

Abstract
Many cell penetrating peptides (CPPs) fold at cell surfaces, adopting α- or β-structure that enable their intracellular transport. However, the same structural folds that facilitate cellular entry can also elicit potent membrane-lytic activity limiting their use in delivery applications. Further, a distinct CPP can enter cells via many mechanisms, often leading to endosomal entrapment. Here, we describe an intrinsically disordered peptide (CLIP6) that exclusively employs non-endosomal mechanisms to cross cellular membranes, while being remarkably biocompatible and serum stable. We show the presence of a single anionic glutamate residue is responsible for maintaining the peptide’s disordered bioactive state, defines its mechanism of cellular entry and is central to its biocompatibility. CLIP6 can deliver membrane impermeable cargo directly to the cytoplasm of cells, suggesting its broad utility for delivery of drug candidates limited by poor cell permeability and endosomal degradation.
Keywords: cell-penetrating peptide, translocation, gene expression, drug delivery, live-cell microscopy
Graphical Abstract
Disorder imparts order: CLIP6, an intrinsically disordered peptide, mediates cellular entry through non-endosomal physical translocation across the membrane. This activity, defined by its unstructured state, facilitates the delivery of membrane impermeable cargo to the interior of cells.
Intrinsically disordered proteins (IDPs) lack a well-structured three-dimensional fold in their native state.[1] Certain peptides have also been classified as intrinsically disordered and act mainly by adopting structure in response to a binding event.[1–2] For example, most cell-penetrating peptides (CPPs) exist in solution as an ensemble of random coil conformations but adopt secondary structure upon binding to cell membranes, suggesting that folding is a prerequisite to function (e.g. cellular entry).[3] Herein, we report a constitutively disordered peptide capable of translocating through cell membranes without folding, to gain direct access to the cytoplasm (Figure 1A). Further, our data suggests that the peptide’s intrinsic disorder imparts high cyto- and biocompatibility. Critical to this behavior is the presence of a key glutamate residue that prohibits peptide folding, and allows the CPP to enter cells exclusively through non-endosomal mechanisms. This unimodal entry mechanism is in stark contrast to the majority of reported CPPs, where a single distinct peptide can enter a cell through multiple mechanisms (Figure 1B). This is particularly important for drug delivery, where endosomal localization of ligated cargo leads to its degradation and excretion before reaching an intracellular target. Further, many CPPs are lytic at moderate concentrations due to their self-association in/on the membrane bilayer. These attributes ultimately limit the utility of many CPPs as effective delivery devices.
Figure 1.
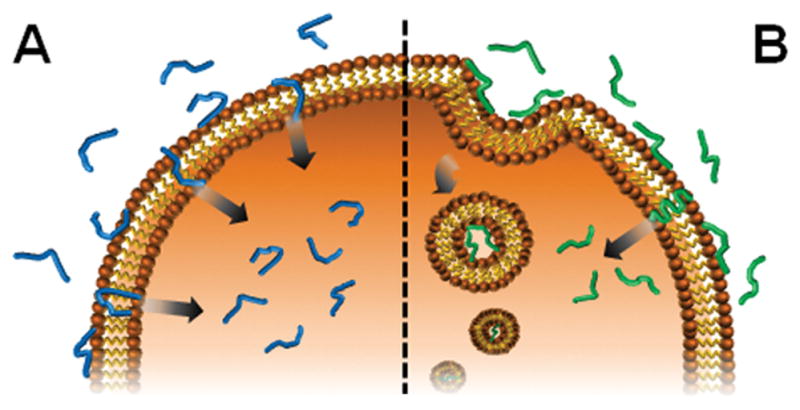
(A) Cellular internalization of the intrinsically disordered CLIP6 peptide occurs exclusively through physical translocation across the cell membrane. (B) In contrast, most CPPs enter cells via a combination of translocation and endocytosis upon membrane-induced folding.
During the study of anticancer peptides,[4] we identified an intrinsically disordered sequence, named cytosol localizing internalization peptide 6 (CLIP6) (KVRVRVRVDPPTRVRERVK-NH2; where DP is D-proline), which can directly translocate across cell membranes. Live-cell microscopy shows that the fluorescein-labeled CLIP6 peptide rapidly enters the cytoplasm of treated HeLa cells (Figure 2A), before subsequently localizing to the nucleus (Figure 2, panels B and C).
Figure 2.

(A–C, Top) Live-cell imaging of HeLa cells as a function of time after addition of 10 μM fluorescein-labeled CLIP6 peptide (scale bar = 10 μm). Associated z-stacks are also shown. (A–C, Bottom) Percentage of total cellular fluorescence measured at the membrane, within the cytoplasm or localized at the nucleus. Mechanism of CLIP6 (D) or TAT (E) uptake into A549, HeLa or OVCAR-3 cells was assessed after a 1 hour incubation with 10 μM of peptide alone (control, gray bars), under ATP depletion (open bars), or with hyperosmolar sucrose (black bars) preconditioning. Uptake results under ATP depletion and hyperosmolar sucrose conditions were compared to direct peptide incubation (gray bars) to determine statistical significance, denoted by * = p ≤ 0.05, or ** = p ≤ 0.01.
To probe the mechanism of CLIP6 uptake we performed a series of flow cytometry experiments in which different cancer cell lines are preconditioned under ATP depletion to globally inhibit endocytosis,[5] or a hyperosmolar sucrose solution to prevent clathrin-dependent mechanisms,[6] before peptide addition. Figure 2D shows that inhibition of endocytosis did not change the overall amount of peptide internalized into cells, indicating that energy-dependent mechanisms, like endocytosis, play no role in CLIP6 cellular uptake. In constrast, TAT (GRKKRRQRRRPPQ-NH2) entered cells through a mixture of physical translocation and endocytosis (Figure 2E). Interestingly, the relative contribution and magnitude of these mechanisms on TAT internalization appears to be cell-type dependent, while CLIP6 did not show any variation in its uptake mechanism across the tested cell lines.
We also observed that cells treated with CLIP6, under both serum-free conditions (Figure 2) and in the presence of serum (Figure S1), did not show punctate fluorescent organelles that would indicate peptide endosomal entrapment. Further, incubation of cells with increasing concentrations of CLIP6 led to a corresponding linear increase in intracellular fluorescence (Figure S2). This indicates that the mechanism of CLIP6 internalization is independent of its concentration, arguing against a receptor-mediated uptake mechanism. Finally, utilizing giant unilamellar vesicles as model membranes,[7] we found that CLIP6 can rapidly permeate across lipid surfaces devoid of endocytic receptors (Figure S3). Collectively, these results strongly suggest that membrane translocation solely mediates intracellular uptake of CLIP6, allowing for immediate bioavailability of the peptide within the cytoplasm of treated cells en route to the nucleus.
Like many other cationic CPPs, CLIP6 contains a large number of arginine residues in its sequence.[8] The importance of arginine on the cell-penetrating ability of the peptide was studied by incubating cells with a family of CLIP6 analogues in which this residue was systematically substituted with lysine (Figure S4 and Table S1). Results show a successive reduction in cellular uptake as arginine content is incrementally decreased, with ~50% of the peptide’s internalization potential lost when all of the arginines are replaced. Although important to cellular transport, high arginine content can also increase the membrane lytic behavior of CPPs.[9] Thus, we next explored the cytotoxic potential of CLIP6 towards A549 lung carcinoma cells (Figure 3A). Remarkably, the peptide was well-tolerated by these cells, even at very high concentrations (IC50 > 1mM). Close examination of CLIP6’s sequence reveals a glutamic acid residue occupying a position that disrupts what would otherwise be a uniform (AB)n amino acid periodicity characteristic of β–sheet rich structures (A and B are polar and non-polar residues, respectively).[10] Importantly, it is well known that many β-sheet amphiphiles have the potential to be membrane lytic.[4, 11] Thus, we surmised that disruption of a possible (AB)n-repeat by the glutamate inhibits the ability of CLIP6 to adopt β-structure, and in turn limits any membrane lytic behavior. To test this, we prepared a control sequence (CLIPΔ) where the glutamate at position 15 is exchanged for valine (Table S1), thereby restoring β-sheet periodicity and creating a uniform hydrophobic face should the peptide fold into an amphiphilic hairpin. This analogue showed marked cytotoxicity, with an IC50 of ~3 μM (Figure 3A). Circular dichroism spectroscopy performed in the presence of model lipid membranes showed that CLIP6 remains disordered, while CLIPΔ displays a strong β-sheet signal characterized by a minima in mean residue elipticity at 218 nm, indicating that the control peptide folds upon binding to the negatively-charged lipid vesicles (Figure 3B). Both CLIP6 and CLIPΔ remain unfolded in buffer alone, or in the presence of neutral vesicles (Figure S5). Taken together, these results suggest that the inability of the glutamate-containing CLIP6 peptide to fold at cellular membranes contributes to its favorable cytocompatability.
Figure 3.

Effect of glutamic-acid on the toxicity and folding of the CLIP6 peptide. (A) Cytotoxicity of CLIP6 (●) and CLIPΔ (○) towards A549 cells after 24 hours of incubation. (B) Circular dichroism spectrum demonstrating that the CLIPΔ (○) peptide displays a β-sheet conformation in the presence of negatively-charged lipid vesicles, while CLIP6 (●) remains unfolded.
Interestingly, mechanistic studies also showed that, like TAT, the toxic CLIPΔ peptide enters cells through a mixture of physical translocation and endocytosis (Figure S6). Thus, CLIP6’s glutamate residue not only limits its membrane-lytic activity, but also reduces the peptide’s propensity to be internalized by endocytic mechanisms. One explanation for this is that binding of cationic CPPs to anionic cell-surface glycans leads to their endocytic uptake following proteoglycan internalization.[5, 12] It is possible that the negatively charged glutamate side chain limits electrostatic binding of CLIP6 to anionic glycans, allowing the peptide to directly access and translocate across the cell membrane.
The exact mechanism by which CLIP6 interpolates into the lipid membrane and enters cells is the subject of on-going work. However, elegant studies by Engelman and co-workers to elucidate the molecular events that govern membrane translocation of the pH (low) insertion peptide (pHLIP) may provide clues.[13] Here, protonation of two aspartic acid residues in the pHLIP sequence, following its adsorption to acidic cancer cell surfaces, increases the peptide’s hydrophobicity and promotes its insertion into the lipid membrane. In the context of CLIP6, a similar mechanism of glutamate protonation may facilitate interpolation of the peptide into the lipid bilayer.
To explore the utility of CLIP6 for delivery applications, we performed a series of in vitro and in vivo compatibility studies, and evaluated the peptide’s serum stability. First, we compared the toxicity and hemolytic potential of CLIP6 to five common cationic CPPs. Table 1 shows that CLIP6 was non-toxic towards both A549 and MCF-7 cells, which tolerated concentrations of the peptide as high as 1 mM, while HeLa cells were somewhat more sensitive. The arginine-rich R8 and TAT sequences were also relatively benign. In contrast, the remaining CPPs were cytotoxic in the low micromolar range. Next, the hemolytic potential of each CPP was evaluated by incubating the peptides with freshly isolated human RBCs. CLIP6 was weakly hemolytic, with an EC50 value ~2–3 folds higher than R8, TAT and Penetratin. Further, CLIP6 was orders of magnitude less hemolytic than both MAP and Transportan. Next, in vivo toxicity of CLIP6 was assessed by measuring its maximum tolerable dose (MTD) in mice. Figure 4A shows that animals tolerated up to 30 mg/kg of CLIP6, beyond which the mice became pale and lethargic. In contrast, 15 mg/kg was determined to be the MTD for both the CLIPΔ peptide and TAT. Animals dosed with these peptides above the MTD immediately experienced spasms. Finally, incubation of CLIP6 in a 25% serum solution for 8 hours resulted in ~60% of the peptide being proteolytically degraded, with a corresponding half-life of 5.0 hours (Figure 4B). Under identical conditions, the TAT peptide was rapidly degraded, with a half-life of approximately 1.7 hours. The significant serum stability of CLIP6, while surprising given the peptide’s intrinsic disorder, is likely due to the D-proline residue contained within its sequence which is well known to enhance the proteolytic stability of peptides.[14]
Table 1.
Toxicity and hemolysis of CPPs.
| CPP | Sequence | IC50 [μM]
|
EC50 [μM]
|
||
|---|---|---|---|---|---|
| A549 | HeLa | MCF-7 | RBCs | ||
| CLIP6 | KVRVRVRVDPPTRVRERVK-NH2 | >1000 | 263.0 | >1000 | 758.6 |
| TAT | GRKKRRQRRRPPQ-NH2 | >1000 | >1000 | 602.6 | 269.2 |
| R8 | RRRRRRRR-NH2 | >1000 | 269.2 | 169.8 | 370.8 |
| Penetratin | RQIKIWFQNRRMKWKK-NH2 | 60.8 | 29.6 | 33.5 | 416.9 |
| MAP | KLALKLALKALKAALKLA-NH2 | 2.1 | 1.0 | 1.8 | 14.2 |
| Transportan | GWTLNSAGYLLGKINLKALAALAKKIL-NH2 | 2.5 | 1.3 | 1.4 | 1.3 |
Figure 4.
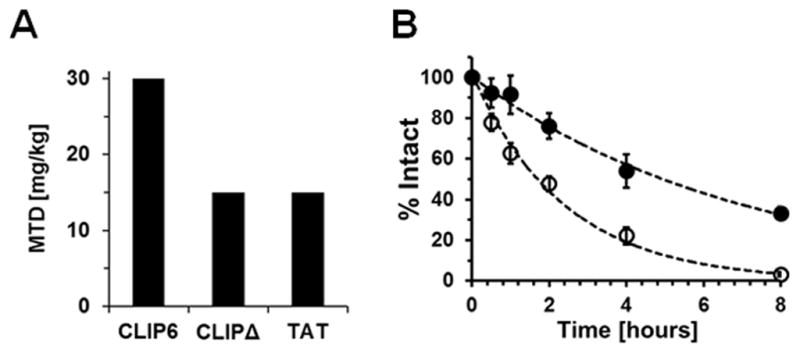
(A) Maximum tolerable dose (MTD) of the CLIP6, CLIPΔ and TAT peptides following intravenous administration to mice. (B) Serum stability of CLIP6 (t1/2 = 5.0h) and TAT (t1/2 = 1.7h) in a 25% serum solution as a function of time.
We next assessed the ability of CLIP6 to deliver membrane impermeable molecules to the cytoplasm of cells employing a ligand-dependent CreER/LoxP-reporter transgenic mouse embryonic fibroblast (MEF) system (Figure 5A).[15] In the abscence of ligand, these cells fluoresce red due to constitutive expression of tdTomato (tdT), and concomitant silence of Enhanced Green Fluorescent Protein (EFGP) expression. However, binding of tamoxifen, or its analogue 4-hydroxycyclofen (4-OHC), to cytoplasmic CreER results in nuclear translocation of the chimeric protein. Ligand-bound CreER then excises the tdT gene leading to expression of the downstream EGFP locus upon site-specific recombination. Thus, this in vitro system provides a simple red-to-green shift in intracellular fluorescence that can be used to monitor cytosolic delivery of a small molecule ligand.
Figure 5.
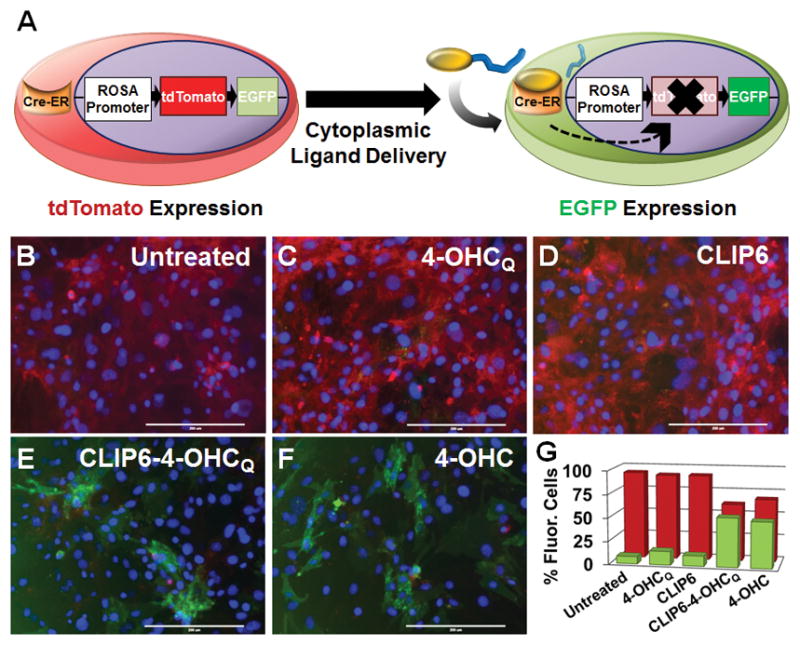
(A) Ligand-mediated CreER activation in Rosa26-driven tdT/EGFP MEF cells. Delivery of a membrane-impermeable 4-OHCQ (yellow) small molecule by CLIP6 (blue) leads to CreER-mediated recombination at LoxP sites (→) and subsequent expression of EGFP. Images of untreated MEFs (B), or those incubated with the cell impermeable 4-OHCQ ligand (C), peptide alone (D), the CLIP6-4-OHCQ conjugate (E), or the 4-OHC membrane permeable positive control (F) (20× magnification, scale bar = 200 μm). Individual fluorescence channels used to compile the merged images can be found in Figure S7. (G) Flow cytometry quantification of the percentage of cells displaying tdT (red) or EGFP (green) intracellular fluorescence following treatment with the indicated compounds. Standard deviation in data shown is <1.5% for each treatment condition.
Importantly, 4-OHC is cell permeable and thus serves as a positive control for cytoplasmic delivery. However, as will be shown, N-quaternized analogues of 4-OHC are not permeable, but are still capable of binding the ER protein. We prepared an N-quaternized analogue of 4-OHC, named 4-OHCQ, and ligated it to the N-terminus of CLIP6 via a cytosol cleavable ester linkage (see Supporting Information). Before testing the CLIP6-4-OHCQ conjugate, we first incubated MEFs with free 4-OHCQ to confirm that it was not cell permeable. As expected, treated cells displayed high levels of tdT fluorescence, with negligible EFGP expression, similar to the negative controls (Figure 5, panels B,C, D and G). However, incubation of MEFs with CLIP6-4-OHCQ led to significant recombination and EGFP expression, with a concominant reduction in tdT fluorescence (Figure 5, panels E and G). This profile was similar to cells treated with the permeable 4-OHC positive control (Figure 5, panels F and G). Collectively, these results demonstrate that CLIP6 is capable of successfully delivering the membrane impermeable 4-OHCQ ligand into the cytoplasm of MEF cells, leading to CreER activation and transcriptional regulation.
In summation, we have identified an intrinsically disordered peptide that exclusively employs non-endosomal mechanisms to enter cells. Critical to its constitutive structural disorder is the presence of an anionic glutamate residue that disrupts an otherwise ideal (AB)n β-repeat and prohibits the peptide from folding at cell surfaces into a lytic conformation. As a result, the peptide is remarkably biocompatible, permitting its use at high concentrations in vitro and in vivo. Furthermore, can CLIP6 can shuttle ligated cargo directly into the cytoplasm of cells, suggesting its broadly utility across multiple delivery applications.
Supplementary Material
Acknowledgments
We thank Drs. Susan Mackem and Jianjian Zhu who established the MEF transgenic cell line. Research funding was provided by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iesmantavicius V, Dogan J, Jemth P, Teilum K, Kjaergaard M. Angew Chem Int Ed. 2014;53:1548–1551. doi: 10.1002/anie.201307712. [DOI] [PubMed] [Google Scholar]
- 3.Eiríksdóttir E, Konate K, Langel Ü, Divita G, Deshayes S. Biochim Biophys Acta Biomembr. 2010;1798:1119–1128. doi: 10.1016/j.bbamem.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Sinthuvanich C, Veiga AS, Gupta K, Gaspar D, Blumenthal R, Schneider JP. J Am Chem Soc. 2012;134:6210–6217. doi: 10.1021/ja210569f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. J Biol Chem. 2003;278:31192–31201. doi: 10.1074/jbc.M303938200. [DOI] [PubMed] [Google Scholar]
- 6.Massodi I, Bidwell GL, III, Raucher D. J Controlled Release. 2005;108:396–408. doi: 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Mishra A, Lai GH, Schmidt NW, Sun VZ, Rodriguez AR, Tong R, Tang L, Cheng J, Deming TJ, Kamei DT, Wong GCL. Proc Natl Acad Sci. 2011;108:16883–16888. doi: 10.1073/pnas.1108795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Nischan N, Herce HD, Natale F, Bohlke N, Budisa N, Cardoso MC, Hackenberger CPR. Angew Chem Int Ed. 2015;54:1950–1953. doi: 10.1002/anie.201410006. [DOI] [PubMed] [Google Scholar]; b) Yamazaki CM, Nakase I, Endo H, Kishimoto S, Mashiyama Y, Masuda R, Futaki S, Koide T. Angew Chem Int Ed. 2013;52:5497–5500. doi: 10.1002/anie.201301266. [DOI] [PubMed] [Google Scholar]; c) Mitchell DJ, Steinman L, Kim DT, Fathman CG, Rothbard JB. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 9.a) Saar K, Lindgren M, Hansen M, Eiríksdóttir E, Jiang Y, Rosenthal-Aizman K, Sassian M, Langel Ü. Anal Biochem. 2005;345:55–65. doi: 10.1016/j.ab.2005.07.033. [DOI] [PubMed] [Google Scholar]; b) Tünnemann G, Cardoso MC, Castanho M. Membrane-Active peptides: methods and results on structure and function. 2009:331–362. [Google Scholar]
- 10.Xiong H, Buckwalter BL, Shieh HM, Hecht MH. Proc Natl Acad Sci. 1995;92:6349–6353. doi: 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Gupta K, Jang H, Harlen K, Puri A, Nussinov R, Schneider JP, Blumenthal R. Biophys J. 2013;105:2093–2103. doi: 10.1016/j.bpj.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kokryakov VN, Harwig SSL, Panyutich EA, Shevchenko AA, Aleshina GM, Shamova OV, Korneva HA, Lehrer RI. FEBS Lett. 1993;327:231–236. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]; c) Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J. Cell Mol Life Sci. 2006;63:1294–1313. doi: 10.1007/s00018-005-5540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Christianson HC, Belting M. Matrix Biol. 2014;35:51–55. doi: 10.1016/j.matbio.2013.10.004. [DOI] [PubMed] [Google Scholar]; b) Madani F, Lindberg S, Langel Ü, Futaki S, Gräslund A. J Biophys. 2011;2011 doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Andreev OA, Karabadzhak AG, Weerakkody D, Andreev GO, Engelman DM, Reshetnyak YK. Proc Natl Acad Sci. 2010;107:4081–4086. doi: 10.1073/pnas.0914330107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Proc Natl Acad Sci. 2008;105:15340–15345. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong SY, Oh JE, Lee KH. Biochem Pharmacol. 1999;58:1775–1780. doi: 10.1016/s0006-2952(99)00259-2. [DOI] [PubMed] [Google Scholar]
- 15.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.