

Abstract
Endoplasmic reticulum is a principal organelle responsible for folding, post-translational modifications and transport of secretory, luminal and membrane proteins, thus palys an important rale in maintaining cellular homeostasis. Endoplasmic reticulum stress (ERS) is a condition that is accelerated by accumulation of unfolded/misfolded proteins after endoplasmic reticulum environment disturbance, triggered by a variety of physiological and pathological factors, such as nutrient deprivation, altered glycosylation, calcium depletion, oxidative stress, DNA damage and energy disturbance, etc. ERS may initiate the unfolded protein response (UPR) to restore cellular homeostasis or lead to apoptosis. Numerous studies have clarified the link between ERS and cardiovascular diseases. This review focuses on ERS-associated molecular mechanisms that participate in physiological and pathophysiological processes of heart and blood vessels. In addition, a number of drugs that regulate ERS was introduced, which may be used to treat cardiovascular diseases. This review may open new avenues for studying the pathogenesis of cardiovascular diseases and discovering novel drugs targeting ERS.
Keywords: endoplasmic reticulum stress, unfolded protein response, ischemic cardiomyopathy, atherosclerosis, hypertension, cardiac hypertrophy, heart failure
Introduction
The endoplasmic reticulum (ER) is a principal organelle responsible for folding, post-translational modifications, and transport of secretory, luminal and membrane proteins and is capable of maintaining cellular homeostasis. Newly synthesized and secretory membrane proteins transfer into the endoplasmic reticulum for modification. The modification processes include protein folding, glycosylation and disulfide bond formation1. The ER quality control system (ERQC) is a system that prevents protein aggregation by promoting correct folding or selective degradation of the improperly folded polypeptide2. This process is regulated by molecular chaperones, foldases, and lectins that maintain the ERQC. ER associates with many processes including apoptosis and autophagy3.
Endoplasmic reticulum stress (ERS) is a condition that is accelerated by the accumulation of unfolded/misfolded proteins after a disturbance in the ERQC owing to a variety of physiological and pathological phenomena4. Nutrient deprivation, altered glycosylation, calcium depletion, oxidative stress, DNA damage and energy disturbance4,5,6 are all the causes of ER environment disturbance, which subsequently lead to ERS7.
Endoplasmic reticulum stress suppresses proteins synthesis at the early stages
At the early stages of ERS, the failure of molecular chaperones break the maintenance of proteostasis8,9. To overcome the deleterious effects of ERS, a series of adaptive and protective strategies are initiated. For example, owing to the abundance of unfolded/misfolded proteins accumulated in the ER, especially the accumulation of unfolded proteins, new protein synthesis is inhibited. In order to clear the accumulated proteins, many chaperone genes are induced at the transcriptional level and activate the ER-associated degradation (ERAD) system, which can translocate and remove misfolded proteins through proteasomal degradation. These processes are identified as the unfolded protein response (UPR)10,11. However, if unresolved, ERS is lethal to cells via what is recognized as ERS-induced apoptosis12.
The UPR is a concerted and complex cellular response that is mediated through three ER transmembrane receptor proteins: double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1, also called ERN1) and activating transcription factor 6 (ATF6)13,14. In addition, glucose-regulated protein 78 kDa (GRP78, also called Bip) also plays an essential role in the UPR.
PERK is a member of the eukaryotic translation initiation factor 2α (eIF2α) kinase subfamily, together with RNA-dependent protein kinase (PKR), general control nondepressible 2 (GCN2) kinase and heme-regulated eIF2α kinase (HRI)15. PERK possesses a luminal domain similar to that of IRE1 and a cytoplasmic portion that manifests protein serine/threonine kinase activity. Moreover, it has a PKR-like catalytic domain, which phosphorylates eIF2α16. eIF2 has three subunits: α, β, and γ. eIF2α is a translation initiation factor that functions in the early steps of protein synthesis by forming a ternary complex with GTP and initiator tRNA. This complex binds to a 40S ribosomal subunit, followed by mRNA binding to constitute a 43S preinitiation complex. Junction of the 60S ribosomal subunit to form the 80S initiation complex is preceded by hydrolysis of the GTP bound to eIF2 and release of an eIF2-GDP binary complex. In order to make eIF2 recycle and then to catalyze another round of initiation, the GDP bound to eIF2 must exchange with GTP by way of the reaction catalyzed by eIF2β. eIF2α phosphorylation stabilizes the eIF2/GDP/eIF2β complex and prevents the GDP/GTP exchange reaction; thus, it impairs the recycling of eIF2 between successive rounds of initiation and leads to global inhibition of translation at early stages of ERS.
IRE1 is an ER transmembrane protein that consists of an N-terminal luminal sensor domain, a single transmembrane domain and a C-terminal cytosolic effector region, which manifests both kinase and endoribonuclease activity17In normal conditions, IRE1 forms a complex with GRP78 in the ER membrane and is inactive. ERS leads to GRP78 release from the complex, which activates IRE1. IRE1 is involved in the activation of ERAD genes via X-binding protein-1 (XBP-1) mRNA splicing and is therefore activated by self-phosphorylation17. XBP-1 is recognized as a transcription factor that participates in phospholipid synthesis other than ERAD and in protein quality control6.
ATF6, which includes two isoforms ATF6α and ATF6β18, is considered a member of a protein family associated with the regulation of genes. ATF6 contains sequences with a site referred to as the cAMP response element (CRE)19. This 90 kDa protein has a bZIP domain for DNA binding after homo- or heterodimerization20.
GRP78, a member of the HSP70 family, is an ER luminal chaperone that works in an ATP-dependent manner and interacts with several ERAD substrates. GRP78 binds to the substrates to enhance the solubility of ERAD proteins and facilitates their targeting. GRP78 acts as a multifunctional modulator for the regulation of PERK, IRE1 and ATF6 in its monomeric form21
All the above receptor proteins are endoplasmic reticulum chaperones. These receptor proteins work together to balance the unfolded protein/chaperone system, which contributes to the homeostasis of ER at early stages of ERS. They also play very important roles in the middle stages of ERS.
Endoplasmic reticulum stress promotes ER homeostasis via the UPR during the middle stages
The processes dealing with ERS are complex. Persistent stimulus brings the UPR into focus; all the mentioned ERS sensor proteins use a unique mechanism to activate transcription factors and upregulate UPR target genes. Three pathways are activated to rescue the damaged ER during the middle stages of ERS.
First, the IRE1 pathway that is the conserved core of the UPR is activated (Figure 1). IRE1 encodes a type 1 ER resident transmembrane protein with a novel lumenal domain and a cytoplasmic portion that contains a protein kinase domain22,23. In response to unfolded proteins, IRE1 activates itself 24,25. Trans-autophosphorylated IRE1 activates its unusual effector function, which causes the precise endonucleolytic cleavage of the only known substrate: an mRNA that encodes a transcription factor named XBP-1 in metazoans26,27. IRE1 cuts the precursor XBP1 mRNA twice, excising an intervening fragment or intron. The 5′and 3′ mRNA fragments then generate a spliced mRNA that translocates to the nucleus and binds to the promoter region of the ER stress-response element (ERSE)28,29, contributing to the rescue of damaged cells.
Figure 1.

Signaling pathways of endoplasmic reticulum stress.
PERK, the second pathway (Figure 1), inhibits protein translation, reduces protein synthesis and prevents accumulation of newly synthesized proteins30. PERK promotes phosphorylation of eIF2α as an immediate response31. Increasing unfolded proteins accumulating in the ER induces dissociation of the GRP78/PERK complex and activation of the kinase domain via autophosphorylation32, leading to eIF2α phosphorylation and activation that decrease mRNA translation, so proteins gather in the ER33. However, the translation of some specific proteins, which include internal ribosomal entry sites such as ATF4, GRP94, and GRP78, is increased via PERK activation34. ATF4 is a bZIP transcription factor involved in the expression of survival genes such as GRP78 and GRP9435. Calcium-binding chaperone calnexin, calreticulin (CRT) and protein-folding enzymes such as protein disulfide isomerase (PDI) and sarco (endo) plasmic reticulum Ca2+ ATPase 2α (SERCA2α) are also increased. In addition, ATF4 upregulates growth arrest and DNA damage-inducible gene 34 (GADD34), promoting translational recovery of eIF2α by dephosphorylation36.
ATF6, the third pathway of the UPR (Figure 1), is another transcription factor that translocates to the nucleus after its cleavage under ERS conditions18,37. ATF6 includes two isoforms: ATF6α and ATF6β. ATF6α has a higher transcriptional activity compared to ATF6β, which gives it a cytoprotective role in several cellular stress models18. After GRP78 release, ATF6 enters coat protein complex II (COPII) vesicles and is targeted to the Golgi lumen, where the cleavage processes take place at site-1 protease (S1P) and site-2 protease (S2P)38. Under ERS conditions, disulfide bonds in the structure of ATF6 reduce the activation and translocation of this protein out of the ER. Conversely, ATF6 could also regulate the expression of genes such as XBP139.
Endoplasmic reticulum stress induces cell apoptosis in the final stages
Although the UPR is primarily a pro-survival response, in the event of prolonged or severe ERS that is not resolved, the unfolded protein response switches to initiate apoptosis in either a UPR-dependent or -independent manner40. ERS-induced apoptosis, which participates in pathological processes of cardiovascular diseases, is mediated by GADD153, also called transcription factor CCAAT enhancer-binding protein (C/EBP), homologous protein (CHOP), IRE1 and caspase 12.
First, CHOP was initially reported as a molecule involved in ERS-induced apoptosis. The expression of CHOP is low under non-stressed conditions, but its expression markedly increases in response to ERS through IRE1-, PERK- and ATF6-dependent transcriptional induction. ATF4 is thought to play a dominant role in inducing CHOP expression in response to ERS because the expression of ATF4 is upregulated when eIF2α is phosphorylated by PERK41. Overexpression of CHOP promotes apoptosis in several cell lines, whereas CHOP-deficient cells are resistant to ERS-induced apoptosis42. CHOP leads to upregulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors and thus promotes the extrinsic apoptotic pathway43,44. Therefore, CHOP plays an important role in the induction of apoptosis.
In addition to CHOP, other ERS-mediated apoptosis mechanisms have been proposed45. For example, ERS activates IRE1, which recruits the adaptor molecule tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) and apoptosis signal-regulating kinase 1 (ASK1); then the IRE1-JNK-TRAF2 complex forms, leading to activation of pro-apoptotic JNK signaling46. Interaction of IRE1–TRAF2 after ERS has also been reported to recruit pro-caspase 12, which has been proposed to be an important mediator of ERS-induced apoptosis47,48.
Caspase 12 is the third protein that regulates the processes of ERS-induced apoptosis49. Procaspase 12 localizes on the ER membrane alone or in complex with TRAF2; severe ERS induces procaspase 12 to dissociate from the ER membrane and become activated. The activated form of procaspase 12 is caspase 1248,50. Two pathways lead to activation of procaspase 12. In the first pathway, elevating Ca2+ in the cytoplasm leads to calpain activation and translocates to the ER membrane. Caspase 12 is initially processed by calpain; active caspase 12 can initiate a positive feedback loop that activates caspase 3 through caspase 9 activation and thus potentiate apoptosis induction51. In the second pathway, the TRAF2-procaspase 12 complex is located on the ER membrane. ERS leads to TRAF2 dissociation from the TRAF2-procaspase 12 complex, which activates caspase 12. At the same time, IRE1-JNK recruits TRAF2 and leads to the formation of the IRE1-JNK-TRAF2 complex. The IRE1-JNK-TRAF2 complex has an effect on ASK1 activation, which may induce JNK phosphorylation and ultimately lead to cell apoptosis52. Signaling pathways of endoplasmic reticulum stress are summarized in Figure 1.
Endoplasmic reticulum stress regulates cardiovascular physiology function
It is knowledgeable that adhesion, proliferation53,54,55, migration56 and autophagy57,58 are the characteristics of cells59,60,61. ERS regulates those functions61. The role of ERS in regulating functions of cardiovascular physiology is still controversial. On the one hand, some researchers have reported that ERS promotes cell adhesion, proliferation, migration and autophagy. For example, Riek et al62 reported that ERS induced monocyte adhesion and migration. Accordingly, suppressed ERS decreased the expression of adhesion markers integrin 1 and 2 and migration receptor chemokine (C-C motif) receptor 2 (CCR2). Nakamura et al63 found that ERS promoted proliferation and migration of human retinal microvascular endothelial cells by increasing the expression of Bip at both the mRNA and protein levels and enhancing the formation of a Bip/T-cadherin immunocomplex. In an oxygen-induced retinopathy (OIR) model in mice, Bip was observed in the retinal microvascular endothelial cells; this finding has many similarities to observations of the endogenous ligand of APJ receptor apelin-1364,65,66. Yang et al67 reported that hypoxia, especially intermittent hypoxia, induced endothelial cell apoptosis, possibly through ERS. Intermittent and persistent hypoxia increased the rate of endothelium apoptosis by increasing the expression of GRP78, CHOP and caspase 12. ERS promoted endothelial cell apoptosis through caspase 12 and the mitochondrial pathway68,69.
On the other hand, other researchers hold the opposite view: that ERS inhibits proliferation and migration of cells. For instance, Nakamura et al63 found that severe ERS induced by 10 μmol/L of the ERS inducers tunicamycin (Tm) and thapsigargin (TG) led to HRMEC apoptosis; however, HRMEC proliferation and migration was inhibited by Tm. Yi et al70 found that pretreatment with Tm significantly inhibited platelet-derived growth factor (PDGF)-BB-induced VSMC proliferation and migration without causing significant apoptosis. Tm increased the expression of the antioxidant heme oxygenase-1 (HO-1) gene at both the transcriptional and translational levels, while reducing phosphorylation and activation of mitogen-activated protein kinases (MAPK).
ERS also has effects on cardiovascular function in vivo. Nakamura et al63 reported that in OIR model mice, ERS accelerated retinal neovascularization. Bip was observed in the pathological vasculature. Yang et al71 showed that ERS led to a compromise of echocardiographic and cardiomyocyte contractile function by increasing intracellular Ca2+. ERS increased the left ventricular end systolic and diastolic diameter and suppressed fractional shortening and whole-heart contractility72. The research on ERS in cardiovascular physiology function is summarized in Table 1.
Table 1. Studies of endoplasmic reticulum stress in cardiovascular physiology function.
| Reference | Protein level | Experiment cell | Effect | Mild ERS | Severe ERS | ERS inducer |
|---|---|---|---|---|---|---|
| Riek et al 62 | β1, β2 integrin | Monocyte | Enhance adhesion; enhance proliferation | Yes | No | Thapsigargin |
| Nakamura et al 63 | Bip/T-cadherin | Human retinal microvascular endothelial cells (HRMEC) | Enhance proliferation; enhance migration | Yes | No | Tunicamycin Thapsigargin |
| Bip/T-cadherin | Human retinal microvascular endothelial cells (HRMEC) | Suppress proliferation; suppress migration | No | Yes | Tunicamycin Thapsigargin | |
| Bip/T-cadherin | OIR model mice | Enhance neovascularization | Yes | No | Tunicamycin Thapsigargin | |
| Yang et al 67 | GRP78, CHOP, caspase 12 | Human umbilical vein endothelial cells (HUVECs) | Induce cells apoptosis | No | Yes | Intermittent/persistent hypoxia |
| Yi et al 70 | XBP1, GRP94, ATF6 | Vascular smooth muscle cells (VSMC); SD rats aortic endothelialcell cells | Suppress abnormal proliferation and migration | No | Yes | Tunicamycin |
Endoplasmic reticulum stress is a major case of cardiovascular diseases
Endoplasmic reticulum stress regulates the processes of atherosclerosis
Animal models of atherosclerosis and human atherosclerotic lesions have provided unmistakable evidence that ERS occurred in atherosclerotic plaques, particularly in the advanced stages73. The number of apoptotic cells depends on the plaque stage; it is generally high in advanced plaques. In this section we briefly elucidate the role of ERS in three major types of cells in atherosclerosis: smooth muscle cells (SMCs)74, macrophages and endothelial cells. The role of sterol regulatory element-binding proteins (SREBPs) in regulating ERS to control atherosclerosis is also discussed.
SMCs within the vascular wall exist in predominantly two phenotypes: a contractile phenotype and a synthetic phenotype. Transformation from the quiescent contractile phenotype to the active synthetic phenotype can be stimulated by numerous growth factors, including PDGF-BB75. The synthetic SMCs phenotype is actively involved in de novo protein synthesis and as such may experience ERS if the protein-folding machinery of the ER is overwhelmed by the demand of newly synthesized proteins76. Generally, in the case of mild ERS, the UPR is responsible for activating molecular chaperones to attenuate cellular dysfunction or damage. However, if ERS is prolonged or too severe, the UPR may result in the activation of apoptosis77. SMCs synthesize most of the interstitial collagen, which stabilizes the fibrous cap of a plaque. Therefore, excessive apoptosis of SMCs in the fibrous cap may break plaque integrity and render it vulnerable to proteolytic attack by inflammatory cells, subsequently leading to plaque rupture78. Pedruzzi et al79 reported that the 7-ketocholesterol (7-Kchol)-activating pathway induced an early triggering of ERS in SMCs, as assessed by transient intracellular Ca2+ oscillations and IRE-1, JNK, and AP-1 expression. The IRE-1/JNK/AP-1 signaling pathway has likewise been shown to promote the expression of NADPH oxidase 4 (Nox-4) and subsequently cause SMC death. Furthermore, 7-ketocholesterol-induced ERS caused macrophage apoptosis. ERS also promoted oxidative stress and enhanced calcification of vascular smooth muscle cells80.
ERS is a prominent mechanism of macrophage apoptosis in advanced atherosclerotic lesions. Solanki et al81 shown that the number of ERS-related apoptotic macrophages was increased by a deficiency of calcium-permeable channel transient receptor potential canonical 3 (TRPC3). TRPC3 reduced scavenging of apoptotic SMCs and macrophages and then allowed cells to undergo secondary necrosis, thereby increasing the thrombogenicity of the plaque82. Suppression of ERS decreased the number of apoptotic macrophages83. Yao et al84 showed that ERS was involved in regulating oxidized low-density lipoprotein (ox-LDL) induced scavenger receptor A1 (SR-A1) upregulation in macrophages. Ox-LDL caused significant SR-A1 upregulation with concomitant activation of ERS as assessed by upregulation of GRP78. Ryan et al85 also detected an increased level of ERS in macrophages. ERS induced by asbestos caused disruption of calcium homeostasis. ERS-induced apoptosis in macrophages may be due to neutral cholesterol ester hydrolase 1 (Nceh1). Sekiya et al86 reported that Nceh1-deficient thioglycollate-elicited peritoneal macrophages (TGEMs) were susceptible to apoptosis induced by oxysterols, particularly 25-hydroxycholesterol (25-HC). Incubation with 25-HC caused massive accumulation of 25-HC ester in the ER due to its defective hydrolysis function, thereby activating ERS signaling proteins such as CHOP. Fatty acid-binding protein-4 (FABP-4), also known as adipocyte FABP (A-FABP)/-adipocyte P2 (aP2), is a cytosolic lipid chaperone that regulates cellular lipid metabolism, receives lipid signals, and promotes atherosclerosis development. FABP-4 links to atherosclerosis also have something to do with ERS. Gao et al87 reported that FABP-4 induced ERS in macrophages. In addition, FABP-4 mediated ERS takes part in the progression of interleukin 17A (IL-17A)-induced atherosclerosis. Erbay et al88 reported that FABP-4 is required in lipid-induced ERS and apoptosis in macrophages from atherosclerotic lesions. Deletion of FABP4 in macrophages increased de novo lipogenesis pathways through LXRα-mediated SCD-1 activation, resulting in production of palmitoleate and resistance to ERS. However, Xu et al89 reported that FABP4/aP2 indirectly controlled macrophage ERS by regulating the expression of uncoupling protein 2 (UCP2).
ERS has also been detected in endothelial cells subjected to atherosclerosis-prone shear stress90. Increased levels of GRP78 were observed in atherosclerotic regions from C57BL6 mice91. Dong et al91 reported that in human umbilical vein endothelial cells and aortic endothelial cells from 5′ adenosine monophosphate-activated protein kinase 2 (AMPK2)-deficient C57BL6 mice, the level of ERS increased. Reduction of AMPK2 expression significantly increased ERS levels in human umbilical vein endothelial cells. In addition, mouse aortic endothelial cells from AMPK2 knockout mice showed higher expression of ERS markers such as XBP1, ATF6, GRP78, p-eIF2α, p-PERK, p-JNK and an increased level of intracellular Ca2+. Li et al92 reported that heat stress activated UPR signaling through the PERK-eIF2α-ATF4, IRE1-XBP1s and ATF6 pathways in human umbilical vein endothelial cells. ERS also mediated aldosterone (Aldo)-induced apoptosis in vascular endothelial cells, as evidenced by increasing expression of GRP78 and CHOP. When CHOP was knocked down, Aldo-mediated apoptosis was attenuated93.
SREBPs, a small family of lipogenic transcription factors, control cholesterol and lipid metabolism and play critical roles during atherosclerosis development. SREBPs are expressed as inactive precursors and reside as integral trans-membrane proteins within the ER membrane, where they bind to the SREBP cleavage-activating protein (SCAP)94. Three SREBP isoforms: SREBP-1a, SREBP-1c and SREBP-2, have been identified in mammalian cells95,96. Usually, SREBPs are activated in response to cholesterol deprivation and control lipid metabolism. ATF6 processing required for S1P and S2P, the enzymes that process SREBPs in response to cholesterol deprivation, demonstrated an important relationship between ERS and SREBPs97. Zeng et al98 reported that ATF6 antagonized SREBP-2 to regulate the homeostasis of lipid and glucose. Hyperhomocysteinemia-induced ERS accompanied by upregulation of SREBP-2 and increased lipid deposits in VSMCs99. Glucosamine accumulated in vascular cells in diabetes. Its accumulation has a primary responsibility for ERS induction in VSMCs from diabetic patients through GRP78 upregulation, and this process may accelerate atherosclerosis100. Increasing evidence indicates that non-alcoholic fatty liver disease (NAFLD) is a major cause of atherosclerosis101. ERS triggered NAFLD by dramatically increasing SREBP-1a and SREBP-1c in vivo102. Fang et al103 demonstrated that ERS may promote lipid synthesis through SREBP-1c in L02 and HepG2 hepatocytes.
In summary, ERS is a key regulator of atherosclerosis, controlling the whole process of atherosclerosis. ERS regulates the progression of atherosclerosis together with several other factors, such as Nox4, TRPC3, SR-A1, and AMPK2. Thus, ERS is a potential target for therapeutic drug design.
Endoplasmic reticulum stress and ischemic cardiomyopathy
ERS mediates the progression of ischemic cardiomyopathy. Ischemia/reperfusion (I/R) injury and myocardial infarction are two major types of ischemic cardiomyopathy. I/R injury is accompanied by contractile dysfunction and cellular damage, contributing to cardiovascular diseases104. I/R injury activates ERS, causing apoptosis by promoting the expression of GRP78, cleaved caspase 3105 and CHOP106. Tao et al107 reported that I/R injury was associated with a time-related increase in ERS-dependent apoptosis through activation of CHOP, caspase 12, and JNK. Liu et al108 reported that myocardial I/R caused ERS and perturbed ER function by increasing the expression of calreticulin, JNK and caspase 12. Ischemic postconditioning (I-postC) is a newly discovered endogenous protective phenomenon. It has the ability to protect myocardium from I/R injury by suppressing ERS through the p38 MAPK and JNK pathways. These reports present the view that I/R injury activates ERS, and, at the same time, ERS exacerbates injury of I/R myocardium. Martindale et al obtained results inconsistent with the above opinions. They suggested that the UPR is activated in the heart during I/R, and as a result, the ATF6 branch of UPR-induced expression of proteins, including GRP78 and GRP94, can reduce I/R injury109. I/R injury rats are accompanied by myocardial infarction (MI), and increased myocardial infarct size and myocardial enzyme activity were observed106. Luo et al110 reported that MI induced ERS and provoked cardiac apoptosis and fibrosis by increasing the expression of ERS markers GRP78 and CHOP, pro-apoptotic proteins Bax and caspase 3, and pro-fibrotic proteins transforming growth factor-beta 1 (TGF-β1) and Smad 2/3, culminating in cardiac rupture and remodeling. Li et al111 reported that ERS contributed to MI through the activation of NADPH oxidase. NADPH oxidase inhibition attenuated an increase in ERS in the remote non-infarcted myocardium and LV remodeling after MI by preventing the increase of GRP78, CHOP and cleaved caspase 12 protein expression in rabbits.
Endoplasmic reticulum stress and hypertension
Hypertension is a major modifiable risk factor for cardiovascular diseases112,113. An increasing amount of evidence indicates that ERS has links to hypertension. Young et al114 first reported that ERS, notably brain ERS, played a key role in chronic hypertension (HTN). The C57BL/6 mouse model of HTN was induced by angiotensin II (Ang II); the circumventricular subfornical organ (SFO), which is a brain region, revealed a robust increase of ERS biomarkers such as p58IPK mRNA, p-PERK, GRP78, CHOP. In addition, ultrastructural abnormalities of the rough ER (RER) are another classic hallmark of ERS. Young et al also detected abnormal RER morphology in SFO neurons in this article. ERS mediated Ang II-dependent hypertension in SFO through nuclear factor (NF)-кB activation115. ERS also contributed to neurogenic hypertension116. Severe hypertension caused neuronal injury through neuronal ERS and apoptosis117. ERS is a key player in endothelial dysfunction during hypertension118. Reactive oxygen species (ROS) overproduction or the increase of oxidative stress impaired endothelial function in hypertension119. ERS may induce oxidative stress to impair endothelial function in the development of hypertension. Liu et al120 reported that carotid arteries exhibited exaggerated acetylcholine-triggered endothelium-dependent contractions (EDCs) and elevated ROS accumulation compared with WKY arteries isolated from spontaneously hypertensive rats (SHRs). In addition, Western blot analysis revealed decreased levels of p-AMPK, and elevated levels of p-eIF2α, ATF3, ATF6, XBP1 and cyclooxygenase 2 (COX-2) in SHR carotid arteries. Chao et al121 found that ERS promoted oxidative stress associated with hypertension in the rostral ventrolateral medulla (RVLM) by increasing the expression of GRP78 and p-eIF2α. ERS also promoted vasoconstriction. Spitler et al122 reported that ERS played a key role in endothelium derived contracting factors (EDCFs), which mediated responses via activation of the cytosolic phospholipase A2 (cPLA2)/COX pathway in the aorta of the SHRs. Suppressed ERS normalized EDCFs-mediated contractions due to reduced cPLA2 phosphorylation, COX-1 expression, and oxidative stress. Kassan et al123 observed that Ang II led mice to develop an increase in blood pressure, cardiac hypertrophy and fibrosis. ERS markers such as ATF4 mRNA, ATF6 mRNA, CHOP mRNA, GRP78, p-eIF2α and caspase 3 increased in the myocardium, dysfunctional aorta and mesenteric resistance arteries (MRA). Hypertension-induced ERS in the aorta and MRA reduced endothelial NO synthase phosphorylation and endothelium-dependent relaxation (EDR) in the aorta and MRA124. Guo et al125 reported that in spontaneously hypertensive rats, MI/R increased the level of ERS via the PERK-eIF2α-ATF4 pathway.
Endoplasmic reticulum stress is involved in the development of cardiac hypertrophy
Cardiac hypertrophy is an adaptive response that is triggered by a series of physiological and pathological conditions. Evidences have shown that several factors, including calcium dysregulation which is characterized by Ca2+ overload, abnormal protein synthesis and apoptosis, are involved in the development of cardiac hypertrophy126,127. However, the potential role of ERS in pathophysiological hearts remains unclear. Park et al128 demonstrated that pressure overload caused by transverse aortic constriction (TAC) induced prolonged ERS, which was performed in the upregulation of ERS chaperones GRP78, p-PERK, p-elF2α, CHOP, caspase 12 and p-JNK, and then contributed to cardiac myocyte apoptosis in hypertrophy. Apart from these changes in TAC model, ERS chaperones GRP78, caspase 12 and JNK increased abdominal aortic constriction (AAC) in rats129.
The ERS contribution to hypertrophy may result from the high level of cytosolic Ca2+ in cardiac myocytes130. Ca2+ mobilizing agents, such as Ang II and phenylephrine, which induced an increase of cytosolic Ca2+ in cardiac myocytes, led to cardiac hypertrophy. These effects appeared to be mediated by calcineurin (CaN) through dephosphorylating the nuclear factor of activated T cells 3 (NF-AT3); CaN translocates to the nucleus and subsequently interacts with promoters of hypertrophic response genes130. Indeed, experiments have shown that a constitutively active NF-AT3 mutant in the hearts of transgenic mice produced ventricular wall fibrosis and cardiac myocytes enlargement in vivo130, which demonstrated the importance of Ca2+, CaN, NF-AT3 pathway in cardiac hypertrophy. In cardiac myocytes, thapsigargin, tunicamycin and Ang II induced rat cardiac hypertrophy by increasing the expression of atrial natriuretic peptide (ANP) or B-type natriuretic peptide (BNP) mRNA, promoting total protein synthesis and enlarging the cell surface area; they have also been shown to induce ERS through increasing ER chaperone expression130. Researchers found that cyclosporine A (CsA) inhibited the development of cardiac hypertrophy. CaN and the transcription factors from the NF-AT family are potential targets for new anti-hypertrophic agents131. Further studies are required to determine the effect of ERS on cardiac hypertrophy.
Endoplasmic reticulum stress and heart failure
Various stimuli, such as hypoxia132,133, Ang II stimulation134, oxidative stress135 and inflammatory factors136, have been proposed to trigger ERS during heart failure (HF). However, the role of ERS in heart failure is still in dispute.
Some researchers reported that ERS promoted heart failure; others hold a contrary view. In heart failure, the crosstalk between the endoplasmic sarcoplasmic reticulum (ER/SR) and juxtaposed mitochondria is altered, leading to malfunction of cardiomyocytes and decline of cardiac function. Ca2+ may be a key regulator between ERS and heart failure. Chaanine et al137 reported that in a rat model of pressure overload-induced heart failure, calcium shifted from the ER to mitochondrial compartments, leading to a decrease in ER calcium content, mitochondrial damage, cells apoptosis and LV interstitial fibrosis, hence contributing to both systolic and diastolic myocardial dysfunction. In systolic heart failure, SERCA2α down-regulation, along with Bcl-2/adenovirus E1B19 kd-interacting protein 3 (BNIP3) upregulation, exacerbate myocardial diastolic and systolic dysfunction. Dysregulation of intracellular Ca2+ cycling is crucial to the pathogenesis of heart failure and central to the decrease of myocyte contractile function. George et al138 also provided some evidence of the importance of Ca2+ in heart failure. When heart failure was induced by daily coronary embolization in chronically instrumented dogs, SERCA, ryanodine receptor (RyR2), Na+-Ca2+ exchanger (NCX1), and Ca2+ storage protein calreticulin (CRT) were disordered. However, when those calcium-handling proteins were restored, heart failure was alleviated. ERS and ERS-induced apoptosis were also observed in a model of acute myocardial infarction139 and chronic myocardial ischemia-induced HF140. Okada et al141 demonstrated that the number of apoptosic cells in cultured cardiac myocytes was significantly increased in mouse hearts 4 weeks after TAC, and CHOP was induced concurrently. ERS may have promoted heart failure via the NF-κB pathway142. NF-κB p65 activity is a nodal point in the transformation of the ERS response from adaptation to cell death in failing myocytes.
Pro-survival and pro-apoptotic are two different effects of ERS on cells. The UPR may rescue patients from heart failure by promoting unfolded or misfolded proteins to fold properly and then decreasing the proteins stored in the cells of heart failure patients. Ling et al143 demonstrated that when the UPR was activated in patients with heart failure, ERS markers XBP-1s and GRP78 mRNA increased. UPR increased the expression of BNP144,145. BNP is mainly secreted in response to stress in the ventricular wall of ventricular hypertrophy. It is detectable at high concentrations in certain circumstances, which often occur in cardiac ischemia and severe heart failure. XBP-1 regulated BNP expression in cultured cardiomyocytes. Thus, UPR increased the expression of BNP to regulate the plasma volume of the heart and kidney, finally rescuing patients from heart failure145. The effects of ERS on diseases are summarized in Table 2 and Figure 2.
Table 2. Endoplasmic reticulum stress is involved in the development of cardiovascular diseases.
| Diseases | Experiment models | Levels | Disease inducer | ERS Lesion | Disease⇄ERS | References |
|---|---|---|---|---|---|---|
| Atherosclerosis | SMCs | IRE-1, JNK, Ca2+ | 7-ketocholesterol | Atherosclerosis→ERS | 79 | |
| Atherosclerosis | SMCs | GRP78/Bip, CHOP, Ca2+ | Atherosclerosis→ERS | 80 | ||
| Atherosclerosis | J774A1 murine macrophages | XBP1 | Tunicamycin | Atherosclerosis→ERS | 81 | |
| Atherosclerosis | RAW264.7 cells | GRP78 | Tunicamycin, thapsigagin | ERS⇄atherosclerosis | 84 | |
| Atherosclerosis | Macrophages | Ca2+ | Asbestos | ERS⇄atherosclerosis | 85 | |
| Atherosclerosis | Macrophages | CHOP | Oxysterols | ERS⇄atherosclerosis | 86 | |
| Atherosclerosis | Macrophages | p-PERK,p-eIF2α,XBP-1,CHOP | FABP4 | ERS⇄atherosclerosis | 88 | |
| Atherosclerosis | Macrophages | Bip, CHOP, XBP1 | ERS⇄atherosclerosis | 89 | ||
| Atherosclerosis | HUVEC, MAEC | p-PERK, p-eIF2α, XBP-1, p-JNK, GRP78, ATF6 | Atherosclerosis→ERS | 91 | ||
| Atherosclerosis | Endothelial cells | PERK, eIF2α, ATF4, IRE1, XBP1, ATF6 | ERS⇄atherosclerosis | 92 | ||
| Atherosclerosis | HUVECs | GRP78, CHOP | Aldosterone (Aldo) | ERS→atherosclerosis | 93 | |
| Atherosclerosis | SMCs | ATF6 | Hyperhomocysteinemia | ERS→atherosclerosis | 98 | |
| Atherosclerosis | SMCs | GRP78 | Glucosamine | ERS→atherosclerosis | 100 | |
| I/R injury | Wistar rats with I/R injury | GRP78, cleaved caspase 3 | Surgery | Myocardium | I/R injury→ERS | 105 |
| I/R injury | Rats | GRP78, CHOP, caspase 12, SERCA | Surgery and TG | Myocardium | I/R injury→ERS | 106 |
| I/R injury | Rats | CHOP, caspase 12, JNK | Surgery | Myocardium | I/R injury→ERS | 107 |
| I/R injury | Rats | calreticulin, JNK and caspase 12 | Surgery | Cardiomyocyte | I/R injury⇄ERS | 108 |
| I/R injury | Mice | GRP78, GRP94 | Surgery | Myocardium | UPR⊣ I/R injury | 109 |
| Myocardial infarction (MI) | Mice | GRP78, CHOP, Bax, caspase 3 TGF-β1, Smad 2/3 | LCA ligated | Heart | MI⇄ERS | 110 |
| Myocardial infarction (MI) | Rabbits | GRP78, CHOP, cleaved caspase 12 | Surgery | Heart | MI⇄ERS | 111 |
| Chronic hypertension (HTN) | HTN in C57BL/6 mice | p58IPK mRNA, p-PERK, GRP78, CHOP | Angiotensin II | Subfornical organ (SFO) | Chronic hypertension⇄ERS | 114 |
| Hypotension | SHR | p-AMPK, p-eIF2α, ATF3, ATF6, XBP1 and COX-2 | Surgery | Carotid arteries | ERS→hypotension | 120 |
| Hypertension | Wistar-Kyoto rats | GRP78, p-eIF2α | Angiotensin II, Tunicamycin | RVLM | 121 | |
| Hypertension | Wistar-Kyoto rats with SHR | ATF4 mRNA, ATF6 mRNA, CHOP mRNA, p-eIF2α, caspase-3 | Angiotensin II | Aorta, MRA | Hypertension→ERS | 123 |
| Hypertension | SHR | PERK, eIF2α, ATF4 | MI/R | Heart tissues | Hypertension⇄ERS | 125 |
| Hypertrophy | C57BL/6 mice model of TAC | GRP78, p-PERK, p-elF2α, CHOP, caspase-12, p-JNK | TAC | Left ventricular | Hypertrophy⇄ERS | 128 |
| Hypertrophy | Rats model of AAC | GRP78, caspase-12, JNK | Left ventricular | Hypertrophy⇄ERS | 129 | |
| Hypertrophy | Transgenic mice | Ca2+, CaN, NF-AT3 | Ang II, phenylephrine | Cardiac myocytes | Hypertrophy⇄ERS | 130 |
| Hypertrophy | Cardiac myocytes | ANP, BNP, CHOP | Thapsigargin, tunicamycin, Ang II | Cell | Hypertrophy⇄ERS | 130 |
| Heart failure | Rat model HF | SERCA2a, Ca2+ | Pressure overload | Left ventricular | Heart failure⇄ERS | 137 |
| Heart failure | Dogs model of coronary embolization induced CHF | SERCA, RyR2, NCX1, CRT | Coronary embolization | Heart | Heart failure⇄ERS | 138 |
| Heart failure | Mouse subject to TAC | CHOP | TAC | Heart | Heart failure⇄ERS | 141 |
| Heart failure | Patients | XBP1s, GRP78 mRNA | Cardiomyocytes | UPR⊣ heart failure | 143 | |
| Heart failure | Cardiomyocytes | ANP, BNP | Heart | UPR⊣ heart failure | 144,145 |
Figure 2.

The relationship between endoplasmic reticulum stress and cardiovascular diseases.
Drugs regulating endoplasmic reticulum stress may be a new choice for cardiovascular disease treatment
Because ERS plays an essential role in cardiovascular diseases, drugs targeting ERS should be studied. Several drugs are briefly described in the following section.
Endoplasmic reticulum stress may be a pathway for metformin to reduce cardiovascular risks
Metformin, a common antidiabetic drug, improves insulin sensitivity and glucose homeostasis and has been demonstrated to activate AMPK in tissues from humans and rodents146,147. AMPK controls systemic energy balance and metabolism and has been found to improve endothelial function and reduce cardiovascular risk in diabetic patients148. Its activation suppresses ERS in endothelial cells149, which is associated with several diseases, including atherosclerosis and obesity. Peroxisome proliferator-activated receptor δ (PPARδ) is ubiquitously expressed, for example, in adipose and endothelial cells. PPARδ protects against atherosclerosis and endothelial dysfunction, in addition to depleting lipid accumulation and reducing obesity150. PPARβ/δ suppress ERS in the heart151. Notably, AMPK is shown to form a transcriptional complex with PPAR and act cooperatively in vivo. Drugs targeting the AMPK/PPAR complex may be a new option for treating obesity-associated cardiovascular diseases. Cheang et al147 reported that metformin restored endothelial function through inhibiting ERS and oxidative stress and increasing NO bioavailability in activation of the AMPK/PPARδ pathway in obese diabetic mice. Thus, metformin may be a novel drug to treat obesity- and diabetes-induced atherosclerosis and other cardiovascular diseases.
Endoplasmic reticulum stress is a pathway for nuclear receptor agonists to ease injury of myocardial ischemia/reperfusion
The vitamin D receptor (VDR), also known as NR1I1 (nuclear receptor subfamily 1, group I, member 1), and liver-X-receptors (LXRs) are members of the nuclear receptor superfamily. VDR has also been reported to regulate a variety of metabolic pathways, such as those involved in kidney diseases, immune responses and cancers. Moreover, recent evidence has demonstrated that VDR has a protective role in cardiac hypertrophy152, heart failure after cardiac remodeling153 and MI/R injury154. LXRs have two receptor subtypes–LXRα (NR1H3) and LXRβ (NR1H2)—that exhibit different expression patterns and perform different functions155. Recent evidence suggested that LXRα/LXRβ are both expressed in the cardiovascular system. However, the two subtypes function differently in the pathogenesis of vascular diseases, such as MI/R injury156, hypertrophy and fibrosis157. Nuclear receptors are major targets for drug discovery, and their modulators regulate ERS to control cardiovascular diseases. Calcitriol and paricalcitol (PC) are natural or synthetic agonists of VDR that are used to treat kidney diseases clinically. Recently, Yao et al154 reported that calcitriol and PC activate VDR to protect against MI/R injury by reducing ERS-induced apoptosis via inhibition of caspase 12 and CHOP expression in mice. 22(R)-hydroxycholesterol [22(R)-HC] and GW3965 are two endogenous or synthetic agonists of LXRs. He et al156 demonstrated that 22(R)-HC and GW3965 activated LXRα, but not the LXRβ subtype, to reduce myocardial infarction and improve contractile function after MI/R by attenuating ERS and the ERS-mediated apoptosis pathway by inhibiting caspase 12 and CHOP expression. ERS may be a novel pathway of nuclear receptor agonists, such as calcitriol, paricalcitol, 22(R)-HC and GW3965, to treat MI/R injury.
Salubrinal blocks endoplasmic reticulum stress to relieve hypertension and myocardial infarction
Salubrinal acts as a specific inhibitor of eIF2α phosphatase and is used in experiments primarily to study stress responses in eukaryotic cells associated with the action of eIF2α. Salubrinal indirectly inhibited eIF2α to suppress the activation of the stress response triggered by oxidative stress or the buildup of unfolded proteins in the ER. Salubrinal currently has a new therapeutic value. Chao et al121 reported that in the RVLM of spontaneously hypertensive rats, salubrinal-mediated ERS stability relieved hypertension. Furthermore, induction of oxidative stress by Ang II induced ERS in the RVLM; induction of ERS by tunicamycin in the RVLM induced the pressor response in normotensive Wistar-Kyoto rats. Autophagy, as reflected by the expression of lysosome-associated membrane protein-2 and microtubule-associated protein 1 light chain 3-II (LC3-II), was significantly increased in the RVLM of spontaneously hypertensive rats and abrogated by salubrinal. Li et al158 reported that salubrinal protected against ERS-induced rat cardiomyocyte apoptosis by suppressing the dephosphorylation of eIF2α in a rat myocardial infarction model. Thus, salubrinal may prove to be a new drug to remedy hypertension and alleviate the damage of myocardial infarction.
Black tea alleviates endoplasmic reticulum stress to protect against hypertension-associated endothelial dysfunction
Hyperhomocysteinemia is a principal complication of hypertension, which has been found to be associated with elevated levels of homocysteine (Hcy) in hypertensive patients. Homocysteine can induce ERS in endothelial cells and result in endothelial dysfunction. Cheang et al observed impaired vasodilatation, upregulated levels of plasma Hcy, reduced Hcy metabolic enzymes and increased ERS markers in aortae from rats with chronic Ang II-induced hypertension. Black tea (BT) extracts and the major theaflavin-3, 3′-digallate (TF3) reversed Hcy-induced endothelial dysfunction and Hcy-elevated ERS in rat aortae. In addition, BT extract treatment normalized plasma Hcy levels; reversed attenuated endothelium-dependent relaxations in aortae, carotid and renal arteries as well as flow-mediated dilatations (FMD) in third-order mesenteric resistance arteries, inhibited ERS, and upregulated the Hcy metabolic enzymes cystathionine-β-synthase (CBS) and cystathionine gamma-lyase (CSE) in the Ang II-infused rats. Moreover, BT mitigated high blood pressure118. BT may not just be a drink anymore, it may become a drug to treat hypertension and hypertension-associated endothelial dysfunction.
Berberine improves hypertension-associated endothelial function by inhibiting endoplasmic reticulum stress
Berberine ([C20H18NO4]+), an isoquinoline alkaloid isolated from many medicinal herbs, such as the Chinese herbs Huanglian, berberis aquifolium, and berberis vulgaris, has long been employed in traditional Chinese medicine to treat various infectious disorders. Recently, its cardiovascular benefits have been investigated159. Berberine has been reported to ameliorate proinflammatory cytokine-induced ERS in human intestinal epithelial cells160, reduce hypoxia/reoxygenation-induced human renal proximal tubular cell injury161, and inhibit the human immunodeficiency virus protease inhibitor-induced inflammatory response in murine macrophages162. Berberine has also been suggested to have an anti-hypertensive property163. Liu et al also reported that berberine improved endothelial function by inhibiting ERS in the carotid arteries of spontaneously hypertensive rats120. Treating hypertension-associated endothelial dysfunction may be another function of berberine.
Apelin-13 may be a new peptide to treat ischemia-reperfusion injury through inhibition of endoplasmic reticulum stress
Apelin-13 is an endogenous ligand of angiotensin receptor-like 1 (APJ), a new G protein-coupled receptor with the typical seven transmembrane domains. It was discovered in 1993 via homology cloning164,165. Apelin-13/APJ system has been identified as a potential therapeutic target for hypertension166 and atherosclerosis167. Our laboratory has demonstrated that apelin-13 induced proliferation of vascular smooth muscle cells66,168 and rat bone marrow-derived mesenchymal stem cells169 through the Jagged-1/Notch3, cyclin D1 and AKT/GSK3β/cyclin D1 pathways. However, other researchers demonstrated that apelin-13 is no longer merely a potential therapeutic target; it may serve as a new peptide to treat I/R injury. Tao et al107 reported that apelin-13 protected against I/R injury by inhibiting ERS-dependent apoptosis activation via the PI3K/Akt, AMPK and ERK pathways.
4-Phenylbutyric acid (PBA) reduces endoplasmic reticulum stress to rescue cardiac hypertrophy and fibrosis
4-Phenylbutyric acid (PBA) was approved by the US Food and Drug Administration (FDA) for use in humans170. PBA is a low molecular weight fatty acid and a non-toxic pharmacological compound that has been found to have a chaperone-like activity. Its physiochemical properties make it possible to stabilize peptide structures and improve luminal folding capacity and traffic of aberrant proteins171. Park et al128 reported that oral administration of PBA to TAC mice reduced the expression of GRP78, p-PERK and p-elF2α. PBA also severely downregulated hypertrophy and fibrosis related genes (TGF-β1, phospho-smad2, and pro-collagen isoforms). Thus, PBA may be a novel therapeutic agent for cardiac hypertrophy and fibrosis through inhibition of ERS.
Cyclosporine A normalizes Ca2+ to regulate hypertrophy through endoplasmic reticulum stress
Cyclosporine A is a CaN inhibitor. High levels of CaN induced ERS in hypertrophy. Fu et al172 found that cyclosporine A decreased ERS by inhibiting CaN activity and reducing protein synthesis in Ang II-stimulated cardiomyocytes. It also downregulated DNA synthesis in Ang II-reated fibroblasts in a dose-dependent manner. At the same time, the mRNA level of hypertrophy genes also decreased. Thus, cyclosporine A may be a new therapeutic agent for cardiac hypertrophy through inhibiting ERS.
Metoprolol and propranolol mediate heart failure through endoplasmic reticulum stress
Metoprolol and propranolol are two major β adrenergic receptor (β-AR) blockers. Several clinical studies have demonstrated that metoprolol and propranolol belong to one of the few classes of drugs that improve cardiac function and reduce mortality in patients with heart failure. However, the mechanisms underlying the therapeutic effects of β-AR blockers on failing hearts is poorly understood. Zhou et al42 found that metoprolol and propranolol suppressed ERS and ERS-mediated apoptosis by decreasing the expression of ERS chaperones GRP78, XBP-1, and calmodulin kinase II (CaMKII) and apoptosis maker CHOP in hypertrophic and failing hearts of rats. Metoprolol also normalized the level of calcium and decreased the expression of p-eIF2α in dogs with heart failure138. So, the mechanism underlying the therapeutic effects of β-AR blockers on failing hearts may be ERS.
Telmisartan and olmesartan have been found to have a novel value related to endoplasmic reticulum stress in hypertrophy and heart failure
Selective Ang II type 1 (AT1) receptor antagonists such as telmisartan and olmesartan are commonly used to treat hypertension clinically. Their new effects were identified recently. For example, Guan et al53 discovered that telmisartan reduced ERS and thereby attenuated both apoptosis and cardiac hypertrophy. Telmisartan significantly reduced LVH and interstitial fibrosis, thus improving left ventricular function compared with AAC alone. ERS markers in the myocardial, such as GRP78, CHOP, caspase 12 and p-JNK, significantly increased in rats with AAC; telmisartan significantly dampened these changes. Sukumaran et al173 reported that olmesartan decreased ERS in rats with heart failure and downregulated the expression of GRP78, caspase 12 and p-JNK in the myocardium. These novel findings improved the therapeutic value of these two drugs.
Heart failure may be another indication of atorvastatin via inhibiting endoplasmic reticulum stress
Atorvastatin, a reductase inhibitor, has the capacity to effectively decrease blood lipids and may provide some advantages compared with other statins174. Sola et al175 found that administration of atorvastatin improved left ventricular ejection fraction and attenuated adverse left ventricular remodeling in patients with nonischemic heart failure. Atorvastatin also dampened the inflammatory process in patients with heart failure176. In a rat model of post-myocardial infarction-induced heart failure, expression of GRP78, caspase 12, and CHOP in myocardial cells increased. Apoptosis was verified in myocardial cells. Atorvastatin down regulated GRP78, caspase 12 and CHOP expression and decreased apoptosis of myocardial cells, suggesting that atorvastatin protected against heart failure through ERS177.
Qi deficiency and blood stasis are the main causes of heart failure according to the traditional Chinese medicine system. Li et al178,179 reported that the Yiqi Huoxue recipe is useful in treating heart failure in laboratory and clinical studies. However, the mechanism of action is not clear. Lou et al180 reported that the Yiqi Huoxue recipe may have improved injured heart function through inhibition of the ERS response pathway by inhibiting the expression of GRP78 and caspase 12 in a rat MI model. ERS may be a pathway targeted by the Yiqi Huoxue recipe to protect the heart from failure. Drugs regulating ERS to treat cardiovascular diseases are summarized in Table 3.
Table 3. Drugs that regulate endoplasmic reticulum stress to treat cardiovascular diseases.
| Name | Molecular formula | Chemical structure | Targets | Target diseases | Reference |
|---|---|---|---|---|---|
| Metoprolol | C15H25NO3 | 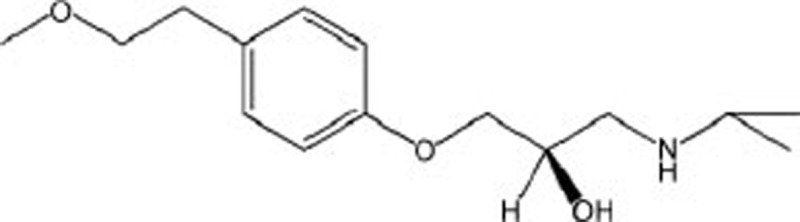 |
GRP78 XBP-1,CaMKII,CHOP,p-eIF2α | Heart failure | 43 |
| Propranolol | C16H21NO2 | 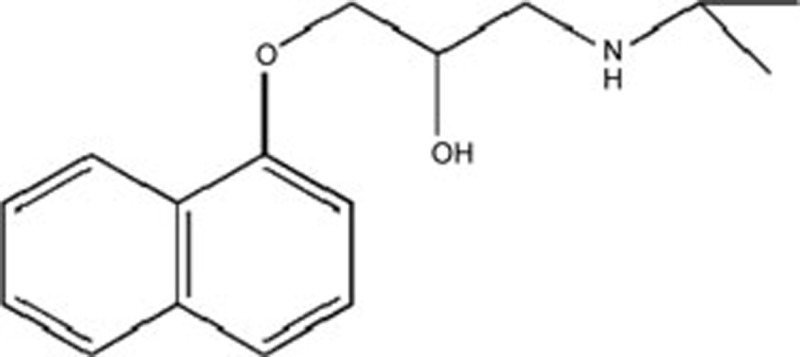 |
GRP78 XBP-1,CaMKII,CHOP | Heart failure | 43 |
| Telmisartan | C33H30N4O2 | 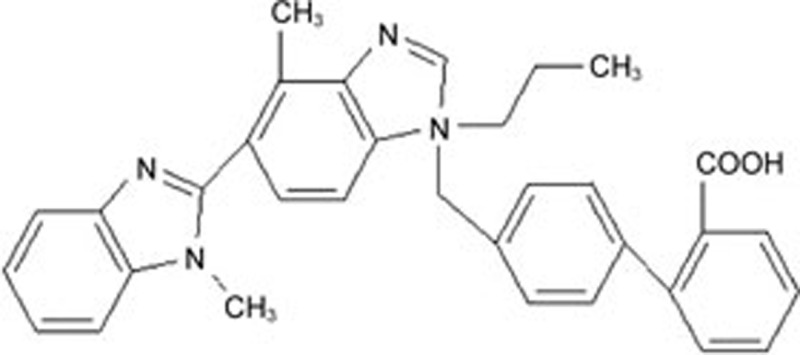 |
GRP78 CHOP Caspase-12 p-JNK | Hypertrophy | 53 |
| Apelin-13 | C69H111N23O16S | 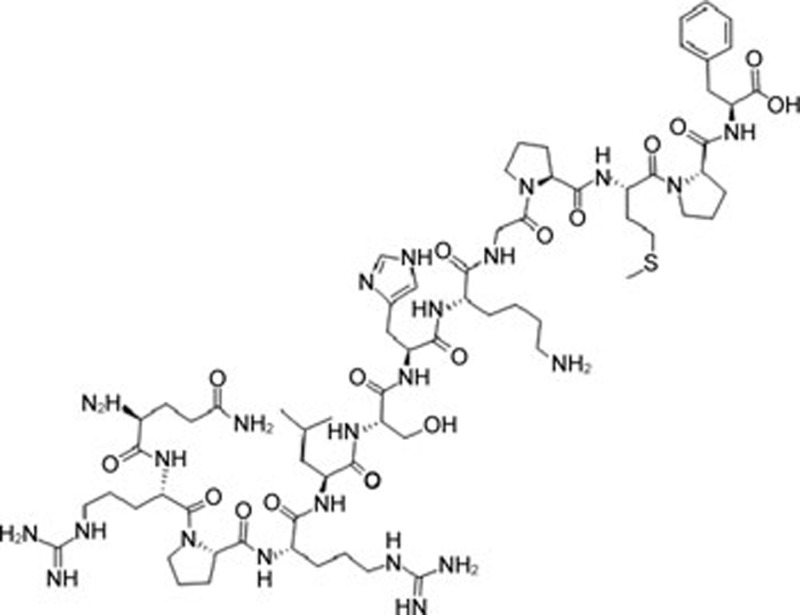 |
CHOP Caspase-12 JNK | Ischemia-reperfusion injury | 107 |
| Theaflavin-3, 3′-digallate (TF3) | C43H32O20 |  |
ATF3 ATF6 p-eIF2α | Hypertension | 118 |
| Berberine | C20H18NO4 | 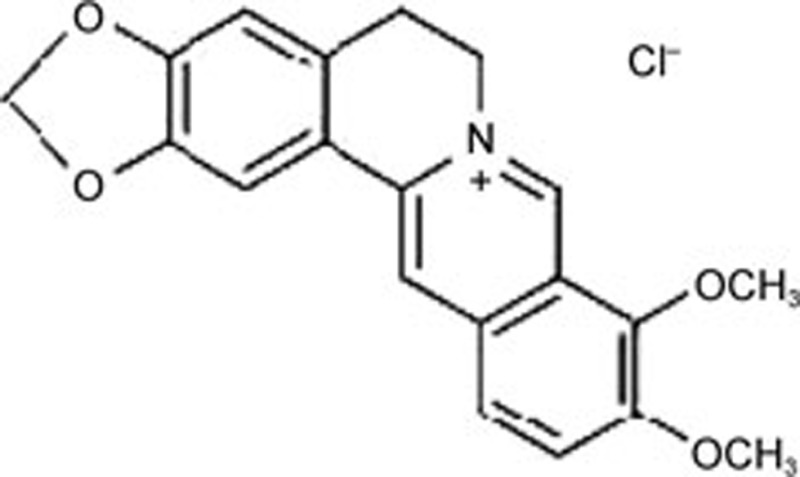 |
ATF3 ATF6 p-eIF2α XBP-1 | Hypertension | 120 |
| 4-Phenylbutyric acid (PBA) | C10H12O2 | 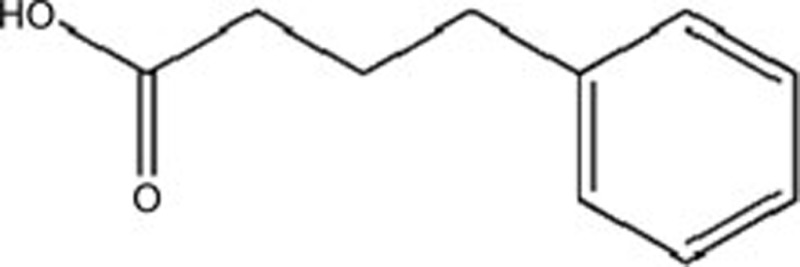 |
GRP78, p-PERK, p-elF2α | Hypertrophy | 128 |
| Metformin | C4H11N5 | 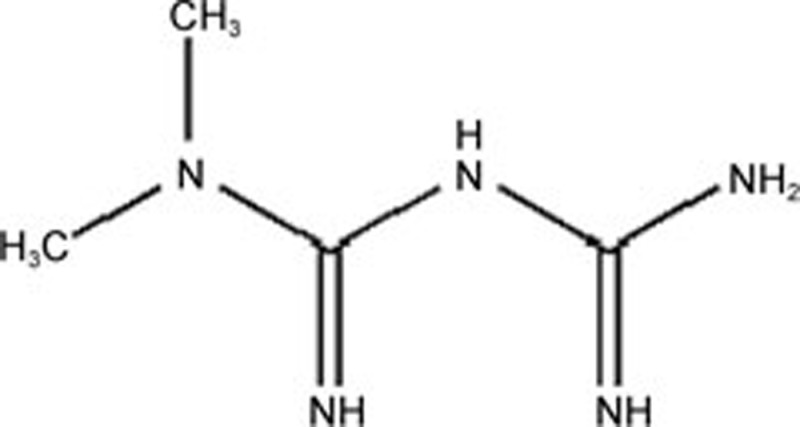 |
ATF6 ATF3 eIF2α | Atherosclerosis | 147 |
| Calcitriol | C27H44O3 | 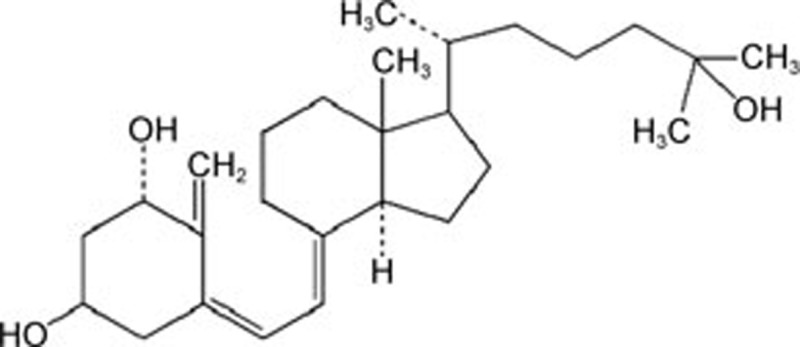 |
Caspase-12, CHOP | Ischemia-reperfusion injury | 154 |
| Paricalcitol (PC) | C27H44O3 | 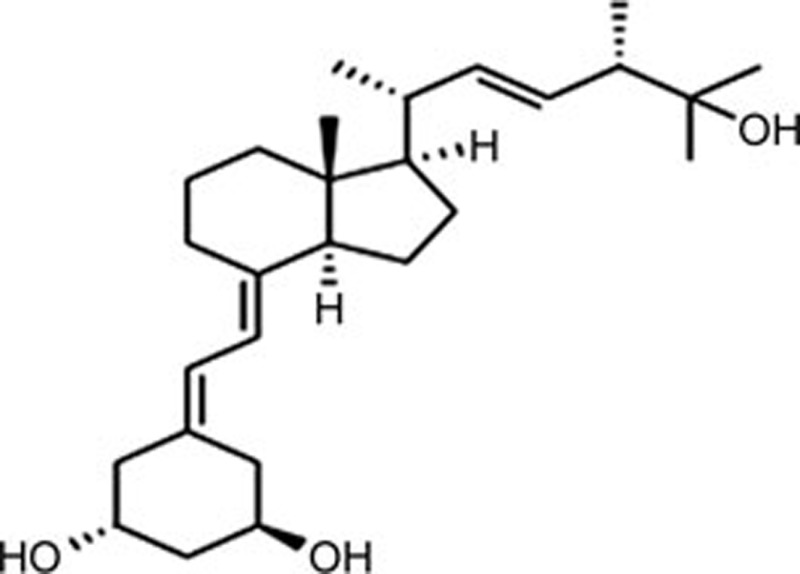 |
Caspase-12 CHOP | Ischemia-reperfusion injury | 154 |
| 22(R)-hydroxy cholesterol [22(R)-HC] | C27H46O2 | 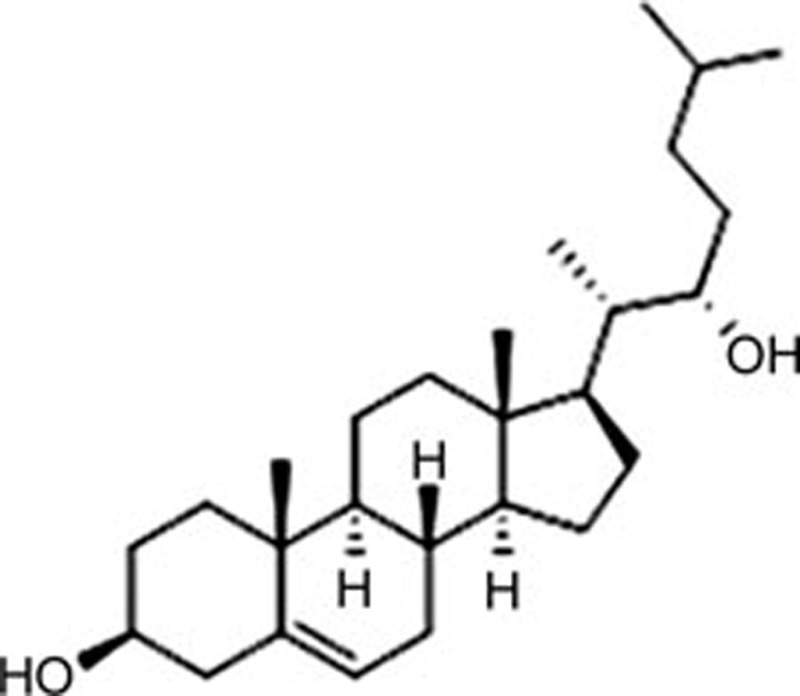 |
Caspase-12, CHOP | Ischemia-reperfusion injury | 156 |
| GW3965 | C33H31CIF3NO3 | 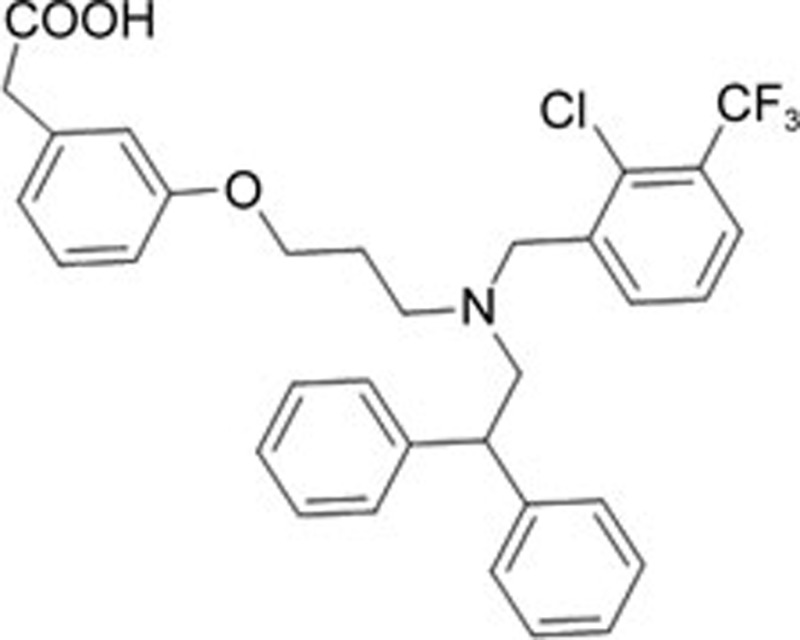 |
Caspase-12, CHOP | Ischemia-reperfusion injury | 156 |
| Salubrinal | C21H17CI3N4OS | 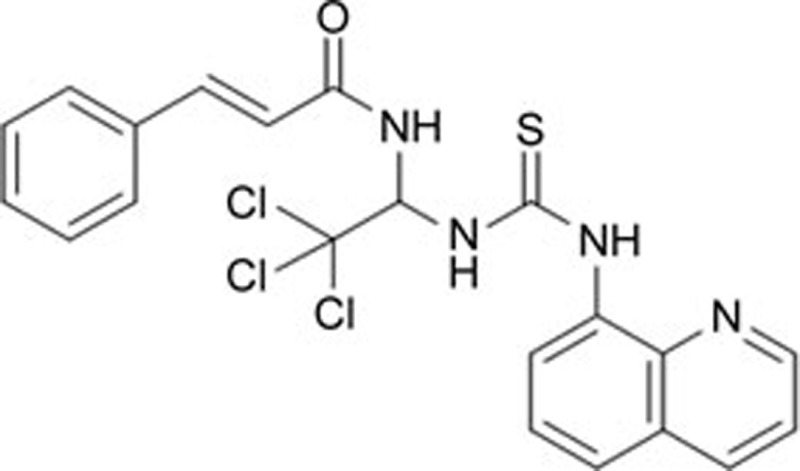 |
eIF2α | Hypertension | 158 |
| Cyclosporine A | C62H111N11O12 | 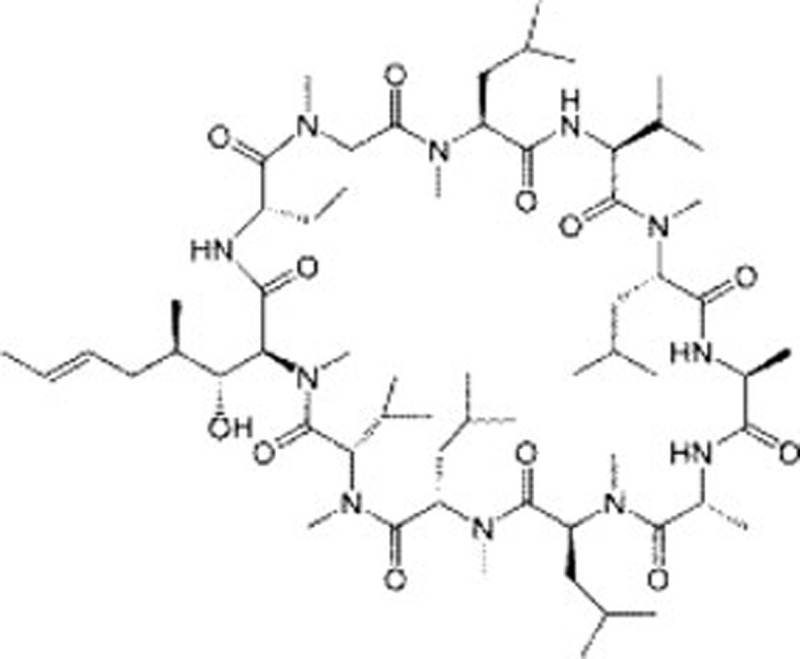 |
CaN Ca2+ | Hypertrophy | 172 |
| Olmesartan | C24H26N6O3 | 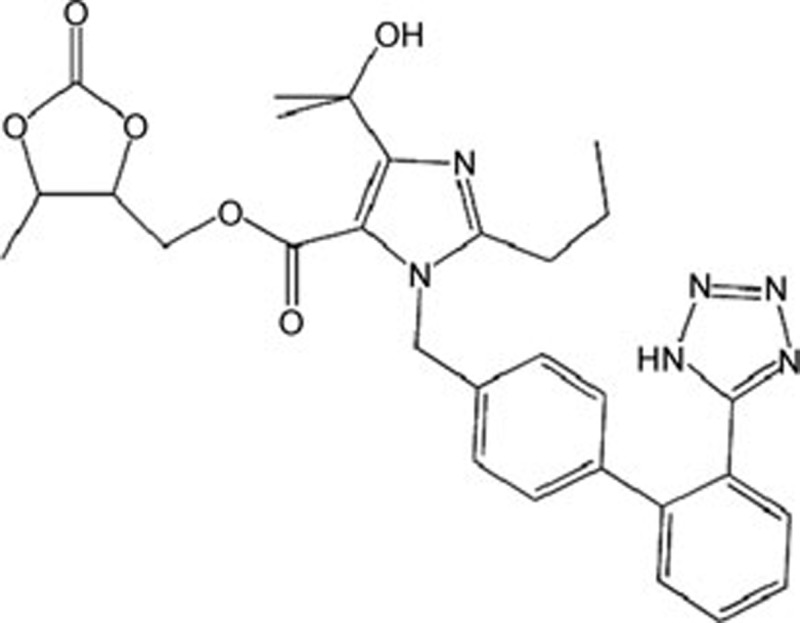 |
GRP78, Caspase-12 p-JNK | Heart failure | 173 |
| Atorvastatin | C33H35FN2O5 | 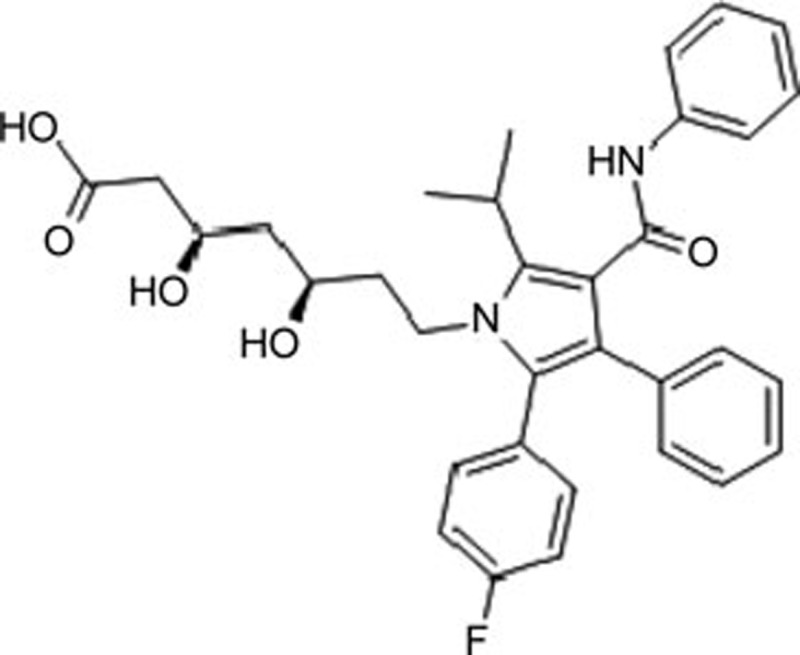 |
GRP78, Caspase-12 CHOP | Heart failure | 177 |
| Yiqi Huoxue Recipe | NO | GRP78, Caspase-12 | Heart failure | 178,179 |
Outlook
ERS-related accumulation of unfolded/misfolded proteins and the unfolded protein response are important signals for the progression of many diseases. Cardiovascular disorders lead to disturbance in the ER. Cardiovascular disorders related to the mechanisms of ERS have been identified, and attention has been paid to them by basic and clinical scientists in recent years. Although many detailed studies have highlighted the mechanisms of ERS in cardiovascular diseases, they have yielded conflicting results and additional studies should be conducted to elucidate these mechanisms. Other factors associated with ERS have also been searched, such as the apelin/APJ system181,182, in which both apelin-13 and ERS can regulate cardiomyocyte proliferation, migration and adhesion183,184. ERS induced ATF4 expression, regulating apelin-13 transcription via a p38 AMPK-dependent pathway. In addition, nuclear receptors that can be activated by ERS are also worthy of attention. Selective nuclear receptor modulators have been reported to regulate ERS to control cardiovascular diseases. Thus, if the relationship between ERS and nuclear receptors in cardiovascular diseases can be understood clearly, it would support more efficient drug discovery.
In this article, we have summarized the relationship between ERS and cardiovascular diseases and discussed drugs that regulate ERS to control these diseases. However, there are several limitations of previous studies. First, although many studies have been conducted on the relationship between ERS and cardiovascular diseases, it is still a controversial topic. Second, because most of the studies were performed in animal models, they do not fully represent human patients. Finally, despite the discovery of drugs that regulate ERS to control cardiovascular diseases, additional studies are needed to determine whether ERS could be an accurate pathway for treating cardiovascular diseases. Therefore, an improved understanding of ERS proteins and pathways involved in cardiovascular diseases is necessary and may lead to identification of potential targets for new therapies.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No 81270420, 81470434, and 81503074), the Hunan Provincial Natural Science Foundation of China (No 14JJ3102), the Construct Program of the Key Discipline in Human Province, the Hunan Province Cooperative Innovation Center for Molecular Target New Drugs Study (Hunan Provincial Education Department, No 2014-405), and the China Postdoctoral Science Foundation (No 2014M560647 and 2015T80875). We especially thank Bing-bing WANG (Assistant Professor, Perinatal Biology Laboratory, Division of Maternal-Fetal Medicine, Rutgers University–Robert Wood Johnson Medical School, USA) for helping with English language correction.
References
- Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Ann Rev Genet 2001; 35: 149–91. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4: 181–91. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Ann Rev Pathol 2015; 10: 173–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008; 7: 1013–30. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 2006; 7: 880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 2005; 115: 2656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan BJ, Krokowski D, Majumder M, Schmotzer CL, Kimball SR, Merrick WC, et al. Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2alpha. J Biol Chem 2014; 289: 12593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SM, Vanslyke JK, Musil LS. Regulation of ubiquitin-proteasome system mediated degradation by cytosolic stress. Mol Biol Cell 2007; 18: 4279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo N. ER and aging-Protein folding and the ER stress response. Ageing Res Rev 2009; 8: 150–9. [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol 2005; 7: 766–72. [DOI] [PubMed] [Google Scholar]
- Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, et al. A thrombospondin-dependent pathway for a protective ER stress response. Cell 2012; 149: 1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng T, Peng L, Chao C, Fu B, Wang G, Wang Y, et al. IRE1alpha-TRAF2-ASK1 complex-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to CXC195-induced apoptosis in human bladder carcinoma T24 cells. Biochem Biophys Res Commun 2015; 460: 530–6. [DOI] [PubMed] [Google Scholar]
- Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell 2011; 22: 3520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 2011; 22: 4390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397: 271–4. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 1998; 18: 7499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334: 1081–6. [DOI] [PubMed] [Google Scholar]
- Thuerauf DJ, Marcinko M, Belmont PJ, Glembotski CC. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J Biol Chem 2007; 282: 22865–78. [DOI] [PubMed] [Google Scholar]
- Hai TW, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Develop 1989; 3: 2083–90. [DOI] [PubMed] [Google Scholar]
- Parker R, Phan T, Baumeister P, Roy B, Cheriyath V, Roy AL, et al. Identification of TFII-I as the endoplasmic reticulum stress response element binding factor ERSF: its autoregulation by stress and interaction with ATF6. Mol Cell Biol 2001; 21: 3220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Folding of VSV G protein: sequential interaction with BiP and calnexin. Science 1994; 266: 456–8. [DOI] [PubMed] [Google Scholar]
- Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993; 73: 1197–206. [DOI] [PubMed] [Google Scholar]
- Mori K, Ma W, Gething MJ, Sambrook J. A transmembrane protein with a CDC2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993; 74: 743–56. [DOI] [PubMed] [Google Scholar]
- Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science 2003; 302: 1533–7. [DOI] [PubMed] [Google Scholar]
- Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 1996; 15: 3028–39. [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002; 415: 92–6. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107: 881–91. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 1998; 273: 33741–9. [DOI] [PubMed] [Google Scholar]
- Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem 2009; 284: 29735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Ann Rev Cell Develop Biol 2002; 18: 575–99. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519–29. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2: 326–32. [DOI] [PubMed] [Google Scholar]
- Raven JF, Baltzis D, Wang S, Mounir Z, Papadakis AI, Gao HQ, et al. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem 2008; 283: 3097–108. [DOI] [PubMed] [Google Scholar]
- Naidoo N. ER and aging–protein folding and the ER stress response. Ageing Res Rev 2009; 8: 150–9. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol cell 2003; 11: 619–33. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 2011; 153: 1011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K, Okada T, Yoshida H, Yanagi H, Yura T, Negishi M, et al. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J 2001; 355: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000; 6: 1355–64. [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988; 332: 462–4. [DOI] [PubMed] [Google Scholar]
- Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res 2007; 67: 10631–4. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13: 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L, Zhou C, Duan Q, Lv J, Fu X, Xia Y, et al. Beta-AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. PloS One 2011; 6: e27294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, et al. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem 2010; 285: 38720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Meng J, Cao W, Li Q, Qiu Y, Sun B, et al. Induction of apoptosis through ER stress and TP53 in MCF-7 cells by the nanoparticle [Gd@C82(OH)22]n: A systems biology study. Methods 2014; 67: 394–406. [DOI] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13: 89–102. [DOI] [PubMed] [Google Scholar]
- Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem 2012; 151: 217–9. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 2001; 276: 13935–40. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase 12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403: 98–103. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275: 90–4. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang Z, Nowicki MJ. Caspase 12 mediates carbon tetrachloride-induced hepatocyte apoptosis in mice. World J Gastroenterol 2014; 20: 18189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Li PP, Jin FS, Yao C, Zhang GH, Zang T, et al. Ursolic acid induces ER stress response to activate ASK1-JNK signaling and induce apoptosis in human bladder cancer T24 cells. Cell Signal 2013; 25: 206–13. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008; 29: 42–61. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang X, Li F, Chen L, Li L, Qin X, et al. 14-3-3 mediates apelin-13-induced enhancement of adhesion of monocytes to human umbilical vein endothelial cells. Acta Biochim Biophys Sin 2010; 42: 403–9. [DOI] [PubMed] [Google Scholar]
- Meng FW, Biteau B. A Sox transcription factor is a critical regulator of adult stem cell proliferation in the Drosophila intestine. Cell Reports 2015; 13: 906–14. [DOI] [PubMed] [Google Scholar]
- Schlosser A, Pilecki B, Hemstra LE, Kejling K, Kristmannsdottir GB, Wulf-Johansson H, et al. MFAP4 promotes vascular smooth muscle migration, proliferation and accelerates neointima formation. Arterioscler Thromb Vasc Biol 2016; 36: 122–33. [DOI] [PubMed] [Google Scholar]
- Li L, Li F, Li F, Mao X, Yang L, Huang H, et al. NOX4-derived reactive oxygen species drive apelin-13-induced vascular smooth muscle cell proliferation via the ERK pathway. Int J Pept Res Ther 2011; 17: 307–15. [Google Scholar]
- Yang L, Su T, Lv D, Xie F, Liu W, Cao J, et al. ERK1/2 mediates lung adenocarcinoma cell proliferation and autophagy induced by apelin-13. Acta Biochim Biophys Sin 2014; 46: 100–11. [DOI] [PubMed] [Google Scholar]
- Xie F, Li L, Chen L. Autophagy, a new target for disease treatment. Sci China Life Sci 2013; 56: 856–60. [DOI] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Zhao Y, Li ZY, Lan XH, et al. Vascular smooth muscle cell apoptosis is an early trigger for hypothyroid atherosclerosis. Cardiovasc Res 2014; 102: 448–59. [DOI] [PubMed] [Google Scholar]
- Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol 2011; 31: 2792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord MS, Chuang CY, Melrose J, Davies MJ, Iozzo RV, Whitelock JM. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol: J Int Society Matrix Biol 2014; 35: 112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek AE, Oh J, Darwech I, Moynihan CE, Bruchas RR, Bernal-Mizrachi C. 25(OH) vitamin D suppresses macrophage adhesion and migration by downregulation of ER stress and scavenger receptor A1 in type 2 diabetes. J Steroid Biochem Mol Biol 2014; 144 Pt A: 172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Takizawa H, Shimazawa M, Hashimoto Y, Sugitani S, Tsuruma K, et al. Mild endoplasmic reticulum stress promotes retinal neovascularization via induction of BiP/GRP78. PloS One 2013; 8: e60517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Su T, Li F, Li L, Qin X, Pan W, et al. PI3K/Akt signaling transduction pathway is involved in rat vascular smooth muscle cell proliferation induced by apelin-13. Acta Biochim Biophys Sin 2010; 42: 396–402. [DOI] [PubMed] [Google Scholar]
- Li L, Li L, He L, Zhang Z, Xie F, Guo Y, et al. Effects of apelin-13 on rat bone marrow-derived mesenchymal stem cell proliferation through the AKT/GSK3β/cyclin D1 pathway. Int J Pept Res Ther 2014; 20: 421–5. [Google Scholar]
- Li L, Li L, Xie F, Zhang Z, Guo Y, Tang G, et al. Jagged-1/Notch3 signaling transduction pathway is involved in apelin-13-induced vascular smooth muscle cells proliferation. Acta Biochim Biophys Sin 2013; 45: 875–81. [DOI] [PubMed] [Google Scholar]
- Yang YY, Shang J, Liu HG. Role of endoplasmic reticular stress in aortic endothelial apoptosis induced by intermittent/persistent hypoxia. Chin Med J 2013; 126: 4517–23. [PubMed] [Google Scholar]
- Gorman AM, Healy SJ, Jager R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther 2012; 134: 306–16. [DOI] [PubMed] [Google Scholar]
- Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arteriosclerosis Thromb Vasc Biol 2011; 31: 2792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi N, Chen SY, Ma A, Chen PS, Yao B, Liang TM, et al. Tunicamycin inhibits PDGF-BB-induced proliferation and migration of vascular smooth muscle cells through induction of HO-1. Anat Rec (Hoboken) 2012; 295: 1462–72. [DOI] [PubMed] [Google Scholar]
- Yang L, Hu N, Jiang S, Zou Y, Yang J, Xiong L, et al. Heavy metal scavenger metallothionein attenuates ER stress-induced myocardial contractile anomalies: Role of autophagy. Toxicol Lett 2014; 225: 333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. Study of the mechanism of heavy metal scavenger metallothionein in myocardial protection [Dissertation]. Xi-an: Fourth Military Medical University; 2014.
- Dai MX, Zheng XH, Yu J, Yin T, Ma MJ, Zhang L, et al. The impact of intermittent and repetitive cold stress exposure on endoplasmic reticulum stress and instability of atherosclerotic plaques. Cell Physiol Biochem 2014; 34: 393–404. [DOI] [PubMed] [Google Scholar]
- Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 2010; 128: 191–227. [DOI] [PubMed] [Google Scholar]
- Bolte C, Ren X, Tomley T, Ustiyan V, Pradhan A, Hoggatt A, et al. Forkhead box F2 regulation of platelet-derived growth factor and myocardin/serum response factor signaling is essential for intestinal development. J Biol Chem 2015; 290: 7563–75. doi: 10.1074/jbc.M114.609487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh A, Subedi K, Kyathanahalli C, Jeyasuria P, Condon JC. Uterine endoplasmic reticulum stress and its unfolded protein response may regulate caspase 3 activation in the pregnant mouse uterus. PLoS One 2013; 8: e75152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickhout JG, Colgan SM, Lhotak S, Austin RC. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome: a balancing act between plaque stability and rupture. Circulation 2007; 116: 1214–6. [DOI] [PubMed] [Google Scholar]
- Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007; 116: 1226–33. [DOI] [PubMed] [Google Scholar]
- Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol 2004; 24: 10703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li X, Qin F, Huang K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J Biol Inorg Chem 2014; 19: 375–88. [DOI] [PubMed] [Google Scholar]
- Solanki S, Dube PR, Tano JY, Birnbaumer L, Vazquez G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am J Physiol Cell Physiol 2014; 307: C521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tano JY, Solanki S, Lee RH, Smedlund K, Birnbaumer L, Vazquez G. Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovasc Res 2014; 101: 138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Shin D, Lim SS, Lee JY, Kang YH. Purple perilla extracts allay ER stress in lipid-laden macrophages. PLoS One 2014; 9: e110581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao ST, Zhao L, Miao C, Tian H, Yang NN, Guo SD, et al. Endoplasmic reticulum stress mediates oxidized low density lipoprotein-induced scavenger receptor A1 upregulation in macrophages. Sheng Li Xue Bao 2014; 66: 612–8. [PubMed] [Google Scholar]
- Ryan AJ, Larson-Casey JL, He C, Murthy S, Carter AB. Asbestos-induced disruption of calcium homeostasis induces endoplasmic reticulum stress in macrophages. J Biol Chem 2014; 289: 33391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya M, Yamamuro D, Ohshiro T, Honda A, Takahashi M, Kumagai M, et al. Absence of Nceh1 augments 25-hydroxycholesterol-induced ER stress and apoptosis in macrophages. J Lipid Res 2014; 55: 2082–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Jiang Y, Dai S, Wang B, Gao F, Guo C, et al. Interleukin 17A exacerbates atherosclerosis by promoting fatty acid-binding protein 4-mediated ER stress in macrophages. Circ Res 2012. doi: 10.1161/CIRCRESAHA.112.272567. [DOI] [PubMed]
- Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 2009; 15: 1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Hertzel AV, Steen KA, Wang Q, Suttles J, Bernlohr DA. Uncoupling lipid metabolism from inflammation through fatty acid binding protein-dependent expression of UCP2. Mol Cell Biol 2015; 35: 1055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas MJ, Kurban W, Shah H, Onstead-Haas L, Mooradian AD. Beta blockers suppress dextrose-induced endoplasmic reticulum stress, oxidative stress, and apoptosis in human coronary artery endothelial cells. Am J Ther 2015 Jan 27. [Epub ahead of print]. [DOI] [PubMed]
- Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha 2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010; 121: 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Tan H, Gu Z, Liu Z, Geng Y, Liu Y, et al. Heat stress induces apoptosis through a Ca2+-mediated mitochondrial apoptotic pathway in human umbilical vein endothelial cells. PLoS One 2014; 9: e111083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JP, Li X, Jin YL, Chen MX. Endoplasmic reticulum stress-mediated aldosterone-induced apoptosis in vascular endothelial cells. J Huazhong Univ Sci Technol Med Sci 2014; 34: 821–4. [DOI] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol 2007; 19: 215–22. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997; 89: 331–40. [DOI] [PubMed] [Google Scholar]
- Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab 2013; 1: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000; 6: 1355–64. [DOI] [PubMed] [Google Scholar]
- Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, et al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J 2004; 23: 950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan SM, Tang D, Werstuck GH, Austin RC. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int J Biochem Cell Biol 2007; 39: 1843–51. [DOI] [PubMed] [Google Scholar]
- Werstuck GH, Khan MI, Femia G, Kim AJ, Tedesco V, Trigatti B, et al. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes 2006; 55: 93–101. [PubMed] [Google Scholar]
- Nahandi MZ, Khoshbaten M, Ramazanzadeh E, Abbaszadeh L, Javadrashid R, Shirazi KM, et al. Effect of non-alcoholic fatty liver disease on carotid artery intima-media thickness as a risk factor for atherosclerosis. Gastroenterol Hepatol Bed Bench 2014; 7: 55–62. [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Zheng Z, Mendez R, Ha SW, Xie Y, Zhang K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol Lett 2012; 211: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Ning B, Shen W, Wan Y. SREBP-1c gene interference decreases lipid accumulation in L02 and HepG2 hepatocytes under endoplasmic reticulum stress. J Third Mil Med Univ 2013; 35: 513–7. [Google Scholar]
- Lowenstein CJ. Myocardial reperfusion injury. N Engl J Med 2007; 357: 2409; author reply 2409–10. [PubMed] [Google Scholar]
- Chien CY, Chien CT, Wang SS. Progressive thermopreconditioning attenuates rat cardiac ischemia/reperfusion injury by mitochondria-mediated antioxidant and antiapoptotic mechanisms. J Thorac Cardiovasc Surg 2014; 148: 705–13. [DOI] [PubMed] [Google Scholar]
- Guo J, Bian Y, Bai R, Li H, Fu M, Xiao C. Globular adiponectin attenuates myocardial ischemia/reperfusion injury by upregulating endoplasmic reticulum Ca2+-ATPase activity and inhibiting endoplasmic reticulum stress. J Cardiovasc Pharmacol 2013; 62: 143–53. [DOI] [PubMed] [Google Scholar]
- Tao J, Zhu W, Li Y, Xin P, Li J, Liu M, et al. Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am J Physiol Heart Circ Physiol 2011; 301: H1471–86. [DOI] [PubMed] [Google Scholar]
- Liu XH, Zhang ZY, Sun S, Wu XD. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress. Shock 2008; 30: 422–7. [DOI] [PubMed] [Google Scholar]
- Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res 2006; 98: 1186–93. [DOI] [PubMed] [Google Scholar]
- Luo T, Kim JK, Chen B, Abdel-Latif A, Kitakaze M, Yan L. Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chemico-Biol Interact 2015; 225: 90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tian J, Sun Y, Xu TR, Chi RF, Zhang XL, et al. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim Biophys Acta 2015; 1852: 805–15. [DOI] [PubMed] [Google Scholar]
- Leibowitz A, Grossman E. The role of the immune system in the pathogenesis of hypertension. Harefuah 2014; 153: 17–18, 65 [PubMed] [Google Scholar]
- Touyz RM. Molecular and cellular mechanisms in vascular injury in hypertension: role of angiotensin II. Curr Opin Nephrol Hypertens 2005; 14: 125–31. [DOI] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, et al. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest 2012; 122: 3960–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Li A, Dong FN, Horwath JA, Clark CG, Davisson RL. Endoplasmic reticulum and oxidant stress mediate nuclear factor-kappaB activation in the subfornical organ during angiotensin II hypertension. Am J Physiol Cell Physiol 2015; 308: C803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, de Queiroz TM, Sriramula S, Feng Y, Johnson T, Mungrue IN, et al. Brain ACE2 overexpression reduces DOCA-salt hypertension independently of endoplasmic reticulum stress. Am J Physiol Regul Integr Comparactive Physiol 2015; 308: R370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li H, Zhou G, Wang Y. Changes of endoplasmic reticulum stress- and apoptosis-related factors in rat cerebral cortex following controlled hypotension. Nan Fang Yi Ke Da Xue Xue Bao 2014; 34: 1804–8. [PubMed] [Google Scholar]
- San Cheang W, Yuen Ngai C, Yen Tam Y, Yu Tian X, Tak Wong W, Zhang Y, et al. Black tea protects against hypertension-associated endothelial dysfunction through alleviation of endoplasmic reticulum stress. Sci Rep 2015; 5: 10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signaling 2014; 20: 164–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liu J, Huang Z, Yu X, Zhang X, Dou D, et al. Berberine improves endothelial function by inhibiting endoplasmic reticulum stress in the carotid arteries of spontaneously hypertensive rats. Biochem Biophys Res Commun 2015; 458: 796–801. [DOI] [PubMed] [Google Scholar]
- Chao YM, Lai MD, Chan JY. Redox-sensitive endoplasmic reticulum stress and autophagy at rostral ventrolateral medulla contribute to hypertension in spontaneously hypertensive rats. Hypertension 2013; 61: 1270–80. [DOI] [PubMed] [Google Scholar]
- Spitler KM, Matsumoto T, Webb RC. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 2013; 305: H344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arteriosclerosis Thromb Vasc Biol 2012; 32: 1652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol 2012; 32: 1652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo XF, Yang XJ. Endoplasmic reticulum stress response in spontaneously hypertensive rats is affected by myocardial ischemia reperfusion injury. Exp Ther Med 2015; 9: 319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Ann Rev Physiol 1997; 59: 551–71. [DOI] [PubMed] [Google Scholar]
- Cooper Gt. Basic determinants of myocardial hypertrophy: a review of molecular mechanisms. Ann Rev Med 1997; 48: 13–23. [DOI] [PubMed] [Google Scholar]
- Park CS, Cha H, Kwon EJ, Sreenivasaiah PK, Kim do H. The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun 2012; 421: 578–84. [DOI] [PubMed] [Google Scholar]
- Guan HS, Shangguan HJ, Shang Z, Yang L, Meng XM, Qiao SB. Endoplasmic reticulum stress caused by left ventricular hypertrophy in rats: effects of telmisartan. Am J Med Sci 2011; 342: 318–23. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998; 93: 215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR. Calcium, calcineurin, and the control of transcription. J Biol Chem 2001; 276: 2313–6. [DOI] [PubMed] [Google Scholar]
- Paschen W. Endoplasmic reticulum dysfunction in brain pathology: critical role of protein synthesis. Curr Neurovasc Res 2004; 1: 173–81. [DOI] [PubMed] [Google Scholar]
- Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 2005; 25: 9554–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. Clin Invest 2003; 111: 1497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Suzuki O, Haruyama T, Akaike T. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. Cell Biochem 2003; 89: 244–53. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005; 54 Suppl 2: S73–8. [DOI] [PubMed] [Google Scholar]
- Chaanine AH, Gordon RE, Kohlbrenner E, Benard L, Jeong D, Hajjar RJ. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ Heart Failure 2013; 6: 572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George I, Sabbah HN, Xu K, Wang N, Wang J. Beta-adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovasc Res 2011; 91: 447–55. [DOI] [PubMed] [Google Scholar]
- Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res 2006; 99: 275–82. [DOI] [PubMed] [Google Scholar]
- Xin W, Li X, Lu X, Niu K, Cai J. Involvement of endoplasmic reticulum stress-associated apoptosis in a heart failure model induced by chronic myocardial ischemia. Int J Mol Med 2011; 27: 503–9. [DOI] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004; 110: 705–12. [DOI] [PubMed] [Google Scholar]
- Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res 2011; 89: 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, Iland H, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 2012; 97: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J Mol Cell Cardiol 2010; 48: 1280–9. [DOI] [PubMed] [Google Scholar]
- Nader L, Lahoud L, Chouery E, Aftimos G, Bois P, Fares NA. B-type natriuretic peptide receptors in hypertrophied adult rat cardiomyocytes. Ann Cardiol Angeiol (Paris) 2010; 59: 20–4. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000; 49: 2063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY, et al. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arteriosclerosis Thromb Vasc Biol 2014; 34: 830–6. [DOI] [PubMed] [Google Scholar]
- Adler AI. Cardiovascular risk reduction in diabetes: underemphasised and overdue. Messages from major trials. Clin Med 2001; 1: 472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, et al. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010; 59: 1386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian XY, Wong WT, Wang N, Lu Y, Cheang WS, Liu J, et al. PPARdelta activation protects endothelial function in diabetic mice. Diabetes 2012; 61: 3285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomer X, Capdevila-Busquets E, Botteri G, Salvado L, Barroso E, Davidson MM, et al. PPARbeta/delta attenuates palmitate-induced endoplasmic reticulum stress and induces autophagic markers in human cardiac cells. Int J Cardiol 2014; 174: 110–8. [DOI] [PubMed] [Google Scholar]
- Chen S, Glenn DJ, Ni W, Grigsby CL, Olsen K, Nishimoto M, et al. Expression of the vitamin D receptor is increased in the hypertrophic heart. Hypertension 2008; 52: 1106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Singh SS, Yu H, Lee JY, Cho BR, Kang PM. Vitamin D signaling pathway plays an important role in the development of heart failure after myocardial infarction. J Appl Physiol 2013; 114: 979–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong H, et al. Vitamin D receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid Redox Signal 2015; 22: 633–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 2012; 13: 213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Pu J, Yuan A, Lau WB, Gao E, Koch WJ, et al. Activation of liver-X-receptor alpha but not liver-X-receptor beta protects against myocardial ischemia/reperfusion injury. Circ Heart Failure 2014; 7: 1032–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon MV, Sillje HH, Sijbesma JW, Vreeswijk-Baudoin I, Ciapaite J, van der Sluis B, et al. Cardiac LXRalpha protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol Med 2015; 7: 1229–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RJ, He KL, Li X, Wang LL, Liu CL, He YY. Salubrinal protects cardiomyocytes against apoptosis in a rat myocardial infarction model via suppressing the dephosphorylation of eukaryotic translation initiation factor 2alpha. Mol Med Reports 2015; 12: 1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XZ, Li XY, Liu J. Recent pharmacological studies on natural products in China. Eur J Pharmacol 2004; 500: 221–30. [DOI] [PubMed] [Google Scholar]
- Hao X, Yao A, Gong J, Zhu W, Li N, Li J. Berberine ameliorates pro-inflammatory cytokine-induced endoplasmic reticulum stress in human intestinal epithelial cells in vitro. Inflammation 2012; 35: 841–9. [DOI] [PubMed] [Google Scholar]
- Yu W, Sheng M, Xu R, Yu J, Cui K, Tong J, et al. Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J Transl Med 2013; 11: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha W, Liang G, Xiao J, Studer EJ, Hylemon PB. Pandak WM Jr, et al. Berberine inhibits HIV protease inhibitor-induced inflammatory response by modulating ER stress signaling pathways in murine macrophages. PloS One 2010; 5: e9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Chan P, Chen YJ, Tomlinson B, Hong SH, Cheng JT. The antihypertensive effect of the berberine derivative 6-protoberberine in spontaneously hypertensive rats. Pharmacology 1999; 59: 283–9. [DOI] [PubMed] [Google Scholar]
- O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, et al. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993; 136: 355–60. [DOI] [PubMed] [Google Scholar]
- Cao J, Li H, Chen L. Targeting drugs to APJ receptor: the prospect of treatment of hypertension and other cardiovascular diseases. Curr Drug Targets 2015; 16: 148–55. [DOI] [PubMed] [Google Scholar]
- Wu D, He L, Chen L. Apelin/APJ system: a promising therapy target for hypertension. Mol Biol Reports 2014; 41: 6691–703. [DOI] [PubMed] [Google Scholar]
- Lv D, Li H, Chen L. Apelin and APJ, a novel critical factor and therapeutic target for atherosclerosis. Acta Biochim Biophys Sin 2013; 45: 527–33. [DOI] [PubMed] [Google Scholar]
- Li F, Li L, Qin X, Pan W, Feng F, Chen F, et al. Apelin-induced vascular smooth muscle cell proliferation: the regulation of cyclin D1. Frontiers Biosci 2008; 13: 3786–92. [DOI] [PubMed] [Google Scholar]
- Li L, Li L, He L, Zhang Z, Xie F, Guo Y, et al. Effects of apelin-13 on rat bone marrow-derived mesenchymal stem cell proliferation through the AKT/GSK3β/Cyclin D1 pathway. Int J Pept Res Ther 2014; 20: 421–5. [Google Scholar]
- Horman SR, To J, Orth AP, Slawny N, Cuddihy MJ, Caracino D, et al. High-content analysis of three-dimensional tumor spheroids: investigating signaling pathways using small hairpin RNA. Nat Methods 2013; 10.
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 2009; 9: 35–51. [DOI] [PubMed] [Google Scholar]
- Fu M, Xu S, Zhang J, Pang Y, Liu N, Su J, et al. Involvement of calcineurin in angiotensin II-induced cardiomyocyte hypertrophy and cardiac fibroblast hyperplasia of rats. Heart Vessels 1999; 14: 283–8. [DOI] [PubMed] [Google Scholar]
- Sukumaran V, Watanabe K, Veeraveedu PT, Gurusamy N, Ma M, Thandavarayan RA, et al. Olmesartan, an AT1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis. Int J Biol Sci 2011; 7: 154–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinowski JM. Atorvastatin: a hydroxymethylglutaryl-coenzyme A reductase inhibitor. Am J Health Syst Pharm 1998; 55: 2253–67. [DOI] [PubMed] [Google Scholar]
- Sola S, Mir MQ, Lerakis S, Tandon N, Khan BV. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. Am Coll Cardiol 2006; 47: 332–7. [DOI] [PubMed] [Google Scholar]
- Tousoulis D, Antoniades C, Vassiliadou C, Toutouza M, Pitsavos C, Tentolouris C, et al. Effects of combined administration of low dose atorvastatin and vitamin E on inflammatory markers and endothelial function in patients with heart failure. Atherosclerosis 2005; 178: 359–63. [DOI] [PubMed] [Google Scholar]
- Song XJ, Yang CY, Liu B, Wei Q, Korkor MT, Liu JY, et al. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci 2010; 8: 564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang Y, Ma J. Effects of yiqi huoxue compound combined with exercise therapy on MMP-1 and collagen type III expressions of cardiac muscle in chronic heart failure rats. Chin J Integr Tradit Western Med 2011; 31: 955–60. [PubMed] [Google Scholar]
- Li DP, Chen Q, Yi L. Effects of yiqi huoxue method on cardiac function in patients with congestive heart failure. Chin J Integr Tradit Western Med 2006; 26: 552–4. [PubMed] [Google Scholar]
- Lou LX, Wu AM. Yiqi Huoxue recipe improves heart function through inhibiting apoptosis related to endoplasmic reticulum stress in myocardial infarction model of rats. 2014; 2014: 745919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Liu W, Feng F, Li X, Yang L, Lv D, et al. A static pressure sensitive receptor APJ promote H9c2 cardiomyocyte hypertrophy via PI3K-autophagy pathway. Acta Biochim Biophys Sin (Shanghai) 2014; 46: 699–708. [DOI] [PubMed] [Google Scholar]
- He L, Xu J, Chen L, Li L. Apelin/APJ signaling in hypoxia-related diseases. Clin Chim Acta 2015; 451: 191–8. [DOI] [PubMed] [Google Scholar]
- Xie F, Lv D, Chen L. ELABELA: a novel hormone in cardiac development acting as a new endogenous ligand for the APJ receptor. Acta Biochim Biophys Sin 2014; 46: 620–2. [DOI] [PubMed] [Google Scholar]
- Lv XR, Zheng B, Li SY, Han AL, Wang C, Shi JH, et al. Synthetic retinoid Am80 up-regulates apelin expression by promoting interaction of RARalpha with KLF5 and Sp1 in vascular smooth muscle cells. Biochem J 2013; 456: 35–46. [DOI] [PubMed] [Google Scholar]