

Corticobasal degeneration (CBD) may be expressed as an atypical parkinsonism with a mean disease survival of about 7 years,1 the shortest reported survival being 24 months.2 We report a patient with corticobasal syndrome (CBS) and pathology-confirmed CBD whose 10-month course was manifested as a frontal alien hand evolving into agrypnia excitata.
Case report.
A 60-year-old man without family history of neurologic illnesses developed falls and worsening dexterity of his right arm over 4 months, followed by severe insomnia and episodes of intermittent truncal tremor triggered by limb movement. By 7 months, he had developed an asymmetric parkinsonism with marked rigidity, high-frequency jerky hand tremor, and hyperreflexia. He had right-hand ideomotor apraxia but no cortical sensory loss. He exhibited picking movements with his right hand, which he did not perceive as alien (video on the Neurology® Web site at Neurology.org).
His medical history included hepatitis C–associated liver cirrhosis, diagnosed 3 years previously, and ribavirin-induced peripheral neuropathy. Brain MRI showed only minimal atrophy of the left posterior frontal and anterotemporal lobes (figure, A). PET with fluorodeoxyglucose showed mild decreased metabolic activity in the posterior parietal lobes and the bilateral thalami (figure, B). Routine CSF studies had normal results, including 14-3-3 protein. Levodopa yielded no benefits. By 8 months, he was bedbound, was unable to fall or remain asleep, and appeared to be in a constant dream-like state (detailed observations by his daughter during this period are available online as supplemental correspondence). He also manifested paranoid ideation and hallucinations. He died within 10 months after symptom onset.
Figure. Selected brain MRI, fluorodeoxyglucose PET, and histopathology images.
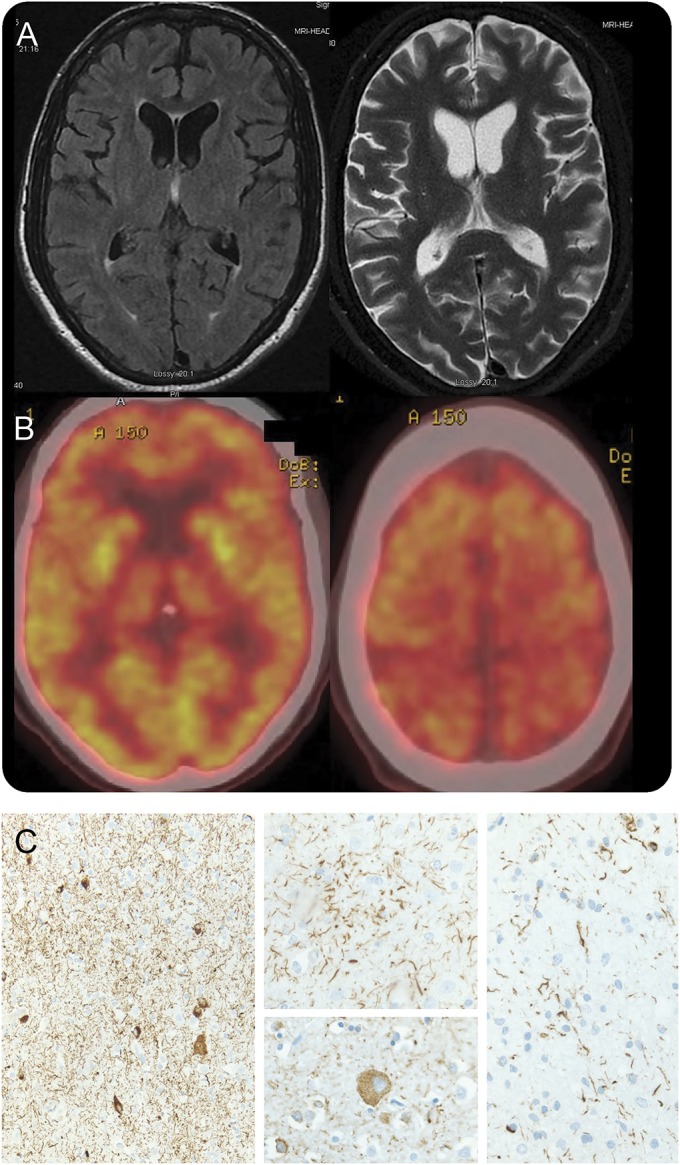
(A) Axial brain MRI demonstrates mild atrophy of the left posterior frontal and anterotemporal lobes, widening the sylvian fissure. There was no abnormal signal in T1 or T2/fluid-attenuated inversion recovery sequences. (B) Fluorodeoxyglucose PET shows mild symmetric decreased metabolic activity in the posterior parietal lobes near the vertex, as well as in the thalami. (C) Histopathology (tau immunohistochemistry) demonstrated abnormal tau deposition within neurons and glia associated with prominent thread-like deposits within gray and white matter. Frontal cortical gray matter (left; original magnification 100×). Astrocytic plaque (center top) and balloon neuron (center bottom; original magnification 400×). White matter with coiled bodies and thread-like deposits (right; original magnification 400×).
Postmortem examination.
The brain weighed 1,275 g. The left hemisphere showed mild frontal and parietal atrophy. There was moderate to severe neuronal cell loss and gliosis in the neocortex, basal ganglia, thalamus, and midbrain. Microvacuolization (spongiform change) was present within the superficial neocortex, most prominent in the middle frontal gyrus, cingulate gyrus, and inferior parietal lobule. No Lewy bodies were identified in the brain with hematoxylin & eosin staining. α-Synuclein staining was negative in the brainstem. 4R tau-immunoreactive neurons and astroglia, thread-like deposits, astrocytic plaques, and coiled bodies were found throughout the cerebral cortex, basal ganglia, subthalamic nucleus, cerebellar dentate nucleus, midbrain, pons, and medulla (figure, C). Within the thalamus, there was severe neuronal and glial tau deposition within the ventral lateral and reticular nuclei, as well as the zona incerta. There was moderate involvement of the dorsomedial nucleus, but relatively sparse involvement of the anterior (anteroventral) nucleus with only a few tau-immunoreactive neurons. Swollen achromatic balloon-like neurons were also documented. There was no associated β-amyloid staining. Tissue submitted to the National Prion Disease Pathology Surveillance Center showed no evidence of abnormal, protease-resistant prion protein. Genetic analysis on frozen tissue did not reveal mutations in MAPT exons 9 through 13. These findings were diagnostic of CBD.
Discussion.
While asymmetric levodopa-unresponsive parkinsonism with ideomotor apraxia suggested CBS, there were 2 main atypical features: (1) very rapid progression, not beyond the 1 year required by current criteria,1 suggesting a prionopathy; and (2) severe insomnia, which evolved into a state of oneiric stupor, characteristic of agrypnia excitata.3
Previous reports of very rapidly progressive CBS (death within a year) have not been due to CBD but rather to Creutzfeldt-Jakob disease (CJD).4,5 Our case suggests that CBS due to CBD pathology may exceptionally have a rapid, nearly fulminant course, leading to death in months. The associated hepatitis C–associated liver cirrhosis probably did not contribute to disease progression: there was no clinical hepatopathy or corresponding imaging or neuropathologic brain changes. The rapid progression could be related to the prominent tau pathology in the thalamus, affecting the thalamo-limbic circuit and severely disrupting sleep. Other rapidly progressive pathologies associated with thalamic involvement include prionopathies (fatal familial insomnia [FFI], CJD, and variant CJD), encephalopathy due to anti-voltage-gated potassium channel complex (VGKC) antibodies, and Wernicke encephalopathy.3
Compromise of the thalamo-limbic circuit may be the underlying cause for sleep-wake alterations characteristic of agrypnia excitata,3 due to either anatomic interruption, as in FFI, or functional blockade caused by VGKC antibodies or delirium tremens.3 A characteristic of agrypnia excitata is the recurrence of stereotyped gestures mimicking simple daily life activities, known as oneiric stupor.3 We speculate that the unusually severe burden of tau pathology in the thalamus, correlating with the hypometabolism documented by PET, may have contributed to our patient's profound insomnia. The pattern of tau deposition within the thalamus, indicating the severity of involvement by CBD, partially overlaps with areas involved by FFI, namely the dorsomedial nucleus. The anteroventral nucleus, typically severely affected in FFI,6 appeared relatively uninvolved in this case. Given the rarity of severe sleep disturbances in CBD, future studies of the relationship between thalamic involvement and sleep disorders in CBD will require a large multisite clinicopathologic study.
Our case highlights the localizing value of the frontal variant of the alien hand syndrome, where the grasping or picking behaviors are not endorsed as truly alien, as is the case with the classic (parieto-occipital) alien hand of CBS, often associated with levitation and cortical sensory deficits7,8 (table e-1).
Supplementary Material
Footnotes
Supplemental data at Neurology.org
Author contributions: Federico Rodriguez-Porcel: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval. Lindsey Lowder: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Rosa Rademakers: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval, study supervision. Thomas Ravenscroft: analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Bernardino Ghetti: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, acquisition of data, study supervision, obtaining funding. Matthew C. Hagen: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Alberto J. Espay: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision.
Study funding: No targeted funding reported.
Disclosure: F. Rodriguez-Porcel and L. Lowder report no disclosures relevant to the manuscript. R. Rademakers receives research support from the NIH (R01 NS080882, R01 NS076471, P50 NS072187, and P50 AG16574), the ALS Therapy Alliance, The Ed and Ethel Moore Alzheimer's Disease Research Program, and the Consortium for Frontotemporal Degeneration Research. Dr. Rademakers received honoraria for lectures or educational activities not funded by industry; she serves on the medical advisory board of the Association for Frontotemporal Degeneration and on the board of directors of the International Society for Frontotemporal Dementia. T. Ravenscroft reports no disclosures relevant to the manuscript. B. Ghetti is supported by NIH grant P30 AG10133. M. Hagen reports no disclosures relevant to the manuscript. A. Espay is supported by NIH grant K23MH092735; has received grant support from CleveMed/Great Lakes Neurotechnologies and the Michael J Fox Foundation; has received personal compensation as a consultant/scientific advisory board member for Abbvie, Chelsea Therapeutics, TEVA, Impax, Merz, Pfizer, Acadia, Cynapsus, Solstice Neurosciences, Eli Lilly, Lundbeck, and USWorldMeds; royalties from Lippincott Williams & Wilkins and Cambridge University Press; and honoraria from UCB, TEVA, the American Academy of Neurology, and the Movement Disorders Society. He serves as Associate Editor of Movement Disorders, Frontiers in Movement Disorders, and Journal of Clinical Movement Disorders, and on the editorial boards of Parkinsonism and Related Disorders and The European Neurological Journal. Go to Neurology.org for full disclosures.
References
- 1.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray R, Neumann M, Forman MS, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007;68:1274–1283. [DOI] [PubMed] [Google Scholar]
- 3.Provini F. Agrypnia excitata. Curr Neurol Neurosci Rep 2013;13:341. [DOI] [PubMed] [Google Scholar]
- 4.Josephs KA, Ahlskog JE, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol 2009;66:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee W, Simpson M, Ling H, McLean C, Collins S, Williams DR. Characterising the uncommon corticobasal syndrome presentation of sporadic Creutzfeldt-Jakob disease. Parkinsonism Relat Disord 2013;19:81–85. [DOI] [PubMed] [Google Scholar]
- 6.Manetto V, Medori R, Cortelli P, et al. Fatal familial insomnia: clinical and pathologic study of five new cases. Neurology 1992;42:312–319. [DOI] [PubMed] [Google Scholar]
- 7.Sarva H, Deik A, Severt WL. Pathophysiology and treatment of alien hand syndrome. Tremor Other Hyperkinet Mov 2014;4:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aboitiz F, Carrasco X, Schroter C, Zaidel D, Zaidel E, Lavados M. The alien hand syndrome: classification of forms reported and discussion of a new condition. Neurol Sci 2003;24:252–257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.