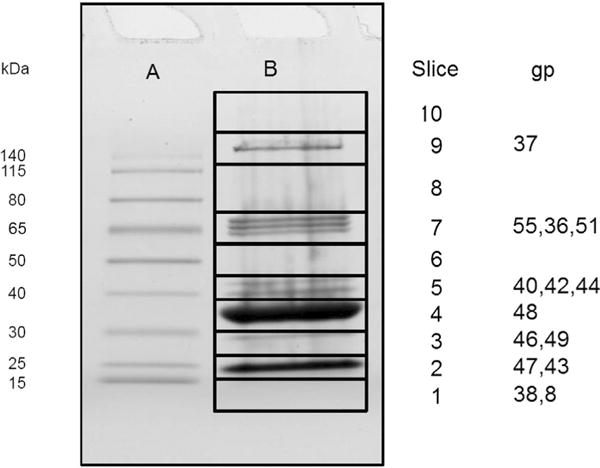
SDS-PAGE of φRIO-1 virions indicating gel position of specific φRIO-1 proteins detected by mass spectrometry. (A) Molecular weight markers. (B) φRIO-1 virions. The slice where maximum number of spectra assigned for each gene product is indicated.
Bioinformatic analysis
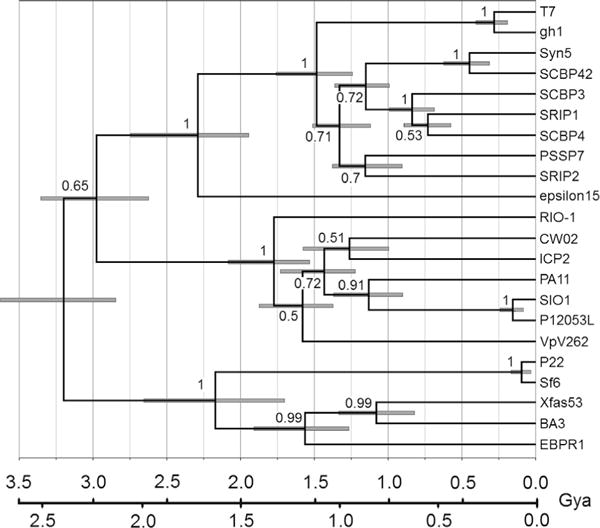
Some of the φRIO-1 structural proteins are easily related to T7 structural proteins through profile searches, while others are not. Of those strategies that might improve the sensitivity of matching up the more difficult proteins, the reduced database strategy requires a prior hypothesis about where the putative homologs are to be found. Hence we did a formal molecular phylogenetic analysis on the more strongly conserved proteins [gp52, large terminase subunit (Fig. 2A); gp51 portal protein (Fig. 2B); and gp48, major capsid protein (not shown)] to establish expectations for the others. In each case a preliminary neighbor joining tree analysis identified the φRIO-1 protein as a member of a clade containing PA11, CW02, ICP2 (Seed et al., 2011), SIO1 [and its sister phage PL12053L (Kang et al., 2012)], and VpV262. The cyanophage Pf-WMP3 (Liu et al., 2008) was found at or near the base of the φRIO-1-like clade for each protein tested. The maximum likelihood trees for all three of these proteins evaluated by MrBayes were roughly congruent in the following properties. Only the pairs (CW02, PA11) and (SIO1, P12053L) are within the range of divergence typically classified as a genus (40% of encoded proteins with E=0.05 by BLASTP; Lavigne et al., 2008), and operationally expected to match up nearly all structural proteins in a BLASTP search. φRIO-1, VpV262, and SIO1 exhibit divergence comparable to different genera within the subfamily Autographivirinae. Matching individual sequences at that distance given the characteristic divergence rates of podoviral tail proteins would be expected to be problematical. However, a profile based approach starting with CW02 and PA11 and employing a reduced database might have a chance of aligning tail genes throughout the φRIO-1-related phages, assuming some level of congruence between the tail and head genes. Such an approach (see methods) was fundamental to converting each of the φRIO-1 structural proteins to an alignment, and an HMM which could then be matched further to T7 and other podoviral structural proteins.
Fig. 2.
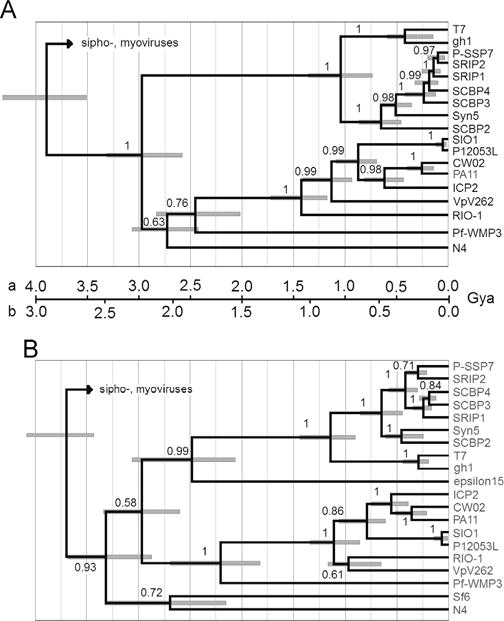
Maximum likelihood tree by Bayesian inference using a relaxed clock. (A) Large terminase subunit. (B) Portal protein. In the T7 clade, gh-1 is among the most divergent phages considered close enough to classify in the same genus as T7, whereas the cyanophages (e.g. Syn5) exemplify more divergent phages considered in the same subfamily as T7, Autographivirinae. Two time scales are proposed with the three major Caudoviral families radiating (a) near the beginning of cellular life; or (b) in early proteobacteria during development of the outer cell envelope. The 95% credibility intervals for node heights are indicated. Posterior probabilities for branch order are given on the face of the tree.
PSI-BLAST searches started from most (but not all) φRIO-1 structural proteins either only find matches in the same φRIO-1 homology group described above, or find matches in that group before extending to find more distant matches outside the group. PSI-BLAST searches from structural proteins of P22, N4, epsilon15, numbers of members of Autographivirinae, and φ29 behave similarly (data not shown, but this behavior is strongly implied by the structure of phage protein families in Pfam, for example). Hence, Podoviridae can be conceptualized as divided into a few core structural homology groups corresponding to those PSI-BLAST groups. Those proteins apparently don’t often engage in horizontal exchanges between phages from different homology groups.
Besides placing φRIO-1 within a homology group, these trees also contain some information about how that group might relate to other phage groups. The relationship between φRIO-1 and T7 is relatively deep, about 3/4 the distance to the diversification of the three tailed phage families. Three proteins from podoviruses not in Autographivirinae or the φRIO-1 homology group were included in the trees in Fig. 2. These were Sf6 (representing P22-like phages), N4, and epsilon15. These show one of two patterns. Either they map at a similar level of divergence as the φRIO-1/T7 split, or they map with the siphoviruses (terminase of P22 or epsilon15, not shown). The latter pattern is consistent with the cladistic association of terminases with kinds of ends produced (Casjens et al., 2005) and marks an interfamily horizontal transfer. The capsid proteins mapped in the same pattern as terminase (not shown). Those tail proteins that are responsible for the podoviral tail morphology by definition cannot engage in interfamily transfers. This creates an expectation that to the extent that proteins in structural groups defined by PSI-BLAST are homologous from one group to another, they probably form clades joining at the same level of depth as the φRIO-1/T7 split in Fig. 2.
Anticipating that the φRIO-1/T7 splits in Fig. 2 could be a useful reference point in the tree for the initial appearance and diversification of the podoviral morphology, we were interested in when that split might have occurred in absolute time. The terminase tree is special among trees made of phage proteins, because the underlying alignment includes all tailed phages (Serwer et al., 2004). Therefore, if an assertion is made about when tailed phages arose, that can be used to scale the tree and all of the nodes within it. Two possible time scales are illustrated on Fig. 2. One (a) is based on the assertion that the tailed phages arose at the earliest time of cellular life on earth. This calibration yields the most ancient possible age for each node on the tree. The second scale (b) is based on the realization that the podoviruses other than picoviruses are heavily concentrated in Gram negative hosts. The major exception is a cluster of cyanobacterial podoviruses (P-SSP7-SCBP2; Fig. 2). The split between the cyanobacterial podoviruses and T7 is long after 3.2 Gya when the hosts would have split (Battistuzzi et al., 2004). The entrance of podoviruses into cyanobacteria must therefore be attributed to horizontal transfer in the form of host range changes. So, scale “b” places the origin of the global T7 homology group of podoviruses in early proteobacteria, and attributes the minor incidence of them in cyanobacteria and Gram positive hosts to horizontal transfer. Placing these two scales on Fig. 2 is meant to convey a plausible range for the ages of each point on the tree that includes both the intrinsic uncertainty in node height and a plausible range of uncertainty in how to calibrate the tree in absolute time.
Tubular tail protein B
The easiest of T7 tail structure proteins to relate to φRIO-1 is tubular tail B. As previously reported (Hardies et al., 2013), the φRIO-1 tubular tail B homolog is split to two polypeptides, gp44 and 40. If these two protein sequences are fused in silico, they match by PSI-BLAST to a similar protein in the immediate φRIO-1 homology group, and then in further iterations to many phage proteins including T7 tubular tail B. The segment interrupting gp40 and gp44 was not a mobile intron, and the two polypeptides migrated separately and as expected according to their size in the SDS-PAGE analysis. The interrupting segment encodes two structural proteins not implicated in tail structure. Other instances of a split of tubular tail B protein were not common in any of the extensive tubular tail B alignments that we constructed. However, VpV262 tubular tail B had an unusual arrangement also indicating some level of plasticity. Its C-terminus was substituted by a sequence that was either non-homologous or diverged beyond recognition, while the sequence homologous to the C-terminus of φRIO-1 tubular tail B was encoded two genes downstream (gp52; Fig. 3).
Fig. 3.
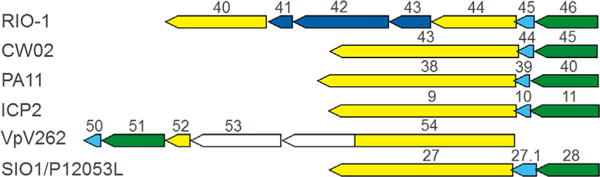
Organization of the genes for tubular tail A and B homologs in the φRIO-1-related genomes. Green—tubular tail A homolog; yellow—tubular tail B homolog; cyan—homolog of RIO-1 gp45 nonstructural gene; blue—inserted three gene module peculiar to φRIO-1; and white—no discernible sequence similarity.
We explored how widely tubular tail B homologs were distributed in Podoviridae. PSI-BLAST is able to draw the φRIO-1 homology group together with the T7 homology group and the epsilon15 homology group. P22 is known to have its gp10 protein in a similar structural position (Lander et al., 2009). HHPRED-style hidden Markov models were constructed for the three above mentioned homology groups and for the P22 gp10 homology group. The HMM–HMM matching scores exhibited a relationship which anticipated the tree joining these groups. Among the individual homology group HMM’s, only epsilon15 and T7 exhibited strong matching (operationally set at E < 10−10 for this experiment). But when a joint alignment and HMM for epsilon15 and T7 was constructed, it strongly matched the other homology groups. However, the HHPRED HMM–HMM matching indicated significant similarity among the homology groups only in a central ~200 residue domain, as indicated in Fig. 4.
Fig. 4.
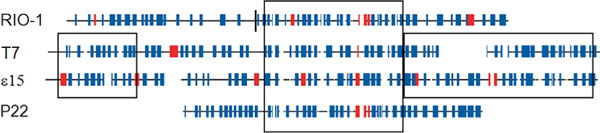
Segments of φRIO-1, T7, epsilon15, and P22 proteins exhibiting significant sequence similarity in the homologs of tubular tail B. The proteins φRIO-1 gp40 plus gp44, T7 gp12, epsilon15 gp12, and P22 gp10 are shown with predicted secondary structural elements as follows: blue—predicted beta strand; red—predicted alpha helix. A vertical line shows the point of fusion of gp44 and gp40 in φRIO-1. The central box encompassing all four sequences is the region of significant alignment as reported by HHPRED’s local alignment option. The coordinates in T7 gp12 are 354–556. Some additional regions of significant similarity between T7 and epsilon15 gp12 are also indicated.
Whether the N- and C-terminal domains are homologous among these homology groups is unclear. Secondary structure prediction indicates that all domains of tubular tail B are predominantly based on beta structure. However, there is significant length variation in the N- and C- terminal domains, and we have no convincing indicator of sequence similarity between the homology groups other than indicated in Fig. 4. Hence the likelihood tree was confined to the central domain. Comparing one family to another within that domain, 63% of residues predicted to be components of a β strand aligned with a β strand residues in the other family. Since Psipred at best correctly predicts 72% of β strand residues (Rost and Eyrich, 2001) and the total density of β strand residues is 34%, 63% correspondence between families would suggest that 76% of residues were correctly aligned in this domain. This agreed with the observation that HHpred aligned the domain when set to reject alignment at less than a 60% posterior alignment threshold and identified a contiguous section of about 100 residues with high posterior alignment probabilities.
The resulting tree (Fig. 5) shares many features in common with the terminase and portal trees (Fig. 2). Specifically, the external tail structure appears to have been generated at a very early time, sorted out into one of several homology groups, and then descended in a relatively uncomplicated fashion thereafter. Given this consistency, we transferred the minimum and maximum absolute time scales from the terminase tree under the assumption that the φRIO-1/T7 split occurred at the same time for both proteins. This result gave rise to two hypotheses: (1) Is there an even greater subrange of the podoviruses that has a homolog to tubular tail B, and (2) do all such podoviruses use a transient tail apparatus?
Fig. 5.
Maximum likelihood tree of the central domain of tubular tail B.
To facilitate asking what subrange of podoviruses have a tubular tail B homolog, first a customized BLAST formatted library was constructed consisting only of proteins encoded by completely sequenced podoviruses named as such in GenBank. The genome accession numbers were inserted in the protein definition lines. This made it simple to obtain a list of phages matched, and subtract it from the list of total podoviruses to generate the list of podoviruses not yet matched. The phages not yet matched were broken into structural homology groups allowing HMM–HMM comparisons to identify additional tubular tail B homologs. In the end, only two major homology groups were identified that did not have identifiable homologs of tubular tail B. Those were the N4-like podoviruses, and the picoviruses.
Further investigation suggested that the lack of similarity to tubular tail B in N4-like phages or picoviruses is unlikely to be overcome by any more sensitive kind of search. In both cases, this conclusion is supported by cryoEM data. The picoviruses do not appear to have internal virion proteins, and their external tail proteins at least superficially seem to be structurally distinct from T7 (Xiang et al., 2006). Hence picoviruses were not considered further in this study. N4 has a sheath protein surrounding a central tube in the analogous position to tubular tail B (Choi et al., 2008). Neither appeared structurally similar or matched in our most advanced HMM searches; the N4-like tube has alpha helical secondary structure instead of the beta sheet-based structure that dominates tubular tail B, and the N4-like sheath is not a conserved component of N4-like phages. Therefore, there seems to be no likelihood that N4 has a diverged version of tubular tail B, even though other N4 components were found to be divergently similar to T7 structural proteins (below).
Table 2 lists the tubular tail B homolog for a representative of various homology groups encountered in this study. Searching further through the full nr and env_nr databases produced a large collection of 898 tubular tail B homologs, which were divided into clades using PAUP. There were essentially three clades that did not fall into one of the four previously discussed homology groups. One had many named phages, and is represented as the F116 homology subgroup in Table 2. One had only a single named phage (HMO-2011) and many marine metagenome sequences. The third had only marine metagenome sequences (not shown). This suggests that sequence analysis of tubular tail B provides a comprehensive system for subdividing the podoviruses based on tail structure.
Table 2.
Divergent tubular tail B and internal virion protein D (IVP-D) homologs and GenBank accession numbers.
| Phage | Subgroup | Tubular tail B | IVP-D | ||
|---|---|---|---|---|---|
| Ralstonia RSB1 | T7 | gp34 | YP_002213723 | gp37 | YP_002213726 |
| Acinetobacter phiAB1 | T7 | gp37 | ADQ12741 | gp40 | ADQ12744 |
| Salmonella epsilon15 | epsilon15 | gp12 | NP_848220 | gp17 | NP_848225 |
| Bordetella BPP-1 | epsilon15 | Bbp13 | NP_958682 | Bbp10 | NP_958679 |
| Burkholderia BcepC6B | epsilon15 | gp12 | YP_024932 | gp17 | YP_024937 |
| Vibrio VP5 | epsilon15 | gp14 | YP_053016 | gp18 | YP_024981 |
| Liberibacter SC1 | epsilon15 | gp10 | YP_007011065 | gp9 | YP_007011064 |
| Phormidium Pf-WMP3 | RIO-1 | ORF30 | YP_001285795 | ORF26 | YP_001285791 |
| Puniceispirillum HMO-2011 | HMO-2011 | gp44 | YP_008320306 | ||
| Pseudomonas F116 | F116 | p52 | YP_164316 | p60 | YP_164324 |
| Pseudomonas H66 | F116 | 0054 | AGC34663 | 0060 | AGC34669 |
| Vibrio douglas 12A4 | F116 | 00068 | YP_007877493 | 00062 | YP_007877487 |
| Enterobacteria VT2-Sakai | F116 | orf61 | NP_050560 | orf71 | NP_050570 |
| Sinorhizobium PBC5 | F116 | p17 | NP_542277 | p23 | NP_542283 |
| Burkholderia BcepMigl | F116 | gp63 | YP_007236809 | gp69 | YP_007236815 |
| Edwardsiella KF-1 | F116 | gp42 | YP_006990472 | gp37 | YP_006990467a |
| Xylella Xfas53 | P22 | gp36 | YP_003344926 | gp40 | YP_003344930a |
| Thalassomonas BA3 | P22 | 0009 | YP_001552278 | 0002 | YP_001552271 |
| Candidatus Accumulibacter phosphatis EBPR | P22 | gp29 | AEI70869 | gp25 | AEI70865a |
Similarity is to P22 gp16 rather than T7 IVP-D.
Tubular tail protein A
The φRIO-1 tubular tail A homolog was not found by standard PSI-BLAST searches. It was established to be φRIO-1 gp46. Homologs of φRIO-1 gp46 were easily found within the φRIO-1 homology group by PSI-BLAST, although a positionally biased approach was required to add the Pf-WMP3 homolog. With a gp46 alignment and HMM in hand, an HHPRED match at E=0.012 was found in a 1:1 HMM–HMM comparison to T7 tubular tail A (gp11). This would not be strong enough to find these as homologs in a general family database search, because there are > 105 total protein families in the library that would typically be searched (for example at the HHPRED web site). However, gp46 is the only candidate structural protein in φRIO-1 with the right size and virion copy number to be a tubular tail A homolog. Under the reduced database concept it is fair to calculate the E value with a database of one if there is only one candidate based on independent criteria. Even with that assertion, E=0.012 is somewhat marginal, so we looked for further support. φRIO-1 gp46 is in syntenous position to T7 tubular tail A. Following on the report that T7 tubular tail A and P22 have structural homology but not sequence similarity (Olia et al., 2011; Cuervo et al., 2013), we applied the same procedure resulting in an E-value of 0.087 relating those two sequences. The HHPRED alignment, when annotated with secondary structure (Fig. 6) is informative. The most conserved elements are the leading and trailing helixes which in the P22 structure are known to form the contacts that stabilize the ring. The middle segment which forms the circumferential face of the structure appears somewhat more plastic. An N4 homolog to tubular tail A has not been previously identified, although proteomics of the virion (Choi et al., 2008) leaves few candidates of which only one (gp67) has the appropriate predicted secondary structure. HMM–HMM matching of T7 or φRIO-1 models to N4 gp46 was unconvincing, however a joint φRIO-1/T7 HMM matched to N4 gp67 with an E value of 0.0076, establishing that protein as the N4 homolog of tubular tail A.
Fig. 6.
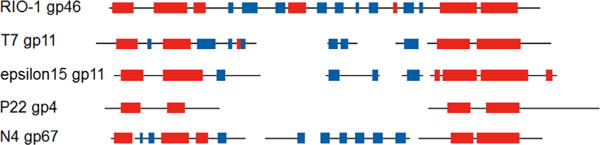
Alignment of putative tubular tail A homologs by HHPRED. Sequence alignments were produced with the HHPRED global alignment option. Predicted secondary structure elements were then graphed in accordance with the sequence alignments. The N-terminal segment dominated the alignment in local alignment mode. Blue—predicted beta strand; Red—predicted alpha helix. The P22 gp4 secondary structure is from X-ray crystallography (Olia et al., 2011).
In the φRIO-1 family, the tubular tail A and B genes maintain close linkage just downstream of the head structure gene module (Fig. 3). In most cases, the gene for tubular tail A precedes the gene for tubular tail B separated by short conserved non-structural gene. In the case of VpV262, the tubular tail A homolog is displaced downstream, and the homolog of the nonstructural gene is displaced with it. This may suggest that the homologs of nonstructural φRIO-1 gp45 play some role in tail morphogenesis, possibly specifically related to assembly involving tubular tail A.
Internal virion proteins
We sought to clarify the presence of homologs of the T7 IVPs in the φRIO-1 structural homology group through sequence analysis. The prototypical T7 IVP operon is a block of four genes, A, B, C, D, immediately downstream of the gene for tubular tail B protein. Internal virion A protein in T7 is a misnomer, in that this protein, although required for morphogenesis, is not retained in the virion (Kemp et al., 2005). In φRIO-1, a conserved block of structural genes of similar size to the T7 IVP set of T7 appears downstream of the gene for tubular tail B (Fig. 7). The relationships among the candidates for homologs to T7 IVP-A through -D are summarized in Fig. 7. The simplest of the IVP candidates of φRIO-1 to establish is IVP-B (φRIO-1 gp38), which was found by HHPRED to match T7 IVP-B with an E value of 5.4 × 10−4. Within the φRIO-1 homology group, there is consistently a small nonstructural gene between the tubular tail B and IVP-B genes. However, rather than this sequence being conserved across the set of φRIO-1-related phages, there are two different sequence families represented. φRIO-1 has a novel sequence which forms a family whose only known members are found at the syntenous positions in PA11 and CW02. The other φRIO-1-like phages have a nonstructural gene in that position that matches as a family at the HHPRED web server as an acetyltransferase. The T7 IVP-A protein also matches as an acetyltransferase. Hence the beginning of the IVP module in the φRIO-1 homology group is like the T7 module, except φRIO-1 itself along with PA11 and CW02 appear to have substituted an alternative protein for IVP-A.
Fig. 7.
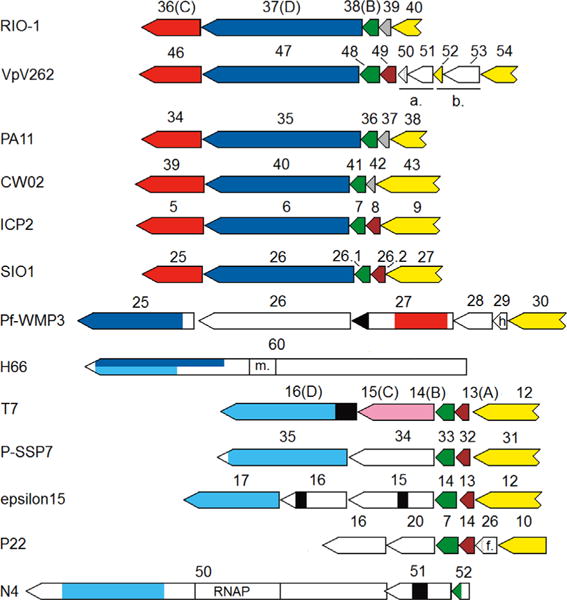
Organization of putative IVP genes in diverse podoviruses. Yellow—gene for tubular tail B, or tubular tail B homology in VpV262 gp52. As discussed in the text, this may reflect reuse of the end of the tubular tail B protein as an adhesin for the tail fiber, which may be composed of gp53 and gp54 in VpV262. SIO1 gp26.2 is an unannotated frame in the SIO1 GenBank entry. Brown—a gene belonging to an acetyltransferase family, including T7 IVP-A. Gray—a homolog of φRIO-1 gp39, which appears to be an alternative to IVP-A. Dark green—homolog of T7 IVP-B. Dark blue—homolog of φRIO-1 gp37. Cyan—homolog of T7 IVP-D. Black—a muralytic domain (murein D,D endopeptidase in the case of Pf-WMP3, and a murein β1,4 hydrolase or transglycosylase in the other cases). Light green—a homolog of epsilon15 gp16. Red—a homolog of φRIO-1 gp36. Rose-T7 IVP-C, and presumptive distant homolog of φRIO-1 gp36. m. (in H66)-a SAM-dependent methylase site. h. (in Pf-WMP3)-homing endonuclease. a.-displayed tubular tail A module in VpV262 consisting of a homolog of φRIO-1 gp45 and the gene for tubular tail A. b.-prospective tail fiber module in VpV262 consisting of a repeated C-terminal domain of the tubular tail B protein and an adjacent frame with similarity to polysaccharide lyase, f. – central fiber.
The strategy of searching for homologs by HMM–HMM comparisons among structural homology groups was then applied to IVP-A and IVP-B. Epsilon15 and P22 have very standard IVP-A and IVP-B homologs in syntenous position between tubular tail B and the genes that will subsequently be considered as IVP-C and -D homologs. The P22 IVP-B protein is known as the product of gene 7, and is an established pilot/DNA injection protein. The F116 subgroup (represented by H66 in Fig. 7) does not have identifiable candidates, and N4 encodes gp52 in the syntenous position for IVP-B that oddly has a C-terminal domain matching the N-terminal half of IVP-B, but has an N-terminal domain predicted to not have helical or beta structure. That portion of N4 gp52 is the only segment of a prospective IVP not predicted to have extensively helical structure.
Homologs of the two large T7 IVP-C and IVP-D proteins are notoriously difficult to identify in divergent podoviruses by sequence similarity. Even in P-SSP7, which is less than half as diverged in other proteins from T7 than is φRIO-1, these genes were assigned based on overall size and synteny rather than sequence similarity (Sullivan et al., 2005). In order to apply the HMM–HMM matching strategy, we found it essential to conduct searches and build HMMs for these proteins in a library of podoviral sequences only. Otherwise, the searches to gather homologs became embroiled in matches to plectin and myosin and other unrelated coiled-coil proteins. All of the candidates for IVP-C or IVP-D homologs are extensively alpha helical by secondary structure prediction, with numerous segments scoring well at the COILS coiled-coil server. Avoiding the problem of being overtaken by extraneous coiled coil matches by limiting the library still leaves the concern that any weak similarity detected may reflect the product of convergent evolution rather than descent from a common ancestor. However, we were able to develop HMMs that matched only one candidate per genome in the divergent homology groups indicating that there was not cross-matching between IVP-D and IVP-C candidates, or cross-matching to other podoviral proteins that are clearly not IVP-C or IVP-D. This provides some confidence that the matches detected are true homologies. Fig. 7 summarizes the state of those matches in a number of different homology subgroups.
IVP-D
Fig. 7 indicates separately the similarity detected starting from either φRIO-1 gp37 (blue), or T7 IVP-D (cyan). In each case, PSI-BLAST in the reduced podovirus-only database was the main tool to define the extent of similarity, and we constructed HMMs of divergent subgroups and did HMM–HMM matching with HHPRED to confirm significance of the match. For example, the first indication that the φRIO-1 gp37 set (Fig. 7, blue) was a divergent version of the T7 IVP-D set (Fig. 7, cyan) was that they both extended into an overlapping region of the F116 homology group represented in Fig. 7 by H66 gp60. As with tubular tail A, confirmation with HMM–HMM matching required first joining some subsets. In this case, separate sets for T7-like phages, φKMV-like phages, and epsilon15-like phages were successively tested for valid similarity and then joined, while the φRIO-1 and F116 homology groups were tested for valid similarity and then joined. These two sets matched in HMM–HMM scoring with E=8×10−19. Besides these sequence similarity results, φRIO-1 gp37 has the appropriate size, alpha helical propensity, and virion copy number to be a homolog of T7 IVP-D. We consider the union of both sets (Fig. 7 blue and cyan) to be an extended T7 IVP-D family.
These searches further clarified IVP-D homologs in most of the other podoviral structural homology groups. In the N4 group, it was the C-terminal domain of gp50, which also carries a virion RNA polymerase. In epsilon15, it was gp17. Epsilon15 is known to form a transient tail tube (Jiang et al., 2006), although the proteins involved are not clearly identified. Gp17, however, is one of the candidates. In H66 (a member of the F116 homology group), it is the very large gp60. This protein contains a small domain in the middle which matches well to the S-adenosyl methionine binding site of DNA methylases, hence it is most commonly annotated as a methylase or SAM-binding protein in GenBank. Indeed, we had taken the expansion of the φRIO-1 gp37 family into this collection of methylases as an indication that PSI-BLAST had lost specificity until the appearance of the GenBank entry clarifying that H66 gp60 is, in fact, a phage structural protein. Hence, H66 gp60 is reminiscent of N4 gp50, being composed of a protein with IVP-D homology attached to a domain that must certainly function intracellularly. Pseudomonas phage H66 gp60 is quite closely related to Podovirus F116 in its structural protein sequences, although the F116 homolog of H66 gp60 is broken into two large genes, more reminiscent of the organization of T7 IVP-C and IVP-D.
There was no IVP-D homolog identified in the P22 group by sequence similarity. P22 has three core proteins thought to be involved in DNA injection (reviewed, Black and Thomas, 2012) encoded in syntenous positions to the T7 IVPs. The first of these was identified as a homolog of IVP-B, and is preceded in the genome by a homolog of IVP-A. The remaining two DNA injection proteins encoded in syntenous positions to IVP-C and -D are predicted to be extensively alpha helical in their structure. They would therefore seem to be homologs of IVP-C and -D that have diverged beyond recognition, or at least alternative proteins with similar structure and function. The selection of phages in Table 2 was searched for homologs of T7 IVP-D or P22 gp16. In each phage one or the other was found, but never both in the same phage. Interestingly, there was some intermixing in the homology groups. Edwardsiella phage KF-1, which is otherwise in the F116 homology group, has the P22-like protein, whereas Thalassomonas phage BA3, which is otherwise in the P22 homology group, has a homolog of IVP-D. So the T7 IVPs and the P22 injection proteins have been able to interchange and segregate as alternative solutions to the DNA injection problem. This is the only interchange we have thus far noticed of non side-fiber tail structure genes between homology groups.
IVP-C
We were generally less successful in demonstrating sequence similarity among prospective IVP-C homologs. Within the T7 homology group, HMM–HMM matching was found between T7 IVP-C and φKMV gp37, confirming the homology suggested by Lavigne et al. (2006) based on size and synteny. However, we were unable to detect sequence similarity of that fused HMM as far away as P-SSP7, even though the P-SSP7 IVP-C candidate closely matches in size and is sandwiched between IVP-B and IVP-D homologs. Similarly, none of the methods that were successful in matching the other structural proteins were able to confirm an IVP-C homolog in the epsilon15 family. We suggest that this difficulty corresponds to an especially rapid divergence within IVP-C. In Table 3, the percent identities are tabulated for a variety of structural proteins between T7 and Pseudomonas phage gh-1, the most divergent of the formally classified T7-like phages in the other structural proteins. If the IVP-C tree is like the trees of the other structural proteins, then IVP-C has diverged to 34% identity in a mere 0.4 Gyr. At this rate, a failure by any method to detect sequence similarity at 2 Gya would hardly be surprising.
Table 3.
Percent identity of selected podoviral structural proteins after a moderate time of evolution.
| Approximate time of ancestral node | T7 × gh-1 0.4 Gya |
CW02 × ICP2 0.6 Gya |
|---|---|---|
| φRIO-1 gp36 homolog | 34 | 20 |
| IVP-D | 41 | 25 |
| Tubular tail B | 56 | 27 |
| Portal | 69 | 53 |
| Large terminase | 62 | 58 |
Given the lack of sequence conservation in IVP-C, there are some other criteria to help identify it. The traditional size and synteny argument is stronger if the candidate is flanked by verified IVP-D and IVP-B homologs. Secondary structure prediction should indicate an extensively alpha helical protein. The case is strengthened if the protein has been shown to be a virion protein with a copy number of about 8. As illustrated by the φKMV family (Lavigne et al., 2006), IVP-C homologs are sometimes associated with tail lysozyme domains. Finally, the finding of rapid divergence within the local homology group could be considered as an additional identifying factor.
In φRIO-1, there is a candidate for IVP-C (gp36) that is downstream of IVP-D instead of between IVP-B and IVP-D. It has an appropriate size, and virion copy number (Table 1). Its secondary structure is extensively alpha helical. The gp36 family was assembled within the φRIO-1 structure homology group mainly with PSI-BLAST, but HMM methods were required to identify a homolog in Pf-WMP3. The gp36 divergence rate was highest among the structural proteins of the φRIO-1 homology group (Table 3), assuming that Pf-WMP3 can be used to establish a comparable time point on each tree. The Pf-WMP3 φRIO-1 gp36 homolog is in the T7 syntenic gene order, although complicated by insertion of Pf-WMP3 gp26. Pf-WMP3 gp26 has an extensively collagen-like sequence, and is presumably yet another non-syntenous tail fiber gene (see discussion of tail fibers in the φRIO-1 homology group below). Finally, the Pf-WMP3 φRIO-1 gp36 homolog has acquired a muralytic domain (a murein D,D endopeptidase) on its C-terminus, reminiscent of the situation in φKMV. Through all of these observations, we believe that φRIO-1 gp36 is an IVP-C homolog that has diverged beyond recognition at the sequence level. Finally, we note that similar observations implicate IVP-C homologs in the other structure homology subgroups: gp 15 and 16 in epsilon15, gp20 in P22, and gp51 in N4.
Tail side fiber
Of the remaining structural proteins of φRIO-1 gp55 has the best match in expected virion copy number to the expectation of 18 for the side fiber. Finding a sequence criterion to confirm this assignment is difficult because of the extensive mosaicism of side fibers. For example, the prototypical side fiber (T7 gp17) is a trimeric protein (Steven et al., 1988) with an extensively coiled-coil structure. However, the cryoEM structure of φRIO-1 (Steven AC, personal communication), shows side fiber structures that do not have the thin tubular appearance of a coiled coil. Hence looking for extensive coiled-coil sequence won’t be helpful, and indeed φRIO-1 gp55 is predicted to be mostly based on beta strand structure by secondary structure prediction. There is a useful trend found in many T7-related phages in that the first 161 residues of T7 gp17 composing the tail tube attachment domain are conserved prior to the onset of mosaic substitutions (Pfam phage_T7_tail, see architectures tab). The only segment of φRIO-1 gp55 with sequence similarity through most of the φRIO-1-related phages is the first 60 residues of the N-terminus (Fig. 8, red). By analogy to T7 gp17, this was evaluated as a putative tail tube attachment domain. There is a weak match to the Pfam phage_fiber_2 domain adjacent to the attachment domain. This domain is found in a variety of phage fibers with trimeric structure. This establishes the gp55 N-terminal domain is a component of trimeric fibers, but doesn’t discriminate between head, side, collar, or central tail fibers. The polypeptides joined to the putative tail tube attachment domains in the other phages of the φRIO-1 homology group are highly mosaic, which is itself encouraging for assignment as the side fiber. The putative φRIO-1 attachment domain does not have sequence similarity to the T7 attachment domain. Hence, the mosaic part of the proteins was examined for evidence that at least some of them are more obvious tail side fibers. It follows that if any of the domains attached through the putative attachment domain are tail side fibers, then all proteins with this putative attachment domain, including φRIO-1 gp55 itself, are tail side fibers.
Fig. 8.
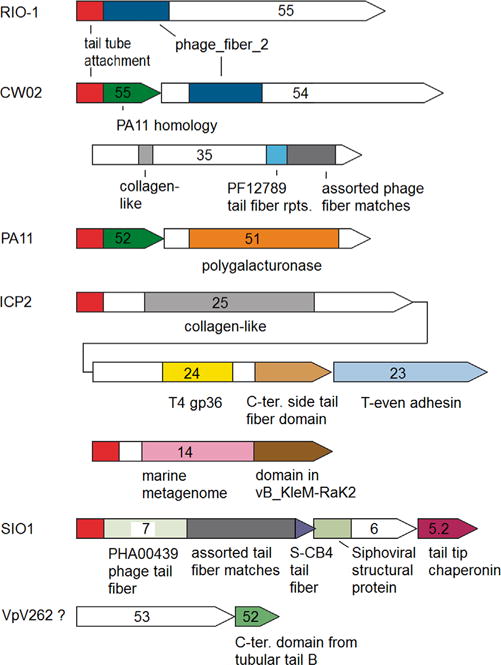
Candidates for tail side fiber assemblies in the φRIO-1 structure homology group of phages. Open segments have no discernible sequence similarity to any other sequence. Most of the other labeled domains are easily recognizable by PSI-BLAST, with the following two exceptions. The red domain clearly matches between φRIO-1 and SIO1, but recognition of it throughout the homology group required the progressive HMM building procedure described in methods. The blue block indicating a similarity between φRIO-1 gp55 and CW02 gp54 when formed to a two sequence profile only weakly matches to the Pfam: phage_fiber_2 family. Pfam: PF12789 is a Pfam protein domain family. CD: PHA00439 refers to an NCBI protein family. SIO1 gp5.2 refers to an unannotated gene between annotated genes for gp5.1 and gp6.
A summary of the various side fiber candidates in the φRIO-1 homology group is given in Fig. 8. The strongest patterns that implicate the gp55-like putative attachment domain as part of a tail fiber are in ICP2 and SIO1. In each case, the putative tail tube attachment domain is followed by a variety of tail fiber domains that are spread out through three adjacently encoded polypeptides. The organization is reminiscent of the long tail fibers of the T-even myoviruses (Cerritelli et al., 1996) including similarity to T4 gp36, and a T-even gp38 homolog in the position of the adhesin (Trojet et al., 2011). There is a second protein encoded in ICP2 (gp14) that also has the putative tail tube attachment domain. This is also reminiscent of the T4 arrangement with multiple adhesins in a complex branching structure (Kostyuchenko et al., 2003). Although most T-even phages have the T-even adhesin in the position of gp38, T4 itself has a different gp38 protein encoded in that position which is nonstructural but required as a chaperonin for the assembly of the fiber (Trojet et al., 2011). In SIO1, a homolog of the T4 gp38 chaperonin (gp5.2) is encoded downstream of two polypeptides, both of which contain domains found in other tail fibers, and the first of which starts with the φRIO-1 gp55-like putative tail tube attachment domain.
An analogy to the branched structure in K1-5 (Leiman et al., 2007) might appear in CW02. Instead of the putative tail tube attachment domain being in a large polypeptide, the small CW02 gp55 in which it appears may be analogous to the adapter in K1-5. If the analogy holds, there are two polypeptides encoded in CW02 (gp54 and gp35) that would bind to the adapter to create a branched fiber with two alternative binding specificities. This arrangement is thought to reflect a host range change in progress. In our tree analysis, Salinivibrio phage CW02 appears much more closely related to Pseudomonas phage PA11, than the host Salinivibrio would be related to Pseudomonas. This requires a relatively recent host range change to have occurred.
The remaining structural proteins (gp42, gp43, and gp47) do not have a proposed role in the φRIO-1 tail.
Discussion
Through a constellation of proteomic and bioinformatic methods, we conclude that a homologous tail and transient tail tube apparatus exists in essentially all of Podoviridae with the exception of the Picoviruses (relatives of φ29). A recent review (Casjens and Molineux, 2012) similarly explored relationships across this wide range of podoviruses, but suffered from the assumption that sequence similarity could not establish relationships among the diverse members of this group and that ascertainment of homology would have to wait for 3D structural determinations. The essence of our study is that specially constructed sequence comparisons can establish sequence similarity among most of the structural components, even addressing homology among some of the transient tail proteins for which structural studies have failed to establish the identities of the constituent proteins. Because of the wide distribution of tail protein homologs within Podoviridae we propose that their implied common ancestor invented a transient tail tube that functioned much like the tail of T7 functions today. We call this group the transient tail structural homology group of Podoviruses, and delineate the major homology subgroups underneath that category as the T7 homology subgroup, the epsilon15 subgroup, the φRIO-1 subgroup, the P22 subgroup, the N4 subgroup, and a new group represented by Pseudomonas phage F116. Essentially all of the podoviruses other than the picoviruses are included. This is a variation of the “T7 supergroup” concept, but dodges the issue of mosaicism by focusing on only the ensemble(s) of proteins reflecting the descent of the ancestral tail function into diverse extant descendants. In the case of the T7 homology subgroup, it is larger than the formally defined T7-like phage genus, and so far corresponds to the subfamily Autographivirinae. However, the designation Autographivirinae is based on content of an RNA polymerase gene (Lavigne et al., 2008), which could potentially reassort differently than the tail structure genes in some phages. Similarly, the P22, N4 and epsilon15 tail structure homology groups are larger than the genera formally named after each of these phages. The tail ensembles include a core structural ensemble derived by relatively vertical descent from the common ancestor, plus some auxiliary components frequently switched out by horizontal transfer to adapt the tail function to specific host bacterial species.
We specifically propose a commonality of the following functions throughout the transient tail structural homology group. (1) The transient tail is formed from IVPs each characterized by highly alpha helical secondary structure, which though highly divergent are related by descent from an ancestral IVP operon. (2) Release and assembly of the tail is triggered by interaction of the adhesin-bearing auxiliary side fibers with homologs of T7 tubular tail A. (3) The tail lysozymes are sometimes integrated into the tail tubes as auxiliary domains. And (4) the tube penetrates the cell membrane and can deliver auxiliary enzymatic domains into the cytoplasm as integrated passengers. Hence, descent from an ancient common ancestor of a stable ensemble of tail and IVPs is proposed wherein the major plasticity is in hosting a highly mosaic tail fiber, and in hosting a variable set of tail lysozyme domains and passenger domains for intracellular function.
Tubular tail A forms a ring just below the portal, interacts with the side fibers, and apparently constitutes the conformational switch by which side fiber engagement causes release of capsid contents. Its functions are of such fundamental necessity to the initiation of infection that it makes great evolutionary sense that it would be present in the ancestor to the family and remain conserved throughout its descendants. Tubular tail B forms a nozzle-like structure mounted below tubular tail A, and is thought to extend the tube through which the DNA will travel. In contrast to tubular tail A, tubular tail B seems more like an adapter for mounting additional functions than an essential component of the virion in its own right. It is the likely point of attachment of the transient tail formed by internal virion proteins (Hu et al., 2013), and this function may have stabilized its presence through most of the podoviruses. It also mounts auxiliary host recognition proteins in P22-like phages, and possibly plays this role in others of the podoviruses. It may also collaborate with tubular tail A in forming a socket for the side fibers. These functions would tend to cause hypervariability in tubular tail B structure.
A puzzle is, if attachment of the transient tail tube stabilizes the presence of tubular tail B, how did N4 substitute a completely different tubular protein below tubular tail A (Choi et al., 2008) even though it apparently also deploys a transient tail tube. There is no information about the consistency of the structure or mode of assembly of the transient tail tubes across the podoviruses. There is also no information about what the phage ancestor preceding the invention of the transient tail tube looked like. It will be of interest as information on these topics comes to light as to how they might clarify the origin of the alternative tail structure of the N4 phages.
Many authors have noted similarities in gene size and order of diverged phages and implied ancient homology that has diverged beyond an ability to confirm with sequence similarity. The reduced database search strategy has converted many such situations into a confirmed sequence similarity, complete with an alignment, a statistical test, a tree to provide some evolutionary context, and a hidden Markov model with which to seek even more distant relationships. This method provides a convenient control against the inadvertent inclusions of false homologs based on weak sequence similarity. If that has happened, the unrelated sequences will appear to be connected by a divergent link on the tree. Any suspect divergent link can be tested by realigning subsets of sequences on each side of the link and conducting a powerful statistical test of whether the two subsets are indeed similar by HMM–HMM comparison using the HHPRED system. The end result of our efforts is that there is a lot of confirmed synteny in the structure and morphogenesis operons of the podoviruses, even more than immediately meets the eye. Although the trees of the different proteins have a crude congruence, there are lots of cases of well supported topology switches. So the synteny is maintained not because of a lack of horizontal exchange and recombination, but in spite of it. Presumably, synteny is maintained because these are essential genes and viable recombinants are going to need one each of them.
On that background, we reflect on how completely differently the side fiber genes behave. It seems that every phage in the φRIO-1 homology group has its side fiber gene(s) in a different location, and there are often two of them in widely different places. It strikes us that the work of Leiman et al. (2007) may be explanatory of this. If it is generally true that phages switch side fibers by passing through an intermediate with dual specificity, then newly acquired side fiber genes will necessarily be forced to new genomic locations. If the framework of syntenous genes is adequately developed, then the property of being a synteny breaker might become an informative characteristic for gene identification.
Materials and methods
Phage purification
φRIO-1 virions were purified as described (Thomas et al., 2007), with the following modifications. A single plaque of bacteriophage φRIO-1 was used to inoculate overlays of marine agar containing the host bacterium Pseudoalteromonas marina. After overnight incubation at 30 °C the overlays were harvested and treated with chloroform (1:50 volume) at room temperature for 30 mins. This mixture was centrifuged at 4300g for 10 min and then 39,000g for 30 min at 4 °C. The phage pellet was suspended in 100 mM NaCl, 10 mM MgSO4, 50 mM Tris–HCl (pH 7.5), 0.01% gelatin, and further purified by CsCl step gradient ultra-centrifugation. φRIO-1 banded at a bouyant density of 1.49 g/ml and was harvested by tube puncture and dialysed against 50 mM Tris–HCl (pH 7.5), 200 mM NaCl and 10 mM MgCl2. The titer of the purified stock was 9.4 × 10−11 pfu/ml.
Proteomics
CsCl purified ϕRIO-1 was subjected to SDS-PAGE on a 12% reducing gel until the proteins were spread over 2 cm. The gel was stained with Coomassie Blue and imaged for densitometric estimation of the relative area and relative copy numbers of the individual bands. The gel was divided into ten slices, each of which was trypsinized and subjected to HPLC-electrospray ionization tandem mass spectrometry according to Thomas et al. (2012). During database searching to assign spectra to φRIO-1 peptides, “semi-trypsin” was specified allowing detection of peptides with non-trypsin ends, such as might be found at the N- or C-termini of the polypeptides, or at posttranslattionally cleaved sites. Additional SDS-PAGE analysis was conducted to refine the quantification as follows: A series of lanes with different loads was quantified; it was found that only the most intense peaks containing gp48 and gp47 plus gp43 were intensely enough stained that a saturation effect was observed. Quantitation of gp47 and gp43 was analyzed separately by comparing the areas of their peak to the gp48 peak in the linear zone of the saturation curve and taking gp48 as the standard at 415 copies per virion as per the canonical T=7 podoviral structure. For other bands, saturation was not an issue, and quantification of replicate gels as well as the agreement between two twelve copy standards gp51 and gp46 were used to estimate the reliability. In general, we concluded that a confidence interval corresponding to750% of each estimate would be enough to encompass the degree of variability observed.
Secondary structure prediction
Secondary structure was predicted with a locally installed version of the PSIPRED server (McGuffin et al., 2000). The underlying PSI-BLAST searches were conducted in small libraries corresponding only to prospective homologs of the respective homology group. Coiled coil prediction was done at the COILs server (Lupas et al., 1991)
Construction of φRIO-1 group profiles
After an initial PSI-BLAST search in the NCBI nr library conducted with a locally implemented version of the BLAST+ package (Camacho et al., 2008), φRIO-1 structural proteins for which a homolog had not yet been found in each of the φRIO-1-related phages were given special treatment as follows. Among the proteins encoded in the syntenous position by φRIO-1-related phages, two or more were found that could be significantly matched and extensively aligned by BLASTP. Finding at least a pair of such sequences was aided by the observation that throughout these operons, SIO1 and P12050L were generally very close in sequence, and PA11 and CW02 were generally moderately close in sequence. Those matching sequences were aligned and converted to a hidden Markov matrix (HMM) by the Sequence Alignment and Modeling system (SAM; Hughey and Krogh, 1996; Karplus et al., 1998). SAM was used instead of some other commonly used alignment systems because it has the capability to reject poorly matching sequences or segments of sequence from the alignment. Then the SAM HMM was used to search the other φRIO-1-related proteomes in the positionally biased search mode of Hardies et al. (2003). In this process, the database is reduced to just the proteins of one phage at a time, and a homolog is considered found if the protein encoded by the syntenous gene scores better (by at least a couple of orders magnitude) than any other protein encoded by the phage. The statistic associated with the biased search mode is that the probability of scoring the syntenic gene highest by chance is 1/n, were n is the number of genes in the viral genome. For these phages, that means that homology can be confirmed at syntenic loci with P<0.05, providing protection against any vulnerabilities of the search algorithm to exaggerate chance matches. The new set of sequences was similarly aligned and converted to an HMM, and the process was iterated if necessary to fill out the alignment of φRIO-1-related proteins.
Profiles of structural proteins from other phage groups
Although profiles for structural proteins of numbers of the prototypical podoviruses are found in Pfam, we were usually more successful at profile–profile matching across phage genera when we made more robust profiles by the following method. The output of a PSI-BLAST search of a joint version of the NCBI nr and env_nr libraries was aligned and converted to an HMM using SAM. The HMM was scored in a reduced database of all proteins encoded by named podoviruses retrieved from GenBank to retrieve a larger collection of sequences which were similarly aligned. If profile–profile matching using a locally installed version of HHPRED (Söding, 2005) indicated a significant match to yet a more diverged phage clade, SAM was used to incorporate the two groups into a single alignment. In a few cases where SAM (which doesn’t have a profile–profile matching mode) refused to incorporate the diverged sequences, a guide alignment derived from the HHPRED match was provided and SAM was constrained not to adjust the guide. Those special cases are referred to as “joint” HMMs in the text. The two large IVP’s and candidates for their homologs required an exception to this process, because for these sequences PSI-BLAST in the nr database was confounded by divergent matches to a variety of unrelated coiled-coil proteins. For these proteins, the initial PSI-BLAST search was limited to a database of only proteins encoded by named podoviruses in GenBank. HHpred HMMs were calibrated against the SCOP database as provided by the software package, but then checked by screening all of the combined databases from the hhpred web site to detect if there was any tendency to assign E valueso1 to families that were not resident in phage genomes (and presumed false positives). Tree construction was also used to be sure that the alignments underlying any two HMMs to be compared did not contain sequences that would more appropriately belong to the other HMM.
Profile–profile matching
Those φRIO-1 structural proteins that were identifiable at a standard web server, including the HHPRED web site, were previously listed (Hardies et al., 2013). The others were subjected to a customized version of HHPRED searching against prototypical podoviruses including an element of database reduction as follows: Based on general size, secondary structure prediction, and association with neighboring genes, one or two plausible candidates for divergent homologs in the target podovirus were proposed. The target protein families were expanded as indicated above. HHPRED-style HMMs were constructed for both φRIO-1 and target families and searched against each other with a database size of 1. Alternative protein families in the target virus were also searched, establishing that the HMM calibration procedure had correctly produced E values around 1 for nonhomologous proteins. The E-value for the proposed homolog, if « 1 was taken as the probability that the proposed homology was a chance association. These searches were done without annotation by predicted secondary structure. The alignment reported by HHPRED was annotated with secondary structure reported by PSIPRED to ask if there was conservation of secondary structure as might be expected from a homologous relationship.
Searching in named podoviruses
The statement “all podoviruses in GenBank” refers to a collection of 423 fully sequenced phages deposited in GenBank and either classified or otherwise identified as podoviruses as of July 28, 2014.
Trees
Trees were calculated using amino acid sequence aligned by SAM as indicated above. Preliminary neighbor joining trees with bootstrap support were calculated with PAUP (Swofford, 2001), and final trees were calculated by Bayesian inference using MrBayes (Ronquist et al., 2012). Gamma distributed rates in four categories were specified. Eight Monte Carlo chains of 300,000 generations each were used with Metropolis coupling, and the results in each case were found to be convergent by replicating the entire process. Among the available substitution matrixes, the blossum matrix was found to produce the maximum model probability. For relaxed clock calculations, the default independent gamma rate parameter was used. Credibility intervals quoted are highest posterior density intervals, computed and displayed using Treeannotator and FigTree from the BEAST 2 package (Bouckaert et al., 2014).
Materials available on request
Alignments and HMMs generated in this study may be acquired by request to S.C.H.
Acknowledgments
We thank Kevin Hakala and Sam Pardo for the excellent mass sepctrometry analyzes that were conducted in the UTHSCSA Institutional Mass Spectrometry Laboratory (supported in part by National Institutes of Health Grant 1S10RR025111-01 for purchase of the Orbitrap mass spectrometer). Computational analyzes were conducted in the University of Texas Health Science Center Bioinformatics facility. We thank C.S. Ahn for help in the laboratory. LWB and JAT were supported by National Institutes of Health Grant R01 AI11676. This work was supported in part by a National Research Foundation of Korea (NRF) Grant (MEST; no. 2011-0012369) and EAST-1 project funded by the Korean Government, and the BK21+ project of the Korean government.
References
- Battistuzzi FU, Feijao A, Hedges SB. A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol Biol. 2004;4:44. doi: 10.1186/1471-2148-4-44. http://dx.doi.org/10.1186/1471-2148-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black LW, Thomas JA. Condensed genome structure. Adv Exp Med Biol. 2012;726:469–487. doi: 10.1007/978-1-4614-0980-9_21. http://dx.doi.org/10.1007/978-1-4614-0980-9_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. http://dx.doi.org/10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M, Kropinski AM. The genome of the Pseudomonas aeruginosa generalized transducing bacteriophage F116. Gene. 2005;346:187–194. doi: 10.1016/j.gene.2004.11.001. http://dx.doi.org/10.1016/j.gene.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2008;10:421. doi: 10.1186/1471-2105-10-421. http://dx.doi.org/10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens SR, Gilcrease EB, Winn-Stapley DA, Schicklmaier P, Schmieger H, Pedulla ML, Ford ME, Houtz JM, Hatfull GF, Hendrix RW. The generalized tranducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J Bacteriol. 2005;187:1091–1104. doi: 10.1128/JB.187.3.1091-1104.2005. http://dx.doi.org/10.1128/jb.187.3.1091-1104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens SR, Molineux IJ. Short noncontractile tail machines: adsorption and DNA delivery by podoviruses. Adv Exp Med Biol. 2012;726:143–179. doi: 10.1007/978-1-4614-0980-9_7. http://dx.doi.org/10.1007/978-1-4614-0980-9_7. [DOI] [PubMed] [Google Scholar]
- Cerritelli ME, Wall JS, Simon MN, Conway JF, Steven AC. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: a hinged viral adhesin. J Mol Biol. 1996;260:767–780. doi: 10.1006/jmbi.1996.0436. http://dx.doi.org/10.1006/jmbi.1996.0436. [DOI] [PubMed] [Google Scholar]
- Ceyssens PJ, Hertveldt K, Ackermann HW, Noben JP, Demeke M, Volckaert G, Lavigne R. The intron-containing genome of the lytic Pseudomonas phage LUZ24 resembles the temperate phage PaP3. Virology. 2008;377:233–238. doi: 10.1016/j.virol.2008.04.038. http://dx.doi.org/10.1016/j.virol.2008.04.038. [DOI] [PubMed] [Google Scholar]
- Chang JT, Schmid MF, Haase-Pettingell C, Weigele PR, King JA, Chiu W. Visualizing the structural changes of bacteriophage Espilon15 and its Salmonella host during infection. J Mol Biol. 2010;402:731–740. doi: 10.1016/j.jmb.2010.07.058. http://dx.doi.org/10.1016/j.jmb.2010.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Weigele P, King J, Chiu W, Jiang W. Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure. 2006;14:1073–1082. doi: 10.1016/j.str.2006.05.007. http://dx.doi.org/10.1016/j.str.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Choi KH, McPartland J, Kaganman I, Bowman VD, Rothman-Denes LB, Rossmann MG. Insight into DNA and protein transport in double-stranded DNA viruses: the structure of bacteriophage N4. J Mol Biol. 2008;378:726–736. doi: 10.1016/j.jmb.2008.02.059. http://dx.doi.org/10.1016/j.jmb.2008.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau AM, Bertrand C, Letarov A, Tétart F, Kirsch HM. Molecular architecture of the T4 phage superfamily: a conserved core genomes and aplastic periphery. Virology. 2007;362:384–396. doi: 10.1016/j.virol.2006.12.031. http://dx.doi.org/10.1016/j.virol.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Cuervo A, Pulido-Cid M, Chagoyen M, Arranz R, González-García VA, Garcia-Doval C, Castón JR, Valpuesta JM, van Raaij MJ, Martin-Benito J, Carrascosa JL. Structural characterization of the bacteriophage T7 tail machinery. J Biol Chem. 2013;288:26290–26299. doi: 10.1074/jbc.M113.491209. http://dx.doi.org/10.1074/jbc.M113.491209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Liu Z, Vago F, Ren Y, Wu W, Wright ET, Serwer P, Jiang W. Visualization of uncorrelated, tandem symmetry mismatches in the internal genome packaging apparatus of bacteriophage T7. Proc Natl Acad Sci USA. 2013;110:6811–6816. doi: 10.1073/pnas.1215563110. http://dx.doi.org/10.1073/pnas.1215563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies SC, Comeau AM, Serwer P, Suttle CA. The complete sequence of marine bacteriophage VpV262 infecting Vibrio parahaemolyticus indicates that an ancestral component of a T7 viral supergroup is widespread in the marine environment. Virology. 2003;310:359–371. doi: 10.1016/s0042-6822(03)00172-7. http://dx.doi.org/10.1016/s0042-6822(03)00172-7. [DOI] [PubMed] [Google Scholar]
- Hardies SC, Hwang YJ, Hwang CY, Jang GI, Cho BC. Morphology, physiological characteristics, and complete sequence of marine bacteriophage φRIO-1 infecting Pseudoalteromonas marina. J Virol. 2013;87:9189–9198. doi: 10.1128/JVI.01521-13. http://dx.doi.org/10.1128/jvi.01521-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Margolin W, Molineux IJ, Liu J. The bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science. 2013;339:576–579. doi: 10.1126/science.1231887. http://dx.doi.org/10.1126/science.1231887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughey R, Krogh A. Hidden Markov models for sequence analysis: extension and analysis of the basic method. Comput Appl Biosci. 1996;12:95–107. doi: 10.1093/bioinformatics/12.2.95. http://dx.doi.org/10.1093/bioinformatics/12.2.95. [DOI] [PubMed] [Google Scholar]
- Jiang W, Chang J, Jakana J, Weigele P, King J, Chiu W. Structure of epsilon 15 phage reveals organization of genome and DNA packaging/injection apparatus. Nature. 2006;439:612–616. doi: 10.1038/nature04487. http://dx.doi.org/10.1038/nature04487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang I, Jang H, Oh HM, Cho JC. Complete genome sequence of Celeribacter bacteriophage P12053L. J Virol. 2012;86:8339–8340. doi: 10.1128/JVI.01153-12. http://dx.doi.org/10.1128/jvi.01153-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. http://dx.doi.org/10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Kemp P, Garcia LR, Molineux IJ. Changes in bacteriophage T7 virion structure at the initiation of infection. Virology. 2005;340:307–317. doi: 10.1016/j.virol.2005.06.039. http://dx.doi.org/10.1016/j.virol.2005.06.039. [DOI] [PubMed] [Google Scholar]
- Kostyuchenko VA, Leiman PG, Chipman PR, Kanamaru S, van Raaij MJ, Arisaka F, Mesyanzhinov VV, Rossmann MG. Three-dimensional structure of the bacteriophage T4 baseplate. Nat Struct Biol. 2003;10:688–693. doi: 10.1038/nsb970. http://dx.doi.org/10.1038/nsb970. [DOI] [PubMed] [Google Scholar]
- Kwan T, Liu J, Dubow M, Gros P, Pelletier J. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol. 2006;188:1184–1187. doi: 10.1128/JB.188.3.1184-1187.2006. http://dx.doi.org/10.1128/jb.188.3.1184-1187.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Khayat R, Li R, Prevelige PE, Potter CS, Carragher B, Johnson JE. The P22 tail machine at subnanometer resolution reveals the architecture of an infection conduit. Structure. 2009;17:789–799. doi: 10.1016/j.str.2009.04.006. http://dx.doi.org/10.1016/j.str.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne R, Noben JP, Hertveldt K, Ceyssens PJ, Briers Y, Dumont D, Roucourt B, Krylov VN, Mesyanzhinov VV, Robben J, Volckaert G. The structural proteome of Pseudomonas aeruginosa bacteriophage φKMV. Microbiology. 2006;152(2):529–534. doi: 10.1099/mic.0.28431-0. http://dx.doi.org/10.1099/mic.0.28431-0. [DOI] [PubMed] [Google Scholar]
- Lavigne R, Seto D, Mahadevan P, Ackermann HW, Kropinski AM. Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol. 2008;159:406–414. doi: 10.1016/j.resmic.2008.03.005. http://dx.doi.org/10.1016/j.resmic.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Battisti AJ, Bowman VD, Stunmeyer K, Mühlenhoff M, Gerady-Schahn R, Scholl D, Molineux IJ. The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J Mol Biol. 2007;371:836–849. doi: 10.1016/j.jmb.2007.05.083. http://dx.doi.org/10.1016/j.jmb.2007.05.083. [DOI] [PubMed] [Google Scholar]
- Liu X, Kong S, Shi M, Fu L, Gao Y, An C. Genomic analysis of freshwater cyanophage Pf-WMP3 infecting cyanobacterium Phormidium foveolarum: the conserved elements for a phage. Microb Ecol. 2008;56:671–680. doi: 10.1007/s00248-008-9386-7. http://dx.doi.org/10.1007/s00248-008-9386-7. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Q, Murata K, Baker ML, Sullivan MB, Fu C, Dougherty MT, Schmid MF, Osburne MS, Chisholm SW, Chiu W. Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat Struct Mol Biol. 2010;17:830–837. doi: 10.1038/nsmb.1823. http://dx.doi.org/10.1038/nsmb.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. http://dx.doi.org/10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. http://dx.doi.org/10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Molineux IJ. The T7 group. In: Calendar R, editor. The Bacteriophages. Second. Oxford University Press; New York, NY: 2006. pp. 227–301. [Google Scholar]
- Olia AS, Prevelige PE, Jr, Johnson JE, Cingolani G. Three-dimensional structure of a viral genome-delivery portal vertex. Nat Struct Mol Biol. 2011;18:597–603. doi: 10.1038/nsmb.2023. http://dx.doi.org/10.1038/nsmb.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer FL, Segall AM, Steward G, Seguritan V, Breitbart M, Wolven F, Azam FF. The complete genomic sequence of the marine phage Roseophage SIO1 shares homology with nonmarine phages. Limnol Ocean. 2000;45:408–418. http://dx.doi.org/10.4319/lo.2000.45.2.0408. [Google Scholar]
- Ronquist F, Teslenko P, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. http://dx.doi.org/10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Eyrich VA. EVA: large-scale analysis of secondary structure prediction. Proteins. 2001:192–199. doi: 10.1002/prot.10051. http://dx.doi.org/10.1002/prot.10051. [DOI] [PubMed]
- Seed KD, Bodi KL, Kropinski AM, Ackermann HW, Calderwood SB, Qadri F, Camilli A. Evidence of a dominant lineage of Vibrio cholerae-specific lytic bacteriophages shed by cholera patients over a 10-year period in Dhaka, Bangladesh. MBio. 2011;2:e00334–e00310. doi: 10.1128/mBio.00334-10. http://dx.doi.org/10.1128/mBio.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwer P, Hayes SJ, Zaman S, Lieman K, Rolando M, Hardies SC. Improved isolation of undersampled bacteriophages: finding of distant terminase genes. Virology. 2004;329:412–424. doi: 10.1016/j.virol.2004.08.021. http://dx.doi.org/10.1002/prot.10051. [DOI] [PubMed] [Google Scholar]
- Shen PS, Domek MJ, Sanz-Garcia E, Makaju A, Taylor RM, Hoggan R, Culumber MD, Oberg CJ, Breakwell DP, Prince JT, Belnap DM. Sequence and structural characterization of great salt lake bacteriophage CW02, a member of the T7-like supergroup. J Virol. 2012;86:7907–7917. doi: 10.1128/JVI.00407-12. http://dx.doi.org/10.1128/JVI.00407-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. http://dx.doi.org/10.1002/prot.10051. [DOI] [PubMed] [Google Scholar]
- Steven AC, Trus BL, Maizel JV, Unser M, Parry DA, Wall JS, Hainfeld JF, Studier FW. Molecular substructure of a viral receptor-recognition protein; the gp17 tail-fiber of bacteriophage T7. J Mol Biol. 1988;200:351–365. doi: 10.1016/0022-2836(88)90246-x. http://dx.doi.org/10.1016/0022-2836(88)90246-x. [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Pro-chlorococcus cyanophage genomes: signature features and ecological interpretations. PloS Biol. 2005:e144. doi: 10.1371/journal.pbio.0030144. http://dx.doi.org/10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed]
- Swofford DL. PAUP: Phylogenetic Analysis using Parsimony (and other Methods) Sinauer Associates; Sunderland: 2001. [Google Scholar]
- Tang J, Lander GC, Olia A, Li R, Casjens S, Prevelige P, Jr, Cingolani G, Baker TS, Johnson JE. Peering down the barrel of a bacteriophage portal: the genome packaging and release valve in P22. Structure. 2011;19:496–502. doi: 10.1016/j.str.2011.02.010. http://dx.doi.org/10.1016/j.str.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Hardies SC, Rolando M, Hayes SJ, Lieman K, Carroll CA, Weintraub ST, Serwer P. Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical Bacillus thuringiensis phage 0305 ϕ;8-36. Virology. 2007;368:405–421. doi: 10.1016/j.virol.2007.06.043. http://dx.doi.org/10.1016/j.virol.2007.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Weintraub ST, Wu W, Winkler DC, Cheng N, Steven AC, Black LW. Extensive proteolysis of head and inner body proteins by a morphogenetic protease in the giant Pseudomonas aeruginosa phage ϕ;KZ. Mol Microbiol. 2012;84:324–339. doi: 10.1111/j.1365-2958.2012.08025.x. http://dx.doi.org/10.1111/j.1365-2958.2012.08025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojet SN, Caumont-Sarcos A, Perrody E, Comeau AM, Krisch HM. The gp38 adhesins of the T4 superfamily: a complex modular determinant of the phages’s host specificity. Genome Biol Evol. 2011;3:674–686. doi: 10.1093/gbe/evr059. http://dx.doi.org/10.1093/gbe/evr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Morais MC, Battisti AJ, Grimes S, Jardine PJ, Anderson DL, Rossmann MG. Structural changes of bacteriophage ϕ;29 upon DNA packaging and release. EMBO J. 2006;25:5229–5239. doi: 10.1038/sj.emboj.7601386. http://dx.doi.org/10.1038/sj.emboj.7601386. [DOI] [PMC free article] [PubMed] [Google Scholar]