

Abstract
Background
Gene isoforms are commonly found in both prokaryotes and eukaryotes. Since each isoform may perform a specific function in response to changing environmental conditions, studying the dynamics of gene isoforms is important in understanding biological processes and disease conditions. However, genome-wide identification of gene isoforms is technically challenging due to the high degree of sequence identity among isoforms. Traditional targeted sequencing approach, involving Sanger sequencing of plasmid-cloned PCR products, has low throughput and is very tedious and time-consuming. Next-generation sequencing technologies such as Illumina and 454 achieve high throughput but their short read lengths are a critical barrier to accurate assembly of highly similar gene isoforms, and may result in ambiguities and false joining during sequence assembly. More recently, the third generation sequencer represented by the PacBio platform offers sufficient throughput and long reads covering the full length of typical genes, thus providing a potential to reliably profile gene isoforms. However, the PacBio long reads are error-prone and cannot be effectively analyzed by traditional assembly programs.
Results
We present a clustering-based analysis pipeline integrated with PacBio sequencing data for profiling highly similar gene isoforms. This approach was first evaluated in comparison to de novo assembly of 454 reads using a benchmark admixture containing 10 known, cloned msg genes encoding the major surface glycoprotein of Pneumocystis jirovecii. All 10 msg isoforms were successfully reconstructed with the expected length (~1.5 kb) and correct sequence by the new approach, while 454 reads could not be correctly assembled using various assembly programs. When using an additional benchmark admixture containing 22 known P. jirovecii msg isoforms, this approach accurately reconstructed all but 4 these isoforms in their full-length (~3 kb); these 4 isoforms were present in low concentrations in the admixture. Finally, when applied to the original clinical sample from which the 22 known msg isoforms were cloned, this approach successfully identified not only all known isoforms accurately (~3 kb each) but also 48 novel isoforms.
Conclusions
PacBio sequencing integrated with the clustering-based analysis pipeline achieves high-throughput and high-resolution discrimination of highly similar sequences, and can serve as a new approach for genome-wide characterization of gene isoforms and other highly repetitive sequences.
Electronic supplementary material
The online version of this article (doi:10.1186/s13040-016-0090-8) contains supplementary material, which is available to authorized users.
Keywords: PacBio, Bioinformatics analysis, Gene isoforms, Repetitive sequences, Major surface glycoprotein, Pneumocystis, NGS, Uclust
Background
Gene isoforms are commonly found in both prokaryotes and eukaryotes organisms, and can be produced from different but closely related genes (known as multi-copy gene families) or from the same gene by alternative splicing. For example, alternative splicing of the human genome can result in greater than 100,000 unique isoforms from only ~20,000 genes [1–4]. Particularly, the single titin gene in humans as well as other mammals can generate millions of alternatively spliced isoforms [2, 5]. The production of these isoforms can increase the structural and functional diversity of gene products, and result in profound effects in cellular processes, developmental states, tissue or cell specificity, and disease conditions [1–5]. In contrast to mammals, low complex organisms, especially microorganisms, have significantly lower rates of alternative splicing but are rich in multi-copy gene families. The most prominent examples include the variant surface glycoprotein (vsg) gene family (~1,000 members) in Trypanosoma brucei [6], the variant surface protein (vsp) gene family (~235–275 members) in Giardia lamblia [7], the var gene family (~60 members) in Plasmodium falciparum [8], and the major surface glycoprotein (msg) gene family (estimated ~80 members) in Pneumocystis [9–11]. All these gene families encode variable surface protein isoforms, which are thought to be employed by the microorganisms to generate antigenic variation in order to evade the host immune response and, in some cases, to maintain persistent infections [12].
While studying the dynamics of gene isoforms is important in understanding biological processes and disease conditions, characterization of the full complement of isoforms within targeted genes or across an entire genome is technically challenging due to the high degree of sequence identity among these isoforms. Traditionally, gene isoforms have been identified primarily by Sanger sequencing of cloned DNA or cDNA fragments (e.g. by PCR or hybridization selection. Fig. 1: Method 2). This approach is very tedious and time-consuming, and has low throughput, thus not suitable for large-scale screening of isoforms across various biological processes or disease conditions. In addition, this approach is not sensitive to low abundant isoforms. Due to these limitations, characterization of gene isoforms in many organisms is limited.
Fig 1.

Comparison of three methods for gene isoform identification. Lines with different colors represent different sequences
Next generation sequencing (NGS) technologies, such as Roche 454 and Illumina platforms, significantly increase the efficiency of sequencing by producing a large number of 100–600 bp reads rapidly and with a high throughput [13]. While these NGS methods are very successful when applied to the sequencing of genomes, exomes and transcriptomes as well as to single nucleotide polymorphism (SNP) discovery [13, 14], accurate characterization of gene isoforms with highly similar sequences remains problematic. The major bottleneck is that NGS read lengths are usually substantially shorter than the length of typical gene isoforms, and thus require assembly, which may result in inaccurate joining of highly similar short reads from different isoforms to form artificial chimeric sequences (exemplified in Fig. 1: Method 2). Most of the currently available assembly software packages (such as 454’s Newbler, Celera Assembler, CLC Bio, MIRA, MINIMUS, Vicuna, Trinity, PredictHaplo, and Cufflinks) [15, 16], were developed for general assembly purposes and have proven to be successful in many NGS studies of diploid/haploid genome assemblies, but their performance is poor in the assembly of highly similar gene isoforms or other repetitive sequences [16–19].
The history of Pneumocystis msg isoform discovery in the past decade reflects the aforementioned challenges for gene isoform identification. We have long been interested in studying the msg genes in P. jirovecii, an opportunistic pathogen of immunosuppressed patients [20, 21]. Using traditional Sanger sequencing of cloned DNA fragments, we identified a limited number of full-length msg gene isoforms in P. jirovecii [22, 23]. Each msg gene is ~3 kb in length. The overall average identity among msg isoforms is 75 %, with a range from 57 % to 91 % [22]. While a limited number of msg genes has been also identified in all other Pneumocystis species examined (reviewed by Stringer [11]), the exact number and sequence diversity of the msg members in any species remains unknown due largely to the lack of an efficient sequencing method. Even in the current genome assemblies of both P. carinii [24] and P. jirovecii [25], which were sequenced by traditional Sanger method and NGS technologies, respectively, the msg genes have been almost entirely excluded due to failure in assembly of highly similar short reads for msg genes. The lack of complete sequences of the msg family has hampered our understanding of its diversity, recombination mechanisms and biological functions.
The newly emerging PacBio sequencing platform, a third-generation sequencer (TGS), is capable of sequencing tens of thousands of individual DNA molecules in parallel and in real time [26, 27]. The most distinctive feature of PacBio is the generation of very long reads (up to ~40 kb), which cover the full length of typical genes and gene families, thus minimizing or eliminating the need for sequence assembly. However, the PacBio long reads are error-prone [28] and cannot be effectively analyzed by standard sequence alignment and assembly programs [27, 29]. In this paper, we describe a unique clustering-based computational pipeline for analyzing the long but error-prone PacBio reads, and demonstrate its capacity to achieve high-throughput and high-resolution profiling of the msg isoforms of P. jirovecii (Fig. 1: Method 3). The key to this approach is that the PacBio long reads enable each targeted isoform (in a mixture of all targeted isoforms amplified by PCR) to be continuously sequenced multiple times at its full length or nearly full length, which eliminates the challenging de novo assembly process required for highly similar short reads generated by other NGS methods. The full-length sequence for each isoform is obtained by an unsupervised clustering-based analysis pipeline during which the randomly distributed PacBio read errors can be largely corrected as demonstrated in the paper (Fig. 2).
Fig 2.
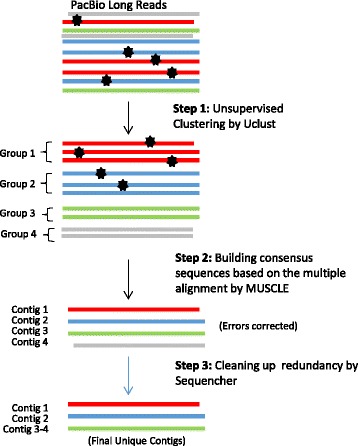
Schema of the clustering-based data analysis procedure using PacBio long reads. The different colors represent the reads belonging to different isoforms. The black stars represent sequencing errors. In this example, contig 3 and 4 are merged because they are the plus and minus strands of the same isoform
Methods
Two benchmark isoform-admixture samples containing known, cloned P. jirovecii msg genes
Ten plasmids, representing different msg gene isoforms (~3 kb each) previously identified from a P. jirovecii strain by traditional Sanger sequencing of cloned PCR amplicons [22, 29], were mixed together to form an artificial admixture of highly similar isoforms with 81–91 % identity (Fig. 3). This admixture served as a benchmark sample mimicking a gene family in vivo. Degenerate PCR primers were then used to amplify an ~1.5 kb portion of the msg isoforms in a bulk PCR reaction as described in an earlier paper [22]. The bulk PCR product was sequenced in parallel by the Roche 454 and PacBio platforms. The ~1.5 kb fragment was chosen as it was the optimal size for PacBio sequencing at the time our work began (see Additional file 3 fasta sequence file). As the PacBio system improved, we tested its ability to sequence the full-length msg isoforms (~3 kb) using another benchmark admixture containing 22 of the 24 plasmid clones representing unique msg gene isoforms previously cloned from a different P. jirovecii strain [22]. This plasmid admixture was amplified by PCR targeting the full-length msg genes (~3 kb each) using a pair of degenerate primers JSG.f1, 5’-TGGCGCGGGCGGTYAAGCGG-3’, and JSG.r1, 5’-YRCCCCYCTCATCAC-3’, and conditions described elsewhere [22].
Fig 3.

High sequence identity of 10 msg isoforms of P. jirovecii previously identified by Sanger sequencing of plasmid-cloned PCR products [22]. Text labels on the left side and the top represent plasmid ID. Colors indicate different degrees of identity from low (blue) to high (red). Plasmids containing these 10 isoforms were mixed together to form a benchmark admixture, which was amplified by PCR and sequenced in parallel by PacBio and 454 sequencing
Genomic DNA from a clinical strain of P. jirovecii
Human subject experimentation guidelines of the National Institutes of Health were followed in the conduct of these studies. P. jirovecii genomic DNA was extracted from the same patient lung sample used in a previous study to clone 24 msg isoforms [22], which were used to make the second benchmark admixture as described above. This DNA sample was used to amplify the full-length coding regions of msg genes. To maximize the coverage of msg genes, two pairs of primers were designed from highly conserved regions at the 5’ and 3’ ends of ~30 known P. jirovecii msg genes [22, 23], including the primer pair JSG.f1 and JSG.r1 described above and another pair GK126a (5’-GGCGCGGGCGGTGGCGCGGGCGGT-3’) and GK452 [22]. These two pairs of primers were used in separate PCR reactions following conditions described elsewhere [22]. Equal amount of PCR products were pooled from each reaction and sequenced by the PacBio sequencing platform as described below.
Roche 454 and PacBio sequencing
For 454 sequencing, the input DNA samples (PCR amplicons) were fragmented using a Covaris sonicator before library construction. A standard shotgun procedure was used for all steps of 454 library construction and deep sequencing according to the manufacturer’s standard shotgun protocols. For PacBio sequencing, the input DNA samples (PCR amplicons) were used directly (without fragmenting) for circular consensus sequences (CCS) library construction (with a 1.5 kb CCS library for the ~1.5 kb amplicon and a 3 kb CCS library for the ~3 kb amplicon). All CCS libraries were sequenced following the manufacturer’s standard protocols, using either XL or P4 polymerase chemistry, and 90 minute movies.
Bioinformatics analysis of 454 reads
Raw 454 sff files were converted to fastq files using the standard 454 software package. The 454 reads were filtered with QC criteria of read length ≥ 50 bp, average quality score ≥ 30, and total Ns ≤ 3. The key bioinformatics analysis was de novo assembly of the short reads (50–600 bp) into the full length sequences (~1.5 or ~3 kb) of the input amplicons (Fig. 1: Method 2). In order to obtain the best possible results, the qualified reads were subjected to de novo assembly using various software packages including 454’s Newbler, Celera Assembler, CLC Bio, MIRA, MINIMUS, Trinity, Vicuna, PredictHaplo, and Cufflinks [15]. These assembly programs cover the popular assembly algorithms including the de Bruijn graph algorithm and overlap-to-consensus [15]. We first tested these packages with the default parameters, and then adjusted the parameters from lower to higher stringencies. The resulting contigs from various programs and parameters were compared to the known msg sequences in the benchmark isoform-admixture sample.
A unique clustering-based analysis pipeline for PacBio long reads
PacBio raw movie files were converted into CCS reads in fastq format using the standard PacBio SMRT software package. The reads were filtered with QC criteria of read length ≥ 1 kb and < 3.5 kb. Given that the PacBio reads should represent the full length or nearly full length of the input amplicons, the key process was to classify these reads rather than assemble them as required with the 454 reads. The unique clustering-based analysis pipeline consisted of the following steps (Fig. 2 and Additional file 2: Table S2 for details):
Firstly, all raw CCS reads were clustered into different groups of reads based on their global similarities using the Uclust [30] program (similarity threshold set to 0.9). Uclust is a sequence clustering program mainly used in phylogenetic analysis to congregate similar sequences into groups [30] and has not been used previously for NGS data analysis. We are the first group to adopt it for PacBio sequencing analysis and found Uclust’s speed to be fast enough to sort ~10,000 sequences in several minutes. It is theoretically able to output any number of clusters as driven by the input dataset itself. Of note, the Uclust procedure is completely unsupervised, and relies on classifying full length or near full length reads so that it only concentrates on the unsupervised clustering process without any extra effort to build internal linkages between reads. Thus, this procedure is more suitable for PacBio long reads than for NGS short reads.
Secondly, the input of groups of reads was summarized into each corresponding contig. The reads in each group clustered from the first step were aligned using the MUSCLE [31] program (with default parameters), which resulted in a consensus sequence or contig for each group. At this point, the first and second steps not only reduced redundancy by merging tens of thousands of PacBio reads into a relatively small number of contigs, but also corrected the sequencing errors in individual PacBio reads. The error correction power relied on the procedure of building consensus sequences from multiple alignments since the errors in each individual read were randomly distributed.
Finally, the redundancies of contigs above in either plus or minus strand were further merged into unique contigs. We used the program Sequencher (Gene Codes Corporation, Ann Arbor, MI) (Similarity = 95 % and overlap = 100 bp) to perform the final merging/cleanup step in order to remove redundancies such as plus and minus strands of the same isoform, as well as to filter out singleton contigs. At this point, each contig in the output represented a unique gene isoform. All isoforms identified were supported by sequences from both directions.
Evaluation of contigs
To examine the accuracy of the final contigs corresponding to the known msg isoforms, the NCBI BLAST program was used to match the final contig sequences derived from the various analyses described above with all known P. jirovecii msg genes. Identity (0–100 %) and length (relative to known msg genes) were used to determine the level of accuracy of contigs. To examine the accuracy of contigs containing new msg isoforms identified from the clinical sample, six novel contigs were selected for verification by PCR using isoform-specific primers and original genomic DNA; PCR products were sequenced by Sanger sequencing. All msg isoforms identified in this study have been incorporated into the P. jirovecii RU genome assembly (GenBank under accession no. PRJNA223510) [32].
Results and discussion
Study design
Figure 1 illustrates three approaches for gene isoform characterization, including the traditional Sanger sequencing of plasmid-cloned PCR products (Method 1), 454 sequencing followed by de novo assembly (Method 2) and PacBio sequencing followed by clustering analysis (Method 3). We have previously reported the use of the Method 1 to identify a limited number of P. jirovecii msg isoforms [22]. In the present study, we compared the performance of the latter two methods using two benchmark isoform admixtures containing 10 or 22 previously cloned P. jirovecii msg isoforms. The performance of each method was evaluated by measuring the number, length and sequence identity of msg isoforms reconstructed (relative to the known isoforms in the benchmark isoform admixtures used). The performance of Method 3 was further assessed by applying it to the original clinical sample from which the 22 known msg isoforms were identified previously by the Method 1, with an expectation of identifying all known isoforms as well as additional new isoforms.
Evaluation of method 2 – Roche 454 sequencing followed by de novo assembly using the benchmark sample
The ~1.5 kb amplicon from the benchmark admixture containing 10 known msg isoforms was sequenced using the 454 shotgun protocol, yielding ~47,000 pass-filter reads with an average length of 350 bp (ranging from 50 to 400 bp). By aligning to the 10 known sequences, we found that these reads fully covered the ~1.5 kb amplified region for each of the 10 known isoforms in the admixture. We attempted de novo assembly of these reads using various de novo assemblers, including 454’s Newbler, Celera Assembler, CLC Bio, MIRA, MINIMUS, Vicuna, Trinity, and Cufflinks [15, 18]. As shown in Additional file 1: Table S1, the contigs generated by these assemblers shared the following issues: 1) Only a few (1–2) contigs contained the expected ~1.5 kb sequences though a total of 10 contigs were expected. 2) Most of the remaining contigs were substantially shorter than ~1.5 kb, and showed < 90 % identity to any of the known sequences. Detailed examination of these contigs revealed false joinings, that is, artificial chimeric sequences formed by reads from different isoforms during de novo assembly. Chimeric contigs are a common problem encountered in de novo assembly of highly similar isoforms [17, 19]. Despite adjusting the parameters of the assembly programs in many different ways from lower to higher stringencies, the improvements were limited.
Our experience above is consistent with previous studies showing that the de novo assembly of short, highly similar reads from NGS platforms for gene isoforms is a very difficult task [17, 19]. The challenges come from several aspects [15]: 1) Most of the assembly programs were designed to tolerate a certain degree of read ‘error’, i.e. treating slightly different reads as the same sequence. However, in isoform assembly, such error toleration is problematic as it overlooks true biological variants and may result in compressing slightly different reads from different isoforms into one artificial chimeric contig. Such chimeric contigs cannot be easily eliminated by increasing the stringency through changing the parameters of the assembly programs, due to the difficulty in drawing an appropriate line for the signal-to-noise ratio. Indeed, the line is dynamic from read to read, i.e. reads may have more or less variants depending on the variant distribution along the regions for the given isoforms. 2) Most of the assembly programs were built with the assumption that the sequences are coming from diploid or haploid systems. Thus, they tend to assemble sequence reads into long contigs/scaffolds based on a majority rule without considering the alternatives despite clearly different patterns in minorities in some cases. For example, in order to assemble many reads derived from a bacterial colony, the assemblers tend to generate a single contig while ignoring unmatched reads, which is reasonable under the assumption that one bacteria colony should have only one genome. Thus, such assemblers are generally not able to assemble reads that are related to but different from the majority reads. 3) Some assembly programs (such as Trinity, Vicuna, Cufflinks or PredictHaplo) [19] do indeed consider the possibility of multiple isoforms during de novo assembly and have the ability to produce multiple contigs [33]. However, in our experience and that of others [18], these programs are still unreliable for highly similar isoform characterization. The performance of these assemblers largely depends on factors such as the distribution pattern of sequencing errors (scattered everywhere vs. concentrated in small areas), the proportion of each isoform in the pool, and the level of sequence identity among the isoforms [18].
Due to the failure to reconstruct the ~1.5 kb segment of the 10 known msg isoforms using 454 reads from the benchmark admixture, we did not repeat this experiment using the other benchmark admixture containing 22 known msg isoforms to reconstruct their ~3 kb full-length, which would likely have been even less successful. Our failure to reconstruct the known msg isoforms using the benchmark admixture mirrored the inability of other investigators to identify msg isoforms by assembly of whole genome sequence reads generated by 454 and Illumina platforms for P. jirovecii [25] and by Sanger sequencing for P. carinii [10, 24].
Evaluation of method 3 -PacBio sequencing integrated with a unique clustering-based analysis pipeline using two benchmark samples and one clinical sample
The same ~1.5 kb amplicon used in the Method 2 described above was sequenced using the PacBio platform, yielding ~8,500 pass-filter CCS reads with the length in the range of 0.5 to 1.5 kb. We attempted de novo assembly of these CCS reads using the same set of assembly programs applied in Method 2. Surprisingly, none of these programs was able to reconstruct any one of the 10 expected msg isoforms, with all contigs containing numerous ambiguous sequences resulting from higher errors in individual CCS reads or false joining of CCS reads from different isoforms.
To take advantage of the PacBio long reads and to tolerate their high error rate, we constructed a unique clustering-based analytical pipeline as described in the Methods and illustrated in Fig. 2. When this pipeline was applied to the ~1.5 kb PacBio CCS reads, it generated 10 contigs whose sequences had a one-to-one match with the 10 expected msg isoforms in the benchmark sample at an identity level greater than 99.5 % (Table 1). When it was further applied to the ~3 kb PacBio CCS reads from the benchmark admixture containing 22 of the 24 known msg isoforms, it reconstructed all but 4 of the 22 isoforms in their full length with an identity > 99.4 %; the 4 isoforms that were not reconstructed were present at concentrations ≤1.3 % in the admixture. This finding suggests that this approach has a detection threshold of ~1 % (Fig. 4).
Table 1.
Reconstruction of an ~1.5 kb segment of 10 known msg isoforms of P. jirovecii from the benchmark isoform admixture by PacBio sequencing and clustering-based analysis (see Additional file 2: Table S2 and Additional file 3 for more details)
| Contig no. | Lengtha (bp) | Matched msg isoform | Identity (%) |
|---|---|---|---|
| Contig0001 | 1587 | AR-Cl72 | 100 |
| Contig0002 | 1581 | AR-Cl59 | 99.94 |
| Contig0003 | 1584 | AR-Cl46 | 99.94 |
| Contig0004 | 1584 | AR-Cl14 | 99.87 |
| Contig0005 | 1581 | AR-Cl6 | 99.75 |
| Contig0006 | 1581 | AR-Cl24 | 99.87 |
| Contig0007 | 1593 | AR-Cl20 | 99.94 |
| Contig0008 | 1584 | AR-Cl44 | 99.81 |
| Contig0011 | 1584 | AR-Cl45 | 99.94 |
| Contig0014 | 1583 | AR-Cl58 | 99.87 |
aIdentical between the contigs and known msg isoforms
Fig 4.

The sensitivity of the new approach based on PacBio sequencing and clustering analysis in detection of minor gene isoforms in a mixture containing multiple isoforms. Twenty-two plasmids, representing 22 different P. jirovecii msg isoforms previously cloned [22], were mixed with various concentrations and amplified by PCR for the full-length msg coding region (~3 kb) followed by PacBio sequencing and clustering analysis. The read frequency and concentrations of plasmids (indicated by diamonds) in the mixture are positively correlated. The concentration is the percentage of each plasmid DNA relative to the total amount of plasmid DNA in the mixture. Those 4 msg isoforms, which were not identified, have the lowest concentrations (0.8–1.3 %) in the mixture
The success of the new approach with the two benchmark samples prompted us to directly apply it to the original clinical sample from which the 24 known msg isoforms of P. jirovecii were identified previously [22] by Method 1 as described above. After PCR amplification of the msg repertoire in its full-length (~3 kb) using genomic DNA from this sample, the amplicon was sequenced with the 3 kb PacBio CCS sequencing protocol, yielding ~9,000 pass-filter CCS reads with an length of 1 – 3 kb. These reads were processed by the clustering-based analysis pipeline, resulting in 72 unique contigs, which represent 72 msg isoforms (Table 2; Additional file 3: Sequence Data). All contigs contained the full-length coding regions of the msg genes. Twenty-four of them exhibited one-to-one matches to the 24 known msg isoforms with an identity > 99.4 % (Table 2), supporting the high accuracy and reproducibility of the new approach. The remaining 48 contigs showed high-level sequence identity but were clearly distinct compared to the 24 known msg isoforms, and thus they represented novel msg isoforms. Six new isoforms were chosen for verification by PCR; all of them were successfully amplified from the original P. jirovecii genomic DNA using isoform-specific primers, and the resulting sequences were consistent with the corresponding PacBio contigs, with > 98 % sequence identity (Table 2). The total number of msg isoforms (72) identified from this P. jirovecii strain is within the estimated 70–80 isoforms based on studies of P. carinii [9, 10]. This represents the first report of the complete or near complete set of full-length msg isoforms in any Pneumocystis species. Our findings support the capacity of the new approach to achieve high-throughput and high-resolution profiling of the P. jirovecii msg isoforms in clinical samples.
Table 2.
A partial list of P. jirovecii full-length msg isoforms (~3 kb) identified in a clinical sample by PacBio sequencing and clustering-based analysis
| Contig no. | Length (bp) | Matched msg gene | Status | Identity to the known (%) | PCR verification |
|---|---|---|---|---|---|
| Contig0020 | 3086 | EF371022 | Known | 98.92 | ND |
| Contig0007 | 3086 | EF371023 | Known | 99.87 | ND |
| Contig0021 | 3008 | EF371024 | Known | 99.83 | ND |
| Contig0025 | 3011 | EF371025 | Known | 99.60 | ND |
| Contig0133 | 3068 | EF371026 | Known | 99.71 | ND |
| Contig0022 | 3104 | EF371028 | Known | 99.58 | ND |
| Contig0006 | 3062 | EF371029 | Known | 99.97 | ND |
| Contig0008 | 3041 | EF371030 | Known | 99.87 | ND |
| Contig0026 | 3032 | EF371031 | Known | 98.76 | ND |
| Contig0012 | 3005 | EF371032 | Known | 99.70 | ND |
| Contig0013 | 2996 | EF371033 | Known | 99.87 | ND |
| Contig0011 | 3092 | EF371035 | Known | 99.88 | ND |
| Contig0003 | 3038 | EF371036 | Known | 99.97 | ND |
| Contig0005 | 3002 | EF371038 | Known | 99.90 | ND |
| Contig0010 | 2996 | EF371040 | Known | 99.80 | ND |
| Contig0015 | 3129 | EF371041 | Known | 99.78 | ND |
| Contig0001 | 3002 | EF371042 | Known | 99.37 | ND |
| Contig0027 | 3065 | EF371045 | Known | 99.64 | ND |
| Contig0014 | 3077 | EF371050 | Known | 99.71 | ND |
| Contig0018 | 3044 | EF371051 | Known | 99.70 | ND |
| Contig0009 | 3029 | EF371052 | Known | 99.87 | ND |
| Contig0017 | 3023 | EF371053 | Known | 99.50 | ND |
| Contig0016 | 3050 | EF371055 | Known | 99.84 | ND |
| Contig0004 | 3026 | EF371056 | Known | 99.97 | ND |
| Contig0010b | 3060 | No | Novel | NA | 99.53 |
| Contig0004b | 3086 | No | Novel | NA | 99.45 |
| Contig0015b | 3039 | No | Novel | NA | 99.20 |
| Contig0053b | 3077 | No | Novel | NA | 98.91 |
| Contig0006b | 3062 | No | Novel | NA | 98.50 |
| Contig0054b | 3074 | No | Novel | NA | 98.32 |
| Contig0138b | 3041 | No | Novel | NA | ND |
A total of 72 unique msg isoforms identified in this study, with only 31 of them shown in this table. The first 24 contigs matched in full-length with the 24 previously identified msg genes from the same clinical sample [22] as shown in the third column with GenBank accession no, NA not applicable, ND not determined by PCR. Additional file 4 contains a complete list of sequences for 72 Msg isoforms
In this study, we choose the P. jirovecii msg gene family as the model isoform system since it represents the largest surface protein gene family identified to date in non-protozoan microorganisms [12]. Based on restricted fragment length polymorphism analysis [22, 34], the msg repertoire is different among different P. jirovecii strains. Characterizing the msg repertoire in different P. jirovecii strains will allow us to examine the phylogenetic relationship among these strains, and to determine if there are associations related to pathogenicity or geography. Given the high sequence identity and the large family size and gene length (~3 kb each) of the msg genes, successful characterization of this gene family implies that our approach could be easily adapted to characterize gene isoforms and other highly repetitive sequences in other organisms. Due to the relatively low outputs per run of the PacBio system, a requirement for this approach at present is that the gene families/isoforms must contain highly conserved sequences so that they can be enriched by PCR, hybrid capture [35], or other methods before PacBio sequencing.
Conclusions
PacBio long reads have obvious advantages over the short reads generated by 454 and other NGS platforms for sequencing highly similar gene isoforms because the long reads cover the full-length of typical genes, allowing elimination of the de novo assembly challenges encountered by the NGS short reads. However, such advantages can only be fully exploited for gene isoform characterization when appropriate bioinformatics analyses are applied to distinguish the minor differences and correct the high error rates in PacBio reads. In this study we integrated a clustering-based bioinformatics analysis pipeline with PacBio sequencing, and demonstrated that this approach can efficiently and accurately distinguish very long and highly similarly gene isoforms using the multi-copy msg gene family of P. jirovecii as a model system. This approach could be easily adapted to characterize gene isoforms and other highly repetitive sequences in other organisms. Indeed, for example, we have successfully adapted this approach to characterize msg and kexin gene families in rodent Pneumocystis [32].
Acknowledgements
We thank Biswajit Das, Dun Liang, Jun Yang, Xiaoli Jiao, Xiaojun Hu and Ju Qiu for technical support and discussions throughout the course of this research. This work was supported by the National Cancer Institute, National Institutes of Health (Contract HHSN261200800001E); the Intramural Research Program of the NIH Clinical Center; and the National Institute of Allergy and Infectious Disease. The contents do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Abbreviations
- 454
454 Titanium RS
- PacBio
PacBio sequencing platform
- CCS
circular consensus sequences
- Msg
major surface glycoprotein
- NGS
next generation sequencing
- TGS
third generation sequencing
- PCR
polymerase chain reaction
Additional files
Representative results of failure of de novo assembly of 454 reads for an ~1.5 kb segment of 10 known msg isoforms of P. jirovecii. (DOCX 18 kb)
Explanation of the Uclust-based analysis pipeline in details. (DOC 27 kb)
Benchmark dataset 1 consisting of raw PacBio reads of 10 known MSG plasmid and the final analysis result. (ZIP 14 kb)
72 P. jirovecii msg sequences in fasta format. It contains 24 known MSGs and 48 novel MSGs identified by the described analysis pipeline. (FASTA 218 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LM, CR, MJ, KJ, GK contributed to the experiment implementation; XZ, BR, TS, QS, DWH contributed to data analysis; JK, RL, DWH, LM, RD, BT, RS, TI contributed to the principal design & trouble shooting; DWH, LM, BS, CJ, WL, BZ, CJ contributed to manuscript writing. All authors read and approved the final manuscript.
References
- 1.Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol. 2012;30(1):99–104. doi: 10.1038/nbt.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–5. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31(11):1009–14. doi: 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity--alternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675. doi: 10.1155/2010/753675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309(5733):416–22. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 7.Morrison HG, McArthur AG, Gillin FD, Aley SB, Adam RD, Olsen GJ, et al. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science. 2007;317(5846):1921–6. doi: 10.1126/science.1143837. [DOI] [PubMed] [Google Scholar]
- 8.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keely SP, Renauld H, Wakefield AE, Cushion MT, Smulian AG, Fosker N, et al. Gene arrays at Pneumocystis carinii telomeres. Genetics. 2005;170(4):1589–600. doi: 10.1534/genetics.105.040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keely SP, Stringer JR. Complexity of the MSG gene family of Pneumocystis carinii. BMC Genomics. 2009;10:367. doi: 10.1186/1471-2164-10-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stringer JR. Antigenic variation in Pneumocystis. J Eukaryot Microbiol. 2007;54(1):8–13. doi: 10.1111/j.1550-7408.2006.00225.x. [DOI] [PubMed] [Google Scholar]
- 12.Deitsch KW, Lukehart SA, Stringer JR. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat Rev Microbiol. 2009;7(7):493–503. doi: 10.1038/nrmicro2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buermans HP, den Dunnen JT. Next generation sequencing technology: Advances and applications. Biochim Biophys Acta. 2014;1842(10):1932–41. doi: 10.1016/j.bbadis.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 14.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 15.Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95(6):315–27. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13(1):36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prosperi MC, Yin L, Nolan DJ, Lowe AD, Goodenow MM, Salemi M. Empirical validation of viral quasispecies assembly algorithms: state-of-the-art and challenges. Sci Rep. 2013;3:2837. doi: 10.1038/srep02837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schirmer M, Sloan WT, Quince C. Benchmarking of viral haplotype reconstruction programmes: an overview of the capacities and limitations of currently available programmes. Brief Bioinform. 2014;15(3):431–42. doi: 10.1093/bib/bbs081. [DOI] [PubMed] [Google Scholar]
- 19.Yang X, Charlebois P, Gnerre S, Coole MG, Lennon NJ, Levin JZ, et al. De novo assembly of highly diverse viral populations. BMC Genomics. 2012;13:475. doi: 10.1186/1471-2164-13-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovacs JA, Masur H. Evolving health effects of Pneumocystis: one hundred years of progress in diagnosis and treatment. JAMA. 2009;301(24):2578–85. doi: 10.1001/jama.2009.880. [DOI] [PubMed] [Google Scholar]
- 21.Ma L. Pneumocystis: An atypical fungal pathogen. Ref Module Biomed Sci Elsevier. 2015 [Google Scholar]
- 22.Kutty G, Maldarelli F, Achaz G, Kovacs JA. Variation in the major surface glycoprotein genes in Pneumocystis jirovecii. J Infect Dis. 2008;198(5):741–9. doi: 10.1086/590433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mei Q, Turner RE, Sorial V, Klivington D, Angus CW, Kovacs JA. Characterization of major surface glycoprotein genes of human Pneumocystis carinii and high-level expression of a conserved region. Infect Immun. 1998;66(9):4268–73. doi: 10.1128/iai.66.9.4268-4273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slaven BE, Meller J, Porollo A, Sesterhenn T, Smulian AG, Cushion MT. Draft assembly and annotation of the Pneumocystis carinii genome. J Eukaryot Microbiol. 2006;53(Suppl 1):S89–91. doi: 10.1111/j.1550-7408.2006.00184.x. [DOI] [PubMed] [Google Scholar]
- 25.Cisse OH, Pagni M, Hauser PM. De novo assembly of the Pneumocystis jirovecii genome from a single bronchoalveolar lavage fluid specimen from a patient. mBio. 2012;4(1):e00428–12. doi: 10.1128/mBio.00428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 27.Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol. 2013;14(7):405. doi: 10.1186/gb-2013-14-6-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frey KG, Herrera-Galeano JE, Redden CL, Luu TV, Servetas SL, Mateczun AJ, et al. Comparison of three next-generation sequencing platforms for metagenomic sequencing and identification of pathogens in blood. BMC Genomics. 2014;15:96. doi: 10.1186/1471-2164-15-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiao X, Zheng X, Ma L, Kutty G, Gogineni E, Sun Q, et al. A benchmark study on error assessment and quality control of CCS reads derived from the PacBio RS. J Data Mining Genomics Proteomics. 2013;4(3):136. doi: 10.4172/2153-0602.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 31.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma L, Chen Z, Huang DW, Kutty G, Ishihara M, Wang H, et al. Genome analysis of three Pneumocystis species reveals adaptation mechanisms to life exclusively in mammalian hosts. Nat Commun. 2016;7:10740. doi: 10.1038/ncomms10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McElroy K, Thomas T, Luciani F. Deep sequencing of evolving pathogen populations: applications, errors, and bioinformatic solutions. Microb Inform Exp. 2014;4(1):1. doi: 10.1186/2042-5783-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sassi M, Ripamonti C, Mueller NJ, Yazaki H, Kutty G, Ma L, et al. Outbreaks of Pneumocystis pneumonia in 2 renal transplant centers linked to a single strain of Pneumocystis: implications for transmission and virulence. Clin Infect Dis. 2012;54(10):1437–44. doi: 10.1093/cid/cis217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melnikov A, Galinsky K, Rogov P, Fennell T, Van Tyne D, Russ C, et al. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011;12(8):R73. doi: 10.1186/gb-2011-12-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]