

Abstract
This paper reviews previously published data and presents new results to address the hypothesis that fluorinated aminophosphonates (FAPs), (RO)2P(O)C(CF3)2NHS(O)2C6H5, R = alkyl, inhibit serine esterases by scission of the P-C bond. Kinetics studies demonstrated that FAPs are progressive irreversible inhibitors of acetylcholinesterase (AChE, EC 3.1.1.7.), butyrylcholinesterase (BChE, EC 3.1.1.8.), carboxylesterase (CaE, EC 3.1.1.1.), and neuropathy target esterase (NTE, EC 3.1.1.5.), consistent with P-C bond breakage. Chemical reactivity experiments showed that diMe-FAP and diEt-FAP react with water to yield the corresponding dialkylphosphates and (CF3)2CHNHS(O)2C6H5, indicating lability of the P-C bond. X-ray crystallography of diEt-FAP revealed an elongated (and therefore weaker) P-C bond (1.8797 (13) Å) compared to P-C bonds in dialkylphosphonates lacking α-CF3 groups (1.805–1.822 Å). Semi-empirical and non-empirical molecular modeling of diEt-FAP and (EtO)2P(O)C(CH3)2NHS(O)2C6H5 (diEt-AP), which lacks CF3 groups, indicated lengthening and destabilization of the P-C bond in diEt-FAP compared to diEt-AP. Active site peptide adducts formed by reacting diEt-FAP with BChE and diBu-FAP with NTE catalytic domain (NEST) were identified using peptide mass mapping with mass spectrometry (MS). Mass shifts (mean ± SE, average mass) for peaks corresponding to active site peptides with diethylphosphoryl and monoethylphosphoryl adducts on BChE were 136.1 ± 0.1 and 108.0 ± 0.1 Da, respectively. Corresponding mass shifts for dibutylphosphoryl and monobutylphosphoryl adducts on NEST were 191.8 ± 0.2 and 135.5 ± 0.1 Da, respectively. Each of these values was statistically identical to the theoretical mass shift for each dialkylphosphoryl and monoalkylphosphoryl species. The MS results demonstrate that inhibition of BChE and NEST by FAPs yields dialkylphosphoryl and monoalkylphosphoryl adducts, consistent with phosphorylation via P-C bond cleavage and aging by net dealkylation. Taken together, predictions from enzyme kinetics, chemical reactivity, X-ray crystallography, and molecular modeling were confirmed by MS and support the hypothesis that FAPs inhibit serine esterases via scission of the P-C bond.
Keywords: Acetylcholinesterase (AChE), butyrylcholinesterase (BChE), carboxylesterase (CaE), P-C bond scission, fluorinated aminosulfonates (FAPs), neuropathy target esterase (NTE)
1. Introduction
Organophosphorus (OP) compounds inhibit serine esterases by phosphylation of the active site serine [1,2]. Classical OP inhibitors of serine esterases are normally esters, thiol esters, acid halides, or acid anhydrides of pentavalent phosphorus-containing acids. The general structure of OP inhibitors is R1R2P(O)X, where R1 and R2 are usually simple alkyl or aryl groups that can be connected to phosphorus directly or through an intervening -O-, -S-, or -NH- linkage. Of the three groups attached to phosphorus by sigma bonds, the one most favored for nucleophilic displacement by the esterase serine hydroxyl is denoted by X. In order for X to be a good leaving group, the P-X bond must be relatively labile and the anion of X must be comparatively stable.
Until recently, OP compounds with an α-sp3 carbon atom in a leaving group had been unknown. However, previous studies demonstrated that the P-C bond could be rendered labile by the introduction of electron-withdrawing fluorinated substituents on the α-carbon [3–5]. Furthermore, our preliminary results showed that fluorinated α-aminophosphonates (FAP) containing CF3 groups on the α-carbon progressively and irreversibly inhibited four serine esterases: acetylcholinesterase (AChE, EC 3.1.1.7.), butyrylcholinesterase (BChE, EC 3.1.1.8.), carboxylesterase (CaE, EC 3.1.1.1.), and neuropathy target esterase (NTE, EC 3.1.1.5.), consistent with P-C bond breakage [6,7]. The structure of FAPs and their proposed mode of inhibition and aging of serine esterases are shown in Scheme 1.
Scheme 1.
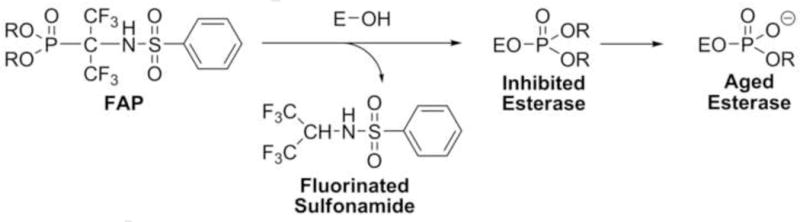
Proposed mode of inhibition of a serine esterase (EOH) by FAP compounds.
We have carried out studies of FAPs and their interactions with serine esterases using kinetics of irreversible enzyme inhibition, chemical reactivity, X-ray crystallography, molecular modeling, and peptide mass mapping with surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS). This paper reviews previously published data and presents new results from these studies to test the hypothesis that FAPs inhibit serine esterases by scission of the P-C bond.
2. Materials and methods
2.1. Chemicals and enzymes
FAPs [6], phenyl valerate, and N,N′ -diisopropylphosphorodiamidofluoridate (mipafox) were synthesized, purified, and characterized in the Institute of Physiologically Active Compounds Russian Academy of Sciences. The purity of all substances was ≥ 99% by NMR, thin-layer chromatography, and elemental analysis. Acetythiolcholine iodide and butyrylthiocholine iodide were from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany). 4-Nitrophenyl acetate was from Acros Organics (Geel, Belgium). Human erythrocyte AChE, horse serum BChE, and porcine liver CaE were from Sigma Chemical Co. (St. Louis, MO). A stable lyophilized preparation of paraoxon-pretreated hen brain membrane fraction [8] was used as the source of NTE. Human recombinant NTE catalytic domain (NEST) incorporated into dioleoylphosphatidylcholine (DOPC)-containing liposomes was prepared as described [9] and obtained from Dr. Paul Glynn, MRC Toxicology Unit (Leicester, UK).
2.2. Enzyme activity and inhibition kinetics
Esterase activities were assayed according to cited methods with slight modifications. AChE and BChE activities were measured spectrophotometrically using acetylthiocholine and butyrylthiocholine, respectively, as substrates (25 °C, 0.1 M phosphate buffer, pH 7.5, λ 412 nm) [10]. CaE activity was determined spectrophotometrically using 4-nitrophenyl acetate as substrate, (25 °C, 0.1 M phosphate buffer, pH 8.0, λ 405 nm) [11]. AChE, BChE, and CaE activities were followed kinetically by following the change in absorbance of each respective chromophore for 0.5 to 10 min depending upon the level of inhibition. NTE activity was assayed spectrophotometrically using phenyl valerate as substrate in a stopped assay of 20 min incubation (25 °C, 0.05 M Tris/0.02 EDTA buffer, pH 8.0, λ 486 nm) following differential inhibition by paraoxon ± mipafox [12,13]. FAPs were dissolved in acetone and the solutions allowed to stand for at least 72 h before dilution in buffer and mixing with enzymes; preliminary studies have shown that full inhibitory potency is not reached before this time, despite no obvious changes in the NMR spectrum. For kinetic studies of irreversible enzyme inhibition, a sample of enzyme was preincubated with inhibitor (4–5 inhibitor concentrations, final acetone concentration 1% v/v from stock solutions, a solvent concentration shown not to affect enzyme activity) for 4–6 different measured times using the same buffers and temperatures employed for the respective assays. The residual enzyme activity was then assayed in duplicate for each experiment. The slopes (k′) of each primary plot of log (% inhibition) versus time were calculated by linear regression. These values of k′ were then plotted against inhibitor concentration [I], and the slope (k″) of the resultant secondary plot was derived by linear regression. The bimolecular rate constant of inhibition (ki) was calculated as a measure of inhibitory potency using the relationship, ki = 2.303k′/[I] = 2.303k″ [14,15]. Each value of k′ was obtained from a line through 4–5 points. Plotting and regression analysis were done using Origin 6.1 software.
2.3. Chemical reactivity
Chemical reactivity was studied by incubation of diMe-FAP or diEt-FAP with water in acetonitrile with or without the presence of catalytic amounts of bases: Et3N, CsF, or KF. Hydrolysis of diEt-FAP to phosphoric acid diethyl ester and the sulfonamide, (CF3)2CHNHS(O)2C6 H5, was followed by 19F and 31P NMR [6]. Hydrolysis of diMe-FAP to phosphoric acid dimethyl ester and the corresponding fluorinated sulfonamide was followed by 19F NMR and 31P NMR; the fluorinated sulfonamide was isolated and its structure characterized by 1H and 19F NMR.
2.4. X-ray crystallography
The crystal structure of diEt-FAP was solved as reported previously [16]. DiEt-FAP was recrystallized from petroleum ether and colorless plates were grown via evaporation from methanol at 22 °C. A 0.60 × 0.42 × 0.40 mm crystal was cut from a larger crystal and mounted on a standard Bruker SMART CCD-based X-ray diffractometer equipped with a LT-2 low temperature device and normal focus Mo-target X-ray tube (λ = 0.71073 Å) operated at 2000 W power (50 kV, 40 mA). X-ray intensities were measured at 113 (2) K with the detector placed 4.980 cm from the crystal. A total of 3030 frames were collected with a scan width of 0.3° in ω and φ and an exposure time of 20 sec/frame. Data integration yielded a total of 20001 reflections to a maximum 2θ value of 56.58° of which 4568 were independent and 4343 were greater than 2 σ(I). The final cell constants were based on the xyz centroids of 6691 reflections above 10 σ(I). For refinement, hydrogen atoms were treated as riding, with N—H distance = 0.88 Å and C—H distances in the range 0.95–0.99 Å with Uiso(H) = 1.2Ueq(N,C), 1.5Ueq(Cmethyl).
2.5. Molecular modeling
A combination of semi-empirical and non-empirical quantum chemical calculations were carried out with full geometry optimization on diEt-FAP, isosteric (EtO)2P(O)C(CH3)2NHS(O)2C6H5 (diEt-AP), which lacks CF3 groups, and related molecules using GAUSSIAN 98 [17]. Methods included parametric model 3 (PM3), density functional theory (DFT) with hybrid exchange-correlation functional B3LYP in basis set 3-21G, and Hartree-Fock theory in basis set 3-21G [18,19]. Two conformations with different values of the torsion angle (O=P-C-N) around the P-C bond were considered and the one possessing the minimal energy was chosen. In both cases the initial conformations for geometry optimization were prepared interactively using PCMODEL [20] and subsequently optimized using the molecular-mechanical force field MMX. In addition, the relative strength of the P-C bond was compared between diEt-AP and diEt-FAP by calculating relative P-C bond energies with the aid of specially constructed isodesmic pseudo-reactions [21,22], as shown in Schemes 3 and 4.
Scheme 3.

Isodesmic reaction with diEt-FAP.
Scheme 4.

Isodesmic reaction with diEt-AP, which lacks CF3 groups.
2.6. Peptide mass mapping with surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS)
Samples were prepared for SELDI-TOF studies in a manner similar to that described by Doorn et al. [23,24]. Horse serum BChE (5 μg) or human recombinant NEST (0.5 μg) were incubated in 20 μL buffer (50 mM ammonium bicarbonate, pH 8.0), for 10 min at 25 °C with diEt-FAP (2.3 μM) or diBu-FAP (2.0 μM), respectively, in order to inhibit the enzymes by approximately 90%. Samples were diluted in buffer up to 1:50 by volume and stored at room temperature for 0 – 72 h to allow time for aging before trypsin digestion. Trypsin (0.5 μL of 0.1 mg/mL in 1 mM HCl) was added at a ratio of 1:10–1:25 μg trysin:μg esterase and incubated 3 – 16 h at 37 °C.
Tryptic digest samples (1 μL) were spotted onto H4 (hydrophobic surface) chips (Ciphergen Biosystems, Inc., Fremont, CA), and allowed to dry at room temperature. Stock matrix was prepared as a saturated solution of α-cyano-4-hydroxycinnamic acid in a 1:1 (v/v) solution of acetonitrile and 1% (w/v) trifluoroacetic acid. Working matrix was prepared by diluting the stock matrix 1:5 (v/v) in 1:1 (v/v) acetonitrile and 1% (w/v) trifluoroacetic acid. Working matrix (1μL) was placed on top of each spot and allowed to dry at room temperature. A PBS-IIc SELDI-TOF MS instrument (Ciphergen Biosystems, Inc., Fremont, CA) equipped with a nitrogen laser (337 nm, 4 ns pulse width) was used to acquire spectra. An acceleration voltage of 20 kV was used, and 150–200 laser shots were averaged for each spectrum.
The sequence of horse serum BChE was acquired from the NCBI nr database (NP_001075319.1) [25]. The predicted tryptic digest peptides and corresponding m/z values (average mass) of their singly charged protonated molecules (MH+) were obtained from ProteinProspector version 5.3.2 [26]. Using these tools, the active site peptide and its m/z value were as follows (active site serine shown underlined): SVTLFGESAGAASVSLHLLSPR (2200.52). Each spectrum was internally calibrated using BChE tryptic digestion peptides and their m/z values (average mass) as follows: 437–452 (2043.22), 248–268 (2222.48), 134–159 (2764.15), and 176–208 (3544.09).
The sequence of human NTE (PLPL6_HUMAN) was obtained from the UniProt/Swiss-Prot database [27], using isoform 2, which is the original sequence published by Lush et al. [28] (Q8IY17-2) and derived from the same cDNA clone (D16) used to produce NEST [9]. The sequence of human recombinant NEST contains residues 727–1216 of human NTE along with an N-terminal T7 tag and a C-terminal His6-tag [9]. Using ProteinProspector version 5.3.2 [26], the sequence and m/z value (average mass) of the tryptic digest active site peptide were determined to be as follows (active site serine shown underlined): ALEEAGVPVDLVGGTSIGSFIGALYAEER (2922.28). External calibration for NEST spectra was carried out using the “All-in-1” peptide standards mix (Ciphergen Biosystems, Inc., Fremont, CA), containing the following peptides with m/z values indicated: [Arg8]-vasopressin (1084.2), somatostatin (1637.9), dynorphin A [209–225] (porcine) (2147.5), ACTH [1–24] (human) (2933.5), insulin B-chain (bovine) (3495.9), insulin (human recombinant) (5807.6), and hirudin BKHV (7033.6).
The theoretical values for positive mass shifts of the peak corresponding to the active site peptide adducted with intact (dialkylphosphoryl) or aged species (monoalkylphosphoryl) were calculated to be as follows (average mass, Da): diethylphosphoryl (136.1), monoethylphosphoryl (108.0), dibutylphosphoryl (192.2), and monobutylphosphoryl (136.1). Calculated mass shifts take into account the mass of the serine hydroxyl proton lost in the organophosphorylation reaction and assume that the aged species is protonated because of the acidic conditions used for MS [29].
3. Results and discussion
3.1. Kinetics of esterase inhibition
Previous kinetics studies showed that all of the tested FAPs (R = Me, Et, iPr, Pr, iBu, Bu, iPe, Pe, and Hx) were irreversible progressive inhibitors for all of the investigated serine esterases (AChE, BChE, CaE, and NTE) [6]. Time courses of esterase inhibition were pseudo-first-order, and the slopes of the semilog plots were proportional to inhibitor concentrations as shown for a typical case in Fig. 1 (inhibition of AChE by diEt-FAP). This type of kinetic behavior is consistent with that exhibited by dialkylphosphates expected to organophosphorylate the active site serine by virtue of possessing candidate leaving groups with labile P-X bonds [7], such as the recently described dialkyl phosphorylated 1-hydroxyperfluoroisopropanols (PFPs) [4]. Thus, the kinetics of inhibition of serine esterases by FAPs is consistent with organophosphorylation of the active site serine, which would require breaking the P-C bond of the inhibitors. As can be seen in Fig. 1, increasing concentrations of inhibitor gave primary kinetic plots with increasing levels of apparent zero-time inhibition, an effect that was observed to some degree across inhibitors and esterases. This effect could arise from a high-sensitivity component or from ongoing inhibition by the inhibitors after addition of substrate [7]; however, it would not have a major impact on the apparent ki values or the conclusions of the study.
Figure 1.
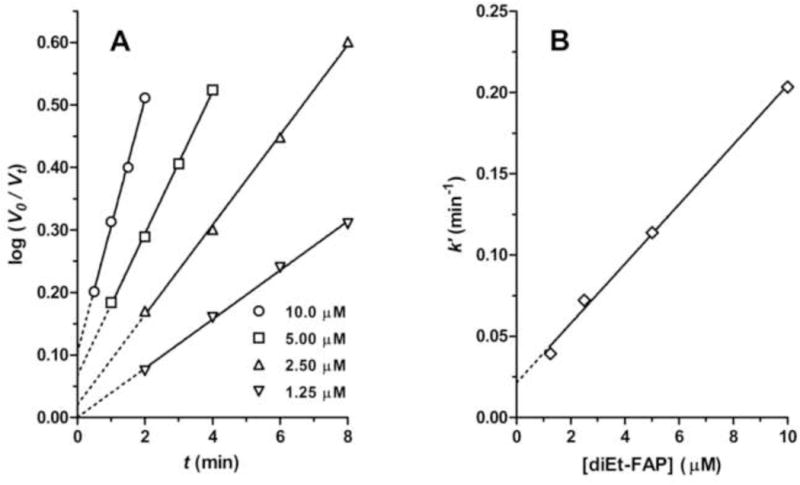
Kinetics of irreversible inhibition of AChE by diEt-FAP. A. Primary kinetic plots showing linear inhibition with time at different inhibitor concentrations. B. Secondary kinetic plot showing linear relationship of slopes of primary plots (k′) as a function of inhibitor concentration.
The derived bimolecular rate constants of inhibition (ki) are presented in Table 1 [6]. Especially with R ≥ Pr, FAPs were strong inhibitors of esterases: ki > 105–106 M−1min−1. Anti-esterase activity was drastically reduced by α-branching; for R = iPr, ki ~ 101 M−1min−1 (AChE and NTE) and ~ 102 M−1min−1 (BChE and CaE). Otherwise, there was a general trend of rising inhibitory potency against all of the esterases with increasing hydrophobicity of R-groups, reaching a maximum around R = Pe. A similar pattern of inhibitory potency against the same esterases was seen for a homologous series of dialkyl PFPs, which are expected to organophosphorylate the active site serine [4].
Table 1.
Bimolecular constants of inhibition (ki, M−1min−1) of AChE, BChE, CaE, and NTE and by FAP compoundsa
| N | R | AChE | BChE | CaE | NTE |
|---|---|---|---|---|---|
| 1 | CH3 (Me) | (2.75 ± 0.12) × 103 | (1.12 ± 0.08) × 104 | (1.75 ± 0.13) × 103 | (4.46 ± 0.41) × 101 |
| 2 | C2H5 (Et) | (4.14 ± 0.08) × 104 | (7.24 ± 0.22) × 105 | (1.06 ± 0.07) × 104 | (1.22 ± 0.04) × 103 |
| 3 | i-C3H7 (iPr) | (3.16 ± 0.27) × 101 | (2.16 ± 0.13) × 102 | (1.27 ± 0.12) × 103 | (3.44 ± 0.15) × 101 |
| 4 | n-C3H7 (Pr) | (1.46 ± 0.09) × 105 | (2.40 ± 0.08) × 106 | (2.13 ± 0.09) × 104 | (7.80 ± 0.26) × 103 |
| 5 | i-C4H9 (iBu) | (1.76 ± 0.09) × 105 | (2.58 ± 0.14) × 106 | (1.89 ± 0.14) × 105 | (1.33 ± 0.05) × 105 |
| 6 | n-C4H9 (Bu) | (5.62 ± 0.21) × 105 | (2.54 ± 0.12) × 106 | (6.64 ± 0.29) × 105 | (1.27 ± 0.05) × 105 |
| 7 | i-C5H11 (iPe) | (4.60 ± 0.11) × 105 | (7.96 ± 0.31) × 106 | (2.52 ± 0.18) × 106 | (1.80 ± 0.09) × 105 |
| 8 | n-C5H11 (Pe) | (1.29 ± 0.07) × 106 | (9.75 ± 0.21) × 106 | (5.66 ± 0.29) × 106 | (1.73 ± 0.12) × 105 |
| 9 | n-C6H13 (Hx) | (1.95 ± 0.10) × 105 | (4.60 ± 0.28) × 106 | (1.20 ± 0.10) × 106 | (1.30 ± 0.11) × 105 |
Data calculated as mean ± SEM, n = 3
3.2. Chemical reactivity
New studies of chemical reactivity showed that a mixture of diMe-FAP and water in acetonitrile was stable during one month, whereas in the presence of bases (Et3N, CsF, or KF), the 19F NMR spectrum showed full hydrolysis in 1 h to the fluorinated sulfonamide and phosphoric acid dimethyl ester (Scheme 2). The fluorinated sulfonamide was isolated and characterized with 1H and 19F NMR. The essential features of the 1H NMR spectrum in CDCl3 (δ, ppm, J, Hz) were as follows: 4.69 d septets (1H, JHF = 6.5, JHH = 10.2, CHCF3); 5.65 d (1H, JHH = 10.2, NH); 7.50 – 7.78 m (3H, o- + p-Har); 7.92 m (2H, m-Har). The essential feature of the 19F NMR spectrum in CDCl3 (δ, ppm, J, Hz) was as follows: 5.67 d (JHF = 6.6). These results are in agreement with those obtained earlier with diEt-FAP [6] and demonstrate the lability of the P-C bond.
Scheme 2.
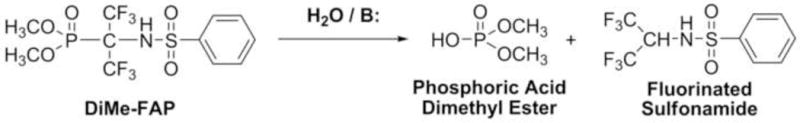
Chemical reactivity of diMe-FAP: base-catalyzed hydrolysis.
3.3. X-ray crystallography
The X-ray crystal structure of diEt-FAP was recently solved [16]. In the crystal, two molecules of diEt-FAP form centrosymmetric hydrogen-bonded dimers via two intermolecular P=O—H-N bonds (Fig. 2). The structure also revealed an intramolecular N-H—O=P hydrogen bond. The hydrogen-bonded dimers offer a potential explanation for the fact that FAPs require at least 72 h in acetone solution before reaching full potency as serine esterase inhibitors. Presumably, the bulky dimers would need to be fully dissociated into monomers before the compounds could gain access to the active site of esterases; however, this contention will require further investigation before it can be accepted or dismissed.
Figure 2.
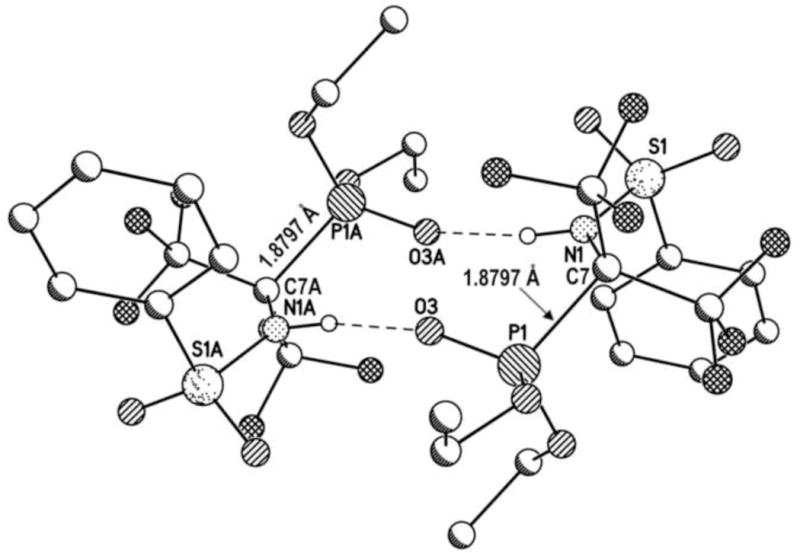
X-ray crystal structure of diEt-FAP showing centrosymmetric hydrogen-bonded dimers and the elongated P-C bond (1.8797 Å).
Notably, the P-C bond in each monomer was elongated (1.8797 (13) Å), and statistically identical in length to the P-C bond in the crystal structure of di-iPe-FAP (1.888 (4) Å) [30]. These values can be compared to the consistently shorter P-C bond lengths (1.803 – 1.822 Å) from crystal structures of a variety of dialkyl OP compounds lacking α-CF3 groups [31–34]. The long P-C bond in diEt-FAP is consistent with its lability toward aqueous hydrolysis and the apparent scission of the bond that would occur to enable this and other FAPs to be irreversible progressive inhibitors of serine esterases.
3.4. Molecular modeling
To gain insight into the unusual lability of the P-C bond in FAPs, semi-empirical and non-empirical quantum chemical calculations of bond length and bond strength were carried out for diEt-FAP and isosteric diEt-AP, which has CH3 groups in place of CF3 groups.
For P-C bond length computations, the initial conformations for geometry optimization for both diEt-FAP and diEt-AP were found using conformational analysis with the molecular-mechanical force field MMX. Two conformations with different values of torsion angle around the P-C bond were explored and that with the zero torsion angle O=P-C-N (which possesses the lowest energy) was chosen. The P-C bond lengths of each compound calculated by three methods are listed in Table 2. All three methods predicted a considerable lengthening of the P-C bond in diEt-FAP compared to diEt-AP. This is in accord with experimental X-ray studies showing a significant increase in P-C bond length in diEt-FAP (1.8797 (13) Å) [16] and di-iPe-FAP (1.888 (4) Å) [30] compared to the standard length (1.803 – 1.822 Å) of P-C bonds in compounds lacking α-CF3 groups [31–34]. The longer P-C bond found in the FAP compounds is weaker than P-C bonds of standard length. In this particular case, the lengthening of the P-C bond and its corresponding weakness can be explained by a strong electrostatic repulsion between the relative negatively charged oxygens at phosphorus and the fluorine atoms in the two CF3 groups. It should be noted that the non-empirical calculations in basis set 3-21G came closer to reproducing the experimental P-C bond lengths than did the semi-empirical PM3 calculations, and the Hartree-Fock method came closer than the DFT approach.
Table 2.
Calculated P-C bond length in diEt-FAP and diEt-AP using quantum chemical methodsa
| Methodb | diEt-FAP, P-C length (Å) | diEt-AP, P-C length (Å) |
|---|---|---|
| PM3 | 2.022 | 1.881 |
| DFT B3LYP/3-21G | 1.923 | 1.868 |
| HF/3-21G | 1.887 | 1.836 |
diEt-FAP = (EtO)2P(O)C(CF3)2NHS(O)2C6H5. diEt-AP = (EtO)2P(O)C(CH3)2NHS(O)2C6H5.
PM3 = parametric model 3; DFT B3LYP/3-21G = density functional theory with hybrid exchange-correlation functional B3LYP in basis set 3-21G; HF/3-21G = Hartree-Fock theory in basis set 3-21G.
The relative strength of the P-C bond was compared between diEt-FAP and diEt-AP by calculating relative P-C bond energies with the aid of specially constructed isodesmic pseudo-reactions [21,22]. Such reactions were first devised as a way of separating bonds so that the number of bonds of a given type remain the same but their relationship to one another changes. The main advantage of isodesmic reaction is that contribution of all factors, which are difficult to compute and which are roughly additive with respect to the number of chemical bonds of certain types, are mutually cancelled. As a result, the difference in enthalpy in isodesmic reaction is approximately equal to the difference in ground-state energy (computable using quantum chemical calculations), while the difference in free energy only correlates with the difference in ground-state energy. However, if the difference of changes in ground-state energy computed for a pair of very close isodesmic reactions is considered, additional entropic contributions are also cancelled, and the difference in free energy changes between such isodesmic reactions can be approximated by the difference of the corresponding changes in ground-state energies (which are computed in quantum chemical calculations) taking place in the course of these reactions.
In the hypothetical isodesmic reaction shown in Scheme 3, the energy change for diEt-FAP was determined to be −9.5 kcal/mol by the HF/3-21G method and −3.2 kcal/mol by the DFT B3LYP/3-21G approach. In contrast, in the hypothetical isodesmic reaction depicted in Scheme 4, the energy change for diEt-AP was predicted to be +3.8 kcal/mol (HF/3-21G) and +2.2 kcal/mol (DFT B3LYP/3-21G). This means that the P-C bond in diEt-FAP is destabilized in comparison with the P-C bond in diEt-AP by 13.4 kcal/mol (HF/3-21G) or 5.3 kcal/mol (DFT B3LYP/3-21G). In spite of some quantitative distinctions between results provided by the Hartree-Fock and DFT approaches, both show the same tendency. Taking into account that the HF/3-21G method reproduces the experimentally determined length of the P-C bond much better than the DFT calculation, it is tempting to speculate that the true value of the P-C bond energy destabilization is closer to that of the HF/3-21G method.
By comparing the pseudo-reactions shown in Schemes 3 and 4, one can see that in both cases an attractive interaction between the oxygen atoms at phosphorus and the hydrogen atom at the NH-group is broken; indeed, the crystal structure of diEt-FAP has an intramolecular N-H—O=P hydrogen bond [16]. Considered in isolation, disruption of an attractive force might account for a positive sign in the energy change of a pseudo-reaction, such as occurred for the reaction shown in Scheme 4. However, because these attractive interactions would be expected to be approximately equivalent in diEt-AP and diEt-FAP, their disruption cannot account for the destabilization of the P-C bond in diEt-FAP. Therefore, the obvious cause of this destabilization is the very strong electrostatic repulsion between the partial negative charges of oxygen atoms at phosphorus and the partial negative charges of the fluorine atoms in the CF3 groups. The strongly negative value of the calculated energy change in the pseudo-reaction shown in Scheme 3 supports this assertion.
Thus, using quantum chemical calculations, the P-C bond in diEt-FAP was predicted to be longer and destablilzed in comparison to the P-C-bond in isosteric diEt-AP; these effects are attributed to the strongly electron-withdrawing CF3 groups on the α-carbon.
3.5. SELDI-TOF MS
To test the hypothesis that FAPs inhibit serine esterases via organophosphorylation of the active site serine and then undergo aging by net loss of an alkyl group as depicted in Scheme 1, adducts on the active site peptide were identified by SELDI-TOF MS in tryptic digests of horse serum BChE treated with diEt-FAP and human recombinant NEST treated with diBu-FAP. Except as specified otherwise, values of m/z refer to average masses (in Da) of singly charged protonated molecules (MH+).
Representative mass spectra of control and treated BChE samples are shown in Fig. 3. Panel A is a spectrum of a control digest. The peak at m/z 2200.4 corresponds to the BChE active site peptide. Panel B is a spectrum of a treated BChE digest. The peak at m/z 2200.2 corresponds to the BChE active site peptide, whose relative intensity has decreased while new peaks have appeared at m/z 2335.6 and 2307.7, corresponding to the active site peak with a dimethylphosphoryl and monoethylphosphoryl adduct, respectively. The new peak at m/z 2357.6 corresponds to a sodium adduct of the dimethylphosphoryl active site peptide where Na+ replaces H+ in the protonated molecule for a net gain in average mass of 22 Da. Sodium adducts are relatively common in SELDI spectra [35]; when observed in the present study, they were associated with peaks corresponding to intact or aged FAP adducts of the active site peptide.
Figure 3.
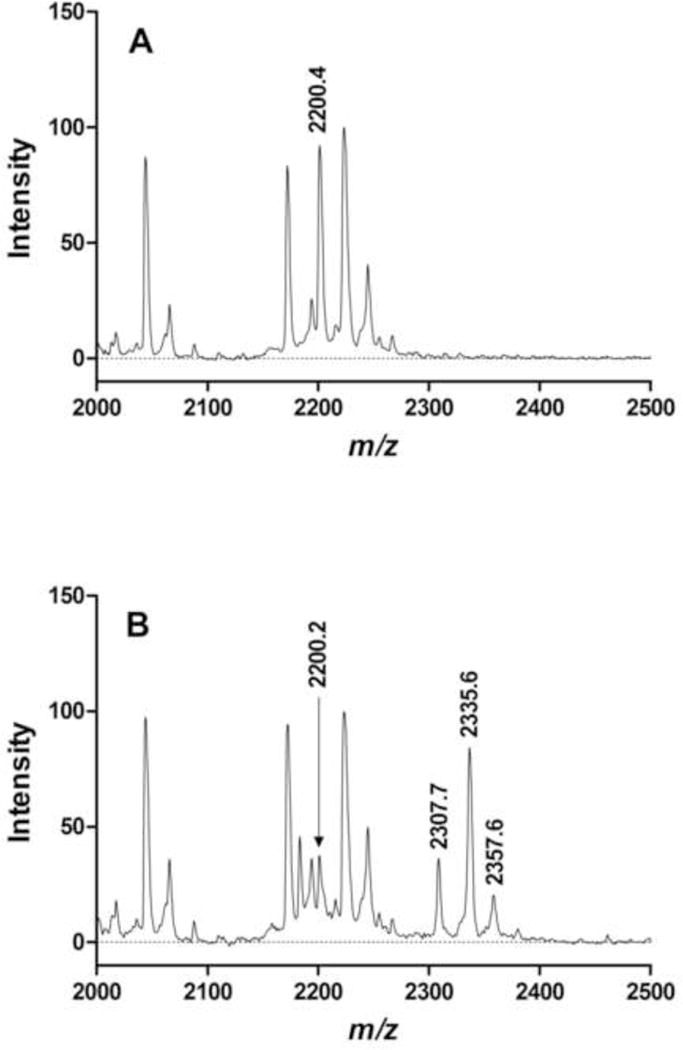
Representative SELDI-TOF mass spectra of BChE digests. A. Control BChE: the peak at m/z 2200.4 corresponds to the active site peptide. B. BChE treated with diEt-FAP: the relative intensity of the peak at m/z 2200.2 corresponding to the active site peptide has decreased, while new peaks at m/z 2335.6 (intact diethylphosphoryl adduct) and 2307.7 (aged monoethylphosphoryl adduct) have appeared. Values of m/z are average masses of singly charged protonated molecules (MH+) in Da. The peak at m/z 2357.6 corresponds to a Na+ adduct (MNa+) of the peak at 2335.6.
Fig. 4 shows representative mass spectra of tryptic digests of NEST samples. Panel A is a spectrum of a control digest. The peak at m/z 2920.4 corresponds to the unmodified active-site peptide; the peaks at m/z 2964.9 and 3203.2 correspond to prominent tryptic digest peptides of NEST. Panel B is a spectrum of a diBu-FAP-treated NEST digest. The peak at m/z 2922.3 corresponds to the active site peptide, whose relative intensity has decreased while new peaks have appeared at m/z 3114.3 and 3058.4, corresponding to the active site peptide with a dibutylphosphoryl and monobutylphosphoryl adduct, respectively. The peaks corresponding to NEST digestion peptides at m/z 2965.4 and 3203.7 remain prominent as they were in the control spectrum.
Figure 4.
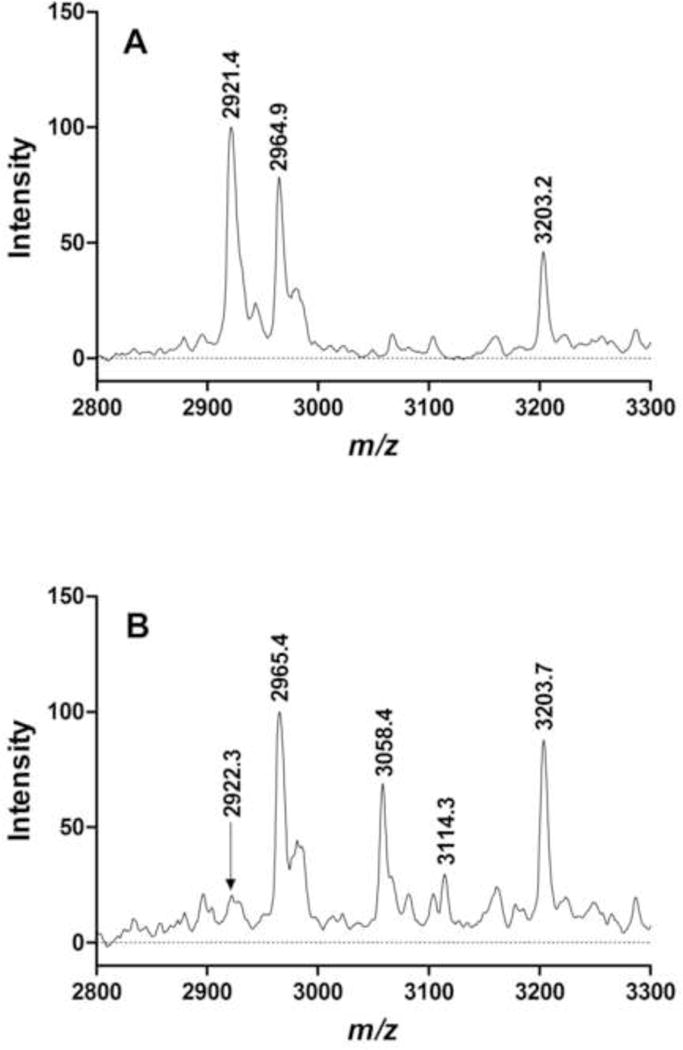
Representative SELDI-TOF mass spectra of NEST digests. A. Control NEST: the peak at m/z 2921.4 corresponds to the active site peptide; peaks at m/z 2964.9 and 3203.2 correspond to prominent NEST digest peptides found in control and treated digests. B. NEST treated with diBu-FAP: the relative intensity of the peak at m/z 2922.3 corresponding to the active site peptide has decreased, while new peaks at m/z 3114.3 (intact dibutylphosphoryl adduct) and 3058.4 (aged monobutylphosphoryl adduct) have appeared. Values are average masses of singly charged protonated molecules (MH+) in Da.
Table 3 summarizes the MS results for the two esterases and FAPs investigated in the current study. The observed and theoretical differences in average mass agree in each case, consistent with formation of a dialkylphosphoryl adduct and aging by net dealkylation to form a monoalkylphosphoryl adduct. Our low-resolution linear-mode MS experiments do not provide unequivocal proof of the structure of the adduct and its location on the active site serine as other recent work using higher-resolution MS and MS sequencing has done for a variety of OP inhibitors of BChE [36]. Nevertheless, our data are certainly consistent with these other studies as well as with our previous work on BChE [37] and NEST [38] inhibited with diisopropylphosphorofluoridate, which is known to organophosphorylate the active site serine of serine esterases and undergo aging by net dealkylation.
Table 3.
Differences in average mass of MH+ ions of active site peptides from intact or aged adducts of BChE with diEt-FAP or NEST with diBu-FAPa
| BChE | NEST | |||
|---|---|---|---|---|
| Data Type | diEt-FAP intact | diEt-FAP aged | diBu-FAP intact | diBu-FAP aged |
| Observed | 136.1 ± 0.1 (9) | 108.0 ± 0.1 (5) | 191.8 ± 0.2 (2) | 135.5 ± 0.1 (2) |
| Theoretical | 136.1 | 108.0 | 192.2 | 136.1 |
Observed data are expressed as means ± SEM (n) of average masses (in Da). All observed values were statistically identical to their respective theoretical values (p > 0.05, one-sample t-test). Calculated mass shifts take into account the mass of the serine hydroxyl proton lost in the organophosphorylation reaction and assume that the aged species is protonated because of the acidic conditions used for MS [29]. Intact adducts are dialkylphosphoryl; aged adducts are monoalkylphosphoryl.
Thus, our MS data support the hypothesis that both BChE and NEST are inhibited by scission of the P-C bond, in agreement with our predictions from enzyme kinetics, chemical reactivity, molecular modeling, and X-ray crystallography studies.
4. Conclusions
P-C bonds are so exceptionally stable in most phosphonates that their cleavage can be accomplished only with the aid of specific enzymes, such as bacterial carbon-phosphorus lyase [39,40]. However, previous work on the X-ray crystal structure of di-iPe-FAP showed that the C-P bond was elongated relative to phosphonates without CF3 groups on the α-carbon [30], and theoretical considerations predicted that this bond would be weaker than customarily found in phosphonates [6]. This finding prompted further work on FAPs, which is presented in the present paper.
Enzyme kinetics studies showed that FAPs were progressive irreversible inhibitors of serine esterases, including AChE, BChE, CaE, and NTE [6]. Chemical reactivity of diMe- and diEt-FAP showed that the P-C bond could be hydrolyzed in the presence of base catalysts to form the corresponding phosphoric acid dialkylesters and the fluorinated sulfonamide. X-ray crystallography of DiEt-FAP revealed an elongated P-C bond as found in the previous work on di-iPe-FAP [16]. Molecular modeling using semi-empirical and non-empirical quantum chemistry methods predicted an elongated and destabilized P-C bond in diEt-FAP relative to isosteric diEt-AP, which lacks CF3 groups on the α-carbon. Finally, SELDI-TOF MS studies showed that diEt-FAP and diBu-FAP reacted with BChE and NEST, respectively, to produce intact dialkylphosphoryl and monoalkylphosphoryl adducts on the active site peptide, consistent with organophosphorylation via C-P bond cleavage and aging via net dealkylation.
Taken together, the data from enzyme kinetics, chemical reactivity, X-ray crystallography, molecular modeling, and MS strongly support the hypothesis that inhibition of serine hydrolases by FAPs proceeds by organophosphorylation of the active site serine, which would require P-C bond cleavage. Additional studies of FAPs are in progress, including corroborative enzyme kinetics experiments, X-ray crystallographic structure determinations, and extended MS investigations.
Acknowledgments
This research was supported in part by CRDF grants RB2-2035 and RB2-2488, ISTC grant 3130, and the Russian Academy of Science Program “Biomolecular and Medicinal Chemistry”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eto M. Organophosphorus Pesticides: Organic and Biological Chemistry. CRC Press; Cleveland: 1974. [Google Scholar]
- 2.Thompson CM, Richardson RJ. Anticholinesterase Insecticides. In: Marrs TC, Ballantyne B, editors. Pesticide Toxicology and International Regulation (Current Toxicology Series) John Wiley & Sons Ltd; Chichester: 2004. pp. 89–127. [Google Scholar]
- 3.Makhaeva GF, Fetisov VI, Sokolov VB, Yankovskaya VL, Goreva TV, Malygin VV, Beznosko BK, Galenko TG, Kolomiets AF, Martynov IV. Interaction of dialkyl(α-carbomethoxy-β,β,β-trifluoroethyl) phosphates with mammalian esterases. Bioorg Khim. 1987;13:33–37. [PubMed] [Google Scholar]
- 4.Makhaeva GF, Serebryakova OG, Boltneva NP, Galenko TG, Aksinenko AY, Sokolov VB, Martynov IV. Esterase profile and analysis of structure-inhibitor selectivity relationships for homologous phosphorylated 1-hydroperfluoroisopropanols. Dokl Biochem Biophys. 2008;423:352–357. doi: 10.1134/s1607672908060094. [DOI] [PubMed] [Google Scholar]
- 5.Makhaeva GF, Aksinenko AY, Sokolov VB, Serebryakova OG, Richardson RJ. Synthesis of organophosphates with fluorine-containing leaving groups as serine esterase inhibitors with potential for Alzheimer disease therapeutics. Bioorg Med Chem Lett. 2009;19:5528–5530. doi: 10.1016/j.bmcl.2009.08.065. [DOI] [PubMed] [Google Scholar]
- 6.Makhaeva GF, Malygin VV, Aksinenko AY, Sokolov VB, Strakhova NN, Rasdolsky AN, Richardson RJ, Martynov IV. Fluorinated α-aminophosphonates – a new type of irreversible inhibitors of serine hydrolases. Dokl Biochem Biophys. 2005;400:831–835. doi: 10.1007/s10628-005-0041-7. [DOI] [PubMed] [Google Scholar]
- 7.Estavez J, Vilanova E. Model equations for the kinetics of covalent irreversible enzyme inhibition and spontaneous reactivation: esterases and organophosphorus compounds. Crit Rev Toxicol. 2009;39:427–448. doi: 10.1080/10408440802412309. [DOI] [PubMed] [Google Scholar]
- 8.Makhaeva GF, Malygin VV. A stable preparation of hen brain neuropathy target esterase for rapid biochemical assessment of neurotoxic potential of organophosphates. Chem-Biol Interact. 1999;119/120:551–557. doi: 10.1016/s0009-2797(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 9.Atkins J, Glynn P. Membrane association of and critical residues in the catalytic domain of human neuropathy target esterase. Biochem J. 2000;275:24477–24483. doi: 10.1074/jbc.M002921200. [DOI] [PubMed] [Google Scholar]
- 10.Ellman GL, Courtney KD, Andres V, Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Ain RJ, Riley CM, Schowen RL. Spectrophotometric assay for porcine liver esterase activity. Anal Biochem. 1995;231:265–267. doi: 10.1006/abio.1995.1531. [DOI] [PubMed] [Google Scholar]
- 12.Johnson MK. Improved assay of neurotoxic esterase for screening organophosphates for delayed neurotoxicity potential. Arch Toxicol. 1977;67:113–115. doi: 10.1007/BF00293860. [DOI] [PubMed] [Google Scholar]
- 13.Kayyali US, Moore TB, Randall JC, Richardson RJ. Neurotoxic esterase (NTE) assay: Optimized conditions based on detergent-induced shifts in the phenol/4-aminoantipyrine chromophore spectrum. J Anal Toxicol. 1991;15:86–89. doi: 10.1093/jat/15.2.86. [DOI] [PubMed] [Google Scholar]
- 14.Aldridge WN, Reiner E. Enzyme Inhibitors as Substrates, North Holland Publishing Co, Amsterdam. 1972:37–52. [Google Scholar]
- 15.Clothier B, Johnson MK, Reiner E. Interaction of some trialkyl phosphorothiolates with acetylcholinesterase. Characterization of inhibition, aging and reactivation. Biochim Biophys Acta. 1981;660:306–316. doi: 10.1016/0005-2744(81)90175-3. [DOI] [PubMed] [Google Scholar]
- 16.Wijeyesakere SJ, Nasser FA, Kampf JW, Aksinenko AY, Sokolov VB, Malygin VV, Makhaeva GF, Richardson RJ. Diethyl [2,2,2-trifluoro-1-phenylsulfonylamino-1-(trifluoromethyl) ethyl]phosphonate. Acta Cryst. 2008;E64:o1425. doi: 10.1107/S1600536808020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaussian 98. Revision A.9. Gaussian, Inc; Pittsburgh PA: 1998. [Google Scholar]
- 18.Kim K, Jordan KD. Comparison of Density Functional and MP2 calculations on the water monomer and dimer. J Phys Chem. 1994;98:10089–10094. [Google Scholar]
- 19.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab Initio Calculation of vibrational absorption and circular dichroism spectra using Density Functional force fields. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 20.PCMODEL. version 7.0. Serena Software; Bloomington, IN: 1998. [Google Scholar]
- 21.Hehre WJ, Ditchfield R, Radom L, Pople JA. Molecular orbital theory of the electronic structure of organic compounds. V. Molecular theory of bond separation. J Am Chem Soc. 1970;92:4796–4801. [Google Scholar]
- 22.Bickelhaupt FM, Hermann HL, Boche G. Stabilization of carbanions: fluorine is more effective than the heavier halogens. Angew Chem Int Ed Engl. 2006;45:823–826. doi: 10.1002/anie.200501633. [DOI] [PubMed] [Google Scholar]
- 23.Doorn JA, Gage DA, Schall M, Talley TT, Thompson CM, Richardson RJ. Inhibition of acetylcholinesterase by (1S,3S)-isomalathion proceeds with loss of thiomethyl: Kinetic and mass spectral evidence for an unexpected primary leaving group. Chem Res Toxicol. 2000;13:1313–1320. doi: 10.1021/tx000184v. [DOI] [PubMed] [Google Scholar]
- 24.Doorn JA, Thompson CM, Christner RB, Richardson RJ. Stereoselective interaction of Torpedo californica acetylcholinesterase by isomalathion: Inhibitory reactions with (1R)- and (1S)-isomers proceed by different mechanisms. Chem Res Toxicol. 2003;16:958–965. doi: 10.1021/tx030026e. [DOI] [PubMed] [Google Scholar]
- 25.http://www.ncbi.nlm.nih.gov/protein/126352540
- 26.http://prospector.ucsf.edu/prospector/mshome.htm
- 27.http://www.uniprot.org/uniprot/Q8IY17
- 28.Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J. 1998;332:1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jennings LL, Malecki M, Komives EA, Taylor P. Direct analysis of the kinetic profiles of organophosphate-acetylcholinesterase adducts by MALDI-TOF mass spectrometry. Biochemistry. 2003;42:11083–11091. doi: 10.1021/bi034756x. [DOI] [PubMed] [Google Scholar]
- 30.Chekhlov AN, Aksinenko AY, Sokolov VB, Martynov IV. Crystal and molecular structure and synthesis of O,O-diisopentyl-1-benzenesulfonamido-1-trifluorimethyl-2,2,2-trifluoroethylphosphonate. Dokl Chem. 1995;345:296–299. [Google Scholar]
- 31.Chen C, Jin W, Li X. Diethyl [hydroxy(2-nitrophenyl)methyl]phosphonate. Acta Cryst. 2008;E64:o144. [Google Scholar]
- 32.Guo YC, Wang XF, Ding Y. Dimethyl [1-(1-allyl-5-iodo-1H-indol-3-yl)-3-hydroxypropyl]phosphonate. Acta Cryst. 2008;E64:o384. doi: 10.1107/S1600536807068171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kachkovskyi GO, Kolodiazhnyi OI. α-Acylaminophosphonates possessing epoxyisoindolone moiety. Tetrahedron. 2007;63:12576–12582. [Google Scholar]
- 34.Liu XL, Zhou Y, Li WZ, Fan Z, Miao FM, Mao LJ, Chen RY. Dimethyl [[alpha]-(Benzylamino)-p-chlorobenzyl]phosphonate. Acta Cryst. 1995;C51:2350–2352. [Google Scholar]
- 35.Malyarenko DI, Cooke WE, Adam BL, Malik G, Chen H, Tracy ER, Trosset MW, Sasinowski M, Semmes OJ, Manos DM. Enhancement of sensitivity and resolution of surface-enhanced laser desorption/ionization time-of-flight mass spectrometric records for serum peptides using time-series analysis techniques. Clin Chem. 2005;51:65–74. doi: 10.1373/clinchem.2004.037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilley C, MacDonald M, Nachon F, Schopfer LM, Zhang J, Cashman JR, Lockridge O. Nerve agent analogues that produce authentic soman, sarin, tabun, and cyclohexyl methylphosphonate-modified human butyrylcholinesterase. Chem Res Toxicol. 2009;22:1680–1688. doi: 10.1021/tx900090m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kropp TJ, Richardson RJ. Mechanism of aging of mipafox-inhibited butyrylcholinesterase. Chem Res Toxicol. 2007;20:504–510. doi: 10.1021/tx600310y. [DOI] [PubMed] [Google Scholar]
- 38.Kropp TJ, Glynn P, Richardson RJ. The mipafox-inhibited catalytic domain of human neuropathy target esterase ages by reversible proton loss. Biochemistry. 2004;43:3716–3722. doi: 10.1021/bi049960e. [DOI] [PubMed] [Google Scholar]
- 39.Quinn JP, Kulakova AN, Cooley NA, McGrath JW. New ways to break an old bond: the bacterial carbon-phosphorus hydrolases and their role in biochemical phosphorus cycling. Environ Microbiol. 2007;9:2392–2400. doi: 10.1111/j.1462-2920.2007.01397.x. [DOI] [PubMed] [Google Scholar]
- 40.Adams MA, Luo Y, Hove-Jensen B, He SM, van Staalduinen LM, Zechel DL, Jia Z. Crystal structure of PhnH: an essential component of carbon-phosphorus lyase in Escherichia coli. J Bacteriol. 2008;190:1072–1083. doi: 10.1128/JB.01274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]