

Abstract
Genotyping tumor tissue in search of somatic genetic alterations for actionable information has become routine practice in clinical oncology. Although these sequence alterations are highly informative, sampling tumor tissue has significant inherent limitations; tumor tissue is a single snapshot in time, is subject to selection bias resulting from tumor heterogeneity, and can be difficult to obtain. Cell-free fragments of DNA are shed into the bloodstream by cells undergoing apoptosis or necrosis, and the load of circulating cell-free DNA (cfDNA) correlates with tumor staging and prognosis. Moreover, recent advances in the sensitivity and accuracy of DNA analysis have allowed for genotyping of cfDNA for somatic genomic alterations found in tumors. The ability to detect and quantify tumor mutations has proven effective in tracking tumor dynamics in real time as well as serving as a liquid biopsy that can be used for a variety of clinical and investigational applications not previously possible.
INTRODUCTION
Fragmented DNA is found in circulation in the cell-free component of whole blood. Initially reported by Mandel and Metais1 in 1948, the clinical utility of circulating cell-free DNA (cfDNA) in the serum and plasma has been an area of active research in many disciplines of medicine. Evaluation of fetal DNA in the circulation of expecting mothers has seen the most success.2–4 Investigation of fetal DNA can now uncover germline fetal changes weeks after conception, including point mutation and aneuploidy, and is likely to become part of the standard of care in prenatal assessment in high-risk patients.5,6
Investigation of cfDNA has included other clinical scenarios such as exercise, end-stage renal failure, stroke, myocardial infarction, surgery, and trauma.7–20 These studies have demonstrated that circulating cfDNA exists at steady-state levels and increases, sometimes dramatically, with cellular injury or necrosis.17
In oncology, detection of cfDNA derived from tumors, also known as circulating tumor DNA (ctDNA), has been challenging for three primary reasons, which include: discrimination of ctDNA from normal cfDNA; presence of sometimes extremely low levels of ctDNA; and the accurate quantification of the number of mutant fragments in a sample.
Discriminating ctDNA from normal cfDNA is aided by the fact that tumor DNA is defined by the presence of mutations. These somatic mutations, commonly single base-pair substitutions, are present only in the genomes of cancer cells or precancerous cells and are not present in the DNA of normal cells of the same individual. This juxtaposition assures ctDNA exquisite biologic specificity as a biomarker. Accordingly, all DNA sequencing methodologies that identify somatic variants could be used easily to identify ctDNA if tumor DNA fragments were abundant in the circulation of patients with cancer. Unfortunately, detection of cfDNA derived from tumors carries substantial challenges, largely because ctDNA often represents a small fraction (< 1.0%) of total cfDNA.17,21,22 Therefore, standard sequencing approaches like Sanger sequencing or pyrosequencing can only detect tumor-derived mutant fragments in patients with heavy tumor burden and high levels of ctDNA.
Investigation of cfDNA in patients with cancer has recently increased, largely because of digital genomic technologies that allow for enumeration of rare mutant variants in complex mixtures of DNA. Before the introduction of techniques like digital polymerase chain reaction (PCR),23 beads, emulsion, amplification, and magnetics (BEAMing),24 or pyrophosphorolysis-activated polymerization (PAP),25 detection of cfDNA derived from tumors was inconsistently detected,26–29 with several reports suggesting that ctDNA measurement was inferior to that of other biomarkers, such as circulating tumor cells30–32 (Fig 1). In advanced tumors, digital genomic approaches have high sensitivity, with the mutation identified in the tumor tissue matching the mutation in the ctDNA fraction in virtually every case.17,21,39 Recently, PCR-based digital approaches have been updated with techniques that use next-generation sequencing (NGS) to identify rare mutant variants in complex mixtures of DNA (Table 1).37,40–42 These techniques have expanded the ability to detect a single point mutation, and now multiple genes of interest can be investigated in one sample. Amplifications, rearrangements, and aneuploidy may now be detectable as well17,41,43–45 (Fig 1).
Fig 1.

Methodologies for detecting circulating tumor DNA (ctDNA). Sanger sequencing (dideoxy-terminator sequencing),33 amplification refractory mutation system (ARMS),34,35 pyrosequencing,36 pyrophosphorolysis-activated polymerization (PAP),25 tagged-amplicon deep sequencing (TAM-Seq),37 digital polymerase chain reaction (PCR),23 and beads, emulsion, amplification, and magnetics (BEAMing).38
Table 1.
Applications of Liquid Biopsy
| Application |
|---|
| Early detection |
| Assessment of molecular heterogeneity of overall disease |
| Monitoring of tumor dynamics |
| Identification of genetic determinants for targeted therapy |
| Evaluation of early treatment response |
| Monitoring of minimal residual disease |
| Assessment of evolution of resistance in real time |
The ability to detect and enumerate ctDNA creates a wide array of practical clinical applications that are not possible with routine sequencing of tumor tissue or with other circulating biomarkers (Table 1). This review will highlight some of these applications, in addition to the biology of ctDNA, and discuss promising future applications to solve unmet clinical needs.
MECHANISMS OF TUMOR DNA SHEDDING
In general, patients with cancer have much higher levels of normal circulating cfDNA than healthy individuals.46–54 As the tumor increases in volume, so too does the cellular turnover and hence the number of apoptotic and necrotic cells.55,56 Under normal physiologic circumstances, apoptotic and necrotic remains are cleared by infiltrating phagocytes. This does not happen efficiently within the tumoral mass, leading to the accumulation of cellular debris and its inevitable release into the circulation (Fig 2).
Fig 2.
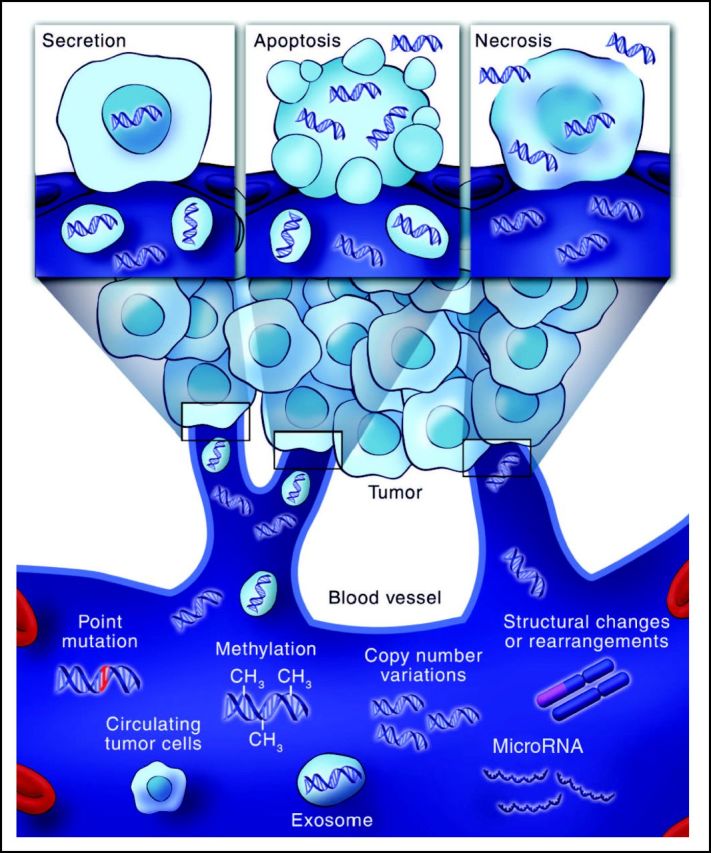
Genetic alterations detectable in circulating cell-free tumor DNA. Tumor cells release small fragments of cell-free DNA into circulation by multiple mechanisms. Cancer-associated genetic alterations such as point mutations, copy number variations, chromosomal rearrangements, and methylation patterns can be detected in circulating cell-free DNA.
When the length of cfDNA strands are measured, they often assume the classic ladder pattern in integer multiples of 180 base pairs,57 characteristic of the apoptotic process.55,58,59 In fact, most cfDNA fragments measure between 180 and 200 base pairs, suggesting that apoptosis likely produces the majority of cfDNA in circulation.6,17,21,55 The passive release of cfDNA into the bloodstream from apoptotic or necrotic cells is dependent on the location, size, and vascularity of the tumor, perhaps accounting for the variability in cfDNA levels often observed.
Salient to this review, it is evident that a proportion of fragmented DNA in circulation is derived directly from the tumor. Multiple methods have shown that the fraction of circulating DNA contributed from the tumor varies greatly, between 0.01% and more than 90%.17 As discussed later in this review, the amount of ctDNA is related to the tumor burden and is therefore expected to vary significantly among individuals with different clinical histories.17,43
TISSUE VERSUS LIQUID BIOPSY
Many of the major recent advances in targeted therapies have relied on the acquisition of tumor tissue via biopsy before initiation of therapy or after the onset of resistance. The availability of tissue for molecular analysis has been instrumental in understanding the primary mechanism of action of agents such as trastuzumab, imatinib, cetuximab, and vemurafenib. Likewise, access to tumor tissue after clinical resistance has helped to define mechanisms of secondary resistance to these targeted agents, often in the very pathway or gene involved in their responsiveness.60
Although tumor tissue is the gold standard for clinical and investigational sequencing, major barriers exist in terms of acquisition and utility. Biopsies are an inconvenience from a scheduling perspective; they also increase the cost of patient care and are yet another uncomfortable, invasive procedure for patients that often do not influence outcome. Finally, and most importantly, biopsies are not without clinical complications. A review of the investigative biopsies at MD Anderson Cancer Center reported adverse event rates of 17.1% and 1.6% for thoracic biopsies and abdominal/pelvic sampling, respectively.61
In addition to the issues related to tissue acquisition, sample preservation and tumor heterogeneity also hamper the use of tumor tissue for cancer sequencing.22 Most tumor tissue is preserved in formalin-fixed paraffin-embedded (FFPE) blocks, which crosslink DNA and in some cases can result in FFPE samples being inadequate for molecular analysis. Although this has improved for individual gene mutations and targeted gene sets, there are still limitations for whole genome and exome analyses. Furthermore, the quantity of tumor cells in each biopsy varies and is largely dependent on the tumor cellularity (percent tumor) and size of the specimen acquired. This is further compounded by small tissue amounts from fine-needle aspirates or core-needle biopsies, which often result in smaller amounts of tumor tissue for molecular analysis in comparison with surgically resected specimens.
Arguably, the major limitation of tissue biopsy is heterogeneity, which characterizes most advanced cancers.62,63 Cancers are heterogeneous, with different areas of the same tumor showing different genetic profiles (ie, intratumoral heterogeneity); likewise, heterogeneity exists between metastases within the same patient (ie, intermetastatic heterogeneity). A biopsy or tissue section from one part of a solitary tumor will miss the molecular intratumoral as well as intermetastatic heterogeneity.
To overcome the limitations of tissue biopsies, less invasive techniques capable of capturing tumor heterogeneity and the molecular changes cancer cells undergo when they are exposed to therapy are needed.44,64,65 Circulating tumor DNA can in principle provide the same genetic information as a tissue biopsy necessary to interrogate key companion diagnostics. Accessing the bloodstream has clear advantages. For one, it is a source of fresh DNA, unhampered by preservatives. Sampling the blood from a needle stick is minimally invasive and avoids the dangers of biopsies. Furthermore, blood can be drawn at any time during the course of therapy and allow for dynamic monitoring of molecular changes in the tumor rather than relying on a static time point.
What information can be derived from ctDNA? Circulating tumor DNA fragments contain genetic defects identical to those of the tumors themselves; these DNA alterations span the types of genomic alterations identified in the tumor and include point mutations (EGFR and KRAS), rearrangements (EML4-ALK), amplifications (HER2 and MET), and even aneuploidy (Fig 2). Moreover, investigating plasma from patients can account for molecular heterogeneity, because ctDNA fragments are collected from all tumors in a patient's body through circulation (Fig 2).44,65 Accordingly, examination of ctDNA for genetic alterations present in the tumor tissue is, in reality, a liquid biopsy. Although current evidence suggests that ctDNA likely represents a molecular proxy of the overall disease, it remains to be formally proven that multiple metastatic lesions located in different organs shed ctDNA homogeneously.
Although much of the current data for ctDNA are investigational, the sensitivity of these liquid biopsies for patients with stage IV disease seems to be approaching 100%.17,21,39 Despite the promising results in advanced disease, the sensitivity of measuring ctDNA is reliant on biologic and technical factors. The abundance of tumor cells represented by tumor stage or overall tumor burden can dictate sensitivity. Lower-stage tumors and even advanced cases involving low-level micrometastatic disease have lower numbers of ctDNA fragments.21 Technically, sensitivity is limited by the error rate of DNA polymerase, which is generally considered to be 0.01%.38 Therefore, if the fraction of ctDNA in a sample is at or below 0.01%, it is considered negative for ctDNA.17,38 Newer approaches aimed at increasing overall sensitivity using barcoding strategies in the context of NGS are being investigated.40 This will be important because false-negative results, especially when ctDNA results are used for therapeutic decision making or in detecting occult disease, could be detrimental. The absolute sensitivity for ctDNA analysis in a variety of clinical scenarios will have to be evaluated to best understand the limitations and advantages of this new technology.
MONITORING TUMOR BURDEN
Liquid biopsies may also be useful in monitoring tumor burden, a central aspect in the management of patients with cancer that is typically assessed with imaging. Circulating biomarkers have played an increasingly important role in assessing disease burden, especially in circumstances where imaging delivers indeterminate results. PSA, cancer antigen (CA) 19-9, carcinoembryonic antigen, and CA-125 are examples of protein biomarkers that aid in assessment of therapeutic response. Unfortunately, many malignancies do not have a reliable protein biomarker, and even in those diseases with useful biomarkers, these markers often lack specificity and may be elevated as a result of clinical situations not related to tumor growth or progression. Furthermore, most protein biomarkers persist in circulation for weeks, thereby only allowing accurate assessment over weeks to months.37,66–68
There are clear advantages to measuring ctDNA as a marker of tumor dynamics over conventional protein biomarkers or even imaging studies. For one, ctDNA has a comparatively short half-life (approximately 2 hours), allowing for evaluation of tumor changes in hours rather than weeks to months.17 Changes in ctDNA can predate those seen in imaging studies or using protein biomarkers by weeks to months.17,43 Furthermore, ctDNA is exquisitely specific for an individual's tumor, because by definition somatic cancer mutations are identified by their presence in tumor DNA and absence in matched normal DNA. This bypasses the issues related to confusing false-positive results often encountered with other circulating biomarkers and imaging studies.
Several investigational studies have shown that ctDNA can be a surrogate for tumor burden and that much like viral load changes (eg, HIV viral load), levels of ctDNA correspond with clinical course. Studies in melanoma, ovarian, breast, and colon cancers have solidified the potential utility of this approach to more precisely define tumor dynamics during therapy for patients with advanced disease.17,28,37,43,69 There are rapid increases in ctDNA levels with disease progression and corresponding declines in levels after successful treatment with pharmacologic therapy or resective surgery.17,28,37,43
Two potential processes have been described for performing ctDNA analysis. The first requires identifying a specific mutation or mutations in the tumor tissue to target and quantify the level of ctDNA at a particular time point.17 The alternative approach is scanning regions of DNA extracted from plasma or serum for mutations of interest in a blinded manner because the tumor tissue was not initially assessed.37,40,43 In both cases, the genotype is identified, and the mutation is quantified and represented in a number of mutant fragments per mL or as a mutant fraction (%) in an individual sample.
Clinical applications for this technology include monitoring tumor response to therapy and potentially defining ambiguous clinical scenarios like stable disease or mixed responses.37 Furthermore, changes in ctDNA may predict for treatment response early in the course of therapy, which may allow for real-time modification of the treatment regimen, rather than waiting weeks or months to monitor response to therapy. This question is currently under investigation. All of these potential indications will have to be evaluated in the context of clinical trials also testing the benefit of novel therapeutic agents.
MINIMAL RESIDUAL DISEASE
Another potential application of ctDNA is the detection of minimal residual disease after surgery or therapy with curative intent. In these scenarios, as is the case in breast or colon cancer, resective surgery alone cures a large fraction of patients with localized disease. However, we have no effective means presently to identify which patients are cured and which have residual disease that will result in disease recurrence. Furthermore, those groups of patients with high-risk clinical and pathologic criteria are indiscriminately treated with adjuvant chemotherapy despite the fact that a large portion are cured and do not need such potentially toxic therapy.
Currently, predicting which patients are disease free after surgery (ie, cured) and those who have residual disease depends largely on clinical and pathologic parameters. The most important criterion has been the TNM staging system. This system stratifies patients by risk for recurrence and possible benefit from adjuvant chemotherapy. Recently, novel molecular features such as gene expression profiles have been used to improve the predictive power of the TNM staging system.70 Assessed in the resected tumor specimen, these markers are biologic surrogates for phenotypes such as tumor age and aggressive biology. However, they do not address whether residual tumor is present after surgical resection.
Circulating tumor DNA is a potential marker of residual disease after resection and may determine which patients will experience recurrence. Optimally, ctDNA should be measured after surgery but before initiation of adjuvant therapy (in general, 6 to 8 weeks after surgery) to best facilitate therapeutic decision making.
The best example to date has been in a cohort of patients with colorectal cancer undergoing resection with curative intent.17 A mutation profile was determined from each patient's resected tumor, and this personal and unique molecular signature was then used to create a set of mutation-specific probes for each patient. These probes were used to detect and quantify ctDNA after surgery in these patients undergoing potentially curative resection, and the patients were subsequently observed over the course of 2 to 5 years. In this study, ctDNA was shown to be sufficiently sensitive to detect minimal residual disease after surgical resection.17 All patients with detectable postoperative levels of ctDNA experienced recurrence, whereas all patients with undetectable postoperative levels of ctDNA remained disease free. Similar studies have been performed blindly, examining KRAS mutations in patients with resected colorectal cancer, and shown a strong correlation between postoperative detection of mutant DNA in circulation and recurrence.58
Future studies evaluating postoperative levels of ctDNA could offer personalized markers based on the unique mutational profiles of resected tumors and thus having exquisitely high specificity. Sensitivity will rely on the ability to detect low levels of ctDNA released from micrometastatic deposits not detectable by imaging or other diagnostic modalities. Applications of such an approach have the potential to affect a broad array of tumors treated with curative intent.
MONITORING OF MOLECULAR RESISTANCE AND HETEROGENEITY
The emergence of clinical resistance to a previously effective antineoplastic therapy results from the acquisition of molecular alterations in genes or pathways that govern the mechanisms of action of the newly ineffective therapy. Defining these mechanisms of resistance to a targeted agent is often done using preclinical models (cell lines or xenografts in mice), because it is generally more difficult to identify and confirm these findings in clinical samples. In principle, every patient enrolled onto a therapeutic clinical trial should undergo a tissue biopsy before initiation of the experimental therapy and after progression. Molecular tools, such as NGS, can be implemented to find genetic differences between the tissue collected before and after therapy. This will offer a snapshot of the predominant resistant clone of a portion of the lesion under examination.
Recent studies have shown that liquid biopsies can be used effectively to monitor the emergence of multiple resistance clones during the course of treatment (Fig 3). The genetic bases of secondary resistance to various targeted drugs have been elucidated in great detail.71,72 The most extensive analyses are those involving patients with Philadelphia chromosome–positive chronic myeloid leukemia treated with the tyrosine kinase inhibitor imatinib. Emergence of ABL kinase domain mutations have been extensively implicated in the pathogenesis of acquired resistance to imatinib.73
Fig 3.

Detection of tumor-specific DNA mutations in the blood of patients to monitor response and relapse with targeted therapies. This schematic depicts a representation of a patient with metastatic colorectal cancer. At the time of presentation, DNA from the primary tumor is used to identify the baseline mutation profile; in this case, the tumor is found to be APC mutant and KRAS wild type (WT). At baseline, evaluation of the patient's plasma DNA only identifies KRAS WT fragments. This patient is treated with an anti–epidermal growth factor receptor (EGFR) monoclonal antibody, experiences a clinical response, and has a corresponding decrease in APC mutation level, further indicating a decrease in tumor burden. Continuous monitoring of plasma DNA shows the emergence of KRAS and NRAS mutations and/or MET amplification, indicative of the emergence of multiple different resistance clones. Clinical resistance becomes manifest at a later time point. Test tubes represent samples of plasma from which circulating free DNA is extracted and used to monitor the presence of cancer-specific aberrations.
More recently, the mechanisms of acquired resistance to anti–epidermal growth factor receptor (EGFR) therapies have been defined in lung and colorectal cancers. Acquired resistance to gefitinb or erlotinib in approximately 50% of patients with lung cancer develops through the emergence of EGFR T790M variants.44,71 The mutation at residue 790 increases the affinity of EGFR for ATP and so out-competes binding of the inhibitors. These results were initially obtained in examining biopsies from patients who relapsed during anti-EGFR therapy and were later confirmed through analyses of plasma, providing the first example that resistance to targeted therapies of solid tumors can be detected noninvasively in the blood of patients.74 Analogously, secondary resistance to the EGFR-blocking monoclonal antibodies cetuximab and panitumumab is associated with the emergence of KRAS mutations or MET amplification (Fig 3).60,65 Detection of KRAS variants in cfDNA of patients receiving anti-EGFR therapies can identify relapse months before radiologic examination (Fig 3). In this case, the discovery of the mechanisms of resistance to the targeted agent was simultaneously accomplished in tissue and liquid biopsies.60,65 Another noteworthy aspect of these studies was the emergence of multiple different resistance mutations in the same patient.
These studies were based on candidate gene analyses and required prior knowledge of the mechanisms of resistance. Considering the accessibility of plasma from patients undergoing therapy, it is likely that future studies tracking the emergence of resistant clones will be unbiased. For example, an unbiased approach has been applied to study the acquisition of secondary resistance and to monitor clonal evolution in the blood of patients. In a comprehensive study by Murtaza et al,44 it was shown that genetic markers of resistance can be noninvasively tracked throughout the course of treatment in breast, ovarian, and lung cancers.
Analysis of ctDNA in plasma samples obtained before and after treatment can ultimately provide a global picture of the molecular genetics of a patient's tumor. This genetic picture includes the dynamic changes in the mutation profile that occur during therapy as well as the heterogeneity that emerges as a result of this therapeutic selective pressure. This understanding of the mechanisms of acquired resistance to targeted agents at the molecular level can be used to plan combinatorial treatments with drugs that will suppress the expansion of the clones that are responsible for most of the current failures of medical treatment (Fig 3). This knowledge could result in the early adoption of alternate therapies before clinical resistance is detected.
NOVEL ADAPTATIONS
Changes in the DNA sequence (eg, somatic point mutations or deletions) are the most frequent class of variants associated with the development of solid tumors and span common oncogenic events, such as KRAS, PIK3CA, EGFR, and BRAF mutations.63 In addition to point mutations, there are several other alterations in nucleic acids present in cancer cells, including amplifications, rearrangements, methylated DNA, mutant mitochondrial DNA, and aneuploidy.63,75–79 Many have been successfully detected in circulation in patients with cancer, and these studies are further described in our article (Fig 2).
CHROMOSOMAL ALTERATIONS: AMPLIFICATIONS, REARRANGEMENTS, AND ANEUPLOIDY
In addition to using somatic point mutations as markers for the detection of tumor DNA, strategies to detect tumor-derived rearrangements and chromosomal copy number changes (ie, amplifications) in the plasma of patients with cancer have been developed (Fig 2).41,45,79–81 Two novel methods to quantitate levels of tumor DNA in circulation include personalized analysis of rearranged ends (PARE) and digital karyotyping.41,45,82 PARE is an approach to identifying DNA rearrangements in human tumors and using these alterations for development of tumor biomarkers. Digital karyotyping is a genome-wide method for detection of copy number alterations associated with such chromosomal changes.83 Initial analyses have demonstrated that the sensitivity of this approach (ie, ability to detect tumor DNA in a mixture of tumor and normal DNA) is lower than 0.001%. Furthermore, this approach can be modified to detect amplifications across the genome based on the fact that amplified segments of genomic DNA result in chromosomal rearrangements.41,45 Therefore, when whole-genome sequencing of plasma DNA is performed, paired-end sequence data may reveal aberrantly mapped paired-end reads. The breakpoints of these regions are mapped to genes of interest. Tag counts in the aberrantly mapped regions and pooled normal plasma from healthy individuals are then compared to determine copy number variations in amplified regions.
Approaches like PARE and digital karyotyping using NGS allow for detection of tumor-specific rearrangements, amplification, and aneuploidy in circulation. If initially identified in tumor tissue, these alterations can be tracked in the blood much like point mutations, and levels in ctDNA correspond with tumor burden. Furthermore, these approaches can be used in a blind fashion as liquid biopsies to detect tumor-derived rearrangements and amplifications for genotyping purposes. For example, HER2/neu (ERBB2) amplifications or ALK rearrangements could be detected using blood samples without the need to examine tumor tissue for these purposes.45 Although the cost of such approaches is high at this time, as the price of NGS decreases, these types of analyses will become more readily available for investigative and clinical purposes.
EPIGENETIC CHANGES
Several reports have demonstrated that methylation changes present in the tumor genome are also present in the DNA fragments detected in circulation. Techniques such as methyl-BEAMing, methylation-specific PCR, and quantum dot–based fluorescence resonance energy transfer (QD-FRET) have been adapted to detect methylated fragments of DNA extracted from plasma.78,84–86 Much like measuring mutations in ctDNA, methylated DNA fragments from the tumor found in circulation are similar to the degree of methylation in the tumor itself and may also correspond to tumor burden. The overall specificity of measuring methylated DNA is lower as compared with genomic alterations, potentially because of the fact that there are often overlapping methylation changes in the tumor and surrounding normal tissue.78 Furthermore, methylation is not entirely a tumor-specific process; the epigenetic regulation present in the tumor may be active in other nontumor tissue, and often changes increase in frequency in an age-dependent fashion. Accordingly, it is imperative to select the appropriate candidate genes if DNA methylation is to be used as a circulating biomarker. Even with these differences compared with mutations, methylation changes in ctDNA can be useful markers of dynamic changes in tumor burden and noninvasively identify methylation changes that predominate in the tumor. Interestingly, despite the lower specificity, the sensitivity of methylated ctDNA is higher even for early-stage disease, because DNA methylation is often an early event in carcinogenesis. This characteristic of DNA methylation in cancer may speak to its potential use as a screening tool for early detection of malignancy.78
DISCUSSION
The analysis of circulating tumor DNA is a promising area of investigation that allows for interrogation of tumor-specific molecular alterations in the circulation. One of the key advantages of ctDNA analyses is the high degree of specificity offered, because mutations found in cfDNA are in essence integral agents of an individual's cancer and are defined by their presence in tumor DNA and absence in matched normal DNA. In terms of sensitivity, levels of ctDNA are abundant in most patients with advanced cancer, allowing for the assessment of molecular heterogeneity, monitoring of tumor dynamics, identification of genetic determinants for therapy, tracking of genomic evolution, and development of acquired resistance. However, more research is needed to establish if these applications will be feasible in tumor types beyond those already studied. In scenarios where only low levels of ctDNA are present (eg, early-stage disease and minimal residual disease), novel genomic methodologies, based on digital PCR, have made such applications possible. New approaches using NGS have increased the throughput necessary for detection of chromosomal abnormalities in ctDNA and will open up the possibility of screening for ctDNA as a tool for the early detection of cancer.
Acknowledgment
We thank Nilofer Azad, MD, and Giulia Siravegna, PhD, for their critical review of this article.
Footnotes
Supported by the European Union Seventh Framework Programme of the Colon Therapy Research Consortium (Grant No. 259015); by the Associazione Italiana per la Ricerca sul Cancro (AIRC) Investigator Grant No. 12812; by AIRC 2010 Special Program Molecular Clinical Oncology 5 per mille, Project No. 9970; by Fondazione Piemontese per la Ricerca sul Cancro 5 per mille 2010 Ministero della Salute; Ministero dell'Istruzione, dell'Università e della Ricerca (Progetti di Ricerca di Interesse Nazionale), and by Swim Across America.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Luis A. Diaz Jr, Personal Genome Diagnostics (U) Consultant or Advisory Role: Luis A. Diaz Jr, Amgen (C) Stock Ownership: Luis A. Diaz Jr, Personal Genome Diagnostics Honoraria: None Research Funding: None Expert Testimony: None Patents: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Financial support: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Mandel P, Metais P: Les acides nucleiques du plasma sanguin chez l'homme [in French] C R Seances Soc Biol Fil 142:241–243,1948 [PubMed] [Google Scholar]
- 2.Lo YM, Chiu RW: Genomic analysis of fetal nucleic acids in maternal blood Annu Rev Genomics Hum Genet 13:285–306,2012 [DOI] [PubMed] [Google Scholar]
- 3.Lo YM Corbetta N Chamberlain PF, etal: Presence of fetal DNA in maternal plasma and serum Lancet 350:485–487,1997 [DOI] [PubMed] [Google Scholar]
- 4.Lo YM Hjelm NM Fidler C, etal: Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma N Engl J Med 339:1734–1738,1998 [DOI] [PubMed] [Google Scholar]
- 5.Papageorgiou EA Karagrigoriou A Tsaliki E, etal: Fetal-specific DNA methylation ratio permits noninvasive prenatal diagnosis of trisomy 21 Nat Med 17:510–513,2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan HC Blumenfeld YJ Chitkara U, etal: Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood Proc Natl Acad Sci U S A 105:16266–16271,2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beiter T Fragasso A Hudemann J, etal: Short-term treadmill running as a model for studying cell-free DNA kinetics in vivo Clin Chem 57:633–636,2011 [DOI] [PubMed] [Google Scholar]
- 8.Atamaniuk J Kopecky C Skoupy S, etal: Apoptotic cell-free DNA promotes inflammation in haemodialysis patients Nephrol Dial Transplant 27:902–905,2012 [DOI] [PubMed] [Google Scholar]
- 9.García Moreira V de la Cera Martínez T Gago González E, etal: Increase in and clearance of cell-free plasma DNA in hemodialysis quantified by real-time PCR Clin Chem Lab Med 44:1410–1415,2006 [DOI] [PubMed] [Google Scholar]
- 10.Korabecna M Opatrna S Wirth J, etal: Cell-free plasma DNA during peritoneal dialysis and hemodialysis and in patients with chronic kidney disease Ann N Y Acad Sci 1137:296–301,2008 [DOI] [PubMed] [Google Scholar]
- 11.Tovbin D Novack V Wiessman MP, etal: Circulating cell-free DNA in hemodialysis patients predicts mortality Nephrol Dial Transplant 27:3929–3935,2012 [DOI] [PubMed] [Google Scholar]
- 12.Antonatos D Patsilinakos S Spanodimos S, etal: Cell-free DNA levels as a prognostic marker in acute myocardial infarction Ann N Y Acad Sci 1075:278–281,2006 [DOI] [PubMed] [Google Scholar]
- 13.Chang CP Chia RH Wu TL, etal: Elevated cell-free serum DNA detected in patients with myocardial infarction Clin Chim Acta 327:95–101,2003 [DOI] [PubMed] [Google Scholar]
- 14.Destouni A Vrettou C Antonatos D, etal: Cell-free DNA levels in acute myocardial infarction patients during hospitalization Acta Cardiol 64:51–57,2009 [DOI] [PubMed] [Google Scholar]
- 15.Jing RR Wang HM Cui M, etal: A sensitive method to quantify human cell-free circulating DNA in blood: Relevance to myocardial infarction screening Clin Biochem 44:1074–1079,2011 [DOI] [PubMed] [Google Scholar]
- 16.Shimony A Zahger D Gilutz H, etal: Cell free DNA detected by a novel method in acute ST-elevation myocardial infarction patients Acute Card Care 12:109–111,2010 [DOI] [PubMed] [Google Scholar]
- 17.Diehl F Schmidt K Choti MA, etal: Circulating mutant DNA to assess tumor dynamics Nat Med 14:985–990,2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macher H Egea-Guerrero JJ Revuelto-Rey J, etal: Role of early cell-free DNA levels decrease as a predictive marker of fatal outcome after severe traumatic brain injury Clin Chim Acta 414:12–17,2012 [DOI] [PubMed] [Google Scholar]
- 19.Ohayon S Boyko M Saad A, etal: Cell-free DNA as a marker for prediction of brain damage in traumatic brain injury in rats J Neurotrauma 29:261–267,2012 [DOI] [PubMed] [Google Scholar]
- 20.Tsai NW Lin TK Chen SD, etal: The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke Clin Chim Acta 412:476–479,2011 [DOI] [PubMed] [Google Scholar]
- 21.Diehl F Li M Dressman D, etal: Detection and quantification of mutations in the plasma of patients with colorectal tumors Proc Natl Acad Sci U S A 102:16368–16373,2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holdhoff M Schmidt K Donehower R, etal: Analysis of circulating tumor DNA to confirm somatic KRAS mutations J Natl Cancer Inst 101:1284–1285,2009 [DOI] [PubMed] [Google Scholar]
- 23.Vogelstein B, Kinzler KW: Digital PCR Proc Natl Acad Sci U S A 96:9236–9241,1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dressman D Yan H Traverso G, etal: Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations Proc Natl Acad Sci U S A 100:8817–8822,2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Q, Sommer SS: Pyrophosphorolysis-activated polymerization (PAP): application to allele-specific amplification Biotechniques 29:1072–1076,2000 1078, 1080 [DOI] [PubMed] [Google Scholar]
- 26.Daniotti M Vallacchi V Rivoltini L, etal: Detection of mutated BRAFV600E variant in circulating DNA of stage III-IV melanoma patients Int J Cancer 120:2439–2444,2007 [DOI] [PubMed] [Google Scholar]
- 27.Kimura H Kasahara K Kawaishi M, etal: Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer Clin Cancer Res 12:3915–3921,2006 [DOI] [PubMed] [Google Scholar]
- 28.Shinozaki M O'Day SJ Kitago M, etal: Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy Clin Cancer Res 13:2068–2074,2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan SR Whiteley J Donald E, etal: Comparison of KRAS mutation assessment in tumor DNA and circulating free DNA in plasma and serum samples Clin Med Insights Pathol 5:15–22,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maheswaran S Sequist LV Nagrath S, etal: Detection of mutations in EGFR in circulating lung-cancer cells N Engl J Med 359:366–377,2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuang Y Rogers A Yeap BY, etal: Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer Clin Cancer Res 15:2630–2636,2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punnoose EA Atwal S Liu W, etal: Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: Association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib Clin Cancer Res 18:2391–2401,2012 [DOI] [PubMed] [Google Scholar]
- 33.Jänne PA Borras AM Kuang Y, etal: A rapid and sensitive enzymatic method for epidermal growth factor receptor mutation screening Clin Cancer Res 12:751–758,2006 [DOI] [PubMed] [Google Scholar]
- 34.Milbury CA, Li J, Makrigiorgos GM: PCR-based methods for the enrichment of minority alleles and mutations Clin Chem 55:632–640,2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newton CR Graham A Heptinstall LE, etal: Analysis of any point mutation in DNA: The amplification refractory mutation system (ARMS) Nucleic Acids Res 17:2503–2516,1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogino S Kawasaki T Brahmandam M, etal: Sensitive sequencing method for KRAS mutation detection by pyrosequencing J Mol Diagn 7:413–421,2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forshew T Murtaza M Parkinson C, etal: Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA Sci Transl Med 4:136ra68,2012 [DOI] [PubMed] [Google Scholar]
- 38.Li M Diehl F Dressman D, etal: BEAMing up for detection and quantification of rare sequence variants Nat Methods 3:95–97,2006 [DOI] [PubMed] [Google Scholar]
- 39.Higgins MJ Jelovac D Barnathan E, etal: Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood Clin Cancer Res 18:3462–3469,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinde I Wu J Papadopoulos N, etal: Detection and quantification of rare mutations with massively parallel sequencing Proc Natl Acad Sci U S A 108:9530–9535,2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leary RJ Kinde I Diehl F, etal: Development of personalized tumor biomarkers using massively parallel sequencing Sci Transl Med 2:20ra14,2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taly V Pekin D Benhaim L, etal: Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients Clin Chem [epub ahead of print on August 12, 2013] [DOI] [PubMed] [Google Scholar]
- 43.Dawson SJ Tsui DW Murtaza M, etal: Analysis of circulating tumor DNA to monitor metastatic breast cancer N Engl J Med 368:1199–1209,2013 [DOI] [PubMed] [Google Scholar]
- 44.Murtaza M Dawson SJ Tsui DW, etal: Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA Nature 497:108–112,2013 [DOI] [PubMed] [Google Scholar]
- 45.Leary RJ Sausen M Kinde I, etal: Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing Sci Transl Med 4:162ra154,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delgado PO Alves BC Gehrke Fde S, etal: Characterization of cell-free circulating DNA in plasma in patients with prostate cancer Tumour Biol 34:983–986,2013 [DOI] [PubMed] [Google Scholar]
- 47.Hashad D Sorour A Ghazal A, etal: Free circulating tumor DNA as a diagnostic marker for breast cancer J Clin Lab Anal 26:467–472,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.No JH Kim K Park KH, etal: Cell-free DNA level as a prognostic biomarker for epithelial ovarian cancer Anticancer Res 32:3467–3471,2012 [PubMed] [Google Scholar]
- 49.Park JL Kim HJ Choi BY, etal: Quantitative analysis of cell-free DNA in the plasma of gastric cancer patients Oncol Lett 3:921–926,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salvianti F Pinzani P Verderio P, etal: Multiparametric analysis of cell-free DNA in melanoma patients PLoS One 7:e49843,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwarzenbach H Alix-Panabières C Müller I, etal: Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer Clin Cancer Res 15:1032–1038,2009 [DOI] [PubMed] [Google Scholar]
- 52.Schwarzenbach H Müller V Milde-Langosch K, etal: Evaluation of cell-free tumour DNA and RNA in patients with breast cancer and benign breast disease Mol Biosyst 7:2848–2854,2011 [DOI] [PubMed] [Google Scholar]
- 53.Schwarzenbach H Stoehlmacher J Pantel K, etal: Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer Ann N Y Acad Sci 1137:190–196,2008 [DOI] [PubMed] [Google Scholar]
- 54.Fleischhacker M, Schmidt B: Circulating nucleic acids (CNAs) and cancer: A survey Biochim Biophys Acta 1775:181–232,2007 [DOI] [PubMed] [Google Scholar]
- 55.Jahr S Hentze H Englisch S, etal: DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells Cancer Res 61:1659–1665,2001 [PubMed] [Google Scholar]
- 56.Stroun M Lyautey J Lederrey C, etal: About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release Clin Chim Acta 313:139–142,2001 [DOI] [PubMed] [Google Scholar]
- 57.Wyllie AH: Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation Nature 284:555–556,1980 [DOI] [PubMed] [Google Scholar]
- 58.Mouliere F Robert B Arnau Peyrotte E, etal: High fragmentation characterizes tumour-derived circulating DNA PLoS One 6:e23418,2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan KC Zhang J Hui AB, etal: Size distributions of maternal and fetal DNA in maternal plasma Clin Chem 50:88–92,2004 [DOI] [PubMed] [Google Scholar]
- 60.Misale S Yaeger R Hobor S, etal: Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer Nature 486:532–536,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Overman MJ Modak J Kopetz S, etal: Use of research biopsies in clinical trials: Are risks and benefits adequately discussed? J Clin Oncol 31:17–22,2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerlinger M Rowan AJ Horswell S, etal: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing N Engl J Med 366:883–892,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vogelstein B Papadopoulos N Velculescu VE, etal: Cancer genome landscapes Science 339:1546–1558,2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Mattos-Arruda L Cortes J Santarpia L, etal: Circulating tumour cells and cell-free DNA as tools for managing breast cancer Nat Rev Clin Oncol 10:377–389,2013 [DOI] [PubMed] [Google Scholar]
- 65.Diaz LA Jr Williams RT Wu J, etal: The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers Nature 486:537–540,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ito K Hibi K Ando H, etal: Usefulness of analytical CEA doubling time and half-life time for overlooked synchronous metastases in colorectal carcinoma Jpn J Clin Oncol 32:54–58,2002 [DOI] [PubMed] [Google Scholar]
- 67.Yoshimasu T Maebeya S Suzuma T, etal: Disappearance curves for tumor markers after resection of intrathoracic malignancies Int J Biol Markers 14:99–105,1999 [DOI] [PubMed] [Google Scholar]
- 68.Riedinger JM Wafflart J Ricolleau G, etal: CA 125 half-life and CA 125 nadir during induction chemotherapy are independent predictors of epithelial ovarian cancer outcome: Results of a French multicentric study Ann Oncol 17:1234–1238,2006 [DOI] [PubMed] [Google Scholar]
- 69.Bidard FC Madic J Mariani P, etal: Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma Int J Cancer [epub ahead of print on August 12, 2013] [DOI] [PubMed] [Google Scholar]
- 70.Goncalves R, Bose R: Using multigene tests to select treatment for early-stage breast cancer J Natl Compr Canc Netw 11:174–182,2013quiz 182 [DOI] [PubMed] [Google Scholar]
- 71.Pao W Miller VA Politi KA, etal: Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain PLoS Med 2:e73,2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Antonescu CR Besmer P Guo T, etal: Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation Clin Cancer Res 11:4182–4190,2005 [DOI] [PubMed] [Google Scholar]
- 73.Branford S Rudzki Z Walsh S, etal: Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis Blood 102:276–283,2003 [DOI] [PubMed] [Google Scholar]
- 74.Taniguchi K Uchida J Nishino K, etal: Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas Clin Cancer Res 17:7808–7815,2011 [DOI] [PubMed] [Google Scholar]
- 75.Azad N Zahnow CA Rudin CM, etal: The future of epigenetic therapy in solid tumours: Lessons from the past Nat Rev Clin Oncol 10:256–266,2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.He Y Wu J Dressman DC, etal: Heteroplasmic mitochondrial DNA mutations in normal and tumour cells Nature 464:610–614,2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holland AJ, Cleveland DW: Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis Nat Rev Mol Cell Biol 10:478–487,2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li M Chen WD Papadopoulos N, etal: Sensitive digital quantification of DNA methylation in clinical samples Nat Biotechnol 27:858–863,2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beck J Urnovitz HB Mitchell WM, etal: Next generation sequencing of serum circulating nucleic acids from patients with invasive ductal breast cancer reveals differences to healthy and nonmalignant controls Mol Cancer Res 8:335–342,2010 [DOI] [PubMed] [Google Scholar]
- 80.McBride DJ Orpana AK Sotiriou C, etal: Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors Genes Chromosomes Cancer 49:1062–1069,2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan KC Jiang P Zheng YW, etal: Cancer genome scanning in plasma: Detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing Clin Chem 59:211–224,2013 [DOI] [PubMed] [Google Scholar]
- 82.Wang TL Diaz LA Jr Romans K, etal: Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients Proc Natl Acad Sci U S A 101:3089–3094,2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang TL Maierhofer C Speicher MR, etal: Digital karyotyping Proc Natl Acad Sci U S A 99:16156–16161,2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weaver KD, Grossman SA, Herman JG: Methylated tumor-specific DNA as a plasma biomarker in patients with glioma Cancer Invest 24:35–40,2006 [DOI] [PubMed] [Google Scholar]
- 85.Bailey VJ Keeley BP Zhang Y, etal: Enzymatic incorporation of multiple dyes for increased sensitivity in QD-FRET sensing for DNA methylation detection Chembiochem 11:71–74,2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bailey VJ Zhang Y Keeley BP, etal: Single-tube analysis of DNA methylation with silica superparamagnetic beads Clin Chem 56:1022–1025,2010 [DOI] [PMC free article] [PubMed] [Google Scholar]