

Abstract
Neurodegenerative disorders represent the culmination of numerous insults including oxidative stress. The long etiology of most of these disorders suggests that lessening the effects of one of more of the insults could significantly delay disease onset. Antioxidants have been tested as possible therapeutics for neurodegenerative disorders, but with little success. Here we report that serotonin acts as a scavenger of hypochlorous acid (HOCl) in the brain. Serotonin was shown to prevent the oxidation of 2-thio-5-nitrobenzoate by HOCl in a biphasic manner. The first phase was a partial scavenging that occurred at concentrations of serotonin that exceeded those of HOCl. 1H-NMR studies indicated that HOCl chlorinates both the aryl and akyl nitrogen atoms of serotonin. Thus, the residual but lower oxidation of 2-thio-5-nitrobenzoate that occured during the first phase of scavenging is likely to due to formation of serotonergic chloramines. A second phase of scavenging occurred at concentrations of HOCl that exceeded those of serotonin. Under these conditions, the chlorinated serotonin polymerized and formed inert aggregates. Serotonin was further shown to prevent the loss of cells and cellular α-ketoglutarate dehydrogenase activity caused by HOCl. Extracellular concentrations of serotonin in the brain can be elevated with selective serotonin reuptake inhibitors and suggests that such compounds could be used to increase the cerebral antioxidant capacity. Acute administration of selective serotonin reuptake inhibitors to mice treated with endotoxin partially mitigated sickness behavior and protein chlorination in the brain. These observations suggest that serotonin may act to suppress chlorinative stress in the brain.
Introduction
Most neurodegenerative disorders develop over several years and occur later in life. These features suggest that even small changes in the trajectories of these diseases will significantly lessen their impact. Oxidative stress has been implicated in the etiology of pathologies such as Alzheimer Disease, Parkinson Disease and Amyotrophic Lateral Sclerosis [1]. Consequently, there have been numerous attempts to mitigate oxidative stress with antioxidants and thereby treat neurodegenerative disorders. These attempts have been largely unsuccessful [2–4].
Serotonin or 5-hydroxytryptamine (5HT) is a neurotransmitter [5] and is not typically considered an antioxidant. When 5HT has been evaluated in the context of redox chemistry, it was as the precursor of tryptamine-4,5-dione [6, 7]. Dryhurst and his colleagues proposed that tryptamine-4,5-dione results from the reaction of 5HT and superoxide [8], whereas Kettle and his group invoked the oxidation of 5HT by myeloperoxidase at sites of vascular injury [7, 9]. These sites satisfy the necessary conditions for the formation of the dione including 5HT at concentrations that effectively compete with chloride for oxidation by myeloperoxidase [9]. While 5HT is the better myeloperoxidase substrate [9], the physiological amounts of chloride ensure the enzymatic oxidation of this ion to hypochlorous acid (HOCl) [10].
In addition to vascular lesions, myeloperoxidase is active in the brain regions affected by Alzheimer Disease [11, 12], as well as paradigms of this disorder [13, 14] and of Parkinson Disease [15, 16]. Myeloperoxidase is not produced in the parenchyma of normal brain [11, 12]. The presence of this enzyme in neurodegenerative diseases may therefore reflect a crucial pathological transition, especially since HOCl is a powerful 2-electron oxidant [10]. 5HT is unlikely to be a myeloperoxidase substrate in the brain because the cerebral concentrations of this neurotransmitter are less than 10−8 M [17] while those of chloride are 10−1 M. Vesicular 5HT is highly concentrated [17], but is not available to myeloperoxidase which is predominantly found within activated glia [11, 12]. HOCl, however, is freely diffusible and can therefore access sites distal to its site of production. Indeed HOCl-modified proteins have been observed within neurons at the inflamed regions of parkinsonian brains [16]. HOCl is highly reactive and as a consequence thought to have limited diffusion through most tissues. The oxidation of neuronal proteins by HOCl in parkinsonian brains, though, is explicable in neurons abutting inflamed microglia given the relative paucity of HOCl scavengers in interstitial fluid [18]. These considerations suggested the possibility that in the diseased brain 5HT reacts with HOCl rather than myeloperoxidase. This possibility is bolstered by the observations that the 2-electron redox potentials of myeloperoxidase and HOCl are similar (1.16 V [19] and 1.48V [20], respectively) and that many compounds that act as substrates for myeloperoxidase are also oxidized by HOCl [21].
Oxidation of 5HT by myeloperoxidase compound II or III produces 5HT radicals [9]. The same radicals can also be formed by the reaction of superoxide and 5HT [9]. These radicals can react further to produce tryptamine-4,5-dione [9] and a dimer, 5,5’-dihydroxy-4,4’-bitryptamine [9]. The dimerization consumes superoxide and 5HT radicals and led Huether and his colleagues to propose that 5HT was a radical scavenger [22]. Dimers represent the simplest polymers and in our preliminary experiments we noted that the reaction of 5HT and HOCl produced precipitates indicative of polymer formation [23]. These observations suggested the possibility that 5HT acted as a scavenger of HOCl, which is supported by the studies of Sariahmetoglu et al. [24].
The aforementioned expression of myeloperoxidase in neurodegenerative disorders highlights the need for effective scavengers of HOCl and its reactive derivatives. Increasing the antioxidant capacity of the brain has so far proved to be a daunting task [2–4]. Selective 5HT reuptake inhibitors (SSRIs) increase the extracellular concentration of 5HT [25]. Thus, the actions of 5HT as a scavenger of HOCl could be augmented by increasing brain 5HT concentration using SSRIs. Given these considerations, we sought in this initial report to assess the scavenging of HOCl by 5HT and the protection afforded by SSRIs in a model of neuroinflammation.
Results
HOCl alters the UV-visible spectrum of 5HT
HOCl and 5HT react together as indicated by changes in the UV-vis spectrum of 5HT (Fig 1). The spectrum of 5HT consists of two non-symmetric peaks of absorption between 195 to 245 nm and 245 to 320 nm (Fig. 1A). Addition of an equimolar amount of HOCl, such that the final concentration of reactants was 50 µM, caused a uniform decrease in the absorbance between 195 to 245 nm with little change in the absorbances between 245 to 310 nm, and the evolution of a shoulder at 310 nm and beyond (Fig. 1A). In contrast, doubling the ratio of HOCl to 5HT produced a pronounced and uniform increase in the absorbances between 245 and 600 nm, with minor modifications of the absorbances between 195 and 245 as compared to spectrum formed by the 1:1 combination of these reactants (c.f. Fig. 1A & B). Higher ratios of HOCl to 5HT (3:1, Fig. 1C and 4:1, Fig 1D) diminished the absorbance between 195 to 245 nm with little change in the 245 to 600 nm absorbance.
Figure 1. HOCl alters the UV-visible spectrum of 5HT.


The spectrum of 50 µM 5HT in PBS is presented along with spectra obtained 15 seconds following the addition of HOCl at final concentrations of 50 (A), 100 (B) and 200 (C) µM at 22°C. Absorbances due to 5HT are represented by grey traces whereas those due to the addition of HOCl are shown as black traces. Each trace represents the mean of at least four spectra that did not vary by more than 5% of the mean absorbance at all wavelengths. Spectra were also recorded 64 minutes following the addition of HOCl and the absorbance changes at 218 and 274 nm are depicted in panels E and F, respectively, for the indicated ratios of 5HT to HOCl (mean ± SEM of three independent observations).
Fig. 1E & F show the HOCl induced changes in the absorbance spectrum at 218 nm and 274 nm, representing peaks in the 195 to 245 nm and 245 to 320 nm regions, respectively. These two panels also show the changes in absorbance obtained 15 seconds and 64 minutes after combining HOCl and 5HT. Allowing the reaction to proceed for an additional h produced no further changes in the absorbance at 218 nm (Fig. 1E), indicating that the reaction between HOCl and 5HT is essentially complete within seconds. The absorbances at 274 nm, however, decreased to a modest extent with increasing reaction time (Fig. 1B). Finally, increasing the ratio of HOCl to 5HT from 4:1 to 5:1 cause no further changes in absorbance at these wavelengths (Fig. 1E & F), which was also evident at the other wavelengths (not shown).
N-chlorination of 5HT by HOCl
The initial products of the reaction of HOCl and 5HT were in examined by 1H-NMR (Fig. 2). These reactions were carried out in the presence of deuterated methanol to prevent the precipitation of the reaction products in aqueous buffers [26]. The 1H NMR spectrum of 10 mM 5HT in deuterated methanol is shown in Figure 2A. Addition of HOCl to 5HT at equimolar concentrations gave rise to a set of new resonances in both the aromatic and aliphatic regions of the NMR spectrum (marked with primes in Fig. 2B & C). While these new resonances were shifted relative to the parent compound, no change was observed in their relative intensity or coupling patterns, indicating that chlorination of the ring carbons had not occurred. In the aliphatic region HOCl caused a downfield shift of the methylene resonances from protons adjacent to the -NH group (11’) and an upfield shift of the methylene protons closest to the ring (10’). This is consistent with chlorination of the ring and aliphatic nitrogens of 5HT. At equimolar HOCl and 5HT concentrations, the new resonances appear to comprise ~10% of the total NMR signal (Figure 2B), while at 2-fold excess of HOCl, serotonin and the chlorinated product are present at about equal concentrations (Figure 2C). As the HOCl concentration is further increased, the original 5HT resonances disappear completely and a number of new resonances appear indicating sample decomposition (not shown).
Figure 2. N-Chlorination of 5HT by HOCl.

The 500 MHz 1H-NMR spectrum of 10 mM 5HT in deuterated methanol is shown (A). The low and high field portions of the spectrum are shown separated by the structure in which the resonant protons are numbered. The top two traces show the spectra of 10 mM serotonin mixed (B) 1:1 and (C) 1:2 with HOCl. New resonances appearing in these spectra are labeled with the prime sign and are assigned on the structure in the center. The addition of HOCl induces a chemical shift in the new resonances, but no change in the relative intensity of the coupling pattern, indicating that the chlorination takes place on the nitrogens and not on the ring.
5HT scavenges HOCl
The oxidation of 2-thio-5-nitrobenzoate by the reaction products of HOCl and 5HT was used to assess the reactivity of these compounds. This method is commonly used to evaluate the reactivity of chlorinating agents and is based on the avidity with which these compounds react with thiol moieties [27]. For example, the second order rate constant for the pseudohalide hypothiocyanous acid and 2-thio-5-nitrobenzoate is 4.4 × 105 M−1.s−1 at pH 7.4 [27]. The reaction of HOCl and 2-nitro-5-thiobenzoate begins with the chlorination of the thiol group to generate a sulfenyl chloride compound, which is then reduced by another dye molecule to yield 5,5’-dithiobis-(2-nitrobenzoate) (Fig. 3A). Thus, two molecules of 2-nitro-5-thiobenzoate are consumed in the reduction of HOCl to H2O. This stoichiometry was confirmed in Fig. 3B for reactions in which increasing amounts of HOCl were incubated in PBS at 22°C for either 15 seconds or 64 minutes and then combined with 2-nitro-5-thiobenzoate (R2 = 0.99 & 1.00 for regressions of the 15 second and 64 minute data, respectively). The same stoichiometry was not observed when 0.2 mmol 5HT was incubated with 0.05 to 0.5 mmol of HOCl (Fig. 3B). Instead, less 2-nitro-5-thiobenzoate was consumed as compared to reactions containing HOCl alone indicating scavenging of HOCl by 5HT. The scavenging reaction was biphasic in that it could be fitted to two linear expressions corresponding to 0 to 0.2 mmol and 0.2 to 0.5 mmol HOCl. At 15 sec, the slopes of these lines are 1.05 and 0.58 mole dye consumed per mole 5HT (R2 = 0.97 & 0.98, respectively) and indicate significant scavenging of HOCl by 5HT, particularly at ratios of HOCl to 5HT greater than 1:1 (the upper axis in Fig. 3B depicts the ratios of HOCl to 5HT). Scavenging was even more pronounced in the 64 minute incubations with HOCl and 5HT (Fig. 3B). As noted for Fig. 1 E & F, the higher wavelengths of the HOCl-treated 5HT spectra continued to change over time indicative of ongoing rearrangements of the reaction products. Such changes might account for the increased HOCl scavenging by 5HT over time.
Figure 3. 5HT scavenges HOCl.

Panel A depicts the scheme for the reaction of 5-thio-2-nitrobenzoate and HOCl. The 2:1 stoichiometry for this reaction was confirmed for mixtures 5-thio-2-nitrobenzoate and HOCl sampled at 15 seconds ( ) and 64 minutes (
) and 64 minutes ( ) post mixing at 22°C (B). In contrast, mixtures of 5HT and HOCl at 15 seconds () and 64 minutes (
) post mixing at 22°C (B). In contrast, mixtures of 5HT and HOCl at 15 seconds () and 64 minutes ( ) consumed less 5-thio-2-nitrobenzoate indicative of HOCl scavenging. The final concentration of 5HT in these experiments was 200 µM for the reaction performed in one mL. HOCl was added at indicated amounts and ratios. The mean ± SEM of three separate trials are shown. In some cases, the magnitude of the SEM values is obscured by the symbols.
) consumed less 5-thio-2-nitrobenzoate indicative of HOCl scavenging. The final concentration of 5HT in these experiments was 200 µM for the reaction performed in one mL. HOCl was added at indicated amounts and ratios. The mean ± SEM of three separate trials are shown. In some cases, the magnitude of the SEM values is obscured by the symbols.
The reaction of HOCl and 5HT produces inert aggregates
Changes observed in the UV-visible spectra of chlorinated 5HT at 274 nm and higher wavelengths are indicative of the formation of polymers based on our observations of other chlorinated aromatic neurotransmitters [23]. The possibility that chlorinated 5HT forms polymers and aggregates, was suggested by the precipitation observed in aqueous mixtures of 10−2 M amounts of 5HT and HOCl and confirmed by light microscopy (data not shown). These aggregates which were examined further using flow cytometry [23].
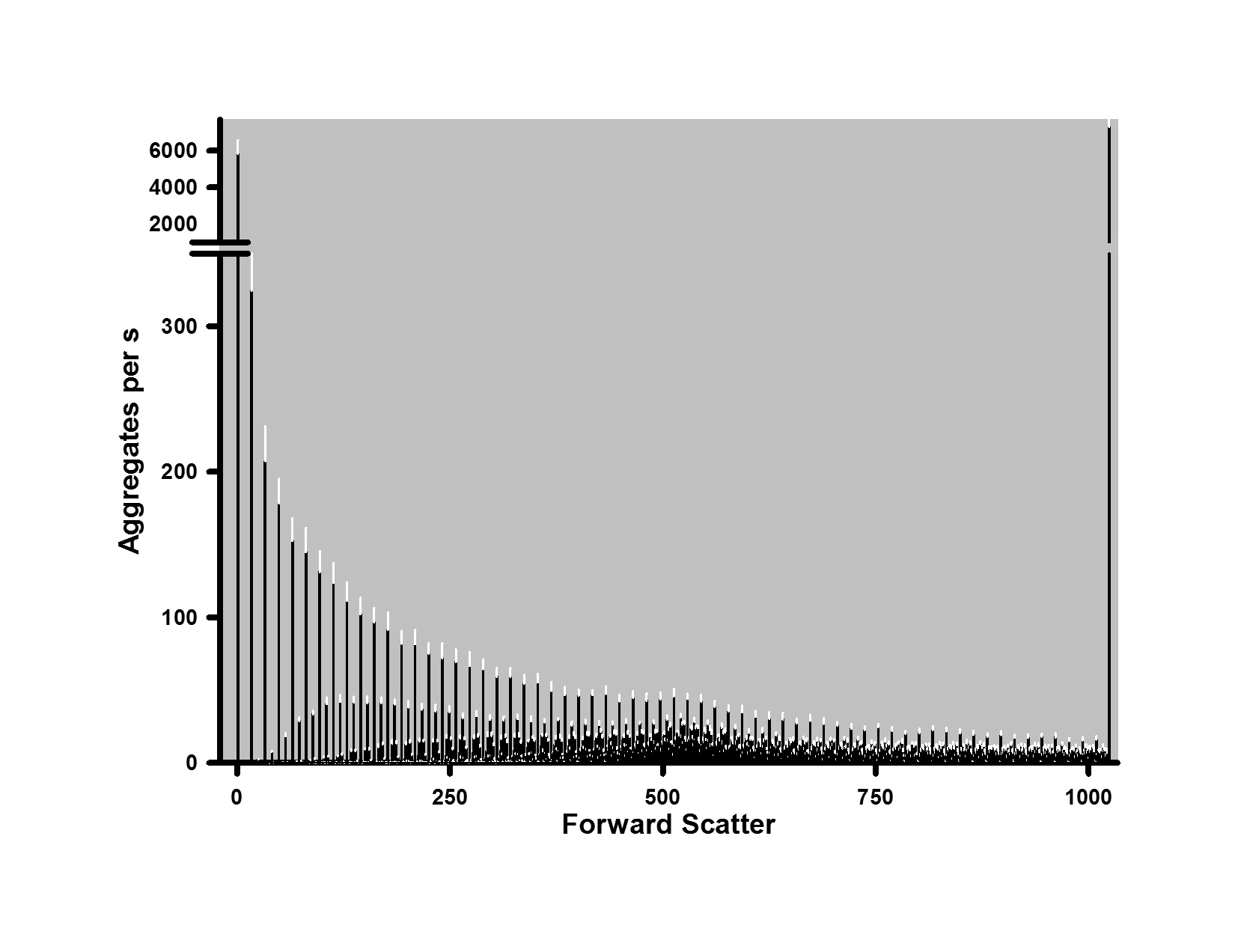
For the flow cytometric studies, 5HT and HOCl were combined at a final concentration of 5 mM each in PBS at 22°C and the resulting aggregates measured over time from 5 to 160 minutes (Fig. 4A–C). Significant amounts of aggregates were formed under these conditions within 5 min. The sizes of aggregates ranged between 0.05 and 6.2 mm based on the peak positions of calibrated beads (Fig. 4A & B). Two distributions of aggregates are evident in Fig. 4A & B: a skewed distribution in favor of smaller-sized aggregates, as well as a broad peak with a maximum at 529 Forward Scatter units. This maximum also corresponds to the maximum of 1.5 mm calibration beads.
Figure 4. Distribution and kinetics of aggregate formation by chlorinated 5HT.


The distribution of aggregates as a function of light scattering or size at 5 min (A) and 160 min (B) after the reaction of 5 mM HOCl and 5 mM in PBS at 22°C is shown. Light scattering (forward scatter) was measured by flow cytometry. Inverted triangles at the top of plots for panels A and B indicate the peak positions for the distributions of 0.05, 1.5, and 6.2 mm size marker beads. The distribution in panel A represents the mean of 11 separate experiments while that of panel B represents the mean of six separate determinations. These distributions are also reproduced in the Supplementary Fig. 1 together with the relevant SEM values. Panel C shows the concentrations of the total aggregate population and that of the 1.5 mm aggregates as a function of time. Aggregate concentrations are expressed as number of aggregates collected per seconds with the experimental conditions the same as above. Shown are the means ± SEM of between six and 14 separate experiments. Panel D shows the LDH activity released by 1.25 × 105 differentiated THP-1 cells after 24 h incubation with 6.75 µg of aggregates produced by the chlorinated dopamine or serotonin. The means ± SEM of four individual experiments are shown with significant difference indicated by ** for p < 0.01
The data in Fig. 4 is reported as aggregates collected per second at each sample incubation time to account for samples with low aggregation formation as described in the Methods. Both the concentration and distribution of different sized aggregates varied as a function of time (Fig. 4C). After an initial increase in the total number of aggregates, this number falls by 47% at 20 minutes (Fig. 4C). This decrease was due to smaller aggregates coalescing into larger structures and this is illustrated by an increased concentration of 1.5 mm-sized aggregates at 20 minutes (Fig. 4C). The 1.5 mm-sized aggregates represented the apex of a broad peak that evolved from 20 to 160 minutes (Fig. 4C). Increases in this peak after 20 minutes broadly followed the increases in total aggregate concentrations (Fig. 4C)
The toxicity of the aggregates resulting from the reaction of HOCl and 5HT was tested by measuring the release of LDH from differentiated THP-1 cells (Fig. 4D). These cells phagocytosed the aggregates no increase in released LDH was observed. In contrast, the addition of the same amount of chlorodopamine aggregates to THP-1 cell resulted in cell death and LDH release consistent with our previous studies [23].
5HT protects SH SY5Y cells from the toxicity of HOCl
The ability of 5HT to protect cells from the toxicity of HOCl was investigated using retinoic acid-differentiated SH SY5Y cells, which exhibit some of the characteristics of dopaminergic neurons. HOCl was added to these cells in the absence of scavengers for a period of 15 minutes and survival assessed 24 h later. Exposure of the cells to 50 µM HOCl under these conditions led to the death of 84% of the cells (Fig. 5). This loss could be prevented by the addition of 5HT. Concentrations of 5HT as low as 25 µM, mitigated the cell loss due to 50 µM HOCl, increasing cell survival from 16% to 53%. Higher concentrations of 5HT produced correspondingly greater gains in survival; for example, 800 µM 5HT decreased the cell loss due to 50 µM HOCl to 26%. The difference in protection due to 25 and 800 µM 5HT was statistically significant (p < 0.05, t-test). As expected, 5HT also protected cells from the toxicity of 25 µM HOCl (Supplemental Fig. 2). 5HT, however, did not improve survival above 20% for cells treated with 100 µM HOCl (Supplemental Fig. 2). Fifteen minute exposures of the cells to 5HT at concentrations up to 800 µM alone did not affect their viability (data not shown). Thus, 5HT did not provide complete protection against the tested concentrations of HOCl.
Figure 5. 5HT protects SH SY5Y cells from the toxicity of HOCl.

SH SY5Y cells were exposed to HOCl and serotonin alone and in various combinations for 15 min at 37°C after which the cells were lysed and KGDH complex activity determined. Shown are the mean ± SEM of between four and 14 separate determinations. Significant differences from control are indicated by ** for p < 0.01 whereas significant differences from 50 µM HOCl are indicated as # and ## for p < 0.05 and p < 0.01, respectively.
The α-ketoglutarate dehydrogenase (KGDH) complex is sensitive to inactivation by HOCl and thought to account, in part, for toxicity of HOCl to neuronal cells [28]. We assessed the ability of 5HT to protect this complex from inactivation by HOCl (Fig. 6). Exposure of SH SY5Y cells to 50 µM HOCl for 15 minutes caused a 76% reduction in KGDH complex activity, which was restored by 35% by the addition of 50 µM 5HT. Higher concentrations of 5HT further preserved KGDH complex activity.
Figure 6. 5HT mitigates the inactivation of cellular KGDH complex by HOCl.

SH SY5Y cells were exposed to 50 µM HOCl and 5HT alone and in various combinations for 15 min at 37°C after which the cells were lysed and KGDH complex activity determined. The means ± SEM of six individual experiments expressed as % control are shown. Control KGDH complex activity in SH SY5Y cells were 254 ± 3 nmol/min/mg (mean ± SEM, n = 6). Significant differences from control are indicated by ** for p < 0.01 whereas significant differences from 50 µM HOCl are indicated as ## for p < 0.01.
It is important to note that 5HT administered at 800 or 400 µM the highest concentrations in Fig. 5 and Fig. 6, respectively, had no adverse effects of either cell survival or KGDH complex activity (not shown). Furthermore, incubating the cells with 800 µM 5HT for two h either before or after the administration of HOCl did not alter the toxicity of the latter compound (not shown). These observations indicate the effects reported in Fig. 5 & 6 are due to the scavenging of HOCl by 5HT rather than an action of 5HT on the cells.
Fluoxetine attenuates sickness behavior in LPS-treated mice
The results of in vitro and cell culture studies suggested the notion that 5HT might scavenge HOCl in the brain. This hypothesis was tested by determining the effect of the SSRI fluoxetine on a model of cerebral inflammation. The model relies on the intraperitoneal (i.p.) injection of lipopolysaccaride (LPS) into mice. These injections stimulate the innate immune system to produce pro-inflammatory cytokines, which in turn promote a comparable expression of cytokines in the brain [29]. Expression of these cytokines in brain causes an exaggerated local inflammation and sickness behavior [29]. SSRIs antagonize the uptake of 5HT through the 5HT transporter [25]. In mammals, this transporter is expressed in platelets, placenta, and brain [30]. Thus, the brain represents the major tissue affected by both the i.p. administration of LPS and SSRIs.
The cerebral inflammation resulting from systemic endotoxemia is accompanied by increased myeloperoxidase activity [31]. This observation was corroborated by demonstration of greater expression of myeloperoxidase in the brains of mice 24 h after the administration of LPS (Table 1). The increase in cerebral myeloperoxidase was not prevented by three days prior treatment with the SSRI fluoxetine (Table 1). Thus, the actions of fluoxetine on LPS-induced sickness behavior may involve the interaction between 5HT and myeloperoxidase-catalyzed products. In contrast, the NSAID ibuprofen prevented the expression of myeloperoxidase in the brain consistent with general suppression of inflammation [32]. The actions of ibuprofen are therefore unlikely to include reactions with chlorinating species.
Table 1.
Fluoxetine and ibuprofen mitigate LPS-induced sickness behavior in mice
| Parameters | Saline a | LPS a | SSRI a | NSAID a |
|---|---|---|---|---|
| Myeloperoxidase b | 0 ± 0 (3) | 8.26 ± 5.09 (3) c | 12.4 ± 8.12 (3) c | 0.34 ± 0.22 (3) |
| Body weight change (g) d | 0.6 ± 0.24 (3) | 3.77 ± 0.13 (5) c | 3.22 ± 0.12 (9) c,e | 3.79 ± 0.17 (7) c |
| Distance (m) f | 13.1 ± 1.21 (3) | 0.92 ± 0.06 (5) c | 1.29 ± 0.15 (9) c,e | 1.52 ± 0.21 (7) c,e |
| −93.0 | −90.2 | −88.4 | ||
| Number of moves (# × 100) f | 21.0 ± 0.55 (3) | 3.89 ± 0.14 (5) c | 5.07 ± 0.80 (9) c,e | 6.52 ± 0.96 (7) b, e |
| −81.5 | −75.9 | −69.0 | ||
| Move time (min) f | 96.4 ± 4.83 (3) | 13.0 ± 0.48 (5) c | 17.2 ± 2.40 (9) c,e | 22.17 ± 3.13 (7) c,e |
| −86.5 | −82.2 | −77.0 |
The mice received either i.p. injections of saline, fluoxetine (10mg/Kg), or ibuprofen (10 mg/Kg) every day for three days and then LPS (5mg/Kg) or saline on the fourth day. Twenty four h later the animals were sacrificed and their brains collected for the measurement of MPO. The sickness behavior measurements are depicted on a grey background. Shown are the means ± SEM (n) values and the percent changes in italics.
Values represent the density measurement of myeloperoxidase immunoreactivity as determined by Western blotting and normalized against the β-actin density in each blot.
Different from Saline group, p <0.05 (ANOVA with Kruskal Wallis post hoc analysis).
’Change in weight’ reflect the difference in weight measured just prior the LPS injection and 24 h later at the time of sacrifice.
Different from LPS group, p <0.05 (ANOVA with Kruskal Wallis post hoc analysis).
The behavioral measurement were recorded between 20 and 24 hour post LPS (4 h).
HOCl reacts with tyrosyl residues to produce 3-chlorotyrosine, which serves as a unique biomarker for the activation of myeloperoxidase [12, 14, 16]. Therefore, staining for 3-chlorotyrosine was used to confirm the activation of myeloperoxidase in the brains of mice treated with LPS. A commercial antibody raised against 3-chloro-4-hydroxybenzoic acid bound to keyhole limpet hemocyanin recognizes 3-chlorotyrosine in histological sections, including brain. We reasoned that this antibody might also recognize 3-chlorotyrosine in Western blot, if the proteins were extracted in the presence of excess 3-chlorotryosine to competitively inhibit dehalogenases specific for 3-chlorotyrosine [33]. Under these conditions, enhanced staining for 3-chlorotyrosine was evident for proteins at 25, 30, 50 and 150 kDa extracted from mice treated with LPS but not saline or LPS and ibuprofen (Supplementary Fig. 3). The latter result is important because ibuprofen prevented the expression of myeloperoxidase in the brain (Table 1). 3-Chlorotyrosine staining was markedly decreased in the brains from animals treated with fluoxetine prior to LPS, consistent with a possible scavenging of HOCl by 5HT.
Sickness behavior manifests as weight loss and decreased ambulation [29]. Significant changes in both of these parameters were observed in the current study (Table 1). Movement was measured in terms of the number of movements made, the time spent moving, and the distance traveled between 20 and 24 h post-LPS (Table 1). Fluoxetine, which increases 5HT in the brain [25], mitigated the weight loss and the decreases in normal movements induced by LPS (Table 1). The extents of mitigation, while significant, were modest. For example, the distance travelled improved from a 93% loss due to LPS to a 90% loss with fluoxetine pretreatment (Table 1). The largest change was observed in the number of movements made by the mice in the four h sampling period. In this case, LPS decreased the number of movement by 82% and 76% in the absence and presence of fluoxetine pretreatment, respectively (Table 1). The magnitude of these changes, however, is consistent with magnitude of changes brought about by ibuprofen. In general, the effects of ibuprofen were greater than those of fluoxetine and presumably reflect the ability of this NSAID to suppress inflammation throughout the body. Unlike fluoxetine, however, ibuprofen did not alter the loss of body weight due to LPS (Table 1).
Discussion
Despite the recognized need to quell reactive oxygen species in the brain, clinical trials have yet to identify antioxidants capable of stemming oxidative damage in the brain and halting neurodegenerative diseases [2–4]. Here we describe the possibility of scavenging a particularly noxious and powerful class of oxidants – hypohalous acids – by elevating the cerebral 5HT content. 5HT reuptake inhibitors act to increase the amounts of 5HT within minutes of the application of these drugs [34–38]. The studies presented here indicate that each molecule of 5HT readily scavenges multiple molecules of HOCl. Moreover, the scavenging capacity of 5HT increases with the reaction of more than one molecule HOCl per 5HT. These indications suggest that even small increases in 5HT will scavenge HOCl in the brain. Our studies with fluoxetine administered to mice demonstrate that SSRIs blunt both chlorinative stress in the brain and some of the physiological effects of LPS on the mice.
The scavenging of HOCl by HOCl was biphasic as assessed by the oxidation of 2-nitro-5-thiobenzoate (Fig. 3). The first phase is characterized by a partial scavenging of HOCl that occurs at 5HT concentrations greater or equal to the concentration of HOCl, i.e., HOCl:5HT ≤ 1:1. Given that all of the HOCl was consumed in these reactions, the reaction products must contain species capable of oxidizing 2-nitro-5-thiobenzoate. Based on the propensity for hypohalous acid to react with amines [39, 40], the most likely oxidant formed by the reaction of HOCl and 5HT is a chloramine (Fig. 2). The formation of this specie was confirmed in the present study by 1H NMR which also demonstrated the chlorination of the nitrogen atom on the indole ring (Fig. 2). Mixtures for which HOCl:5HT ≤ 1:1 oxidized half the amount of 2-nitro-5-thiobenzoate oxidized by the corresponding concentrations of HOCl (Fig. 3). This stoichiometry suggests that at least half of chloride atoms from HOCl were incorporated as serotonergic chloramines.
Formation of serotonergic chloramine also suggests that the reaction HOCl and 5HT may occur in vivo. Hypohalous acids react with amines at rates between 103 and 105 M−1.s−1 [39, 40]. These rates are sufficient fast to occur in vivo, particularly in the brain where the interstitial fluid has significantly less HOCl-scavenging capacity than blood. This conclusion is based on the scavenging capacity of cerebrospinal fluid [18], which reflects the contents of both the ventricles and interstitial fluid. The formation of chloramines at all ratios of HOCl to 5HT is also suggested by the results of the toxicity studies, where it was shown that 5HT did not completely protect cells from the toxic actions of HOCl, even when 5HT was present at greater concentrations than HOCl. Chloramines are toxic to cells, but less so than HOCl [28]. The toxicity of serotonin chloramine is unlikely to relevant in vivo where numerous scavengers exist.
The second phase of the biphasic scavenging of HOCl by 5HT is characterized by a plateau of 2-nitro-5-thiobenzoate oxidation that occurs when the concentration of HOCl exceeds that of 5HT (Fig. 3). In other words, the products still contained species capable of oxidizing 2-nitro-5-thiobenzoate, but the amounts was limited despite the excess of HOCl. This excess led to degradation of 5HT as evidenced by UV-vis and 1H-NMR spectrometry (Fig. 1 & 2). The degraded molecules polymerized and aggregated, as shown in Fig. 4, and consistent with our earlier studies of the reactions of dopamine and HOCl [23]. Polymerization could remove chloride from 5HT thereby detoxifying the chlorinated 5HT. Aggregation also conceals a proportion of the residual chloramine moieties thereby contributing further to the detoxification of the chlorinated polymers. Indeed aggregated polymers formed by the reaction of HOCl and 5HT did not kill phagocytic cells (Fig. 4D).
The experiments demonstrating the formation of aggregates by HOCl and 5HT utilized 10−2 M concentrations of the reactants. In our previous study, we demonstrated that the aggregation observed at 10−2 M reactant concentrations were comparable in size to the aggregation due to 10−8 M reactant concentration, albeit occurring at a slower rate [23]. Thus, it is likely that polymers and aggregates of chlorinated 5HT were formed in the experiments depicted in Fig. 1 and 3–6.
Studies with 5HT analogs lend support to the hypothesis that oxidized 5HT polymerizes in vivo. 5HT conjugated to gadolinium chelates acted as substrates for myeloperoxidase-catalyzed polymerization thus improving their ability to act as contrast agents for magnetic resonance imaging [41, 42]. The polymerization was thought to proceed by oxidation of 5HT to a radical by the complex I form of myeloperoxidase. This is plausible given that the one-electron redox potential of complex I is 1.35 V [19] and that in this state myeloperoxidase oxidizes 5HT at a rate of 2 × 107 M−1.s−1[43]. Complexes II and III also catalyze one-electron oxidations of 5HT [9, 43] and 5HT and tyrosine radicals readily condense to form dimers [7, 9] and polymers [44]. Kettle and his coworkers have argued that at the concentrations of 5HT expected in vivo, myeloperoxidase will oxidize both chloride ions and 5HT [9]. This is also possible for the gadolinium-chelate 5HT conjugates [41] and suggests that a portion of the polymerization of these constructs in vivo could result from reaction with HOCl. This hypothesis suggests the exciting possibility of detecting HOCl production in the brain using imaging agents bound to 5HT.
The ability of 5HT to scavenge double or more the molar equivalent of HOCl is remarkable since this stoichiometry is better than the majority of the reactions of HOCl and its scavengers. Thiols are among the best scavengers of hypohalous acids based on the rate constants for these reactions [27, 40]. The stoichiometry for HOCl scavenging by thiols is 1:2 is less than the >2:1 ratio for the reaction of HOCl and 5HT reported here. Glutathione is the major thiol-bearing scavenger of hypohalous acid in the body [45]. HOCl oxidizes the thiol moiety of this tripeptide to either a disulfide or a sulfonyl chloride [45, 46]. The oxidation to the sulfonyl chloride, however, effectively destroys glutathione. Vitamins C and E are thought to react with HOCl with a 1:1 stoichiometry, but the rates of consumption of these antioxidants by HOCl again suggests a negligible role in the scavenging of this oxidant in vivo [47, 48]. In contrast, the stoichiometry for reaction of HOCl and 5HT suggests that this may be a particularly efficient mechanism for the scavenging of hypohalous acids in the brain.
Thiols are good scavengers of chlorinating species and it is possible to increase thiol levels in the brain using either cysteamine or cystamine. These increases, however, occurs in specific regions of the brain and require the sustained administration of these drugs at high doses [49, 50]. Cysteamine is approved for use in humans who find it unpleasant to consume [51, 52]. In contrast, drugs like fluoxetine are well tolerated and efficacious in treatment of depression [36–38]. Thus, the chronic administration of SSRIs may represent a more efficient mechanism for augmenting the oxidant defenses of the brain. This idea was tested using a paradigm of neuroinflammation based on the systemic administration of LPS, which causes pro-inflammatory cytokines to be expressed in the periphery and brain [29]. One consequence of this expression is the recruitment and activation of neutrophils in the brain [53]. Neutrophils are the major source of myeloperoxidase in the body and thus the recruitment and activation of these cells results in the deposition of this enzyme in the brain. The normal and inflamed brain produces significant amounts of hydrogen peroxide [54, 55], which in the presence of myeloperoxidase leads to the oxidation of chloride ions to HOCl [10]. The concurrence of myeloperoxidase and HOCl production has been established by measurements of the enzyme and 3-chlorotyrosine in the brain [12, 14, 16]. The presence of myeloperoxidase due to administration of LPS was also confirmed in the current study (Table 1). Similarly, evidence for the chlorination of proteins in the brain of LPS was also obtained using an antibody to 3-chlorotyrosine in Western blots. Staining with this antibody was contingent on the presence of myeloperoxidase (Table 1) and occurred at a low antibody titer relative to recommended histological methods, suggesting that the staining was specific for protein-bound 3-chlorotyrosine. This suggestion is further corroborated by the observation that ibuprofen which prevented the appearance of myeloperoxidase in the brain also negated staining due to LPS treatment.
The systemic administration of LPS induces sickness behavior, which in part, reflects the effect of inflammatory mediators in the brain [29]. Pretreatment with fluoxetine mitigated two indices of sickness behavior due to LPS: weight loss and the decreases in ambulation. In contrast, ibuprofen only attenuated the decreases in movements caused by LPS. This difference may be due to fluoxetine acting mainly in the brain whereas ibuprofen acts throughout the body. Fluoxetine selectively increases 5HT in the brain brain [25]. Thus, the mitigation of LPS-induced sickness behavior may have been the result of scavenging of HOCl. Fluoxetine has numerous inflammatory actions on the brain [56–58] and the actions of 5HT could include the scavenging of hydrogen peroxide [24], as well as the inhibition of myeloperoxidase [9, 59]. Establishing the contribution of HOCl scavenging by SSRI-induced rise in extracellular 5HT will, therefore, require further investigations. These investigations will be facilitated by the observation that HOCl scavenging by 5HT leads to polymerization of this indole. This polymerization might be observed using the gadolinium-chelate 5HT conjugates [41], as discussed above.
The changes in sickness behavior brought about by either fluoxetine or ibuprofen were relatively modest. Even so, these changes were larger than those reported by Yirmiya et al. [60] in their study of the effects of fluoxetine on LPS-induced sickness behavior. Martin et al. [61] have argued that sickness behavior is a crucial survival mechanism and therefore resistant to intervention. If this is the case, then the changes reported in this study may be more significant than indicated by their magnitudes. Berg et al. [62] reported profound attenuation of LPS-induced sickness behavior using α-tocopherol. These effects depended on the doses of both α-tocopherol and LPS, as well as the durations of α-tocopherol pretreatments. Fluoxetine and other SSRIs may therefore mitigate sickness behavior to a greater extent, if given for longer periods and in other models of neuroinflammation. In future studies, we intend to analyze the inflammation induced by direct injections of LPS, 1-methyl-4-phenylpyridinium, or quinolinic acid into the brain.
HOCl is produced in the brains of patient with AD or PD. The long durations over which these pathologies develop suggests that even small changes in the trajectories of AD and PD would stem the onset and extent of these diseases. Based on these considerations, HOCl scavenging through increased 5HT release has the potential to alter the course of PD or AD. The use of drugs with a history of safe and efficacious use in humans to scavenge HOCl makes this an exciting and real possibility.
Materials and Methods
Materials
The ELISA kit for Tumor Necrosis Factor-α (TNFα) was obtained from BD Biosciences (San Jose, CA). Pre-cast 15% gels were bought from Bio-Rad (Hercules, CA) and Immbilon PDVF membranes from Millipore (Millipore, Billerica, MA). The Halt protease inhibitor cocktail mix was obtained from Thermo Scientific (Pittsburgh, PA). Thermo Scientific supplied the 1.5 and 6.2 µm size markers (Duke Standards). The chemiluminescence kit was obtained from GE Healthcare (Pittsburgh, PA) and film from Denville Scientific (South Plainfield, NJ). Reagent grade sodium hypochlorite and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO). Ultrapurified water was used in the preparations of stock solutions and the concentrations of HOCl were determined using 5-thio-2-nitrobenzoic acid [63]. The latter reagent was generated by reduction of 5,5’-dithiobis-(2-nitrobenzoic acid) by sodium borohydride in sodium phosphate buffer (pH 7.9) as described by Jeitner et al. [63].
Animals
Adult male C57/Bl6 mice aged 8–10 weeks old (20–26g) were housed individually. Mice were acclimated to the institution’s Animal Care Facility for 1 week prior to the experiments. All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of Winthrop University Hospital and were compliant with NIH guidelines for ethical treatment of animals.
Cell culture
The human monocytic cells THP-1 were maintained in a medium containing RPMI medium, bovine calf serum and 10,000 U/mL penicillin/streptomycin (v:v:v, 89:10:1), at between 2.5 × 105 and 106 cells per mL. For each experiment, the cells were diluted at a density of 2.5 × 105 per mL and treated with 100 nM phorbol 12-myristate 13-acetate to induce differentiation to phagocytic phenotype. Immediately following the addition of the phorbol ester, the cells were seeded in a volume of 0.5 mL per well in 24 multiwell plates to yield 6.25 × 104 cells per cm2. Forty eight hours later, the medium was removed and replaced with either 0.3 mL fresh medium or medium containing graded amounts of serotonergic aggregates. After a further 24 h of incubation, 0.25 mL of the medium bathing the cells was removed and assessed for lactate dehydrogenase activity as a measure of cell viability. Care was taken to ensure to not disturb the settled aggregates while removing the medium. In one group of cells treated with medium alone, the medium was removed and replaced with 0.3 mL RIPA buffer to lyse the cells to quantify the intracellular lactate dehydrogenase activity. The activity present in the medium was expressed as a function of the total cellular activity.
Neuroblastoma-derived SH SY5Y cells were maintained in medium composed of F12/MEM (1:1), bovine calf serum and penicillin/streptomycin (89:10:1). In preparation for the experiments, the cells were diluted to a density of 6.0 × 104 per mL and seeded in a volume of 0.5 mL per well of 24 multiwell plates to yield 3 × 104 cells per cm2. The next day the medium was replaced with medium containing 10 µM retinoic acid to induce cellular differentiation. This treatment was repeated four more times every other day for a total of five treatments over 10 days. The differentiated cells were used between six and 48 h of the last retinoic acid treatment. Retinoic acid was prepared as a 10 mM solution in ethanol and stored at −20°C.
UV Spectroscopy
Spectra were recorded using a UV-1601PC spectrophotometer (Shimadzu, Kyoto, Japan). Scans were performed in PBS at pH 7.4 and 20°C in a quartz cell. Concentrations of HOCl ranged from 50 to 200µM with 5HT at 50µM. The recordings were done over the wavelength range of 190–700nm for 150 m.
1D 1H-NMR
1D 1H-NMR spectra were obtained on a Brucker 500 MHz spectrometer using the following acquisition parameters: 90° pulse, 32 transients of 32K data points were collected with a sweep width of 6,000 Hz (12 ppm), 8 second relaxation delay during which presaturation of the water signal was applied for 2 sec. The total acquisition time was 6 minutes. Data were analyzed using Mnova Lite 5.2.5 software (Mestrelab Research, Santiago de Compostela, Spain).
Reactions of Chlorinated 5HT and 5-thio-2-nitrobenzoic acid
5HT (200 µM) was incubated with HOCl (100µM – 1mM) in PBS and a final volume of 1 mL at 20°C for 15s and 64 m. Five mL aliquots of the reaction mix were then combined with 1 mL 50 µM 5-thio-2-nitrobenzoic acid for 15 m, after which absorbance was measured at 412nm. The residual amounts of 5-thio-2-nitrobenzoic acid were calculated using the Beer-Lambert equation and an extinction coefficient of 14.15 mM.cm−1 [63].
Preparation of aggregates of chlorinated 5HT or dopamine
Particles were prepared in 10 ml aliquots of PBS containing 10 mM HOCl and either 20 mM dopamine or 5HT. The reaction of dopamine and HOCl was carried out for 24 hours at 20°C with rotation of the 15 mL reaction tubes every 2 s. 5HT and HOCl were reacted similarly but for different durations as indicated in Fig. 4. After the reaction, the precipitates were collected with centrifugation at 600 × g for 15 minutes at 4°C. Nine ml of the supernatant resulting from this centrifugation was discarded and the remainder used to transfer the particles to 1.5 ml centrifuge tubes for centrifugation at 10,000 × g for 15 minutes at 4°C. The resulting pellet was washed three times with H2O and centrifugations at 10,000 × g. Following this centrifugation, the supernatant was discarded and the pellet resuspended with 1.5 ml ice-cold H2O. Aliquots were withdrawn for the determination of the particle mass. For addition to cells, the samples collected by centrifugation and resuspended in tissue culture media.
Flow cytometry of chlorinated 5HT aggregates
Flow cytometry was performed using a BD FACSArray with the flow rate adjusted to 0.5 mL/m and the power of the laser adjusted to the lowest values that distributed the aggregates as a diagonal in a dot plot of Forward Scatter and Side Scatter. A total of 105 light scattering events were analyzed for each sample. The maximum time for sample collection, however, was limited to 200 seconds to avoid exceeding the sample volume per well. This meant that some samples contained less than 105 events. Thus, the data was normalized by dividing the number of events by the collection time in seconds for each sample.
The consistency of these recordings was evaluated by assessing the peak distribution of 50 nm gold nanoparticles and 1.5 µm size marker beads as per the manufacturer’s instruction. The peak of the 1.5 µm size beads consistently occurred at 529 forward scatter units.
Cell viability assays
The release of lactate dehydrogenase (LDH) into the medium conditioned by cells was used to measure the toxicity of aggregates to THP-1 cells as described in [23, 26]. These experiments began with replacement of the culture medium with 0.3 mL fresh medium containing 6.75 µg aggregates per well. Twenty four h later 0.25 mL of the medium was removed and combined with 2.5 µL 10% Triton X-100. Two hundred µL of this solution was assayed for LDH activity as described by Beutler [64].
The toxicity of HOCl to SH SY5Y cells was also assessed by the measuring the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in cultures in 24-multiwell plates These experiments were adapted from those of Whitman et al. [65] and began with washing the cells twice with Hank’s Buffered Saline Solution (HBSS) at 37°C. HOCl, serotonin, or combinations thereof were added to the cells for 15 minutes in a total volume of 0.5 mL HBSS per well and incubated under tissue culture conditions. Stocks of HOCl and serotonin were prepared as 100 mM solutions in H2O on ice and diluted into HBSS at 37°C immediately prior to their application. The incubations were terminated with the addition of 1 mL of spent media that served to scavenge any HOCl present. The mixture of HBSS and spent media was replaced with 0.5 mL fresh media per well and the cells cultured for a further 24 h. This incubation was followed by the replacement of the media with 250 µL of 1 mg/mL MTT in HBSS per well for 15 minutes and incubated culture conditions. The dye solution was then aspirated from the wells and replaced with 200 mL dimethyl sulfoxide to dissolve the formazan products in the wells. These products were quantified at 510 nm.
KGDH complex assay
KGDH complex activity was measured by monitoring the reduction of NAD+ by this complex extracted from SH SY5Y cells cultured on 60 mm diameter dishes. The cells were washed twice with HBSS at 37°C, prior to the addition of HOCl, 5HT or combinations thereof in HBSS for 15 minutes under tissue culture conditions. HOCl and 5HT were prepared as 100 mM solutions in H2O on ice and diluted into HBSS at 37°C immediately prior to their addition to the cells in a total volume of 10 mL per dish. These compounds were removed at the end of the reactions and the cells then washed with ice-cold HBSS. Following this wash, a lysis solution consisting of 20 mM Tris-HCl (pH 7.5), 1 mM dithiothreitol, 1 mM EDTA, 0.2% Triton X-100, and 1 µL/mL Halt™ protease inhibitors was added at a ratio of 150 µL per dish. The lysed material was then collected with scraping and assayed for KGDH complex activity as described by Gibson et al. [66]. This assay utilized a reaction mixture of 50 mM MOPS (pH 8.0), 1.2 mM MgCl2, 1.2 mM CaCl2, 0.16 mM coenzyme A, 1 mM α-ketoglutarate, 0.2% Triton X-100, and 40 µM rotenone. The latter compound was prepared fresh as a 250 mM solution in dimethyl sulfoxide and added with vigorous mixing. Lysates were preincubated with the reaction components except NAD+ for 7 minutes at 30°C, after which NAD+ was added to initiate the reaction. The reduction of NAD+ to NADH was recorded at 340 nm over three minutes and quantified using extinction coefficient 6.22 mM.cm−1.
Treatments and Lipopolysaccharide Injections
Mice were assigned to two control groups, one receiving saline followed by LPS (n =6), and one receiving saline alone (n =5), an ibuprofen group (10mg/kg, n = 7) and a fluoxetine group (10mg/kg, n =9). Mice received i.p. injections of their respective treatments for three days. On the fourth day, mice received LPS i.p. injections (5mg/kg) and were sacrificed the following day.
Markers of Inflammation
After euthanasia, the mouse brains were excised, flash frozen and homogenized in ice-cold lysis buffer (100 mg tissue/ml) consisting of 25 mM Tris, 0.25M sucrose, 2 mM EDTA, 10 mM EGTA, 1% Triton X-100, 1 mM 3 chlorotyrosine and an aliquot of the Halt protease inhibitor cocktail mix (as per the manufacturer’s instructions). Given that the brain is a rich source of dehalogenases [33], we reasoned that these enzymes might remove chloride from the tyrosyl residues and thereby obviate the immunoreactivity to 3-chlorotyrosine antibody during the preparation of samples for SDS-PAGE. Consequently, free 3-chlorotyrosine was included in our tissue extraction buffers to inhibit chloride-specific dehalogenase activity in lysed tissues [33]. The samples were centrifuged at 12,000 rpm for 20 minutes and supernatants were collected. Protein concentrations were determined with the Bradford assay. We assessed the extent of neuroinflammation by measuring TNFα, myeloperoxidase and the presence of chlorinated adducts via 3-chlorotyrosine. TNFα levels were measured with the mouse TNFα ELISA kit. Myeloperoxidase and 3-chlorotyrosine were measured by Western blot analysis using 50 µg protein per lane separated on 4–15% gradient SDS-PAGE gels and probed after transfer with polyclonal rabbit myeloperoxidase antibody (1:1000 #ab45977, Abcam, Cambridge, MA) or polyclonal rabbit 3-chlorotyrosine antibody (1:250; Hycult Biotech, Plymouth Meeting, PA). These antibodies were visualized using chemiluminescence and film. Band density was measured with a Kodak 4000 camera (Kodak, Rochester, NY) using 0.3 second exposure and net pixel density was calculated using the Carestream MI program (Kodak, Rochester, NY). Density analysis was performed with the auto-background option enabled, and with the area for pixel density measurement held constant for each blot. The net pixel density from each band of interest was analyzed and normalized to the net pixel density of the corresponding β-actin band.
Open field behavioral measurements
Mice were weighed daily and placed in an open field system. Subjects were recorded for four hours beginning from an h prior to the onset of the dark phase. Locomotor behavior was assessed through the Coulbourn Tru Scan Activity System (Whitehall, PA). Weight and gauges of locomotor behavior (distance, number of moves and movement time) were compared the day before and the day after LPS administration across treatment groups.
Statistics
Significance was determined by one-way ANOVA followed by Fisher LSD or Kruskal Wallis post hoc analysis and t-test where appropriate. Graphical fittings were performed using the SigmaPlot program (Systat Software).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
HOCl chlorinates the nitrogen atoms of serotonin
The reaction products have lower oxidation potentials than HOCl
Each molecule of serotonin can scavenge multiple molecules of HOCl by forming polymers
Serotonin protects cells from the toxicity of HOCl
Serotonin reuptake inhibitors mitigate endotoxin-induced sickness behavior in mice
Acknowledgments
This project was funded by RO3-NS074286 and the Theresa Pantnode Santmann Foundation Award, both of which were awarded to TMJ. The authors are grateful to Jim Mathew, B.Sc. for his assistance with the experiments and also Sean Arlauckas, Ph.D. and Anatoliy Popov, Ph.D. for their assistance in the NMR experiments.
Abbreviations
- 5HT
5-Hydroxytryptamine (serotonin)
- HOCl
Hypochlorous acid
- KGDH
α-Ketoglutarate dehydrogenase
- LDH
Lactate dehydrogenase
- LPS
Lipopolysaccharide
- SSRI
Selective serotonin reuptake inhibitor
- TNFa
Tumor Necrosis Factor a
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta. 2010;1802:122–134. doi: 10.1016/j.bbadis.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012;3:CD007176. doi: 10.1002/14651858.CD007176.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2012;11:CD002854. doi: 10.1002/14651858.CD002854.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grodstein F, O’Brien J, Kang JH, Dushkes R, Cook NR, Okereke O, et al. Long-term multivitamin supplementation and cognitive function in men: a randomized trial. Ann Intern Med. 2013;159:806–814. doi: 10.7326/0003-4819-159-12-201312170-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–366. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang XR, Wrona MZ, Alguindigue SS, Dryhurst G. Reactions of the putative neurotoxin tryptamine-4,5-dione with L-cysteine and other thiols. Chem Res Toxicol. 2004;17:357–369. doi: 10.1021/tx020084k. [DOI] [PubMed] [Google Scholar]
- 7.Kato Y, Peskin AV, Dickerhof N, Harwood DT, Kettle AJ. Myeloperoxidase catalyzes the conjugation of serotonin to thiols via free radicals and tryptamine-4,5-dione. Chem Res Toxicol. 2012;25:2322–2332. doi: 10.1021/tx300218f. [DOI] [PubMed] [Google Scholar]
- 8.Jiang XR, Wrona MZ, Dryhurst G. Tryptamine-4,5-dione, a putative endotoxic metabolite of the superoxide-mediated oxidation of serotonin, is a mitochondrial toxin: possible implications in neurodegenerative brain disorders. Chem Res Toxicol. 1999;12:429–436. doi: 10.1021/tx9801615. [DOI] [PubMed] [Google Scholar]
- 9.Ximenes VF, Maghzal GJ, Turner R, Kato Y, Winterbourn CC, Kettle AJ. Serotonin as a physiological substrate for myeloperoxidase and its superoxide-dependent oxidation to cytotoxic tryptamine-4,5-dione. Biochem J. 2010;425:285–293. doi: 10.1042/BJ20090776. [DOI] [PubMed] [Google Scholar]
- 10.Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxidants & redox signaling. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds WF, Rhees J, Maciejewski D, Paladino T, Sieburg H, Maki RA, et al. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp Neurol. 1999;155:31–41. doi: 10.1006/exnr.1998.6977. [DOI] [PubMed] [Google Scholar]
- 12.Green PS, Mendez AJ, Jacob JS, Crowley JR, Growdon W, Hyman BT, et al. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J Neurochem. 2004;90:724–733. doi: 10.1111/j.1471-4159.2004.02527.x. [DOI] [PubMed] [Google Scholar]
- 13.Calingasan NY, Huang PL, Chun HS, Fabian A, Gibson GE. Vascular factors are critical in selective neuronal loss in an animal model of impaired oxidative metabolism. J Neuropathol Exp Neurol. 2000;59:207–217. doi: 10.1093/jnen/59.3.207. [DOI] [PubMed] [Google Scholar]
- 14.Ryu JK, Tran KC, McLarnon JG. Depletion of neutrophils reduces neuronal degeneration and inflammatory responses induced by quinolinic acid in vivo. Glia. 2007;55:439–451. doi: 10.1002/glia.20479. [DOI] [PubMed] [Google Scholar]
- 15.Chang CY, Song MJ, Jeon SB, Yoon HJ, Lee DK, Kim IH, et al. Dual functionality of myeloperoxidase in rotenone-exposed brain-resident immune cells. Am J Pathol. 2011;179:964–979. doi: 10.1016/j.ajpath.2011.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC, et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J Neurosci. 2005;25:6594–6600. doi: 10.1523/JNEUROSCI.0970-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Audhya T, Adams JB, Johansen L. Correlation of serotonin levels in CSF, platelets, plasma, and urine. Biochim Biophys Acta. 2012;1820:1496–1501. doi: 10.1016/j.bbagen.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Alho H, Leinonen JS, Erhola M, Lönnrot K, Aejmelaeus R. Assay of antioxidant capacity of human plasma and CSF in aging and disease. Restor Neurol Neurosci. 1998;12:159–165. [PubMed] [Google Scholar]
- 19.Arnhold J, Furtmüller PG, Regelsberger G, Obinger C. Redox properties of the couple compound I/native enzyme of myeloperoxidase and eosinophil peroxidase. Eur J Biochem. 2001;268:5142–5148. doi: 10.1046/j.0014-2956.2001.02449.x. [DOI] [PubMed] [Google Scholar]
- 20.style=“font-size:11.0pt s, line-height:115%, font-family:”Times New Roman” s, mso-fareast-font-family:Calibri, mso-fareast-theme-font:minor-latin, mso-ansi-language:EN-US et al. CRC Handbook of Chemistry & Physics. Boca Raton, Fl, USA: CRC Press; 2012. Electrochemical Series; pp. 5.80–5.90. [Google Scholar]
- 21.Jeitner TM, Lawrence A. Pulmonary autotoxicity and inflammation. In: Cohen MD, Zelikoff JT, Schlesinger RB, editors. Pulmonary Immunotoxicology. Norwell, MA, USA: Kluwer Academic Publishers Group; 2000. pp. 153–180. [Google Scholar]
- 22.Huether G, Fettkötter I, Keilhoff G, Wolf G. Serotonin acts as a radical scavenger and is oxidized to a dimer during the respiratory burst of activated microglia. J Neurochem. 1997;69:2096–2101. doi: 10.1046/j.1471-4159.1997.69052096.x. [DOI] [PubMed] [Google Scholar]
- 23.Jeitner TM, Kalogiannis M, Patrick PA, Gomolin I, Palaia T, Ragolia L, et al. Inflaming the diseased brain: a role for tainted melanins. Biochim Biophys Acta. 2015;1852:937–950. doi: 10.1016/j.bbadis.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sariahmetoglu M, Wheatley RA, Cakýcý Y, Kanzýk Y, Townshend A. Evaluation of the antioxidant effect of melatonin by flow injection analysis-luminol chemiluminescence. Pharmacol Res. 2003;48:361–367. doi: 10.1016/s1043-6618(03)00177-4. [DOI] [PubMed] [Google Scholar]
- 25.Fuller RW. Uptake inhibitors increase extracellular serotonin concentration measured by brain microdialysis. Life Sci. 1994;55:163–167. doi: 10.1016/0024-3205(94)00876-0. [DOI] [PubMed] [Google Scholar]
- 26.Jeitner TM, Kalogiannis M, Patrick PA, Ragolia L, Palaia T, Brand D, et al. Inflaming the diseased brain: a role for tainted melanins. Biochimica et Biophysica Acta. 2015;1852:937–950. doi: 10.1016/j.bbadis.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy P, Jameson GN, Winterbourn CC. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem Res Toxicol. 2009;22:1833–1840. doi: 10.1021/tx900249d. [DOI] [PubMed] [Google Scholar]
- 28.Jeitner TM, Xu H, Gibson GE. Inhibition of the alpha-ketoglutarate dehydrogenase complex by the myeloperoxidase products, hypochlorous acid and mono-N-chloramine. J Neurochem. 2005;92:302–310. doi: 10.1111/j.1471-4159.2004.02868.x. [DOI] [PubMed] [Google Scholar]
- 29.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nature reviews Neuroscience. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- 31.Gavins FN, Hughes EL, Buss NA, Holloway PM, Getting SJ, Buckingham JC. Leukocyte recruitment in the brain in sepsis: involvement of the annexin 1-FPR2/ALX anti-inflammatory system. FASEB J. 2012;26:4977–4989. doi: 10.1096/fj.12-205971. [DOI] [PubMed] [Google Scholar]
- 32.Griffin É, Skelly DT, Murray CL, Cunningham C. Cyclooxygenase-1-dependent prostaglandins mediate susceptibility to systemic inflammation-induced acute cognitive dysfunction. J Neurosci. 2013;33:15248–15258. doi: 10.1523/JNEUROSCI.6361-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mani AR, Ippolito S, Moreno JC, Visser TJ, Moore KP. The metabolism and dechlorination of chlorotyrosine in vivo. J Biol Chem. 2007;282:29114–29121. doi: 10.1074/jbc.M704270200. [DOI] [PubMed] [Google Scholar]
- 34.Bosker FJ, Folgering JH, Gladkevich AV, Schmidt A, van der Hart MC, Sprouse J, et al. Antagonism of 5-HT(1A) receptors uncovers an excitatory effect of SSRIs on 5-HT neuronal activity, an action probably mediated by 5-HT(7) receptors. J Neurochem. 2009;108:1126–1135. doi: 10.1111/j.1471-4159.2008.05850.x. [DOI] [PubMed] [Google Scholar]
- 35.Crespi F. SK channel blocker apamin attenuates the effect of SSRI fluoxetine upon cell firing in dorsal raphe nucleus: a concomitant electrophysiological and electrochemical in vivo study reveals implications for modulating extracellular 5-HT. Brain Res. 2010;1334:1–11. doi: 10.1016/j.brainres.2010.03.081. [DOI] [PubMed] [Google Scholar]
- 36.Henry G, Williamson D, Tampi RR. Efficacy and tolerability of antidepressants in the treatment of behavioral and psychological symptoms of dementia, a literature review of evidence. Am J Alzheimers Dis Other Demen. 2011;26:169–183. doi: 10.1177/1533317511402051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang HY, Deng M, Zhang YH, Chen HZ, Chen Q, Ruan B. Specific serotonin reuptake inhibitors prevent interferon-α-induced depression in patients with hepatitis C: a meta-analysis. Clin Gastroenterol Hepatol. 2014;12:1452–1460. e3. doi: 10.1016/j.cgh.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 38.Qin B, Zhang Y, Zhou X, Cheng P, Liu Y, Chen J, et al. Selective serotonin reuptake inhibitors versus tricyclic antidepressants in young patients: a meta-analysis of efficacy and acceptability. Clin Ther. 2014;36:1087–1095. e4. doi: 10.1016/j.clinthera.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Pattison DI, Davies MJ, Hawkins CL. Reactions and reactivity of myeloperoxidase-derived oxidants: differential biological effects of hypochlorous and hypothiocyanous acids. Free Radic Res. 2012;46:975–995. doi: 10.3109/10715762.2012.667566. [DOI] [PubMed] [Google Scholar]
- 40.Storkey C, Davies MJ, Pattison DI. Reevaluation of the rate constants for the reaction of hypochlorous acid (HOCl) with cysteine, methionine, and peptide derivatives using a new competition kinetic approach. Free Radic Biol Med. 2014;73:60–66. doi: 10.1016/j.freeradbiomed.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 41.Chen JW, Pham W, Weissleder R, Bogdanov A., Jr Human myeloperoxidase: a potential target for molecular MR imaging in atherosclerosis. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2004;52:1021–1028. doi: 10.1002/mrm.20270. [DOI] [PubMed] [Google Scholar]
- 42.Chen JW, Querol Sans M, Bogdanov A, Jr, Weissleder R. Imaging of myeloperoxidase in mice by using novel amplifiable paramagnetic substrates. Radiology. 2006;240:473–481. doi: 10.1148/radiol.2402050994. [DOI] [PubMed] [Google Scholar]
- 43.Dunford HB, Hsuanyu Y. Kinetics of oxidation of serotonin by myeloperoxidase compounds I and II. Biochem Cell Biol. 1999;77:449–457. [PubMed] [Google Scholar]
- 44.Michon T, Chenu M, Kellershon N, Desmadril M, Guéguen J. Horseradish peroxidase oxidation of tyrosine-containing peptides and their subsequent polymerization: a kinetic study. Biochemistry. 1997;36:8504–8513. doi: 10.1021/bi963168z. [DOI] [PubMed] [Google Scholar]
- 45.Yang YT, Whiteman M, Gieseg SP. Intracellular glutathione protects human monocyte-derived macrophages from hypochlorite damage. Life Sci. 2012;90:682–688. doi: 10.1016/j.lfs.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 46.Harwood DT, Kettle AJ, Brennan S, Winterbourn CC. Simultaneous determination of reduced glutathione, glutathione disulphide and glutathione sulphonamide in cells and physiological fluids by isotope dilution liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3393–3399. doi: 10.1016/j.jchromb.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 47.Hazell LJ, Stocker R. Alpha-tocopherol does not inhibit hypochlorite-induced oxidation of apolipoprotein B-100 of low-density lipoprotein. FEBS Lett. 1997;414:541–544. doi: 10.1016/s0014-5793(97)01066-1. [DOI] [PubMed] [Google Scholar]
- 48.Carr AC, Hawkins CL, Thomas SR, Stocker R, Frei B. Relative reactivities of N-chloramines and hypochlorous acid with human plasma constituents. Free Radic Biol Med. 2001;30:526–536. doi: 10.1016/s0891-5849(00)00495-0. [DOI] [PubMed] [Google Scholar]
- 49.Pinto JT, Van Raamsdonk JM, Leavitt BR, Hayden MR, Jeitner TM, Thaler HT, et al. Treatment of YAC128 mice and their wild-type littermates with cystamine does not lead to its accumulation in plasma or brain: implications for the treatment of Huntington disease. J Neurochem. 2005;94:1087–1101. doi: 10.1111/j.1471-4159.2005.03255.x. [DOI] [PubMed] [Google Scholar]
- 50.Pinto JT, Khomenko T, Szabo S, McLaren GD, Denton TT, Krasnikov BF, et al. Measurement of sulfur-containing compounds involved in the metabolism and transport of cysteamine and cystamine. Regional differences in cerebral metabolism. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3434–3441. doi: 10.1016/j.jchromb.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleta R, Gahl WA. Pharmacological treatment of nephropathic cystinosis with cysteamine. Expert opinion on pharmacotherapy. 2004;5:2255–2262. doi: 10.1517/14656566.5.11.2255. [DOI] [PubMed] [Google Scholar]
- 52.Gibrat C, Cicchetti F. Potential of cystamine and cysteamine in the treatment of neurodegenerative diseases. Progress in neuro-psychopharmacology & biological psychiatry. 2011;35:380–389. doi: 10.1016/j.pnpbp.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 53.Ji KA, Yang MS, Jeong HK, Min KJ, Kang SH, Jou I, et al. Resident microglia die and infiltrated neutrophils and monocytes become major inflammatory cells in lipopolysaccharide-injected brain. Glia. 2007;55:1577–1588. doi: 10.1002/glia.20571. [DOI] [PubMed] [Google Scholar]
- 54.Patel JC, Rice ME. Classification of H(2)O(2)as a neuromodulator that regulates striatal dopamine release on a subsecond time scale. ACS chemical neuroscience. 2012;3:991–1001. doi: 10.1021/cn300130b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spanos M, Gras-Najjar J, Letchworth JM, Sanford AL, Toups JV, Sombers LA. Quantitation of hydrogen peroxide fluctuations and their modulation of dopamine dynamics in the rat dorsal striatum using fast-scan cyclic voltammetry. ACS chemical neuroscience. 2013;4:782–789. doi: 10.1021/cn4000499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung ES, Chung YC, Bok E, Baik HH, Park ES, Park JY, et al. Fluoxetine prevents LPS-induced degeneration of nigral dopaminergic neurons by inhibiting microglia-mediated oxidative stress. Brain research. 2010;1363:143–150. doi: 10.1016/j.brainres.2010.09.049. [DOI] [PubMed] [Google Scholar]
- 57.Novio S, Nunez MJ, Amigo G, Freire-Garabal M. Effects of Fluoxetine on the Oxidative Status of Peripheral Blood Leucocytes of Restaint-Stressed Mice. Basic Clin Pharmacol Toxicol. 2011 doi: 10.1111/j.1742-7843.2011.00736.x. [DOI] [PubMed] [Google Scholar]
- 58.Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain, behavior, and immunity. 2012;26:469–479. doi: 10.1016/j.bbi.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 59.Galijasevic S, Abdulhamid I, Abu-Soud HM. Melatonin is a potent inhibitor for myeloperoxidase. Biochemistry. 2008;47:2668–2677. doi: 10.1021/bi702016q. [DOI] [PubMed] [Google Scholar]
- 60.Yirmiya R, Pollak Y, Barak O, Avitsur R, Ovadia H, Bette M, et al. Effects of antidepressant drugs on the behavioral and physiological responses to lipopolysaccharide (LPS) in rodents. Neuropsychopharmacology. 2001;24:531–544. doi: 10.1016/S0893-133X(00)00226-8. [DOI] [PubMed] [Google Scholar]
- 61.Martin SA, Pence BD, Greene RM, Johnson SJ, Dantzer R, Kelley KW, et al. Effects of voluntary wheel running on LPS-induced sickness behavior in aged mice. Brain Behav Immun. 2013;29:113–123. doi: 10.1016/j.bbi.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berg BM, Godbout JP, Kelley KW, Johnson RW. Alpha-tocopherol attenuates lipopolysaccharide-induced sickness behavior in mice. Brain Behav Immun. 2004;18:149–157. doi: 10.1016/S0889-1591(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 63.Jeitner TM, Kalogiannis M, Mathew J. Preparation of 2-nitro-5-thiobenzoate for the routine determination of reagent hypochlorous acid concentrations. Anal Biochem. 2013;441:180–181. doi: 10.1016/j.ab.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beutler E. Red Cell Metabolism. New York: Churchill Livingstone; 1986. [Google Scholar]
- 65.Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, et al. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun. 2005;326:794–798. doi: 10.1016/j.bbrc.2004.11.110. [DOI] [PubMed] [Google Scholar]
- 66.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, et al. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.