

Summary
BACE1 is the major drug target for Alzheimer's disease, but we know surprisingly little about its normal function in the CNS. Here, we show that this protease is critically involved in semaphorin 3A (Sema3A)-mediated axonal guidance processes in thalamic and hippocampal neurons. An active membrane-bound proteolytic CHL1 fragment is generated by BACE1 upon Sema3A binding. This fragment relays the Sema3A signal via ezrin-radixin-moesin (ERM) proteins to the neuronal cytoskeleton. APH1B-γ-secretase-mediated degradation of this fragment stops the Sema3A-induced collapse and sensitizes the growth cone for the next axonal guidance cue. Thus, we reveal a cycle of proteolytic activity underlying growth cone collapse and restoration used by axons to find their correct trajectory in the brain. Our data also suggest that BACE1 and γ-secretase inhibition have physiologically opposite effects in this process, supporting the idea that combination therapy might attenuate some of the side effects associated with these drugs.
Graphical abstract
Introduction
Beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) and γ-secretase are responsible for the generation of amyloid-beta (Aβ) peptide from Aβ precursor protein (APP) in Alzheimer's disease (AD) (De Strooper et al., 1998; Vassar et al., 1999). Remarkably little is known about the physiological functions of these proteases, and a major clinical trial with γ-secretase inhibitors has dramatically failed for unexplained reasons (De Strooper, 2014). Whereas optimism about the tractability of BACE1 in the clinic prevails, Bace1−/− mice show complex cognitive and behavioral phenotypes, suggesting the existence of other important substrates besides APP (Dominguez et al., 2005; Harrison et al., 2003; Laird et al., 2005; Savonenko et al., 2008; Willem et al., 2006). One BACE1 substrate, neuregulin-1 type III, has been implicated in myelination (Hu et al., 2006; Willem et al., 2006) and muscle spindle development (Cheret et al., 2013), but this observation cannot fully explain the complex cognitive phenotypes in Bace1−/− adult mice. Recently, a long list of potential BACE1 substrates was discovered, several of which are involved in axonal guidance, neurite outgrowth, and synapse formation (Kuhn et al., 2012; Zhou et al., 2012).
Although now most of the physiological substrates of BACE1 are likely identified, it should be mentioned that, for none of the substrates, including neuregulin-1 type III, it is known how BACE1 processing is exactly linked to function, i.e., whether it ends a signaling function, whether any of the fragments are involved in function, or whether the BACE1 processing is only important for the turnover and membrane shedding of the substrates. Additionally, we do not know whether BACE1 and γ-secretase are active in similar physiological pathways. This question is especially important when considering combinatory therapies. To address those questions, we focus here on the processing of the neural cell adhesion molecule close homolog of L1 (CHL1). CHL1 is of particular interest because both Chl1−/− and Bace1−/− mice present abnormal axonal guidance of hippocampal mossy fibers and olfactory sensory neurons in adult brain (Cao et al.,2012; Hitt et al., 2012; Rajapaksha et al., 2011). Additionally, both mice show impaired cognitive function, aberrant emotional reactivity, and defective sensorimotor coordination (Dominguez et al., 2005; Montag-Sallaz et al., 2002; Pratte et al., 2003).
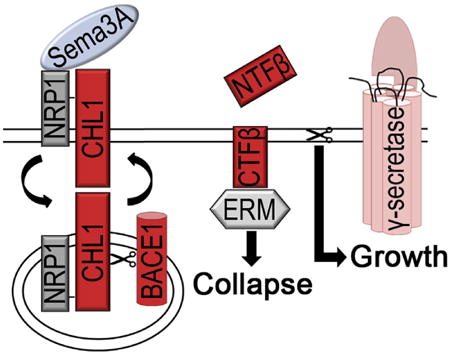
Abnormal projections of thalamocortical circuits are associated with the lack of response of Chl1−/− thalamic neurons to semaphorin 3A (Sema3A) (Schlatter et al., 2008; Wright et al., 2007). Sema3A is an axonal guidance molecule that belongs to the class III secreted semaphorins known to induce growth cone collapse in culture and to affect axon guidance, neuronal migration, and synapse formation in diverse brain circuits (Bagnard et al., 2001; Sahay et al., 2005; Wright et al., 2007). Sema3A binds to the neuropilin 1 (NRP1)/plexin A receptor, which recruits CHL1 (Wright et al., 2007). How exactly the extracellular binding of Sema3A induces intracellular cytoskeleton alterations and growth cone collapse remains elusive. In the present study, we show that the consecutive processing of CHL1 by BACE1 and APH1B-γ-secretase regulates growth cone collapse and recovery. The CHL1CTFβ produced in the course of this process links Sema3A binding to actin alterations via its interactions with ERM (ezrin, radixin, and moesin) proteins. APH1B-γ-secretase is needed to degrade this fragment and to prepare the growth cone for a new round of axonal pathfinding. Thus, BACE1 and γ-secretase work co-operatively but in an opposite way in this signaling pathway.
Results
BACE1 Is Required for Sema3A-Induced Growth Cone Collapse in Thalamic Neurons
CHL1 is well known as a subunit of the Sema3A receptor, NRP1/ plexin A (Wright et al., 2007). Sema3A induces growth cone collapse of thalamic neurons and is crucial for thalamocortical axon pathfinding (Bagnard et al., 2001; Wright et al., 2007). To investigate the participation of BACE1 in this process, we studied the effect of Bace1 genetic deletion and inhibition on directed Sema3A-mediated thalamic axonal outgrowth. Thalamic explants were co-cultured with Sema3A-secreting aggregates of COS-1 cells, and the response to Sema3A was evaluated by the axonal growth pattern on the proximal (P) and distal (D) side of the thalamic explant toward the COS-1 aggregate (Figure 1A1). In WT thalamic explants, axons grew primarily from the distal side of the Sema3A-secreting aggregate, indicating strong sensitivity to this repellant (Figures 1A2 and 1B). Surprisingly, Bace1−/−, Chl1−/−, and BACE1-inhibitor-treated thalamic explants showed comparable proximal and distal axonal growth in this assay (Figures 1A4–1A6 and 1B). Thus, BACE1 and CHL1 are both required for directed Sema3A-mediated axonal growth of thalamic neurons. Note that the P/D ratio measured for WT axons growing in the presence of non-transfected COS-1 aggregated is slightly higher than 1 (Figure 1B). This phenomenon was observed before and associated with possible unknown attractive forces between the COS cells and the growing axons (Romi et al., 2014).
Figure 1. BACE1 Is Required for Sema3A-Induced Growth Cone Collapse in Co-culture Explants and Thalamic Neurons.
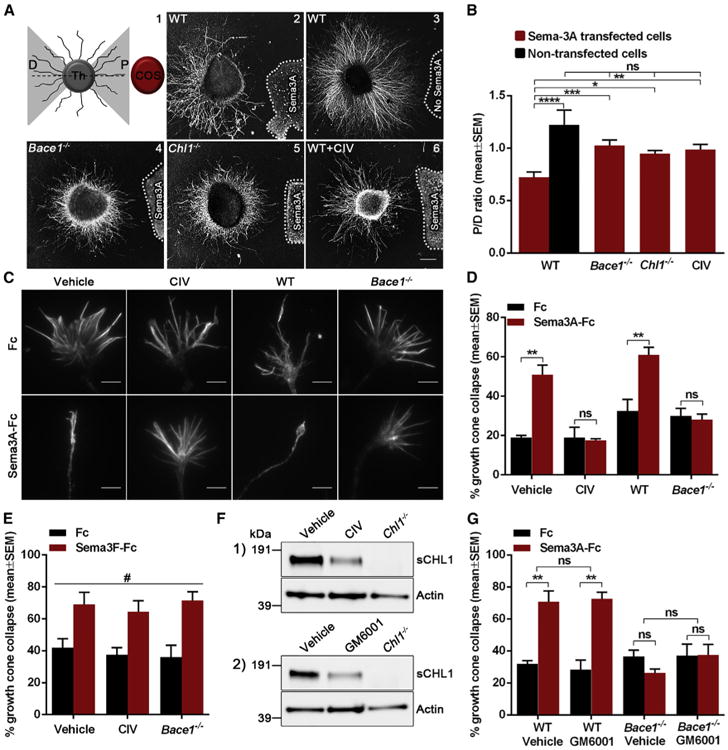
(A and B) Analysis of axonal sensitivity to Sema3A where WT (n = 17; A2), Bace1−/− (n = 18; A4), Chl1−/− (n = 15; A5), and BACE1-inhibitor-treated (1 mM CIV; n = 15; A6) thalamic explants were co-cultured with Sema3A-secreting aggregates of COS-1 cells. WT thalamic explants were also co-cultured with non-transfected COS-1 aggregates (n = 7; A3). The level of Sema3A-induced repulsion is represented by a P/D ratio, which compares axonal growth on proximal (P) and distal (D) side of the thalamic explant toward the COS aggregate (A1).
(C and D) Analysis of Sema3A-induced growth cone collapse in WT, BACE1-inhibitor-treated (1 μM CIV), and Bace1−/− thalamic neurons.
(E) Quantification of Sema3F-induced growth cone collapse in WT, BACE1-inhibitor-treated (1 μM CIV), and Bace1−/− thalamic neurons.
(F) Western blot analysis of WT thalamic neurons treated with 1 μM CIV (1) or with 50 μM GM6001 (2).
(G) Quantification of Sema3A-induced growth cone collapse in WT and Bace1−/ − thalamic neurons treated with 50 μM GM6001.
Results are presented as mean ± SEM. The scale bars represent 200 μm (explants) and 5 μm (growth cones). See also Figure S1.
Next, we treated primary cultures of thalamic neurons with Sema3A-Fc and visualized growth cones using phalloidin staining (Figures 1C and 1D). The robust growth cone collapse in WT neurons was abolished after treatment with the BACE1 inhibitor (CIV) or after genetic deletion of BACE1 expression, suggesting an essential role for BACE1 in Sema3A-induced growth cone collapse (Figures 1C and 1D). In contrast, BACE1 was not needed for semaphorin 3F (Sema3F)-induced growth cone collapse (Figure 1E), which is mediated by neuropilin 2 independently of CHL1 (Sahay et al., 2003).
In BACE1-inhibitor-treated (CIV) and Bace1−/− thalamic neurons, proteolytic shedding of CHL1 was reduced (Figures S1A–S1D), whereas NRP1 and plexin A expression was not affected (Figure S1A2). Because CHL1 is also cleaved by ADAM8 and other metalloproteases at different cleavage sites (Naus et al., 2004), we wondered whether blocking this processing would have similar effects to BACE1 inhibition on the Sema3A-induced growth cone collapse. Although a comparable decrease in the levels of CHL1 soluble fragments was seen in thalamic neurons treated with the metalloprotease inhibitor GM6001 and with the BACE1 inhibitor CIV (Figure 1F), GM6001 had no effect on Sema3A-mediated growth cone collapse (Figure 1G). Therefore, BACE1, which is located in presynaptic endosomal compartments together with CHL1 (Hitt et al., 2012), is specifically required for Sema3A-mediated growth cone collapse in thalamic neurons.
The Processing of CHL1 by BACE1 Is Required for Sema3A-Induced Growth Cone Collapse in Thalamic Neurons
The similarity between Bace1 and Chl1 knockout growth cone collapse phenotypes suggests that deficient BACE1 cleavage of CHL1 results in CHL1 loss of function. First, we investigated whether BACE1 processing of CHL1 was induced by Sema3A. The levels of soluble CHL1 were indeed increased after 1 hr treatment with Sema3A-Fc compared to control Fc whereas the processing of APP by BACE1 was not affected (Figures 2A–2D), indicating that CHL1 cleavage by BACE1 is induced by Sema3A. Next, we investigated to what extent misprocessing of CHL1 by BACE1 would lead to defective Sema3A-induced growth cone collapse. As shown previously (Wright et al., 2007), Chl1−/− thalamic neurons displayed a deficient growth cone collapse response to Sema3A-Fc that was rescued by expressing full-length CHL1 (Fl-Chl1) (Figures 2F and 2G). However, this rescue was not observed in the presence of BACE1 inhibitor (Figures 2F and 2G). Furthermore, introducing a previously characterized processing mutation at the BACE1 cleavage site of CHL1 (Zhou et al., 2012) (Chl1D1062H; Figure 2E) neutralized the rescuing effect of Fl-Chl1 (Figures 2F and 2G). This mutant showed a dominant-negative effect when transfected into WT thalamic neurons (Figures S2A and S2B) probably by replacing WT-CHL1 in the NRP1/plexin A/CHL1 complex (Wright et al., 2007) and supporting the conclusion that Fl-CHL1 has to be cleaved by BACE1 to play its role in this process.
Figure 2. The Processing of CHL1 by BACE1 Is Required for Sema3A-Induced Growth Cone Collapse in Thalamic Neurons.
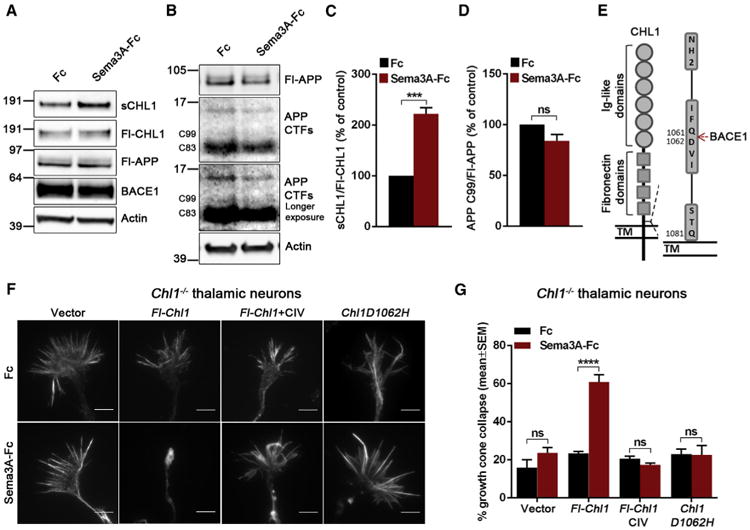
(A and B) Western blot analysis of the conditioned medium and cell lysates from WT thalamic neurons in response to 5 nM Sema3A-Fc.
(C and D) Quantification of the western blot analysis for soluble CHL1 (sCHL1) and APP C-terminal fragment β (C99).
(E) Schematic representation of BACE1 cleavage site on CHL1 (Gln1061/Asp1062).
(F and G) Analysis of Sema3A-induced growth cone collapse in Chl1−/− thalamic neurons transfected with empty vector, Fl-Chl1, and Fl-Chl1 and treated with 1 μM CIV or with non-cleavable Chl1D1062H.
Results are presented as mean ± SEM. The scale bars represent 5 μm. See also Figure S2.
CHL1CTFβ Fragment Is Required for Sema3A-Induced Growth Cone Collapse in Thalamic Neurons
We next transfected Chl1−/− neurons with CHL1 constructs encoding the proteolytic products generated by BACE1 processing, i.e., the secreted CHL1NTFβ (N-terminal fragment generated by BACE1) or the membrane-bound CHL1CTFβ (C-terminal fragment generated by BACE1). Surprisingly, the collapse response was rescued with Chl1CTFβ to the same extent as Fl-Chl1 (Figures 3A and 3B), whereas Chl1NTFβ had no effect. In Bace1−/−, Fl-Chl1 did not rescue the deficiency whereas Chl1CTFβ expression alone was sufficient to restore Sema3A-induced growth cone collapse (Figures 3A and 3C). We confirmed these rescue experiments in WT neurons treated with BACE1 inhibitor (Figures 3A and S3A). These constructs had no effect on Sema3A-induced growth cone collapse of WT neurons (Figures 3A and S3B). Thus, membrane-bound CHL1CTFβ is necessary and sufficient for Sema3A-induced growth cone collapse in thalamic axons.
Figure 3. CHL1CTFβ Fragment and Its ERM Recruitment Domain Are Required for Sema3A-Induced Growth Cone Collapse in Thalamic Neurons.
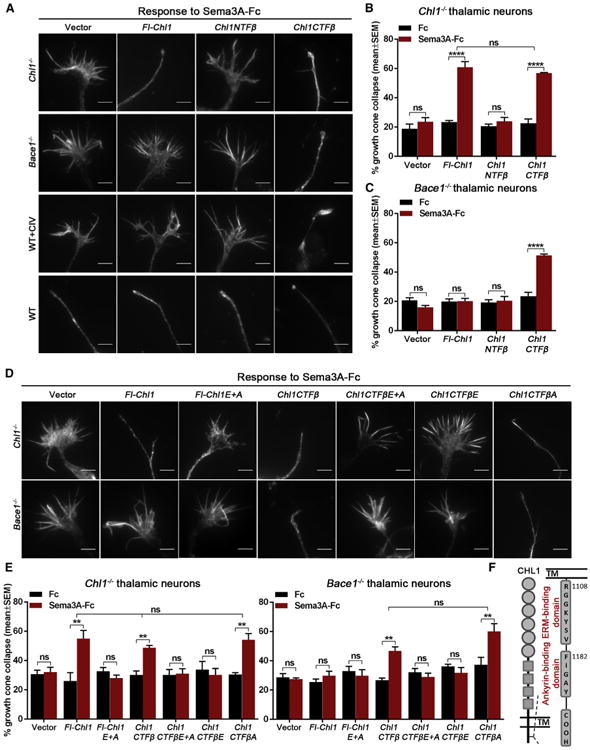
(A) Representative images of Chl1−/−, Bace1−/−, BACE1 inhibitor-treated (1 μMCIV), and WT growth cones co-transfected with GFP and empty vector, Fl-Chl1, Chl1NTFβ, or Chl1CTFβ in response to 5 nM Sema3A-Fc.
(B and C) Quantification of Sema3A-induced growth cone collapse in Chl1−/− (B) and Bace1−/− thalamic neurons co-transfected with empty vector, Fl-Chl1, Chl1NTFβ, or Chl1CTFβ (C).
(D and E) Analysis of Sema3A-induced growth cone collapse in Chl1−/− and Bace1−/− thalamic neurons co-transfected with GFP and empty vector, Fl-Chl1, CHL1 ERM and ankyrin double mutant (Fl-Chl1 E+A), Chl1CTFβ, CHL1CTFβ ERM and ankyrin double mutant (Chl1CTFβE+A), CHL1CTFβ ERM (Chl1CTFβE), or CHL1CTFβ ankyrin (Chl1CTFβA).
(F) Schematic representation of CHL1 C-terminal ERM- and ankyrin-binding sites.
Results are presented as mean ± SEM. The scale bars represent 5 μm. See also Figure S3.
The ERM Recruitment Domain of CHL1CTFβ Is Required for Sema3A-Induced Growth Cone Collapse in Thalamic Neurons
ERM (ezrin, radixin, and moesin) proteins directly influence actin dynamics in growth cone filopodia by regulating the actin polymerization state (Ramesh, 2004). In order to understand the pathway by which CHL1 influences cytoskeletal growth cone dynamics, we mutated the known binding sites for the ERM (E) and ankyrin (A) domains in Fl-CHL1 (Fl-Chl1E+A; Figure 3F) and in CHL1CTFβ (Chl1CTFβE+A) as described (Schlatter et al., 2008). In contrast to WT Fl-CHL1 and CHL1CTFβ, the double mutants were unable to rescue the Sema3A-induced growth cone collapse response in Chl1−/− neurons (Figures 3D and 3E). The ankyrin domain single mutant (Chl1CTFβA) rescued this response, but the ERM domain single mutant (Chl1CTFβE) was not effective (Figures 3D and 3E). Moreover, Chl1CTFβA, but not Chl1CTFβE, rescued the defect in Bace1−/− neurons. Taken together, the results suggest that ERM, but not ankyrin, binding to CHL1CTFβ is required for Sema3A-induced growth cone collapse in thalamic neurons.
CHL1CTFβ Levels Are Modulated by γ-Secretase
The question arises as to how the growth cone collapse signal emitted by CHL1CTFβ generation is terminated. We found that CHL1CTFβ levels are modulated by γ-secretase activity (Figure 4A). Similar to APP CTFs, a strong accumulation of CHL1CTFβ levels was observed when primary mixed brain neuronal cultures were treated with a γ-secretase inhibitor (DAPT), suggesting that CHL1CTFβ might be a γ-secretase substrate. Moreover, inhibition of γ-secretase strongly enhanced the Sema3A-induced but also the basal growth cone collapse of thalamic neurons (Figures 4B and 4C). We next tested the γ-secretase inhibitor in Chl1−/− and Bace1−/− thalamic neuronal cultures, which do not produce CHL1CTFβ. Addition of the γ-secretase inhibitor to these neurons did not alter their growth cone collapse response (Figure 4D). As expected, transfection of Chl1−/− and Bace1−/− thalamic neurons with Chl1CTFβ restored the response toward γ-secretase inhibition (Figure 4D). These results proof that γ-secretase inactivates the CHL1CTFβ signals that regulate growth cone collapse. This evidence was validated by live-cell imaging experiments monitoring Sema3A-induced growth cone collapse and recovery in the presence or absence of the γ-secretase inhibitor. In these studies, Sema3A was added to cultured WT thalamic neurons for 30 min to induce growth cone collapse and neurite retraction and was then removed. Within 30 min of Sema3A washout, WT thalamic neurite growth began to recover. In contrast, neurons treated with the γ-secretase inhibitor continued to retract their neurites (Figures 4F and 4G; Movies S1 and S2).
Figure 4. APH1B-γ-Secretase Is Required to Stop the Sema3A-Induced Growth Cone Collapse.
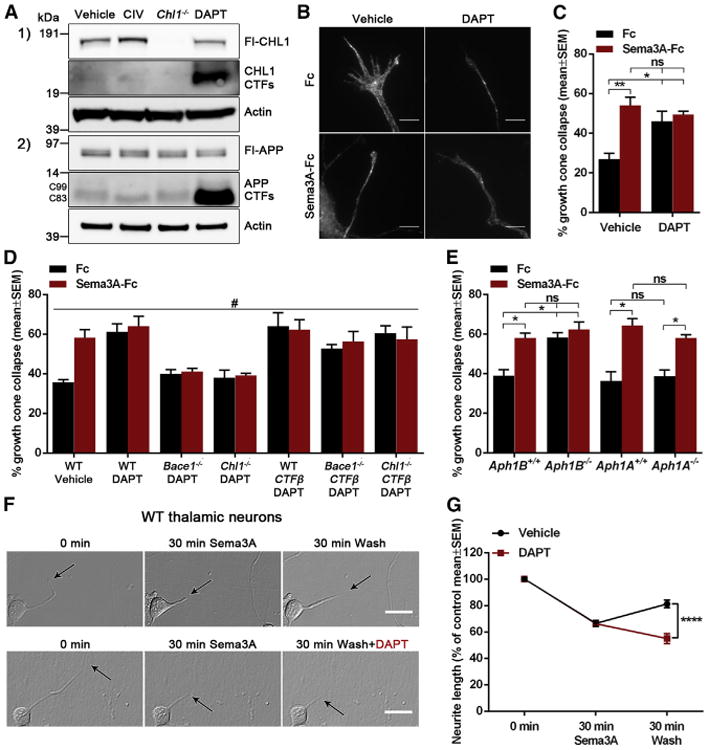
(A) Western blot analysis of cell lysates from WT primary mixed brain neuronal cultures treated with 1 μM CIV or 10 μM DAPT and Chl1−/−.
(B and C) Analysis of Sema3A-induced growth cone collapse in γ-secretase-inhibitor-treated (10 μM DAPT) thalamic neurons.
(D) Quantification of Sema3A-induced growth cone collapse in WT thalamic neurons treated with 10 μM DAPT; Bace1−/ − and Chl1−/− thalamic neurons treated with 10 μM DAPT; and WT, Bace1−/−, and Chl1−/− thalamic neurons transfected with Chl1CTFβ and treated with 10 μM DAPT.
(E) Quantification of Sema3A-induced growth cone collapse in Aph1Afl/fl and Aph1BCfl/fl thalamic neurons transfected with pCMV-GFP or pCMV-GFP-ires-Cre.
(F and G) Analysis of neurite length changes in WT thalamic neurons monitored by live-cell imaging at 0 and 30 min after Sema3A treatment and at 30 min after recovery in absence and presence of 10 μM DAPT.
Results are presented as mean ± SEM. The scale bars represent 5 μm (growth cones) and 20 μm (live-cell imaging).
APH1B-γ-Secretase Is Required to Stop the Sema3A-Induced Growth Cone Collapse
Different γ-secretase complexes containing different presenilin (PSEN1 and PSEN2) or APH1 (APH1A and APH1B) protein sub-units exist in humans, and several evidences indicate that each protease complex has different biochemical properties and exert distinct biological functions (Acx et al., 2014; Fazzari et al., 2014; Serneels et al., 2005, 2009). To investigate which γ-secretase complexes regulate growth cone recovery after Sema3A-induced growth cone collapse, we performed the collapse assay in Aph1A−/− andAph1BC−/− thalamic neurons. In order to obtain Aph1A- andAph1BC-deficient neurons, the neurons from conditional knockout mice, Aph1Afl/fl and Aph1BCfl/fl, were transfected with pCMV-GFP-ires-Cre. We observed that Aph1B−/− thalamic neurons show an exacerbation of Sema3A-induced and basal growth cone collapse whereas Aph1A−/− neurons present a normal response to Sema3A (Figure 4E). Therefore, APH1B-γ-secretase complexes are responsible for the recovery of the growth cone after Sema3A-induced growth cone collapse.
Different Dose-Dependent BACE1 Inhibition Effects on Aβ Levels and Sema3A-Induced Growth Cone Collapse
We wondered what our findings imply for the safe use of BACE1 inhibitors in patients and whether a therapeutic window can be defined. Thus, we performed dose response analysis of BACE1 activity and Sema3A-iduced growth cone collapse upon BACE1 inhibition. Effects on Aβ were already significant at the lowest concentration used (0.003 μM CIV; Figure 5A), whereas in the growth cone collapse assay, they only became significant at 100-fold higher concentration (0.3 μM CIV; Figure 5B), suggesting the existence of a safe therapeutic window for BACE1 inhibition.
Figure 5. BACE1 Inhibitor Dose Curve Response in Thalamic Neurons.
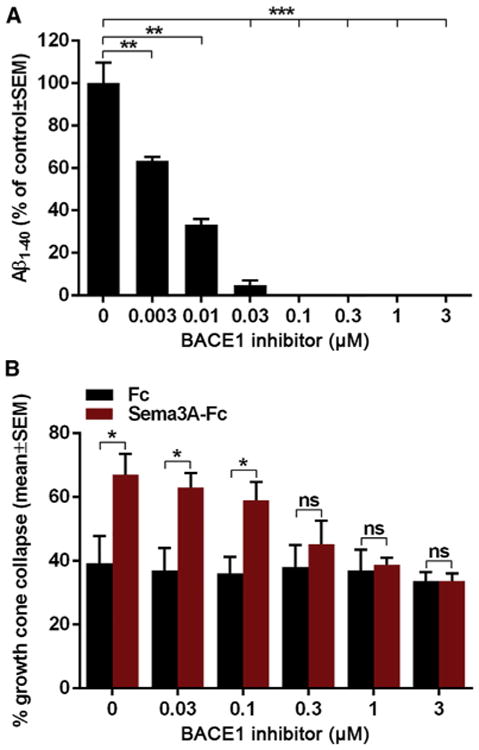
(A) Aβ1–40 levels in the conditioned medium of WT thalamic neurons after treatment with BACE1 inhibitor (0, 0.003, 0.01, 0.03, 0.1, 0.3, 1, and 3 μM CIV).
(B) Quantification of Sema3A-induced growth cone collapse in WT thalamic neurons after treatment with BACE1 inhibitor (0, 0.03, 0.1, 0.3, 1, and 3 μM CIV).
Results are presented as mean ± SEM.
Discussion
How deficient proteolytic activity of BACE1 can result in axonal guidance phenotypes in the adult brain is unknown. To further understand the physiological function of BACE1 in this context, we focused on the processing of CHL1 and its role in axonal guidance and growth cone collapse (Schlatter et al., 2008; Wright et al., 2007). We show here that thalamic brain explants, thalamic neurons, and hippocampal neurons, when treated with β-secretase inhibitor or when derived from Bace1−/− animals, do not longer respond to Sema3A (Figures 1 and S4). Wild-type CHL1, but not a cleavage-resistant mutant (Chl1D1062H; Zhou et al., 2012), was able to rescue Chl1−/− deficits (Figure 2) whereas a membrane-bound carboxy-terminal fragment of CHL1 (CHL1CTFβ) was necessary and sufficient to restore Sema3A-induced growth cone collapse in Bace1−/− and Chl1−/− neurons (Figure 3). This unequivocally demonstrates that BACE1 cleavage of CHL1 is necessary for this process. The binding of Sema3A to the NRP1/plexin A receptor in complex with CHL1 induces BACE1 cleavage of CHL1 (Figure 2), allowing a signal to be transmitted to the actin cytoskeleton. Intriguingly, we found that the signal is ended by APH1B-γ-secretase processing of CHL1CTFβ (Figure 4). This cleavage closes the cycle and prepares the growth cone for another round of pathfinding.
The requirement for BACE1 in this process is exquisitely specific. Although CHL1 can be cleaved by metalloproteases (Figure 1), metalloprotease inhibitors had no effect on Sema3A-induced growth cone collapse, suggesting that only BACE1 is cleaving CHL1 in the appropriate cellular compartment for this signaling event to occur. Sema3A induces growth cone collapse by dynamic remodeling of actin filaments; however, how this link is established remained unclear (Takahashi et al., 1999; Tamagnone et al., 1999). Here, we show that the interaction between CHL1CTFβ and ERM proteins is needed for Sema3A-induced growth cone collapse. ERM proteins are known to directly influence actin dynamics in growth cone filopodia by regulating the actin polymerization state (Ramesh, 2004).
Additionally, we observed that APH1B-γ-secretase inhibition caused an exacerbation of the growth cone collapse. Although the physiological functions of APH1B and APH1A-γ-secretase complexes are not yet fully discovered, several observations indicate that each complex exerts distinct functions. Aph1A−/− mice display an embryonic lethal phenotype resembling the Notch1-null mice phenotypes, whereas Aph1B−/− mice survive until adulthood, are fertile, and display a rather normal phenotype (Serneels et al., 2005). Specific inactivation of the APH1B-γ-secretase in a mouse model of AD led to improvements of AD-related phenotypes without any Notch-related side effects (Serneels et al., 2009). APH1B-γ-secretase complexes favor the generation of longer Aβ peptides and therefore are considered to play a major role in AD pathology (Acx et al., 2014). More recently, we showed that APH1B-γ-secretase complexes regulate hippocampal dendritic spine formation via NRG1 processing (Fazzari et al., 2014). To date, no axonal guidance functions have been linked to APH1B and APH1A-γ-secretase complexes. To understand whether the exacerbation of the collapse was dependent on CHL1, we tested the γ-secretase inhibitor in Chl1−/− and Bace1−/− thalamic neuronal cultures and performed genetic rescue experiments expressing CHL1CTFβ. In Chl1−/− and Bace1−/− neurons, the effect of γ-secretase inhibitors was gone, whereas transfection with Chl1CTFβ restored the effect (Figure 4). These results confirm that the effect of γ-secretase inhibition on growth cone collapse is dependent on CHL1CTFβ.
Thus, our work reveals how BACE1 and γ-secretase act together to regulate growth cone collapse and recovery, a process crucial for axonal pathfinding. Temporal separation of the two cleavage processes, needed to provide the active signal by cleaved CHL1, is probably achieved by subcellular trafficking of the NRP1/plexin A receptor in complex with CHL1 upon Sema3A binding toward the endocytic compartment. Sema3A binding is known to induce endocytosis of NRP1 (Fournier et al., 2000; Mintz et al., 2008), which might bring CHL1 into early endosomes, where BACE1 is available to cleave. Upon cleavage, CHL1CTFβ may alter actin polymerization through ERM signaling either in the endosome or after recycling to the plasma membrane, resulting in growth cone collapse and neurite retraction. Subsequent γ-secretase activity serves to terminate the signal and sensitizes the growth cone for the next axonal guidance cue (see graphical abstract). It is intriguing that two proteases, β- and γ-secretase, so closely involved in the pathogenesis of AD, also appear to coordinately regulate a physiological process of axonal guidance. Although the functional role of Sema3A and CHL1 in the adult brain is not clearly established, it is tempting to speculate that the continued release and expression of these molecules in hippocampus and olfactory bulb, where synaptic plasticity is highly dynamic, is important for modulation and maintenance of brain circuits in adult CNS (Pozas et al., 2001; Sahay et al., 2005). Genetic deficiency of BACE1 and CHL1 affects circuitry in the adult olfactory bulb and/or hippocampus (Cao et al., 2012; Hitt et al., 2012; Rajapaksha et al., 2011), and we find that Sema3A-induced growth cone collapse is also altered in hippocampal neurons treated with BACE1 or γ-secretase inhibitors (Figure S4).
Finally, it should be noted that this work indicates a critical function for the membrane-bound part of CHL1. The function of this β-secretase-generated fragment is a rare example of protein “shedding,” where the membrane-bound fragment and not the shed ectodomain is important.
The discovery of this unexpected function of BACE1 calls at least for additional caution while testing BACE1 inhibitors in humans. Finding a safe therapeutic window (Figure 5) will be crucial to avoid another setback in therapy because of not understood side effects in a phase III clinical trial for AD (De Strooper, 2014). It is also intriguing to see how BACE1 and γ-secretase have opposite effects in this process. This suggests that the defective generation of CHL1CTFβ by BACE1 inhibition can be compensated by a clever additional dosing of γ-secretase inhibitor. The clinical relevance of this finding needs further investigation.
Experimental Procedures
Mice
Bace1−/− (Dominguez et al., 2005), Chl1−/− (Montag-Sallaz et al., 2002), Aph1Afl/fl, and Aph1BCfl/fl mice (Serneels et al., 2005) were crossed to obtain E14 and E18 embryos. All experiments were approved by the ethics committee of the University of Leuven and carried out according to the Belgian and European Union regulations.
Preparation of Primary Neuronal Cultures
Primary cultures were prepared from E14 (thalamic and mixed brain) or E18 (hippocampal) mouse embryos as described (Fazzari et al., 2014; Leyva-Díaz et al., 2014; Wright et al., 2007; Zhou et al., 2012). See Supplemental Experimental Procedures for further information.
Western Blot Analysis
After 48 hr in culture, the thalamic or mixed brain neuronal cultures were treated overnight with 1 μM β-secretase inhibitor, 50 μM metalloprotease inhibitor, or 10 μM γ-secretase inhibitor. Samples were analyzed by western blot to detect full-length or C-terminal fragments of the proteins of interest (full-length-CHL1 [AF2147; R&D Systems], CHL1CTFs [antibody generated by Thermo Fisher Scientific using peptide DGSFIGAYTGAKEKGSVE], APP [B63 antibody; Annaert et al., 2001], NRP1 [AF566; R&D Systems], plexin A [MAB5856; R&D Systems], BACE1 [D10E5; Cell Signaling], and actin [A5441; Sigma]). Results are presented as mean ± SEM of the immunoblot band intensity for each protein measured in three independent experiments. The levels of the full-length proteins were normalized to actin levels. The bands intensity was quantified using the AIDA image analyzer software.
Thalamic Explants in Collagen
Thalamic explants were prepared as described (Leyva-Díaz et al., 2014). See Supplemental Experimental Procedures for more information.
Growth Cone Collapse Assay
To measure growth cone collapse, thalamic or hippocampal neurons were plated into 8-well glass Lab-Tek IICC2 chamber slide (154941/ W2495X; Fisher Scientific) pre-coated with 0.5 mg/ml poly-L-lysine. After 48 hr, cells were treated with either human Fc (4460-MG-100; R&D Systems), semaphorin 3A fused to human Fc(5926-S3; R&D Systems), or semaphorin 3F fused to human Fc (3237-S3-025; R&D Systems) for 30 min at 37°C. Cells were fixed with 4% paraformaldehyde (PFA) in PBS, permeabilized with 0.2% Triton X-100, and labeled with Alexa Fluor 555-phalloidin. Growth cone morphology was analyzed using a Zeiss ELYRA S1 (SR-SIM) super-resolution microscope. For genetic rescue experiments, neurons were co-transfected with GFP and the plasmids of interest (Fl-Chl1, Chl1D1062H, Chl1-CTFβ, Chl1-NTFβ, Fl-Chl1E+A, Chl1CTFβE+A, Chl1CTFβE, and Chl1CTFβA; see Supplemental Experimental Procedures). Transfections were performed using Lipofectamine 2000 reagent (11668-019; Invitrogen) according to the manufacturer's protocol. After 48 hr, cells were treated overnight with 0.03, 0.1, 0.3, 1, or 3 μM β-secretase inhibitor IV (CIV), 50 μM metalloprotease inhibitor (GM6001), or 10 μM γ-secretase inhibitor (DAPT). Only cells expressing GFP were included in the analysis. Results are presented as mean ± SEM of the percentage of collapsed axonal growth cones from three independent experiments. In each experiment, we counted the number of collapsed and non-collapsed axonal growth cones, and based on the total number of cells analyzed, the percentage of collapsed growth cones was determined. In Figures 1C, 1D, 4B, 4C, and 4E (only phalloidin staining), all cells from ten different areas of each well were analyzed. For the other experiments (genetic rescue experiments where the different constructs were co-transfected with GFP), all GFP-positive cells in each well were analyzed.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (Prism; GraphPad Software). Results are presented as mean ± SEM.
Growth Cone Collapse Assay
Two-way ANOVA was used to detect a significant difference between the different genotypes and treatments in response to Sema3A-Fc, Sema3F-Fc, or control Fc. Tukey's multiple comparisons test was used to compare the different groups. In case there was only the effect of one analyzed variable (no interaction), the significant difference was expressed differently (#).
Western Blot Analysis
Student's t test was used to detect a significant difference of Fl-CHL1, sCHL1, and APP protein levels between WT, BACE1-inhibitor-treated, and Bace1−/− thalamic neurons.
Thalamic Explants
One-way ANOVA was used to detect a significant difference between the P/D ratios of the different genotypes and treatments. Tukey's multiple comparisons test was used to compare the different groups.
Live-Cell Imaging of Thalamic Neurons
Two-way ANOVA was used to detect a significant difference between the different treatments in response to Sema3A-Fc across three different time points. Tukey's multiple comparisons test was used to compare the different groups.
Growth Cone Collapse Assay BACE1-Inhibitor Dose Curve Response
Student's t test was used to detect a significant difference between Sema3A-Fc and control Fc for the different doses of BACE1 inhibitor. p < 0.05 was set as a statistically significant level. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, #p < 0.05, ##p < 0.01, and ns is statistically not significant.
Please check Supplemental Experimental Procedures for additional information.
Supplementary Material
Highlights.
BACE1 and γ-secretase act in a cycle that regulates axonal outgrowth
CHL1CTFβ production by BACE1 is essential for Sema3A-induced growth cone collapse
Subsequent APH1B-γ-secretase is needed to terminate the growth cone collapse
Acknowledgments
Dr. Joris De Wit is thanked for critical discussion and providing Sema3A-RFP expression construct. This work was supported by the European Research Council (ERC) grant ERC-2009-StG_20081210 to G.L-B. and ERC-2010-AG_268675 to B.D.S., the Fonds voor Wetenschappelijk Onderzoek (FWO), the KU Leuven and VIB, a Methusalem grant of the KU Leuven/Flemish Government to B.D.S., and a Fundação para a Ciência e a Tecnologia (FCT) POPH/FSE/FCT/SFRH/BD/71949/2010 grant to S.B. G.L.-B. is an EMBO YIP Investigator. B.D.S. is supported by the Bax-Vanluffelen Chair for Alzheimer's Disease and “Opening the Future” of the Leuven Universiteit Fonds (LUF). Confocal microscope equipment was acquired through a Hercules Type 1 AKUL/09/037 to W. Annaert. The Zeiss ELYRA S1 (SR-SIM) super-resolution microscope was acquired through a CLME grant from minister Lieten to the VIB Bioimaging Core facility. Mouse experiments were supported by Infra-mouse. B.D.S. is a consultant for Janssen Pharmaceutica, Forum Pharmaceuticals, and Remynd NV.
Footnotes
Accession Numbers: The accession number for the DNA sequence of mouse CHL1 reported in this paper is GenBank: BC131670.1 (cDNA clone MGC: 150204).
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, four figures, and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.07.059.
Author Contributions: B.D.S. and S.B. conceived the study and planned experiments with the help of G.L-B. and P.F.M. G.L-B and P.F.M. contributed equally to this work. S.B. performed most of the experiments with the help of A.G., E.L-D., G.D., S.M., and T.V. All authors interpreted data. B.D.S. and S.B. wrote the initial version of the manuscript, and all authors read critically and helped rewrite the final version of the manuscript.
References
- Acx H, Ch´vez-Gutiérrez L, Serneels L, Lismont S, Benurwar M, Elad N, De Strooper B. Signature amyloid β profiles are produced by different γ-secretase complexes. J Biol Chem. 2014;289:4346–4355. doi: 10.1074/jbc.M113.530907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert WG, Esselens C, Baert V, Boeve C, Snellings G, Cupers P, Craessaerts K, De Strooper B. Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron. 2001;32:579–589. doi: 10.1016/s0896-6273(01)00512-8. [DOI] [PubMed] [Google Scholar]
- Bagnard D, Chounlamountri N, Püschel AW, Bolz J. Axonal surface molecules act in combination with semaphorin 3a during the establishment of corticothalamic projections. Cereb Cortex. 2001;11:278–285. doi: 10.1093/cercor/11.3.278. [DOI] [PubMed] [Google Scholar]
- Cao L, Rickenbacher GT, Rodriguez S, Moulia TW, Albers MW. The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep. 2012;2:231. doi: 10.1038/srep00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheret C, Willem M, Fricker FR, Wende H, Wulf-Goldenberg A, Tahirovic S, Nave KA, Saftig P, Haass C, Garratt AN, et al. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J. 2013;32:2015–2028. doi: 10.1038/emboj.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Lessons from a failed γ-secretase Alzheimer trial. Cell. 2014;159:721–726. doi: 10.1016/j.cell.2014.10.016. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- Fazzari P, Snellinx A, Sabanov V, Ahmed T, Serneels L, Gartner A, Shariati SA, Balschun D, De Strooper B. Cell autonomous regulation of hippocampal circuitry via Aph1b-γ-secretase/neuregulin 1 signalling. eLife. 2014;3:e02196. doi: 10.7554/eLife.02196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, Nakamura F, Kawamoto S, Goshima Y, Kalb RG, Strittmatter SM. Semaphorin3A enhances endocytosis at sites of receptor-F-actin colocalization during growth cone collapse. J Cell Biol. 2000;149:411–422. doi: 10.1083/jcb.149.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH, Shilliam CS, Hughes ZA, Dawson LA, et al. BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol Cell Neurosci. 2003;24:646–655. doi: 10.1016/s1044-7431(03)00227-6. [DOI] [PubMed] [Google Scholar]
- Hitt B, Riordan SM, Kukreja L, Eimer WA, Rajapaksha TW, Vassar R. β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J Biol Chem. 2012;287:38408–38425. doi: 10.1074/jbc.M112.415505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A, et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31:3157–3168. doi: 10.1038/emboj.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyva-Díaz E, del Toro D, Menal MJ, Cambray S, Susín R, Tessier-Lavigne M, Klein R, Egea J, López-Bendito G. FLRT3 is a Robo1-nteracting protein that determines Netrin-1 attraction in developing axons. Curr Biol. 2014;24:494–508. doi: 10.1016/j.cub.2014.01.042. [DOI] [PubMed] [Google Scholar]
- Mintz CD, Carcea I, McNickle DG, Dickson TC, Ge Y, Salton SR, Benson DL. ERM proteins regulate growth cone responses to Sema3A. J Comp Neurol. 2008;510:351–366. doi: 10.1002/cne.21799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag-Sallaz M, Schachner M, Montag D. Misguided axonal projections, neural cell adhesion molecule 180 mRNA upregulation, and altered behavior in mice deficient for the close homolog of L1. Mol Cell Biol. 2002;22:7967–7981. doi: 10.1128/MCB.22.22.7967-7981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naus S, Richter M, Wildeboer D, Moss M, Schachner M, Bartsch JW. Ectodomain shedding of the neural recognition molecule CHL1 by the metalloprotease-disintegrin ADAM8 promotes neurite outgrowth and suppresses neuronal cell death. J Biol Chem. 2004;279:16083–16090. doi: 10.1074/jbc.M400560200. [DOI] [PubMed] [Google Scholar]
- Pozas E, Pascual M, Nguyen Ba-Charvet KT, Guijarro P, Sotelo C, Chédotal A, Del Río JA, Soriano E. Age-dependent effects of secreted semaphorins 3A, 3F, and 3E on developing hippocampal axons: n vitro effects and phenotype of semaphorin 3A (−/−) mice. Mol Cell Neurosci. 2001;18:26–43. doi: 10.1006/mcne.2001.0999. [DOI] [PubMed] [Google Scholar]
- Pratte M, Rougon G, Schachner M, Jamon M. Mice deficient for the close homologue of the neural adhesion cell L1 (CHL1) display alterations in emotional reactivity and motor coordination. Behav Brain Res. 2003;147:31–39. doi: 10.1016/s0166-4328(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Rajapaksha TW, Eimer WA, Bozza TC, Vassar R. The Alzheimer's β-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener. 2011;6:88. doi: 10.1186/1750-1326-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh V. Merlin and the ERM proteins in Schwann cells, neurons and growth cones. Nat Rev Neurosci. 2004;5:462–470. doi: 10.1038/nrn1407. [DOI] [PubMed] [Google Scholar]
- Romi E, Gokhman I, Wong E, Antonovsky N, Ludwig A, Sagi I, Saftig P, Tessier-Lavigne M, Yaron A. ADAM metalloproteases promote a developmental switch in responsiveness to the axonal repellant Sema3A. Nat Commun. 2014;5:4058. doi: 10.1038/ncomms5058. [DOI] [PubMed] [Google Scholar]
- Sahay A, Molliver ME, Ginty DD, Kolodkin AL. Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J Neurosci. 2003;23:6671–6680. doi: 10.1523/JNEUROSCI.23-17-06671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Kim CH, Sepkuty JP, Cho E, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci. 2005;25:3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci USA. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatter MC, Buhusi M, Wright AG, Maness PF. CHL1 promotes Sema3A-induced growth cone collapse and neurite elaboration through a motif required for recruitment of ERM proteins to the plasma membrane. J Neurochem. 2008;104:731–744. doi: 10.1111/j.1471-4159.2007.05013.x. [DOI] [PubMed] [Google Scholar]
- Serneels L, Dejaegere T, Craessaerts K, Horré K, Jorissen E, Tousseyn T, Hébert S, Coolen M, Martens G, Zwijsen A, et al. Differential contribution of the three Aph1 genes to gamma-secretase activity in vivo. Proc Natl Acad Sci USA. 2005;102:1719–1724. doi: 10.1073/pnas.0408901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Van Houtvin T, Esselmann H, Paul S, Schäfer MK, Berezovska O, et al. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell. 1999;99:59–69. doi: 10.1016/s0092-8674(00)80062-8. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wright AG, Demyanenko GP, Powell A, Schachner M, Enriquez-Barreto L, Tran TS, Polleux F, Maness PF. Close homolog of L1 and neuropilin 1 mediate guidance of thalamocortical axons at the ventral telencephalon. J Neurosci. 2007;27:13667–13679. doi: 10.1523/JNEUROSCI.2888-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Barão S, Laga M, Bockstael K, Borgers M, Gijsen H, Annaert W, Moechars D, Mercken M, Gevaert K, De Strooper B. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem. 2012;287:25927–25940. doi: 10.1074/jbc.M112.377465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.