

Abstract
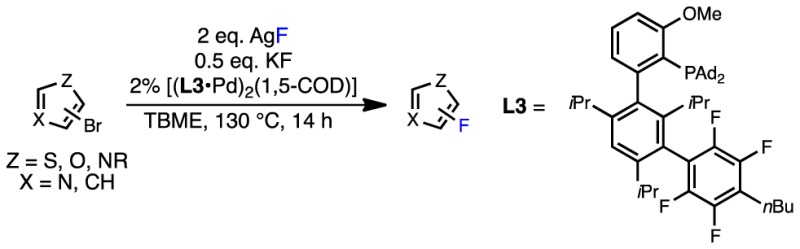
A thorough investigation of the challenging Pd-catalyzed fluorination of five-membered heteroaryl bromides is presented. Crystallographic studies and density functional theory (DFT) calculations suggest that the challenging step of this transformation is C–F reductive elimination of five-membered heteroaryl fluorides from Pd(II) complexes. On the basis of these studies, we have found that various heteroaryl bromides bearing phenyl groups in the ortho position can be effectively fluorinated under catalytic conditions. Highly activated 2-bromoazoles, such as 8-bromocaffeine, are also viable substrates for this reaction.
Introduction
Five-membered heterocycles are widely prevalent in the pharmaceutical industry.1 For example, a number of top-selling drugs, including raltegravier (Isentress),2 sitagliptin (Januvia),3 atorvastatin (Lipitor),4 and resperidone (Risperdal),5 contain at least one five-membered heterocycle (Figure 1, highlighted in blue). The commonality of five-membered heterocycles is due, in part, to their enormous structural diversity and interesting biological and electronic properties.1 Similarly, (hetero)aryl fluorides are frequently employed in medicinal chemistry due to their enhanced metabolic stability and membrane permeability in comparison to nonfluorinated analogues (Figure 1, highlighted in red).6 Indeed, all of the drugs shown in Figure 1 contain both a five-membered heterocyclic core and an aryl fluoride.
Figure 1.
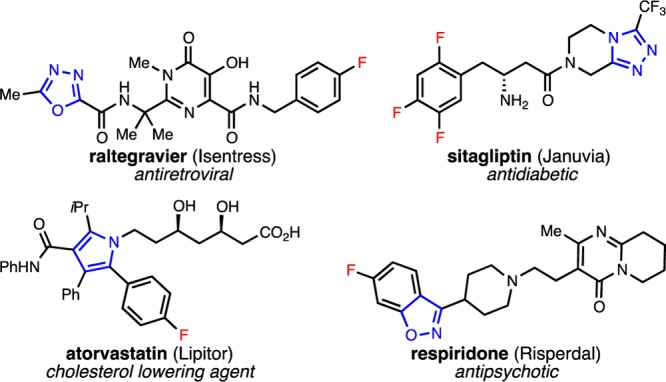
Top-selling pharmaceuticals containing both a five-membered heterocyclic core (blue) and an aryl fluoride (red).
Considering the independent importance of five-membered heterocycles and aryl fluorides in the pharmaceutical industry, there is a surprising lack of five-membered heteroaryl fluorides that have been prepared and studied for potential biological activity.7 This is likely due to the limited methods available for the fluorination of five-membered heteroarenes,8 which include thermal9a or photochemical9b Balz–Schiemann reactions, Halex reactions,10 electrophilic fluorinations of metalated heteroarenes,11 and direct fluorinations with F2.12 All of these methods suffer from severe drawbacks in terms of safety, functional group tolerance, generality, and/or formation of complex mixtures of products, which limit their utility. To date, most of the recently developed transition-metal-mediated methods for aryl fluorination13 have seen limited application to five-membered heteroaryl systems.14 Thus, there remains a strong need for the development of new methods for the fluorination of five-membered heteroarenes.
We15 and others16 have explored the Pd-catalyzed cross-coupling of (hetero)aryl halides with a metal fluoride salt (Figure 2A) as a simple and general method for the synthesis of (hetero)aryl fluorides. Advances in ligand (L1–L3) and precatalyst (P1–P3, Figure 2B) design have allowed us to convert a variety of nitrogen-containing six-membered heteroaryl triflates15a,15d and bromides15a,15c into the corresponding heteroaryl fluorides. Thus, we wondered if this methodology could be extended to the preparation of five-membered heteroaryl fluorides. However, previous stoichiometric and catalytic investigations of cross-coupling reactions involving five-membered heteroaryl halides suggest that reductive elimination is significantly more challenging in these reactions in comparison to that with six-membered aryl halides, likely due to the smaller size and increased electron richness of five-membered heteroaryl groups.17 Considering the already high kinetic barrier for C–F reductive elimination from Pd(II),16b,16c prior to this work it remained unclear if the reductive elimination of five-membered heteroaryl fluorides was feasible under synthetically relevant conditions. As a second challenge, nitrogen-containing heterocycles can inhibit Pd-catalyzed reactions by coordinating to the Pd center.15d,18 Herein, we describe catalytic, stoichiometric, and computational studies aimed toward determining if the Pd-catalyzed fluorination of five-membered heteroaryl bromides is a viable transformation with current catalyst systems.
Figure 2.
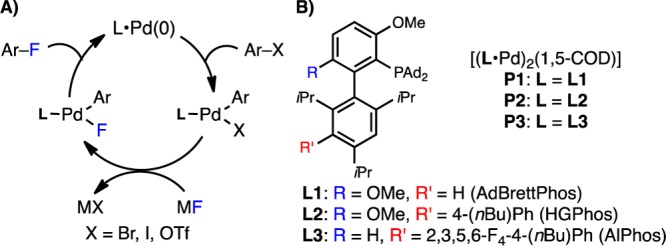
(A) Catalytic cycle for the Pd-catalyzed fluorination of aryl halides. (B) Ligands (L1–L3) and precatalysts (P1–P3) for this reaction.
Results and Discussion
We began our investigation by attempting the Pd-catalyzed fluorination of an array of five-membered heteroaryl bromides (4–13) under the standard reaction conditions used for the fluorination of six-membered heteroaryl bromides15a,15c using P1–P3 as precatalysts (Table 1). Unfortunately, the desired product was not observed in any of these reactions (see Table S1 in the Supporting Information for additional examples). In most cases, the starting material was recovered along with trace amounts of the corresponding reduction (Ar–H) product, as judged by GC/MS analysis of the crude reaction mixtures. Increasing the catalyst loading, reaction temperature, or number of equivalents of AgF/KF did not change the outcome of these reactions. For bromoazoles containing sp2-hybridized nitrogen centers (8–12), catalyst inhibition could account for this observation.18 Indeed, we have found that the addition of various thiazoles and N-substituted (benz)imidazoles to the otherwise high-yielding Pd-catalyzed fluorination of 4-(nBu)PhBr inhibits the desired reaction (see Table S2 in the Supporting Information). However, 1-methyl-1H-pyrazole did not significantly inhibit this reaction, indicating that the unsuccessful fluorinations of 10 and 11 are not necessarily due to catalyst inhibition. Thus, for simple five-membered heteroaryl bromides lacking sp2-hybridized nitrogen centers (e.g., 4–7), as well as bromopyrazoles (10 and 11), at least one of the elementary steps of the catalytic cycle shown in Figure 2 must not be operative under the standard reaction conditions.
Table 1. Selected Examples of Unsuccessful Pd-Catalyzed Fluorinations of Five-Membered Heteroaryl Bromidesa.
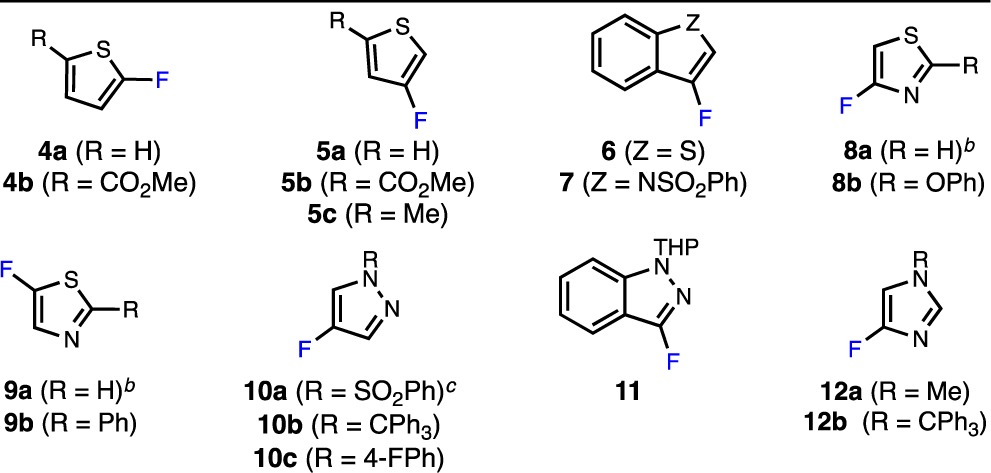
Reaction conditions: ArBr (0.10 mmol), AgF (0.20 mmol), KF (0.05 mmol), P1–P3 (2%), solvent (1.0 mL), 130 °C, 14 h. TBME = tert-butyl methyl ether.
Significant decomposition observed by 19F NMR and GC/MS.
PhSO2F observed by 19F NMR and GC/MS.
On the basis of previous work,15−17 we hypothesized that C–F reductive elimination from Pd(II) was the most challenging step in these reactions. We carried out an in-depth study of this transformation in order to improve its efficiency. To this end, we prepared L1-ligated oxidative addition complexes of 2-bromothiophene (13) and 5-acetyl-2-bromothiophene (14) to study their solid-state structures (Figure 3A).19 Although 13 and 14 proved to be unstable in solution for extended periods of time, single crystals suitable for X-ray diffraction of both complexes could be obtained (Figure 3B).20 Notably, these complexes are among the first biaryl monophosphine-ligated oxidative addition complexes of five-membered heteroaryl halides that have been synthesized and characterized.21 The solid-state structures of 13 and 14 were compared with that of the previously reported complex L1·Pd(4-(CN)Ph)Br (15)17b to analyze the differences that arise upon replacing a six-membered aryl group with a smaller five-membered heteroaryl group (Figure 3C). Consistent with our previous computational studies,17b the Ar–Pd–Br angle is significantly wider in five-membered heteroaryl complexes 13 and 14 (13, 81.48(4)°; 14, 81.2(1)°) than in six-membered aryl complex 15 (79.03(8)°) (Figure 3C). The smaller angle in 15 in comparison to those in 13 and 14 reflects the greater proclivity of this complex to undergo reductive elimination.17b Notably, only small differences were observed in the Pd–Ar and Pd–ipso bond lengths among these complexes (Figure 3C).
Figure 3.
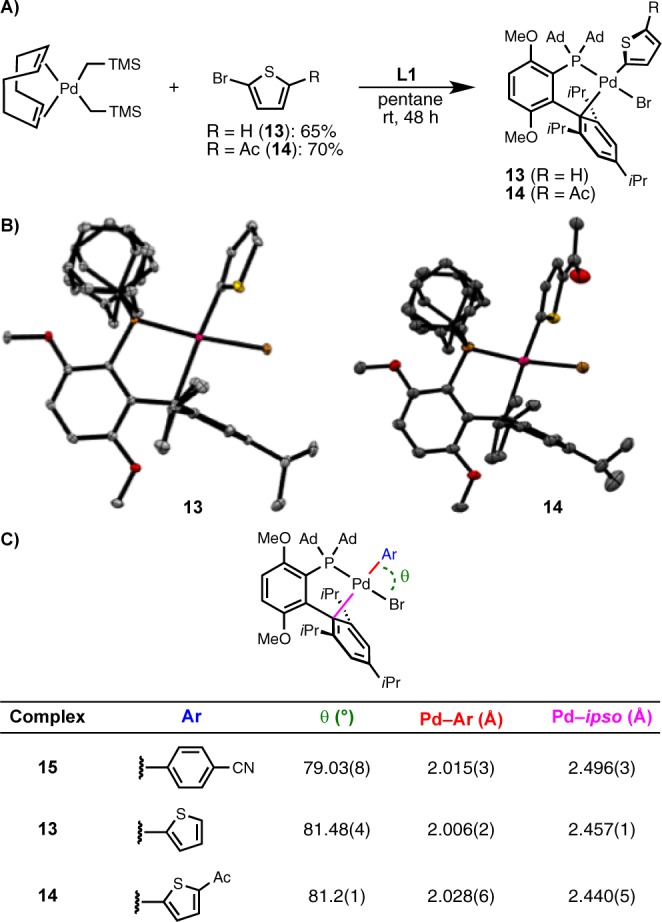
(A) Synthesis of oxidative addition complexes of five-membered heteroaryl bromides 13 and 14. (B) Solid-state structures of 13 and 14 (ellipsoids shown at 50%). (C) Comparison of the structures of 13 and 14 with that previously reported for 15.
Unfortunately, to date, all attempts to prepare L·Pd(Ar)F complexes bearing five-membered heteroaryl groups have been unsuccessful.22 Thus, we carried out density functional theory (DFT) calculations to better understand the structure and reactivity of these species (17–19) in comparison to that of the analogous complex bearing a phenyl group (16); the results of these studies are summarized in Table 2 (see the Supporting Information for optimized ground- and transition-state geometries). Consistent with our initial hypothesis, the barrier to C–F reductive elimination was calculated to be 7.0 kcal/mol higher in energy for the 2-thienyl-substituted complex 17 (27.7 kcal/mol) in comparison to phenyl-substituted complex 16 (20.7 kcal/mol), suggesting that reductive elimination is on the order of 100000 times slower in the former case. Additionally, the ground-state Ar–Pd–F angle was wider in 17 (82.3°) than in 16 (80.7°), which corroborates the X-ray crystallographic findings in Figure 3C. Notably, the calculated Pd–F bond lengths are in line with those that have been observed experimentally for other Ln·Pd(Ar)F complexes.16d The barrier to reductive elimination for the corresponding 3-thienyl complex 18 was 1.8 kcal/mol lower than for 17, which is also consistent with previous experimental and theoretical findings.17c,17d Taken together, these crystallographic (Figure 3) and computational (Table 2) studies confirm that C–F reductive elimination of five-membered heteroaryl fluorides is an extremely challenging process and is therefore most likely the rate-limiting step of the Pd-catalyzed fluorinations presented in Table 1.
Table 2. Computationally Determined Parameters for L3·Pd(Ar)F Complexes 16–19a.
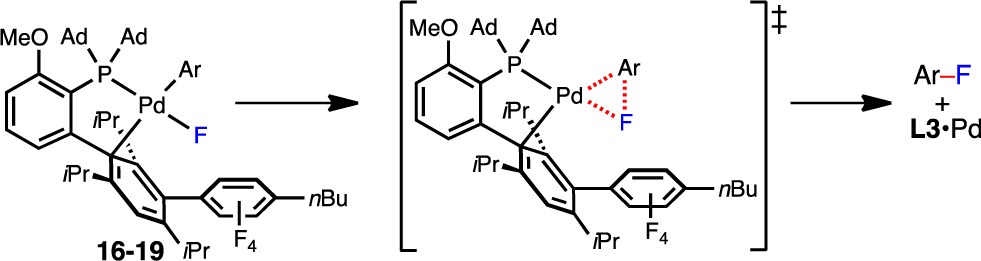

Energies were calculated at the M06/6-311+G(d,p)-SDD/SMD(toluene) level of theory with geometries optimized at the B3LYP/6-31G(d) level. ΔG⧧ values were determined at 25 °C.
Ground-state values.
On the basis of this analysis, we hypothesized that ortho-substituted heteroaryl bromides might be effective substrates for this reaction, due to the known accelerating effect of ortho substituents on reductive elimination.23 Indeed, DFT calculations confirm that the addition of an phenyl group adjacent to the Pd center (19) decreases the barrier of C–F reductive elimination substantially (21.8 kcal/mol) in comparison to 18 (25.9 kcal/mol). Therefore, we investigated the reactivity of 2-substituted-3-bromothiophenes (Table 3), because bromothiophenes tend to be well-behaved in Pd-catalyzed cross-coupling reactions.24 Unfortunately, the desired product was not observed with a methyl group in the 2-position (20a, entry 1). The addition of an additional electron-withdrawing group to further promote reductive elimination (20b, entry 2) was still ineffective.25 However, the corresponding substrate substituted with a bulky phenyl group in the ortho position furnished the desired product 20c, albeit in modest yield (entry 3). This finding represents one of the first transition-metal-catalyzed fluorinations of a five-membered heteroarene. An examination of the solvent and precatalyst employed revealed that tert-butyl methyl ether (TBME) is generally superior to other ethereal (2-MeTHF, cyclopentyl methyl ether, Bu2O) and hydrocarbon (toluene, cyclohexane) solvents and that P3 is consistently superior to P1 and P2(15a) for carrying out this transformation. The incorporation of various electron-withdrawing groups at the 5-position of the heteroaryl bromide further improved the yield of the desired product to synthetically useful levels (entries 4–8).25 Indeed, the presence of an ester (20d), nonenolizable ketone (20e), sulfonamide (20f), or amide (20g) was advantageous at this position, although substrates bearing formyl, acetyl, cyano, and nitro groups underwent significant decomposition during the reaction (see Table S1 in the Supporting Information). It should be noted that isolated products were contaminated with less than 1% of the corresponding reduction product, as judged by GC analysis (see the Supporting Information for details). However, small amounts (<5%) of a second fluorothiophene product, which is likely the regioisomeric product with the fluorine adjacent to the electron-withdrawing group, were detected in the crude reaction mixtures.26 Consistent with this hypothesis, this side product was not observed during the synthesis of 20h (entry 9), wherein the proposed regioisomer and the desired product are identical compounds. Additionally the use of AlPhos (L3) generally affords better selectivity for the desired product in comparison to HGPhos (L2) (as shown for 20d, entries 4 and 5), which is also the case with six-membered-ring substrates.15a In all cases except for 20f, the undesired regioisomer could be chromatographically separated from the desired product.
Table 3. Pd-Catalyzed Fluorination of 2-Substituted 3-Bromothiophenesa.
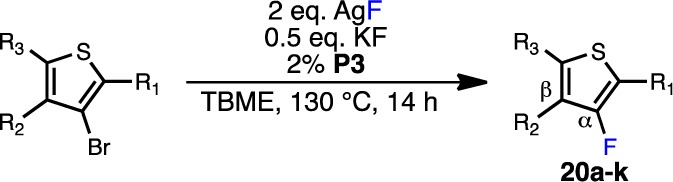
| entry | product | R1 | R2 | R3 | conversn, %b | yield, % (α:β)c |
|---|---|---|---|---|---|---|
| 1 | 20a | Me | H | H | n/d | n/o |
| 2 | 20b | Me | H | CO2Me | n/d | n/o |
| 3 | 20c | Ph | H | H | 45 | 22 (>50:1) |
| 4 | 20d | Ph | H | CO2Me | 95 | 80 (>50:1) |
| 5d | 20d | Ph | H | CO2Me | 95 | 91 (10:1) |
| 6 | 20e | Ph | H | C(O)Ph | 98 | 91 (26:1) |
| 7e | 20f | Ph | H | SO2NEt2 | 100 | 93 (30:1)f |
| 8 | 20g | Ph | H | C(O)NEt2 | 100 | 94 (>50:1)f |
| 9 | 20h | Ph | H | Ph | 95 | 80 |
| 10 | 20i | Ph | Me | Ph | n/d | 20 |
| 11 | 20j | Ph | Ph | Ph | n/d | n/o |
| 12 | 20k | 1-naphthyl | H | H | n/d | n/o |
Reaction conditions unless specified otherwise: ArBr (0.10 mmol), AgF (0.20 mmol), KF (0.05 mmol), P3 (2%), TBME (1.0 mL), 130 °C, 14 h. n/d = not determined. n/o = not observed.
Determined by GC.
Yield determined by 19F NMR comparison to an authentic sample unless otherwise noted.
P2 was used in place of P3.
Toluene as reaction solvent.
Isolated yield, 0.50 mmol scale.
We also investigated whether additional ortho substitution could further promote C–F reductive elimination (entries 10–12). Bromothiophenes bearing additional methyl (20i, entry 10) or phenyl (20j, entry 11) groups adjacent to the bromine atom produced diminished yields in comparison to the corresponding substrate lacking substitution at the 4-position (20h, entry 9). Likewise, the presence of a bulky 1-naphthyl group in the ortho position impeded the formation of 20k (entry 12). The sluggish reactivity of these extremely hindered substrates is likely due to slow oxidative addition of the aryl bromide to the active L3·Pd(0) species. Overall, these studies revealed that only 3-bromothiophenes bearing both phenyl groups in the ortho position and electron-withdrawing groups on the thiophene ring provide synthetically useful yields, which is consistent with our hypothesis that C–F reductive elimination is the challenging process in this transformation.
We next attempted to extend these findings to other five-membered heteroaryl bromides bearing ortho phenyl substituents (Table 4). Consistent with the results highlighted in Table 3, only 2-bromothiophenes bearing an electron-withdrawing group in the 5-position afforded a high yield of the desired product (21a), while those substituted with an electron-neutral phenyl group (21b) or lacking substitution at this position (21c) were less reactive (Table 4). The overall lower yields obtained for these substrates in comparison to those in Table 3 (compare 21b to 20h and 21c to 20c) are consistent with the DFT calculations in Table 2, which show that reductive elimination of 3-thienyl groups is easier than that of 2-thienyl groups, as well as with literature precedent.17c,17d Notably, in the case of 21a, 4% of the corresponding reduction product was isolated along with the desired aryl fluoride.
Table 4. Additional Pd-Catalyzed Fluorinations of Ortho-Substituted Five-Membered Heteroaryl Bromidesa.
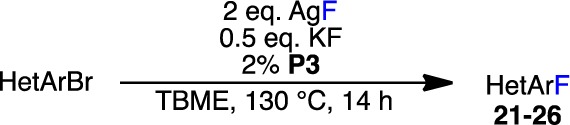
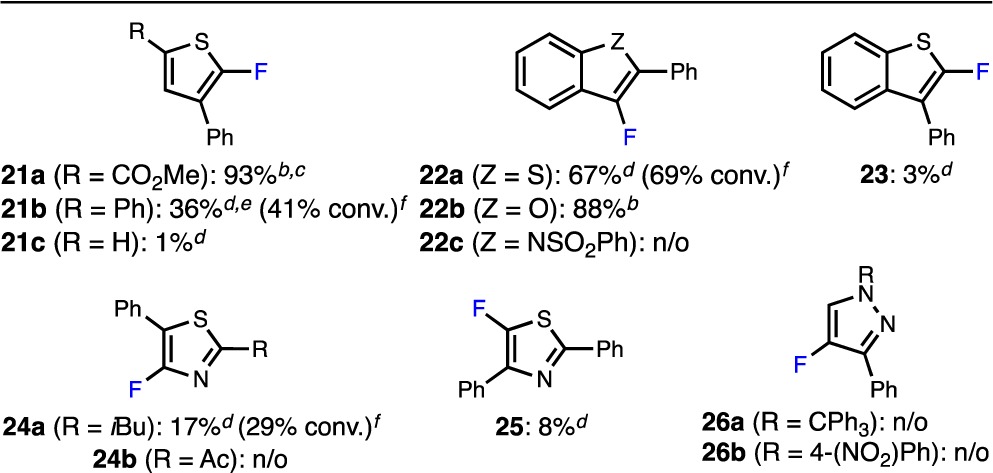
Reaction conditions unless specified otherwise: ArBr (0.10 mmol), AgF (0.20 mmol), KF (0.05 mmol), P3 (2%), TBME (1.0 mL), 130 °C, 14 h. n/o = not observed.
Isolated yield, 0.50 mmol scale.
Contaminated with 4% of the corresponding reduction product.
Yield determined by 19F NMR comparison to an authentic sample.
Toluene as reaction solvent.
Determined by GC.
The fluorinations of ortho-substituted benzofused heteroaryl bromides (22 and 23) afforded similar results. Although 3-bromo-2-phenylbenzo[b]thiophene underwent fluorination only sluggishly, furnishing an inseparable mixture of starting material and 22a, the corresponding benzo[b]furan underwent clean fluorination to give 22b in high yield. The higher reactivity of benzofurans (22b) in comparison to benzothiophenes (22a) likely reflects the stronger inductive electron-withdrawing effect of the O atom in the benzofuran ring.17c,17d,27 Unfortunately, the corresponding 3-bromo-N-sulfonylindole did not undergo fluorination to provide 22c. Consistent with our studies concerning non-benzo-fused bromothiophenes (Tables 3 and 4), the corresponding 2-bromobenzo[b]thiophene bearing an ortho phenyl group provided only a low yield of 23 under the reaction conditions.
We also examined the Pd-catalyzed fluorination of bromoazoles with phenyl groups in the ortho position (24–26, Table 4). Low yields of the desired product were observed with both ortho-substituted 4- (24a,b) and 5-bromothiazoles (25). Thiazoles inhibit the desired reaction, which likely explains the observed decrease in reactivity in comparison to thiophenes (see Table S2 in the Supporting Information). As in previous cases, increasing the catalyst loading did not significantly improve the yield of these reactions. Additionally, none of the desired product was observed with more electron rich 4-bromo-1H-pyrazoles substituted with a phenyl group in the ortho position (26a,b), regardless of the nitrogen protecting group (for additional examples, see Table S1 in the Supporting Information).
To overcome the generally poor reactivity of bromoazoles, we also attempted the fluorination of electron-deficient 2-bromo-1,3-azoles (Table 5). In these cases, significant formation of side products occurred using TBME as the reaction solvent, and so these reactions were carried out in toluene. Although 2-bromothiazole did not provide the desired product (27a) under the reaction conditions, the addition of a phenyl group adjacent to the nitrogen center led to a low yield of 27b. As shown with bromothiophenes (20d–g, Table 3; 21a, Table 4), the presence of an electron-withdrawing group on the thiazole ring was crucial for the isolation of 27c in synthetically useful yield. Notably, less than 5% of 27b,c was observed in the absence of P3, ruling out the possibility of a background Halex process. Although simple N-substituted 2-bromo-1H-imidazoles underwent decomposition (28a) or no reaction (28b) under these conditions, we found that the more activated 8-bromocaffeine could be efficiently converted to 29 in high yield; again, only trace amounts of 29 were observed in the absence of catalyst. Additionally, none of the corresponding reduction product was detected in the purified samples of 27c and 29 (see the Supporting Information for details). It should be noted that benzo-fused 2-bromoazoles, such as 2-bromobenzothiazole and 2-bromo-1-methyl-1H-benzimidazole, underwent significant fluorination in the absence of catalyst, reflecting their proclivity toward Halex processes (not shown). Nevertheless, this methodology may be attractive for the synthesis of 2-fluoroazoles bearing electron-withdrawing groups.
Table 5. Pd-Catalyzed Fluorinations of 2-Bromo-1,3-azolesa.
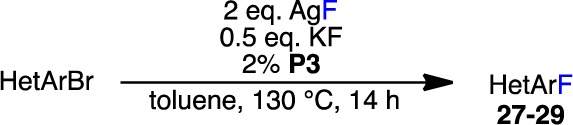
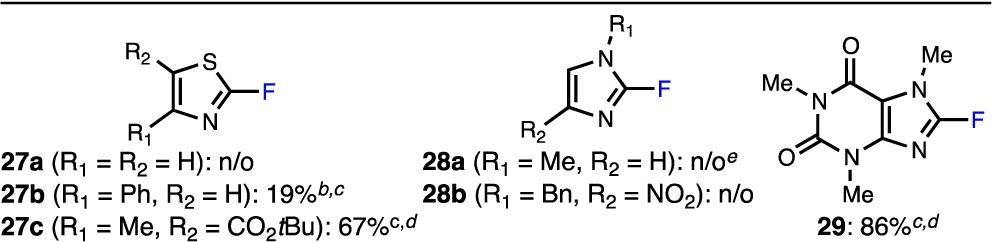
Reaction conditions unless specified otherwise: ArBr (0.10 mmol), AgF (0.20 mmol), KF (0.05 mmol), P3 (2%), toluene (1.0 mL), 130 °C, 14 h. n/o = not observed.
Yield determined by 19F NMR comparison to an authentic sample.
<5% yield observed in the absence of P3.
Isolated yield, 0.50 mmol scale.
Significant decomposition observed by 19F NMR and GC/MS.
Conclusion
By systematically studying substituent effects on the fluorination of five-membered heteroaryl bromides, we were able to identify a number of five-membered heteroaryl fluorides that could be prepared in synthetically useful yields with a catalyst system based on L3. In particular, electron-deficient and ortho-substituted benzo[b]thiophenes, ortho-substituted benzo[b]furans, and highly activated 2-bromo-1,3-azoles are viable substrates for this reaction.28 Despite these advances, the scope of this reaction remains limited, especially with respect to bromoazoles. Although our previous work in this area15d,17a,17b suggests that increasing the steric bulk of the ligand could potentially help overcome these problems, it is probable that a more fundamental change to the reaction, such as a change in mechanism, transition-metal catalyst, or ligand architecture may be needed to access a broader scope of five-membered heteroaryl fluorides. Given the potential importance of five-membered heteroaryl fluorides in medicinal chemistry, this transformation remains an active area of research in our group.
Experimental Section
General Procedure for Pd-Catalyzed Fluorination Reactions
In a nitrogen-filled glovebox, an oven-dried screw-cap reaction tube equipped with a stir bar was charged (in this order) with silver fluoride (26 mg, 0.20 mmol, 2.00 equiv), additive (0.05 mmol, 0.50 equiv), P1–P3 (4.0 mg, 2%), aryl bromide (0.10 mmol, 1.00 equiv), and solvent (1.0 mL). The tube was capped, removed from the glovebox, and placed in an oil bath that had been preheated to 130 °C, and the mixture was vigorously stirred for 14 h. (Caution! Perform behind a barrier such as a blast shield!) At this time, the tube was cooled to room temperature, and 1-fluoronaphthalene (20 μL, 1.55 equiv) was added. The reaction mixture was analyzed directly by 19F NMR. Afterward, the reaction mixture was filtered through a silica gel plug, eluted with EtOAc, and analyzed by GC (or GC/MS).
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award number GM46059. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. P.J.M. thanks the National Science Foundation for a predoctoral fellowship (2010094243). P.J.M. also thanks Amgen for an educational donation, for which we are grateful. One of the X-ray diffractometers used in this work was purchased with the help of funding from the National Science Foundation (Grant CHE 0946721). Dr. Peter Mueller (MIT) is acknowledged for solving the X-ray structures of 13 and 14. Dr. Aaron Sather (MIT) is acknowledged for helpful discussions, assistance with this manuscript, and the generous donation of P3. We thank Prof. Peng Liu (University of Pittsburgh) for help with computational studies. Calculations were performed at the Center for Simulation and Modeling at the University of Pittsburgh.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.5b00631.
The authors declare the following competing financial interest(s): MIT has patents on some of the ligands and precatalysts used in this work, from which S.L.B. and former coworkers receive royalty payments.
Dedication
Dedicated to the memory of Professor Gregory L. Hillhouse: brilliant chemist, great person and friend.
Supplementary Material
References
- a Vitaku E.; Smith D. T.; Njardarson J. T. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; b Baumann M.; Baxendale I. R.; Ley S. V.; Nikbin N. Beilstein J. Org. Chem. 2011, 7, 442–495. 10.3762/bjoc.7.57. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Joule J. A.; Mills K.. Heterocyclic Chemistry, 5th ed.; Wiley: Chichester, U.K., 2010. [Google Scholar]; d Sperry J. D.; Wright D. L. Curr. Opin. Drug Discovery 2005, 8, 723–740. [PubMed] [Google Scholar]
- Crescenzi B.; Gardelli C.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Petrocchi A.; Poma M.; Rowley M.; Scarpelli R.; Summa V.. (IRBM P. Angelitti S.P.A.). N-substituted hydroxypyrimidinone carboxamide inhibitors of HIV integrase. U.S. Patent US7217713, July 24, 2006.
- Edmondson S. D.; Fisher M. H.; Kim D.; MacCoss M.; Parmee E. R.; Weber A. E.; Xu J.. (Merck & Co., USA). Such as 7-((3r)-3-amino-4-(3,4-di.fluorophenyl)butanoyl)-2-(trifluoromethyl)-5,6,7,8 -tetrahydroimidazo(1,2-a)pyrazine dihydrochloride for treatment of insulin resistance disorders. U.S. Patent US6699871 B2, July 5, 2002.
- Roth B. D.(Warner-Lambert Co., USA). Trans-6-[2-(3- or 4-carboxamido-substituted pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis. U.S. Patent US4681893, May 30, 1986.
- Kennis L. E. J.; Vandenberk J.. (Janssen Pharmaceutica N.V.). Psychological disorders. U.S. Patent US4804663A, March 27, 1985.
- a Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. J. J. Med. Chem. 2015, 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]; b Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; c Kirk K. L. Org. Process Res. Dev. 2008, 12, 305–321. 10.1021/op700134j. [DOI] [Google Scholar]; d Müller K.; Faeh C.; Diederich F. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Gakh A. A.; Kirk K. L.. Fluorinated Heterocycles. In Fluorinated Heterocycles; Gakh A. A., Kirk K. L., Eds.; ACS Symposium Series 1003; American Chemical Society: Washington, DC, 2009; pp 3–20. [Google Scholar]
- a Fluorine in Heterocyclic Chemistry; Nenajdenko V., Ed.; Springer: New York, 2014; Vol. 1 (5-Membered Heterocycles and Macrocycles). [Google Scholar]; b Kirk K. L.Fluorinated Five-Membered Nitrogen-Containing Heterocycles. In Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications; Petrov V. A., Ed.; Wiley: Hoboken, NJ, 2009; pp 91–158. [Google Scholar]; c Shermolovich Y.Fluorinated Five-Membered Heterocycles Containing Oxygen, Sulfur, Selenium, and Phosphorus. In Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications; Petrov V. A., Ed.; Wiley: Hoboken, NJ, 2009; pp 159–225. [Google Scholar]
- a Balz G.; Schiemann G. Ber. Dtsch. Chem. Ges. B 1927, 60, 1186–1190. 10.1002/cber.19270600539. [DOI] [Google Scholar]; b Kirk K. L.; Cohen L. A. J. Am. Chem. Soc. 1973, 95, 4619–4624. 10.1021/ja00795a026. [DOI] [PubMed] [Google Scholar]
- a Sun H.; DiMagno S. G. Angew. Chem., Int. Ed. 2006, 45, 2720–2725. 10.1002/anie.200504555. [DOI] [PubMed] [Google Scholar]; b Finger G. C.; Kruse C. W. J. Am. Chem. Soc. 1956, 78, 6034–6037. 10.1021/ja01604a022. [DOI] [Google Scholar]
- For (hetero)aryllithium reagents, see:; a Nagaki A.; Uesugi Y.; Kim H.; Yoshida J.-i. Chem. - Asian J. 2013, 8, 705–708. 10.1002/asia.201201191. [DOI] [PubMed] [Google Scholar]; See also the examples included in the Supporting Information. For (hetero)aryl Grignard reagents, see:; b Yamada S.; Gavryushin A.; Knochel P. Angew. Chem., Int. Ed. 2010, 49, 2215–2218. and references cited therein 10.1002/anie.200905052. [DOI] [PubMed] [Google Scholar]
- Hutchinson J.; Sandford G. Top. Curr. Chem. 1997, 193, 1–43. 10.1007/3-540-69197-9_1. [DOI] [Google Scholar]
- Campbell M.; Ritter T. Chem. Rev. 2015, 115, 612–633. 10.1021/cr500366b. [DOI] [PubMed] [Google Scholar]
- For rare examples, see:; a Wang D.; Sun W.; Chu T. Eur. J. Org. Chem. 2015, 2015, 4114–4118. 10.1002/ejoc.201500338. [DOI] [Google Scholar]; b Ichiishi N.; Canty A. J.; Yates B. F.; Sanford M. S. Org. Lett. 2013, 15, 5134–5137. 10.1021/ol4025716. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Truong T.; Klimovica K.; Daugulis O. J. Am. Chem. Soc. 2013, 135, 9342–9345. 10.1021/ja4047125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sather A. C.; Lee H. G.; De La Rosa V. Y.; Buchwald S. L. Submitted for publication.; b Milner P. J.; Kinzel T.; Zhang T.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 15757–15766. 10.1021/ja509144r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee H. G.; Milner P. J.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 3792–3795. 10.1021/ja5009739. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee H. G.; Milner P. J.; Buchwald S. L. Org. Lett. 2013, 15, 5602–5605. 10.1021/ol402859k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Maimone T. J.; Milner P. J.; Kinzel T.; Zhang Y.; Takase M. K.; Buchwald S. L. J. Am. Chem. Soc. 2011, 133, 18106–18109. 10.1021/ja208461k. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Noël T.; Maimone T. J.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 8900–8903. 10.1002/anie.201104652. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Watson D. A.; Su M.; Teverovskiy G.; Zhang Y.; García-Fortanet J.; Kinzel T.; Buchwald S. L. Science 2009, 325, 1661–1664. 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wannberg J.; Wallinder C.; Ünlsüoy M.; Sköld C.; Larhed M. J. Org. Chem. 2013, 78, 4184–4189. 10.1021/jo400255m. [DOI] [PubMed] [Google Scholar]; b Grushin V. V. Acc. Chem. Res. 2010, 43, 160–171. 10.1021/ar9001763. [DOI] [PubMed] [Google Scholar]; c Yandulov D. V.; Tran N. T. J. Am. Chem. Soc. 2007, 129, 1342–1358. 10.1021/ja066930l. [DOI] [PubMed] [Google Scholar]; d Grushin V. V. Chem. - Eur. J. 2002, 8, 1006–1014. 10.1002/1521-3765(20020301)8:5<1006::AID-CHEM1006>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- a Su M.; Hoshiya N.; Buchwald S. L. Org. Lett. 2014, 16, 832–835. 10.1021/ol4035947. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Su M.; Buchwald S. L. Angew. Chem., Int. Ed. 2012, 51, 4710–4713. 10.1002/anie.201201244. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hooper M. W.; Hartwig J. F. Organometallics 2003, 22, 3394–3403. 10.1021/om030257g. [DOI] [Google Scholar]; d Hooper M. W.; Utsunomiya M.; Hartwig J. F. J. Org. Chem. 2003, 68, 2861–2873. 10.1021/jo0266339. [DOI] [PubMed] [Google Scholar]
- a Düfert M. A.; Billinglsey K. L.; Buchwald S. L. J. Am. Chem. Soc. 2013, 135, 12877–12885. 10.1021/ja4064469. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shen Q.; Hartwig J. F. J. Am. Chem. Soc. 2007, 129, 7734–7735. 10.1021/ja0722473. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shen Q.; Shekhar S.; Stambuli J. P.; Hartwig J. F. Angew. Chem., Int. Ed. 2005, 44, 1371–1375. 10.1002/anie.200462629. [DOI] [PubMed] [Google Scholar]
- The corresponding oxidative addition complex of 3-bromothiophene was also prepared, but it undergoes rapid dearomatization in solution. See ref (15e) as well as:Milner P. J.; Maimone T. J.; Su M.; Chen J.; Müller P.; Buchwald S. L. J. Am. Chem. Soc. 2012, 134, 19922–19934. 10.1021/ja310351e. [DOI] [PMC free article] [PubMed] [Google Scholar]; Thus, a solid-state structure of this complex could not be obtained. See the Supporting Information for details.
- The solid-state structure of 13 consisted of two rotamers of the thiophene ring; only the major rotamer is shown for convenience.
- For other examples, see:; a Verswyvel M.; Steverlynck J.; Mohamed S. H.; Trabelsi M.; Champagne B.; Koeckelberghs G. Macromolecules 2014, 47, 4668–4675. 10.1021/ma500610p. [DOI] [Google Scholar]; b Verswyvel M.; Verstappen P.; Cremer L. D.; Verbiest T.; Koeckelberghs G. J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 5339–5349. 10.1002/pola.25014. [DOI] [Google Scholar]
- Under all studied conditions, treatment of L3-ligated oxidative addition complexes of five-membered heteroaryl bromides with AgF led to incomplete conversion to what we tentatively assign as the corresponding Pd–F complexes. Thus, it is likely that transmetalation is also a challenging elementary step during this transformation.
- Roy A. H.; Hartwig J. F. J. Am. Chem. Soc. 2001, 123, 1232–1233. 10.1021/ja0034592. [DOI] [PubMed] [Google Scholar]
- Gronowitz S.; Hörnfeldt A.-B.. Thiophenes; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Hartwig J. F. Inorg. Chem. 2007, 46, 1936–1947. 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- It should be noted that the addition of tBuOD to these reaction mixtures did not result in deuterium incorporation into aryl fluoride products (see ref (15b)), as judged by 19F NMR and GC/MS analysis. This finding suggests either that regioisomer formation in these cases proceeds through a mechanism different from that outlined in ref (15b), or that the involved Pd–thiophyne intermediate is too short-lived to allow for adequate H/D exchange prior to reprotonation and reductive elimination. Further study of this phenomenon is required.
- A number of other, non-benzo-fused bromofurans were also evaluated, but most underwent significant decomposition under the reaction conditions (see Table S1 in the Supporting Information). Halogenated furans are known to exhibit poor stability (see ref (8a) and the references cited therein for details).
- Unfortunately, to date we have been unable to prepare five-membered heteroaryl bromides bearing secondary or tertiary alkyl groups in the ortho position. For example, attempts to carry out selective Negishi couplings between 2,3-dibromothiophene and secondary or tertiary alkylzinc nucleophiles led to complex mixtures of products.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.