

Abstract
Background
Low-copy-number vectors of potential wide application in biotechnology need to encode stabilization modules ensuring their stable inheritance. The efficiency of stabilization may vary depending on the plasmid host so a thorough analysis of stabilization functions is required before use.
Results
To facilitate such analysis highly unstable, mobilizable, broad-host-range (BHR) vectors based on RK2 replicon were constructed. The vectors are suitable for testing of various stabilization functions, including plasmid and chromosomal partitioning cassettes encoding ParB homologues capable of spreading on DNA. The xylE or lacZ reporter systems facilitate easy monitoring of plasmid segregation.
Conclusion
The range of BHR vectors with different reporter cassettes and alternative mobilization systems expands their application in diverse bacterial species.
Keywords: Broad-host-range, Cloning vector, RK2, lacZ, xylE reporter, Stability functions
Background
The stabilization functions carried by low-copy-number plasmids from a wide range of bacteria ensure their stable inheritance during cell division [1]. Putative stabilization modules (e.g., partitioning operons, toxin-antitoxin systems, restriction-modification mechanisms) are also encoded on bacterial chromosomes [2–6]. Such modules could be used to construct vectors for biotechnological applications. The properties of the stabilization modules may vary depending on the host they are expressed in, so a thorough analysis is required before use.
Several test vectors are available for studying stabilization functions in bacteria. Most of them rely on narrow-host-range replicons and can be used only in certain E. coli strains or other narrowly defined hosts [7, 8]. pALA136 [9] and pOG04 [10] are based on dual pMB1 and P1 or P7 replicons, respectively. The high-copy-number pMB1 replicon requires PolI for replication, so the plasmid is stable in polA+ strains but when transformed into a polA mutant, it depends on the phage vegetative replication system and consequently becomes highly unstable as a single-copy molecule unless a stabilization cassette is included.
The standard method for testing putative stabilization functions relies on a classical segregation test, in which a strain with the plasmid is cultured for a certain number of generations without selection and then the number of cells still carrying the plasmid is estimated by the time-consuming replica plating of colonies or serial dilutions (drop test). Introduction of the reporter gene lacZ in pOU82 [11] simplifies the screening for plasmid loss, but this very useful test vector can only be applied for E. coli and its closely related species since it relies on the narrow-host-range R1 replicon of the IncFII incompatibility group [12].
This paper presents a set of highly unstable broad-host-range plasmids based on the RK2 replicon of IncP-1 group [13] designed to test stabilization functions in diverse bacterial species. pABB35 and its derivatives are single-copy vectors specifying chloramphenicol resistance (CmR). The multiple cloning site (MCS) is flanked by lacO operators serving as binding sites for LacI repressor to build a roadblock for polymerizing ParB-type proteins encoded by the tested partitioning cassettes of type IA [14, 15]. The unstable vectors contain the xylE (klcApRA3-xylE-Tpro/lyzP1) or lacZ (klcApRA3-lacZ-Tpro/lyzP1) reporter gene enabling easy and quick detection of bacterial colonies retaining the plasmid with the potential stabilization cassette. The plasmid segregation process can also be monitored in liquid cultures by a quantitative XylE activity assay. Variants of the unstable vector mobilizable by the RK2 (IncP-1) conjugative system integrated into the E. coli chromosome or by the RA3 (IncU) one integrated into the Pseudomonas putida chromosome are also available.
Results and discussion
Construction of a highly unstable broad-host-range plasmid
The main aim of this project was to engineer an unstable cloning vector suitable for easy monitoring of segregation functions in a wide range of bacteria.
We chose pRK415 [16], a derivative of the RK2 replicon from the IncP-1α incompatibility group, to construct a highly unstable broad-host-range (BHR) test vector.
The pRK415 cloning vector contains four following RK2 fragments: i/ a region encoding Ssb (single-stranded DNA-binding protein), the replication initiator protein TrfA, Upf16.5 of unknown function [13], and TrbA, a regulatory protein of RK2 conjugative transfer operons [17]; ii/ oriVRK2 with eight iterons constituting TrfA binding sites [18, 19]; iii/ part of the central control operon korA-incC encoding KorA, the primary repressor of trfAp [20], since strong trfAp is unclonable when unregulated [21], and iv/ the traJ traK intergenic region with oriTRK2 to facilitate mobilization by the RK2 conjugation system [22]. Additionally, the vector carries a tetracycline resistance cassette (TcR) and a lacp-lacZ fragment with MCS for α-complementation and easy identification of cloned inserts. pRK415 had previously been reported as slightly unstable [16, 23], but our plasmid retention tests showed its almost 100 % stability, since after 40 generations of growth of E. coli DH5α (pRK415) under non-selective conditions in L broth without antibiotics almost 100 % of cells retained the plasmid (Fig. 2a).
Fig. 2.
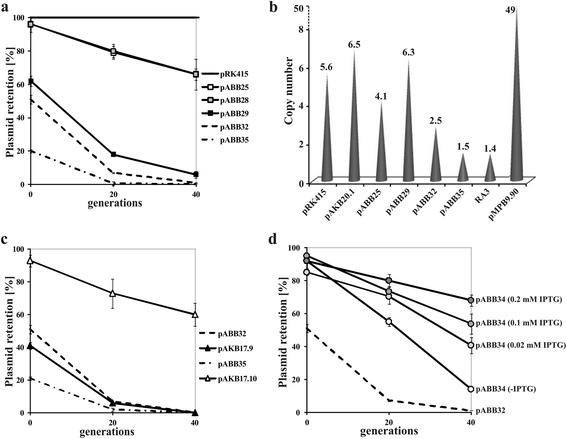
Standard stability tests of constructed vectors. E. coli DH5α transformants were grown overnight with antibiotic (point 0) and for 40 generations without antibiotic. Every 20 generations appropriate dilution was plated on L agar to obtain approximately 100 colonies. The colonies were replica plated to test for chloramphenicol resistance. Plasmid retention was expressed as percentage of CmR colonies. The results shown are average from three experiments with standard deviation. a Retention of constructed vectors. b Plasmid copy number estimated by RealTime qPCR. Plasmid copy relative to the chromosome was assayed in three independent biological samples with three technical replicates each. Average results for plasmids are presented with SD as follows: 0.06; 1.75; 0.21; 0.16; 0.95; 0.32; 0.17; 6.96, respectively. c Stabilizing properties of active partitioning operon from RA3 in pABB32 and pABB35 vectors. d Effect of IPTG-induced expression of P. aeruginosa parA-parB operon on pABB34 plasmid retention. DH5α(pABB34) cultures were grown in L broth with various concentrations of IPTG
The strategy to obtain a truly unstable derivative of the RK2 minireplicon was to limit the expression of trfA, first by introducing a promoter-down mutation in trfAp and, if required, by adding KorB, a second repressor acting cooperatively with KorA on trfAp [24], to the system. It has previously been shown that the T → C mutation in the -10 sequence of trfAp (trfAp-1) decreases the promoter activity at least 10-fold [17]. Site-directed PCR mutagenesis was used to introduce trfAp-1 mutation together with an AatII restriction site into pRK415. Plasmid DNA sequencing confirmed the introduction of the desired mutation into the -10 sequence of trfAp, but also an unexpected deletion of 1974 bp encompassing lacZα with MCS and the traJ-traK region with oriTRK2. The obtained 8716-bp derivative pAKB20.1 (Fig. 1a) was still very stably maintained in E. coli DH5α cells demonstrating 100 % retention after 40 generations of growth under non-selective conditions (data not shown).
Fig. 1.
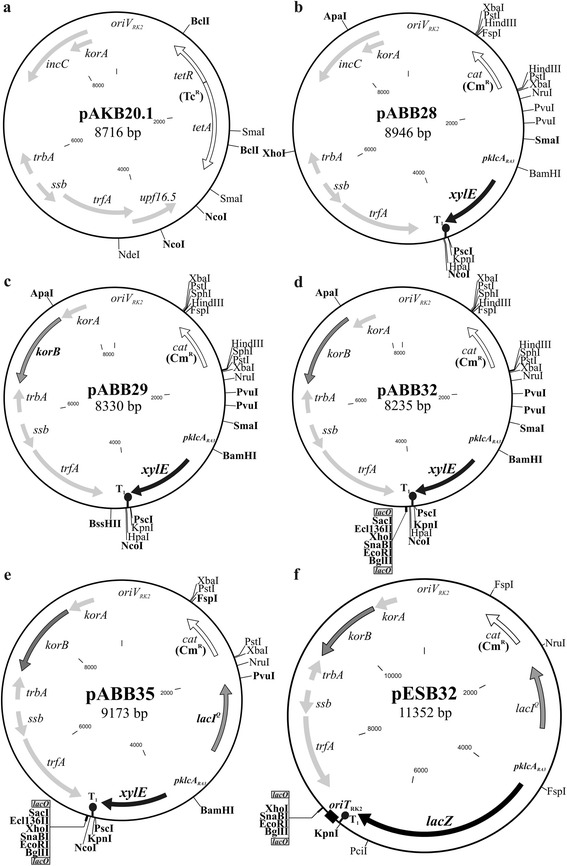
Milestones in construction of unstable BHR vector. Circular maps of intermediate (a, b and c) and final vectors (d, e and f) are drawn to scale. Only intact orfs are indicated. Unique or double restriction sites important for cloning are shown, those described in the text are in bold. T1 marks the divergent transcription terminator sequence Tpro/Tlyz of P1 prophage [49]
To facilitate insertion of potentially large DNA fragments bearing stability cassettes it was required to downsize the cloning vector. Hence, pAKB20.1 was modified by NcoI digestion and self-ligation to delete a 527-bp fragment of the upf16.5 gene of unknown function [13], the last orf in the ssb-trfA-upf16.5 operon present only in one subgroup of IncP-1 plasmids (IncP-1α). Although an Upf16.5 function in copy number control (e.g., efficient replication initiation) has not been reported yet, the new derivative, pABB25, was slightly less stable in E. coli DH5α cells in comparison to pAKB20.1 and after 20 generations of growth without selection 20 % of cells lost the pABB25 plasmid (Fig. 2a). In the next step the tetracycline resistance operon (TcR) of pABB25 was replaced with a shorter cat gene (encoding chloramphenicol acetyltransferase) conferring the CmR phenotype. The resulting plasmid pABB25.1 (CmR) demonstrated stability in E. coli DH5α comparable to that of pABB25 (data not shown).
The initial manipulations did not sufficiently decrease the stability of the RK2 minireplicon, so it was decided to proceed with the addition of korB encoding a co-repressor of trfAp, to the system. It was also important to inactivate incC since IncC and KorB constitute the active partitioning system of RK2 [25, 26]. Two restriction sites, ApaI and XhoI were introduced into pABB25 to facilitate substitution of incC2 orf with korBRK2 and to give pABB27. Before korB cloning, a SmaI-NcoI fragment encoding klcApRA3-xylE-Tpro/lyzP1 was inserted into pABB27 between the trfA and cat genes to give pABB28 (Fig. 1b). klcApRA3 is a strong promoter and xylE encodes catechol 2,3- dioxygenase, whose activity is easy to be monitored following bacteria growth on plates [27] or in liquid cultures (this work). Subsequently, the korBRK2 was inserted into pABB28 between the XhoI and ApaI sites. The obtained pABB29 plasmid (Fig. 1c) demonstrated a high loss rate in E. coli DH5α strain (Fig. 2a). pABB29, comprising a modified RK2 replication system, the korA-korB operon, the klcAp-xylE-Tpro/lyzP1 reporter cassette and the Cm resistance marker, was used in the next step to prepare the final version of the unstable BHR vector for cloning and testing stabilization cassettes in a wide range of bacterial species.
The MCS introduced between the unique NcoI and BssHII (PauI) sites in pABB29 to give pABB32 (Fig. 1d) contained BglII, EcoRI, SnaBI, XhoI, Ecl136 and SacI restriction sites and was surrounded by lacO operator sequences. The binding of LacI repressor to the lacO operators was expected to act as a roadblock [28] for potentially polymerizing ParB partitioning proteins encoded by type IA partition cassettes [14, 15] that might be analyzed using this vector. ParB spreading that follows its binding to parS (centromere-like sequence) may lead to transcriptional silencing of nearby genes and therefore affect results of segregation studies [29–33].
Finally, the lacIq allele [34, 35] was inserted into pABB32 to obtain the final construct, the pABB35 vector (Fig. 1e). The lacIq mutation refers to a change in the −35 motif of lacIp causing overexpression of lacI (superrepressor) [36] and is often used in recombinant strains or vectors to provide tighter control of lacZp (or hybrid tacp) expression in the absence of the IPTG inducer.
The high instability of pABB35 was confirmed by the standard stability test: only approximately 2 % of cells retained the plasmid after 20 generations of growth without selection (Fig. 2a).
Plasmid copy number
The copy number of chosen plasmid constructs described above was determined in E. coli cells by qPCR [37]. The pRK415 derivatives pAKB20.1, pABB25 and pABB29 were present in 4 to 6.5 copies per chromosome, similarly to pRK415 itself (Fig. 2b). The number of pABB32 and pABB35 copies was 1.5 - 2.5 per E. coli chromosome. This lower copy number is due to the tight regulation of trfAp-1 and underlies the instability of these test vectors. The copy number of pMPB9.90 araBADp-trfA, based on pBAD24 [38], was established at 50 copies per chromosome in correlation with a published data [39]. For comparison also a single-copy-number plasmid RA3 of IncU group [40] was used and demonstrated 1–2 copies per chromosome (Fig. 2b).
Plasmidic and chromosomal partitioning cassettes stabilize test plasmids in E. coli
The type IA active-partitioning cassette korA-incC-korB-orf11-parS from RA3 plasmid [40, 41] was chosen to check the applicability of the constructed vectors in a stabilization assay in bacteria. The cassette was cloned into pABB32 and pABB35 vectors to obtain pAKB17.9 and pAKB17.10, respectively, and both plasmids were tested in the standard stabilization assay in E. coli DH5α strain.
The RA3 partitioning cassette did not drastically improve the pABB32 plasmid segregation rate: approximately 10 % of cells retained the pAKB17.9 plasmid after 20 generations of growth without selection (Fig. 2c). Remarkably, the same RA3 fragment cloned into pABB35 exhibited the expected stabilization function and pAKB17.10 was retained in approximately 70 and 60 % of E. coli DH5α cells after 20 and 40 generations of growth without selection, respectively (Fig. 2c). The only difference between pABB32 and pABB35 is the presence of the lacIq allele in the latter (Fig. 1d and e). Since it has been shown previously that KorBRA3 (a ParB homolog) spreads on DNA after binding to parS and silences nearby genes [41], the different stability of pAKB17.9 and pAKB17.10 convincingly demonstrates that overproduction of LacI and its binding to lacO sequences flanking the cloned stabilization cassette blocks effectively the KorB spreading.
The usefulness of the constructed vectors was also checked with a synthetic chromosomal partition cassette lacIq-tacp-parA-parB-parS from P. aeruginosa [29] cloned into pABB32 to give pABB34.
The pABB34 plasmid was present in more than 50 % of E. coli cells after 20 generations of growth without selection, in comparison to only 10 % of cells retaining empty pABB32 (Fig. 2d). After 40 generations pABB34 was still present in 14 % of cells, whereas the retention of pABB32 dropped below 1 %.
The stabilization effect of the RA3 partitioning cassette cloned in pAKB17.10 was stronger than that demonstrated by the parA-parB-parSP.a cassette present in pABB34 (Fig. 2c and d). These differences in the stabilization potential could reflect the individual properties of each cassette, but the rather modest effect of the synthetic parA-parB-parS cassette of P. aeruginosa could also be due to the low amount of partitioning proteins produced since the parA-parB operon in pABB34 is expressed at a low basal level from the strongly repressed tacp. To check which explanation was correct, different concentrations of IPTG were used to boost the production of the partition proteins ParA and ParB. In support of the latter possibility IPTG at 0.02-0.2 mM improved the stability of pABB34 (Fig. 2d). A further increase in IPTG concentration (0.5 mM) did not improve the plasmid stability (data not shown) probably due to the antagonistic effect of IPTG on the action of LacI as the roadblock to ParB, known to spread on DNA starting from parS [29].
Catechol 2,3-dioxygenase activity assay as an estimate of plasmid stability
The results presented in Fig. 2 come from a standard stabilization assay with the use of replica plating to estimate the proportion of colonies retaining the plasmid tested (in this case, conferring resistance to chloramphenicol). The xylE reporter cassette present in pABB32 and pABB35 allows the plasmid segregation to be assayed using a simple plate test to visualize colonies that express the xylE reporter gene and hence must have retained the plasmid.
The cultures of transformants were grown without selection for a certain number of generations, diluted and plated onto L agar without antibiotics to get 100 to 200 colonies. The colonies were sprayed with 10 mM catechol and those derived from cells that had lost the test plasmid with xylE remained opalescent, those in which the test plasmid was stably maintained turned yellow quickly (Fig. 3a), whereas colonies of strains carrying an unstable plasmid with the xylE gene, were in various shades of yellow.
Fig. 3.
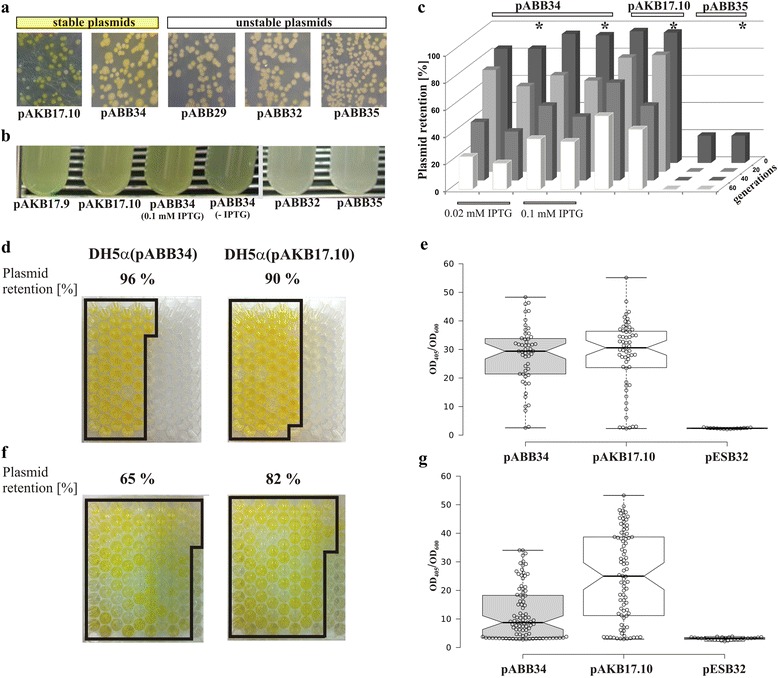
Detection of plasmid retention on plates, in liquid cultures and by high-throughput quantification. a Transformants of E. coli DH5α with stable (pAKB17.10 or pABB34) and unstable (pABB29, pABB32 or pABB35) plasmids were grown for 20 generations without selection and IPTG and then 100 μl of 106-fold dilutions was plated on L agar, sprayed with 10 mM catechol and photographed. b Overnight cultures of E. coli DH5α transformants bearing indicated plasmids were grown for 20 generations in L broth and 0.1 mM IPTG where marked. Catechol was added to 1 mM to the cultures and after 5 min of incubation tubes were photographed. c Comparison of segregation data obtained in the standard replica plating test and visualization test (marked by asterisk) with the use of catechol as in (a). The results shown are representative of three independent experiments. d/f High-throughput quantification of plasmid retention in single-cell subcultures. Overnight cultures of DH5α(pAKB17.10) and DH5α(pABB34) grown in L broth with antibiotic (no IPTG added) were diluted to 5 cells ml−1 in L broth and aliquoted into 100-well plates (200 μl/well). After growth in Bioscreen (ca. 20 generations) the subcultures were diluted 100-fold into a new plate and 1/10 vol of 10 mM catechol was added to each well and photographed (d). Similar tests (f) were carried out after 40 generations of growth without selection (24 h in tubes followed by 24 h in Bioscreen plate). The photographs were taken after 10 min of incubation with catechol. The plasmid retention in liquid cultures (initial and after 20 generations of growth without selection in tubes) corresponds to the percentage of single-cell subcultures turning yellow. e/g The colour development quantified by OD405 after addition of catechol to the single-cell subcultures in the wells from (d) and (f), respectively. DH5α(pESB32) strain was used as a negative control. Plasmid segregation during growth without selection in the wells for 20 generations (e) and for total 40 generations (g) was reflected by variable level of XylE activity (OD405). OD405/OD600 ratio for each culture was plotted using BoxPlotR (boxplot.tyerslab.com; [55]). Boxes indicate the 25th and 75th percentiles and center lines show the medians. Whiskers mark minimum and maximum values in accordance with Spear criteria, and non-overlapping notches indicate that population medians are different with 95 % confidence as determined by R software
A quick semi-quantitative test for plasmid stability can also be performed for liquid cultures directly. Addition of catechol to overnight cultures (2x109 cells ml−1) to 1 mM final concentration clearly distinguishes those in which XylE is produced by the majority of cells (the test plasmid is stably maintained) from the ones where the plasmid is hardly retained (high plasmid loss rate) (Fig. 3b). Care must be taken to determine the initial rate of reaction, i.e., to measure OD405 within a few minutes (2–5) after substrate addition [27, 34, 42].
Comparison of the standard plasmid stability assay with plate catechol 2,3-dioxygenase determination
E. coli DH5α transformants bearing appropriate test plasmids were cultivated in L broth without antibiotics as earlier and tested for plasmid retention after approximately 20, 40 and 60 generations using, in parallel, the standard stabilization assay and the xylE plate test described above. As shown in Fig. 3c the results of plasmid retention estimation are quite similar for the two assays, justifying the use of the quicker xylE test.
High-throughput analysis of plasmid stabilization functions
Plasmid retention was quantified in liquid cultures of DH5α(pAKB17.10) and DH5α(pABB34) using a high-throughput procedure. In this experiment the production of ParA and ParB in DH5α(pABB34) was not induced by IPTG to have two plasmids (pABB34 and pAKB17.10) stabilized to different extent by the various partition cassettes [compare Fig. 2c and graph marked pABB34 (no IPTG) in Fig. 2d]. DH5α(pABB35) and DH5α(pESB32) strains were used as controls. Cultures of the transformants were grown overnight under selection (10 μg ml−1 chloramphenicol) and then diluted to 5 cells ml−1 in L broth and aliquoted into 100-well plates (200 μl per well). The plates were incubated at 37 °C with shaking for ca. 20 generations. In parallel the overnight start cultures were diluted 105-fold and grown in tubes for 20 generations without selection and then diluted, aliquoted and incubated as above (a total of 40 generations without selection). Overnight subcultures derived from single cells and grown in the 100-well plates were diluted 100-fold, OD600 values were measured and after addition of 10 mM catechol to each well (final concentration 1 mM) OD405 was read after 10 min. The results obtained after 20 generations for DH5α(pABB35) showed an almost complete lack of the plasmid indicating its high instability (only ca. 1.5 % of wells showed OD405 above background values obtained for the strain bearing pESB32 without xylE, data not shown). In the case of DH5α(pABB34) and DH5α(pAKB17.10) XylE activity was clearly detectable in 96 and 90 % of wells, respectively, after 20 generations of growth without antibiotic (Fig. 3d). After 40 generations of growth without antibiotic the corresponding values were 65 and 82 % for DH5α(pABB34) and DH5α(pAKB17.10), respectively (Fig. 3f). These results reflected well the differences in stability of the two plasmids observed in the standard and colony visualization assays when DH5α(pABB34) was grown without IPTG (Fig. 2d).
To normalize obtained data the OD405/OD600 ratio was calculated. It was around 2–3 for the control DH5α(pESB32) strain and varied between 2 and 50 for the DH5α(pABB34) and DH5α(pAKB17.10) strains (Fig. 3e and g). When the ratios were plotted and analyzed, the medians reflecting plasmid retention rates in the subcultures were similar for the two plasmids following growth without selection for 20 generations, and substantially higher for DH5α(pAKB17.10) compared with DH5α(pABB34) after 40 generations of growth without selection, as observed before.
The high-throughput approach is obviously more reliable than the standard and colony visualization method since human error is minimized and such a quantitative procedure may help to demonstrate even small, but statistically significant differences in stabilities between various plasmids in a given host, the same plasmid in various hosts, or between variants of the same plasmid.
Modifications of the test vectors to expand their applicability
The test plasmid pABB35 based on the RK2 minireplicon can propagate in a variety of species. Since many bacterial species are not easily transformable, two different oriT regions amplified from BHR conjugative plasmids, RK2 and RA3, were inserted additionally to pABB35 to give pESB30 and pESB31, respectively. Such vectors are mobilizable during conjugation of the recipient with two different bacterial species, E. coli S17-1 with integrated RK2 plasmid [43] or P. putida KT2440 with integrated conjugation module of RA3 (KT2440 traRA3), which may significantly extend the range of recipient strains.
The suitability of pESB30 vector for investigating stability mechanisms other than active partition was in the meantime confirmed by D. Bartosik’s group studying plasmidic toxin-antidote (TA) systems. The use of pESB30 vector with cloned hipAB system (TA) of pKON1 from Paracoccus kondratievae [44] allowed analysis of its stabilization functions in various species of Alphaproteobacteria e.g., P. pantotrophus and Ochrobactrum sp. (Czarnecki and Bartosik, personal communication).
Quick identification of colonies carrying the constructed vectors with the use of the color reaction enabled by xylE cassette could not be applied to some analyzed species e.g., P. aeruginosa, since when PAO1161 was transformed with xylE plasmids it formed yellow colonies due to the intrinsic substrates for catechol 2,3-dioxygenase. An alternative test vector, pESB32 (Fig. 1f), was constructed with the klcAp-lacZ cassette enabling monitoring of plasmid presence by formation of blue colonies in the presence of X-gal.
The partition operon korA-incC-korB-orf11-parS of RA3 was inserted between the EcoRI and XhoI sites in pESB32 to give pESB34. P. aeruginosa PAO1161 was transformed with pESB32 or pESB34 (korA-incC-korB-orf11-parS) and transformants were used to estimate plasmid retention (Fig. 4a). pESB32 or pESB34 were also used to transform E. coli S17-1 and transformants were applied as donors in conjugation with a RifR derivative of A. tumefaciens LBA1010R. The transconjugants were grown under selective conditions, then diluted appropriately and plated on L agar with X-gal. The retention rates of both plasmids assessed by the number of blue colonies are shown in Fig. 4b. pESB32 was less unstable in the both strains tested in comparison with the original pABB35 in E. coli DH5α (Fig. 2a) probably due to variations in the functioning of the copy-number control circuit of the RK2 minireplicon. The presence of the RA3 partition cassette still significantly increased the pESB34 retention in P. aeruginosa PAO1161 strain but had a much lower impact on plasmid stability in A. tumefaciens LBA1010R. The reasons for the observed differences in empty vector stability and the stabilization effects of a given cassette in various bacterial species await elucidation.
Fig. 4.
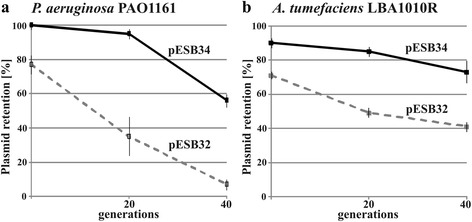
Functionality of test vectors in various bacterial species. a P. aeruginosa PAO1161 was transformed with pESB32 and pESB34 (korA-incC-korB-orf11-parS). Three transformants were grown for 40 generations without selection. The retention of plasmid was estimated by percentage of blue colonies on L agar plates with X-gal. The results shown are average from three experiments with standard deviation. b A. tumefaciens LBA1010R RifR was conjugated with E. coli S17-1(pESB32) and S17-1(pESB34) donor strains. Obtained transconjugants were selected on L agar supplemented with chloramphenicol and rifampicin. Three independent transconjugants were grown for 40 generations without selection. The retention of plasmid was estimated by percentage of blue colonies on L agar plates with X-gal. The results shown are average from three experiments with standard deviation
Conclusions
We have manipulated the broad-host-range RK2 minireplicon pRK415 to obtain a highly unstable pABB35 vector. To facilitate easy monitoring of plasmid retention, a xylE or lacZ reporter system was inserted. A multiple cloning site surrounded by roadblocks for protein spreading enables analysis of various partition cassettes. To broaden applications of the vector to a variety of hosts we added oriT regions from RK2 or RA3 BHR plasmids so they could be mobilized by the widely used S17-1 (with RK2 integrated) and P. putida KT2440 with the tra module of RA3 constructed in this work, respectively.
The constructed vectors were demonstrated to be useful for cloning chromosomal and plasmid partition operons that produce type IA ParB-like proteins able to spread on DNA. Retention of the vectors varied depending on the host so they need to be tested in a given strain prior to application. Analysis of the stabilization properties of the cloned partitioning operons in three different hosts (E. coli, P. aeruginosa, A. tumefaciens) confirmed their variability and at the same time necessity to conduct such experiments.
Estimation of plasmid stability using catechol 2,3-dioxygenase assay in liquid cultures facilitates large scale or high-throughput experiments in bacteria, since hundreds or even thousands of variants can be monitored easily. It can be used to screen for new stabilization cassettes in meta-genomic approaches, to study stability functions in diverse bacteria and to screen mutant proteins affecting plasmid stability as well as inhibitors of the systems.
The test plasmids described here with an easy detection/monitoring system should be useful for studies of various stabilization functions in a wide range of strains in which the RK2 replicon can propagate.
Methods
Bacterial strains and growth conditions
Escherichia coli strains used were DH5α [F-(Φ80dlacZ recA1 endA1 gyrA96 thi-1 hsdR17(rk−mk+) supE44 relA1 deoR Δ(lacZYA-argF)U196] [45] and S17-1 [recA pro hsdR RP4-2-Tc::Mu-Km::Tn7] [43]. DH5α RifR mutant was selected during growth on L agar with 150 μg ml−1 rifampicin. P. putida KT2440 was kindly provided by C.M. Thomas (Birmingham University, Birmingham, United Kingdom). P. putida KT2440 traRA3korC KmR was constructed by integration of pJSB1.28 into a non-coding region of chromosome using homologous recombination (see below). P. aeruginosa strain PAO1161 (leu−, r−, m+) was kindly provided by B.M. Holloway (Monash University, Clayton, Victoria, Australia). Agrobacterium tumefaciens LBA1010R RifR was kindly provided by D. Bartosik (University of Warsaw, Warsaw, Poland). Bacteria were generally grown in L broth [46] at 37 °C or 28 °C (A. tumefaciens). L broth and L agar (L broth with 1.5 % w/v agar) were supplemented with appropriate antibiotics: chloramphenicol (10 μg ml−1 for E. coli, 50 μg ml−1 for A. tumefaciens and 150 μg ml−1 for P. aeruginosa), kanamycin (50 μg ml−1 for E. coli and P. putida) or tetracycline (10 μg ml−1 for E. coli). Strains with plasmids carrying the klcAp-lacZ fusion were tested on L agar with 40 μg ml−1 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal).
Plasmid analysis and PCR amplification
Plasmid manipulations were carried out by standard procedures [47]. All plasmids constructed in this work are listed in Table 1.
Table 1.
Plasmids used in this study
| Plasmids provided by others or reported earlier: | ||
|---|---|---|
| Name | Relevant features | References |
| pABB19 | oriV MB1, ApR, derivative of cloning vector pUC19 with added transcription termination sequence Tpro/Tlyz from P1 | [49] |
| pABB705 | pKRP10 derivative with inactivated NcoI and PvuII sites in CmR cassette | [49] |
| pALA136 | oriV MB1, oriV P1, CmR , dual replicon | [9] |
| pBGS18 | oriV MB1, KmR, cloning vector | [56] |
| pBAD24 | oriV MB1, ApR, araC, araBADp, expression vector | [38] |
| pCM132 | oriV ColE1, oriV RK2, oriT RK2, traJ’, trfA, KmR, promoter-less lacZ, dual replicon | [52] |
| pGBT30 | oriV MB1, ApR, lacI q , tacp expression vector | [34] |
| pGEM-T-Easy | oriV MB1, ApR, cloning vector | Promega |
| pJSB1.24 | pBGS18 tra RA3 korC RA3 (RA3 coordinates 9437- 33857; 3093-3705) | [49] |
| pKLB3 | pGBT30 tacp-parA parB P.a | [29] |
| pKRP10 | oriV MB1, ApR, CmR | [50] |
| pMPB9.90 | pBAD42 araBADp-trfA RK2 | Przyluski M. |
| pPT01 | oriV pSC101, KmR, promoter-less xylE | [10] |
| pRK415 | oriV RK2, TcR, oriT RK2, stable vector | [16] |
| pYC16A | pALA136 with RA3 stabilization region | [41] |
| RA3 | IncU, CmR, SmR, SuR | [40] |
| RK2 | IncP-1α, KmR, ApR, TcR | Thomas C.M. |
| Plasmids constructed during this work: | ||
| Description, relevant features | ||
| pABB18.1 | pPT01 klcAp -xylE, PCR fragment amplified with primers #1 and #2 on RA3 template inserted as SphI-BamHI fragment | |
| pABB18.2 | pABB18.1 cleaved with HpaI and NcoI, filled in and self-ligated to remove 561 bp upstream of xylE | |
| pABB18.3 | pABB19 with klcAp-xylE inserted as SmaI-PscI PCR fragment amplified with primers #3 and #4 on pABB18.2 template | |
| pABB18.4 | pBGS18 with korB RK2 inserted as EcoRI-SalI PCR fragment amplified with primers #5 and #6 on RK2 template | |
| pABB18.5 | pGEM-T-Easy with lacI q gene PCR-amplified primers #11 and #12 on pGBT30 template | |
| pABB25 | pAKB20.1 with 527-bp NcoI fragment removed | |
| pABB25.1 | pABB25 with cat gene (CmR) on BamHI fragment from pABB705 replacing BclI fragment from TcR cassette | |
| pABB26 | pABB25.1 with XhoI restriction site introduced downstream of trbA (PCR directed mutagenesis with primers #20 and #21) | |
| pABB27 | pABB26 with ApaI restriction site downstream of korA gene (PCR directed mutagenesis with primers #22 and #23) | |
| pABB28 | pABB27 with klcp-xylE cassette, SmaI-NcoI fragment from pABB18.3 | |
| pABB29 | pABB28 with korB RK2 gene, ApaI-SalI fragment from pABB18.4 inserted between ApaI and XhoI sites | |
| pABB32 | pABB29 with MCS flanked with lacO operators inserted between BssHII and NcoI sites, unstable vector | |
| pABB33 | pABB32 with lacI q -tacp-parA-parB P.a., DraI-SalI fragment of pKLB3 inserted between SnaBI and XhoI sites | |
| pABB34 | pABB33 with parS P.a., annealed oligonucleotides #9 and #10 inserted into BglII restriction site | |
| pABB35 | pABB32 with lacI q, NruI-PvuI fragment of pABB18.5 cloned between SmaI and PvuI sites, unstable vector | |
| pAKB17.9 | pABB32 with RA3 active partition cassette (korA-incC-korB-orf11-parS), EcoRV-BamHI fragment from pYC16A inserted between Ecl136II and BglII sites | |
| pAKB17.10 | pABB35 with RA3 active partition cassette (korA-incC-korB-orf11-parS), EcoRV-BamHI fragment from pYC16A inserted between Ecl136II and BglII sites | |
| pAKB20.1 | pRK415 TcR with trfAp-1 introduced by PCR mutagenesis with primers #18 and #19 and spontaneous deletion of 1974-bp fragment encompassing MCS, traJ and oriT | |
| pESB3.6 | pUC18 with synthetic RA3 partition cassette (korA-incC-korB-orf11-parS), cloned between EcoRI and SalI sites (RA3 coordinates 5940-9800) | |
| pESB30 | pABB35 with oriT RK2 , 218-bp fragment PCR-amplified on RK2 template using primers #13 and #14, cleaved with PscI and cloned into NcoI site, unstable, RK2 mobilizable vector | |
| pESB31 | pABB35 with oriT RA3, 166-bp fragment PCR-amplified on RA3 template using primers #15 and #16, cleaved with PscI and cloned into NcoI site, unstable, RA3 mobilizable vector | |
| pESB32 | pESB30 with klcAp-lacZ, BglII-NcoI fragment from derivative of pCM132 inserted between BamHI and PscI sites, unstable, RK2 mobilizable vector | |
| pESB34 | pESB32 with synthetic RA3 partition cassette (korA-incC-korB-orf11-parS), EcoRI-SalI fragment from pESB3.6 cloned between EcoRI and XhoI sites | |
| pJSB1.28 | pJSB1.24 Ppu618, 618-nt PCR-amplified fragment of P. putida chromosome, coordinates 58074-58691 | |
Vectors for cloning and analysis of stabilization cassettes are in bold
Standard PCR [48] was performed with pairs of primers listed in Table 2.
Table 2.
Oligonucleotides used in this study
| No | Name | Sequence |
|---|---|---|
| 1 | klcApRA3L | GCGCATGCGGGAGCGTGATCGTTACGGT |
| 2 | klcApRA3R | GCGGATCCATTGCAGCCATACGGCGAGG |
| 3 | klcAsmaF | GGCCCGGGTGCTCGTCTCGTCGGTCTG |
| 4 | xylEPscR | GGACATGTCATCTGCACAATCTCTGCA |
| 5 | KBRK2Apa | CCGAATTCGGGCCCGAAGATGGAGATTTCCCAATGACTGC |
| 6 | KBRK2Sal | CCGTCGACCGCTGTCTTTGGGGATCAGCCCTC |
| 7 | lacOMCS1 | CATGGAATTGTGAGCGCTCACAATTTCAGATCTGAATTCTACGTACTCGAGCTCGGAATTGTGAGCGCTCACAATTTCA |
| 8 | lacOMCS2 | CGCGTGAAATTGTGAGCGCTCACAATTCCGAGCTCGAGTACGTAGAATTCAGATCTGAAATTGTGAGCGCTCACAATTC |
| 9 | parSbg2a | GATCGGTTGCTTGTTCCACGTGGAACAAGGCCG |
| 10 | parSbg2b | GATCCGGCCTTGTTCCACGTGGAACAAGCAACC |
| 11 | NrulacIF | GGTCGCGACTGAATCCGGTGAGAATGG |
| 12 | PvulacIR | CGCGATCGATAAGCTTGCAATTCGCG |
| 13 | oriTRK2Fs | ACGGTCGACACATGTCTGGTTGGCTTGGTTTCATC |
| 14 | oriTRK2Rs | CGGAATTCACATGTTTGCCAAAGGGTTCGTGTAG |
| 15 | oriTRA3F | CGCGTCGACACATGTTTTAGCACAAGCGGCGGCAG |
| 16 | oriTRA3R | CGGAATTCACATGTAGTTAGGGGAAGCCGACGAG |
| 17 | Nco | ACCATGGTCATG |
| Primers used in site-directed mutagenesis | ||
| 18 | muttrfA RK2 | GTCCTTGAGAAAGGAGACgtc gGTTTAGCTA |
| 19 | muttrfA RK2 | CCAATGTTTAGCTAAACc gacGTCTCCTTTC |
| 20 | xhoF8180 | CGGGCCGTCGGCTCGaGCATCATATCGAC |
| 21 | xhoR8180 | CGATATGATGCtCGAGCCGACGGCCCGC |
| 22 | apaF9950 | CTTTCTGGTTGGCCGgGcCCAAAGTTTTtATCGTTTGGTTTCC |
| 23 | apaR9950 | GAAACCAAACGATaAAAACTTTGGgCcCGGCCAACCAGAAAGGC |
| 24 | Ppu618F | CGCTGCAGAGGCCAGACCCCGTGAAATT |
| 25 | Ppu618R | GCAAGCTTGGTCAGCATAGTCCACCTCA |
| Primers used for Real Time qPCR | ||
| 26 | galKF | ATGATCTTTCTTGCCGAGCG |
| 27 | galKR | AGCAGCTTTATCATCTGCCGC |
| 28 | trfAF | GTGAAGATCACCTACACCGGC |
| 29 | trfAR | TGGCAAAGCTCGTAGAACGTG |
| 30 | repBRA3F | CATCGAGAAGCAAAAGGCG |
| 31 | repBRA3R | CCAACTTGCGTAGGTCTTCCAG |
Restriction enzyme recognition sites are in bold, mutated nucleotides are indicated by lowercase, parS palindrome is underlined
PCR-based site-directed in vitro mutagenesis was performed with mutagenic primers (Table 2) as described previously [49]. The PCR mixture after mutagenesis was treated for 1-2 h with 10 U of DpnI restriction enzyme in order to eliminate template DNA and was used for transformation of E. coli DH5α.
All new plasmid constructs were verified by sequencing at the DNA Sequencing and Oligonucleotide Synthesis Laboratory, Institute of Biochemistry and Biophysics, using dye terminator sequencing and an ABI 377 Perkin Elmer automated sequencer. Sequences were analyzed using Clone Manager 9.
Plasmid construction
I/Construction of unstable BHR vector
Site-directed PCR mutagenesis with primers #18 and #19 introducing the T → C mutation in the -10 motif of trfAp together with an AatII restriction site into pRK415 was carried out to give pAKB20.1 (Fig. 1a).
pAKB20.1 was modified further by NcoI digestion and self-ligation to delete a 527-bp fragment of the upf16.5 gene from trfA operon to downsize the vector and to give pABB25.
In the next step the tetracycline resistance operon (TcR) of pABB25 was replaced with a shorter cat gene (encoding chloramphenicol acetyltransferase) conferring the CmR phenotype. The BamHI fragment with the CmR cassette from pABB705 [49], a derivative of pKRP10 [50], with NcoI and PvuII restriction sites eliminated was inserted between the BclI sites to give pABB25.1.
Site-directed mutagenesis was used to introduce a unique XhoI site downstream of trbA (primers #20 and #21) and an ApaI site downstream of korA in the korA-incC operon (primers #22 and #23) to give pABB27. korA overlaps the 5’ end of incC1 (incC encodes two forms of partition protein IncC1/IncC2 with two translational starts [51]) but is translated in a different reading frame, hence the mutagenic primers #22 and #23 introduced a stop codon precluding IncC1 translation.
The new vector pABB27 was further modified by insertion of the reporter gene cassette klcAp-xylE between the trfA and cat genes to give pABB28 (Fig. 1b).
The korBRK2 gene was PCR-amplified with primers #5 and #6 on total DNA from E. coli DH5α(RK2) and inserted as a SalI-ApaI fragment into pABB28 digested with XhoI and ApaI to give pABB29 (Fig. 1c).
The unique NcoI and BssHII (PauI) restriction sites in pABB29 were used to introduce a new MCS sequence made from annealed oligonucleotides #7 and #8, yielding pABB32 (Fig. 1d).
lacIq was amplified on pGBT30 [34] using primers #11 and #12 and inserted as NruI-PvuI fragment into pABB32 between SmaI and PvuI sites to obtain the final pABB35 vector (Fig. 1e).
PCR-amplified 218-bp oriTRK2 on RK2 template (primers #13 and #14) and 166-bp oriTRA3 PCR-amplified on RA3 template (primers #15 and #16), were inserted as PscI fragments into the unique NcoI site of pABB35 to give mobilizable vectors pESB30 and pESB31, respectively.
II/Construction of klcApRA3-xylE and klcApRA3-lacZ reporter cassettes
Two reporter genes, xylE of pWWO from P. putida encoding catechol 2,3-dioxygenase [10], and promoter-less lacZ from pCM132 coding for β-galactosidase [52], were cloned into appropriate vectors.
The strong promoter klcAp [40] was PCR-amplified on RA3 template using primers #1 and #2 and cloned as an SphI-BamHI fragment into pPT0I vector [10] upstream of the xyl operon to yield pABB18.1. Subsequently, pABB18.1 was cut with HpaI and NcoI, filled in with PolI Klenow fragment and self-ligated to remove a 561-bp fragment upstream of the xylE gene (pABB18.2). klcAp-xylE was PCR-amplified from pABB18.2 (primers #3 and #4) and cloned as a SmaI-PscI fragment into pABB19 [49] to provide a cassette with a bi-directional Rho-independent transcription terminator, giving pABB18.3. The role of the transcription terminator inserted at the end of the reporter gene cassette was to prevent transcriptional spillover from the strong klcA promoter and to protect the klcAp-xylE cassette against transcription coming from inserts in the constructed vectors, as a new MCS was planned to be cloned next to the NcoI site. The SmaI-NcoI fragment encoding klcApRA3-xylE-Tpro/lyzP1 was inserted into pABB27 to give pABB28.
pCM132 [52] carrying promoter-less lacZ orf was cut with SphI and ligated with self-annealed oligonucleotide #17 to remove the SphI and insert an NcoI restriction site downstream of lacZ (pCM132Nco). The BglII-NcoI fragment from pCM132Nco carrying a promoter-less lacZ cassette was inserted into pESB30 between the BamHI and PscI sites to replace xylE and transcriptionally fuse lacZ to the strong klcA promoter and to construct pESB32 (Fig. 1f).
III/Cloning the partitioning cassettes into the test vectors
Chromosomal parA-parB operon of P. aeruginosa had been cloned earlier under the control of tacp and lacIq in pKLB3 [29]. The DraI-SalI fragment of pKLB3 bearing lacIq-tacp-parA-parB was re-cloned between the SnaBI and XhoI sites of pABB32 (Fig. 1d), yielding pABB33. A centromere-like sequence parS2/3 [29] made from annealed oligonucleotides #9 and #10 (Table 2) was cloned into BglII-cut pABB33 to give pABB34 with the complete stabilization cassette from P. aeruginosa.
The EcoRV-BamHI fragment of pYC16A carrying the active partition cassette korA-incC-korB-orf11-parS from RA3 plasmid [40, 41] was cloned into pABB32 and pABB35 vectors digested with Ecl136II and BglII to obtain pAKB17.9 and pAKB17.10, respectively. In the case of pESB34, the EcoRI-SalI fragment from pESB3.6 carrying korA-incC-korB-orf11-parS of RA3 was cloned between the EcoRI and XhoI sites of pESB32.
IV/Construction of P. putida KT2440 traRA3korCRA3 KmR helper strain
pJSB1.28 is a high copy number plasmid based on pMB1 replicon unable to replicate in P. putida. It is derivative of pJSB1.24 that carries the conjugative transfer module of plasmid RA3 (RA3 coordinates 9437- 33857) together with the korC gene (RA3 coordinates 3093-3705) encoding an indispensable transcriptional repressor [49]. A short region of P. putida KT2440 chromosome (coordinates 58074-58691) was PCR-amplified with the use of primers # 24 and #25 (Table 2) and cloned between PstI and HindIII sites to facilitate integration of pJSB1.28 into the chromosome by homologous recombination. DH5α (pJSB28) donor strain was conjugated with P. putida KT2440 and integrants were selected on M9 plates [47] with 0.1 % glucose and kanamycin (50 μg ml−1) without thiamine to eliminate DH5α that requires thiamine to grow on minimal medium. The integration of pJSB1.28 into the chromosome of KT2440 traRA3korCRA3 KmR was verified by PCR.
Transformation and conjugation procedures
Competent E. coli and P. putida cells were prepared by the standard CaCl2 method [47]. Competent cells of P. aeruginosa were prepared according to the method of Irani and Rowe [53]. E. coli S17-1 strain was transformed with test plasmids carrying oriTRK2 and such transformants were used as donors in conjugation with recipient A. tumefaciens LBA1010R RifR strain. Overnight cultures of the donor and recipient strains (100 μl each) were mixed on L agar plate and incubated overnight at 28 °C. Bacteria from the plate were suspended in L broth and serial dilutions of the suspension were plated on L agar with rifampicin (150 μg ml−1) and chloramphenicol (50 μg ml−1) and incubated at 28 °C. Alternatively, P. putida KT2440 traRA3-korC KmR strain was transformed with pABB35 derivatives carrying oriTRA3 (e.g., pESB31) and such transformants were used as donor strains.
Standard plate test of plasmid stability
Cultures of hosts carrying various plasmids were grown in L broth with selective antibiotics at 37 °C or 28 °C. Plasmid content in the initial cultures was assessed by plating 100 μl of diluted cultures onto L agar to get approximately 100–200 colonies (usually 106-fold dilution) and then re-streaking 100 colonies onto L agar with the selective antibiotic. Plasmid retention was expressed as the percentage of CmR colonies. The cultures were grown in L broth without antibiotics for up to 60 generations (diluted 105-fold at the start and every 20 generations) and plasmid retention was analyzed as above. No IPTG (isopropyl β-D-1-thiogalactopyranoside) was added to the cultures with the exception of experiments with E. coli DH5α (pABB34) where the effect of IPTG concentration was analyzed.
Rapid plate screening for plasmid retention
Transformants were grown overnight and diluted as described above. Every 20 generations 100 μl of 106-fold diluted culture was plated on L agar (for plasmids with klcAp-xylE fusion) or L agar with X-gal (for plasmids with klcAp-lacZ fusion). Colonies formed by bacteria with plasmids carrying the klcAp-xylE fusion become yellow after being sprayed with 10 mM catechol solution. Plasmid retention was calculated as the percentage of yellow colonies or blue colonies on X-gal for plasmids with klcAp-lacZ fusion.
Catechol 2,3-dioxygenase activity assay
The level of xylE expression from klcAp was determined by an enzymatic assay in extracts from logarithmically growing cultures of E. coli DH5α transformants using a standard method [27]. Protein concentration was assayed by the Bradford method [54]. One unit of catechol 2,3-dioxygenase activity is defined as the amount of enzyme necessary to convert 1 μmol of catechol to the yellow hydroxymuconic semialdehyde in 1 min under standard conditions.
Simplified XylE activity assay was applied to estimate plasmid stability in liquid cultures. Overnight cultures of E. coli DH5α transformants carrying test plasmids with klcAp-xylE (grown without selection for 20 or 40 generations) were diluted 100-fold, the cell densities were estimated by OD600, 1/10 volume of 10 mM catechol was added and absorbance at 405 nm was measured after 5 min. The OD405/OD600 ratio clearly differentiated strains with plasmids of various stability. To quantify plasmid retention a high-throughput procedure with 100-well plates (Labsystems Honeycomb 2 plate) and a Bioscreen C Microbiology Reader Analyser (Labsystems) was used. Details are described in the Results section.
Determination of plasmid copy number by quantitative real-time PCR (qPCR)
The copy number of pRK415 and its derivatives was measured by qPCR using the SYBR® Green JumpStart™ Taq ReadyMix kit (Sigma). The single-copy galK, gene from E. coli chromosome, was used as the chromosomal reference gene (primers #26 and #27) for all strains. The trfA gene of RK2 was used as the plasmid reference gene (primers #28 and #29) for pRK415 derivatives and pMPB9.90 (araBADp-trfARK2) whereas repB gene was amplified for plasmid RA3 (primers #30 and #31) (Table 2). Total DNA was purified from 4 ml of stationary-phase cultures using Genomic Mini purification kit (A&A Biotechnology), treated with an appropriate restriction enzyme to linearize the plasmid DNA and to fragment chromosomal DNA and then used as a template in qPCR. Plasmid copy number (PCN) was calculated relatively to the chromosomal marker on the basis of at least three biological replicates with three technical replicates per strain and average results with standard deviation are reported. The amplification, detection and analysis were carried out in the Laboratory of Genetic Modification Analyses of IBB PAS on an Applied Biosystems 7500 apparatus.
Acknowledgements
This work was supported by the National Science Centre in Poland (grant no 2011/03/B/NZ1/06540) and in part by grant PBZ-MNiSW-04/1/2007. We thank the GMO Laboratory at IBB PAS for Real-Time qPCR analysis.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AAB and GJB designed study. AAB, KG and GJB wrote the manuscript. AAB, AK, EL, JG participated in vectors construction. AAB, KG, AK, EL, AM and JG conducted plasmid stability tests. KG performed high-throughput quantification of plasmid retention. AM prepared templates for plasmid copy number determination. AAB, KG, AK, EL, JG, AM and GJB participated in data analysis. All authors read and approved the final manuscript.
Authors’ information
1 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Department of Microbial Biochemistry, 02-106 Warsaw, Pawinskiego 5A, Poland;
2 Present address: Warsaw University of Technology, Faculty of Chemistry, Institute of Biotechnology, 00-664 Warsaw, Poland;
3 Present address: Warsaw University of Life Sciences, Faculty of Human Nutrition and Consumer Sciences, Laboratory of Food Chemistry, 02-776 Warsaw, Poland;
4 Present address: Pulawska 255A/4, 02-740 Warsaw, Poland.
Contributor Information
Aneta A. Bartosik, Email: anetab2@ibb.waw.pl
Krzysztof Glabski, Email: k_glabski@ibb.waw.pl.
Anna Kulinska, Email: akulinska@ch.pw.edu.pl.
Ewa Lewicka, Email: e.sloniewska@ibb.waw.pl.
Jolanta Godziszewska, Email: jolszar6@wp.pl.
Aleksandra Markowska, Email: alex12marko@gmail.com.
Grazyna Jagura-Burdzy, Phone: 48 22 592 1212, Email: gjburdzy@ibb.waw.pl.
References
- 1.Nordstrom K, Austin SJ. Mechanisms that contribute to the stable segregation of plasmids. Annu Rev Genet. 1989;23:37–69. doi: 10.1146/annurev.ge.23.120189.000345. [DOI] [PubMed] [Google Scholar]
- 2.Gerdes K, Wagner EGH. RNA antitoxins. Curr Opin Microbiol. 2007;10:117–24. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Goeders N, Van Melderen L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 2014;6:304–24. doi: 10.3390/toxins6010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi I. Plasmid Biology. Washington, D.C.: American Society of Microbiology; 2004. Genetic addiction: a principle of gene symbiosis in a genome. [Google Scholar]
- 5.Mierzejewska J, Jagura-Burdzy G. Prokaryotic ParA–ParB–parS system links bacterial chromosome segregation with the cell cycle. Plasmid. 2012;67:1–14. doi: 10.1016/j.plasmid.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Pandey DP. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–76. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayes F. A family of stability determinants in pathogenic bacteria. J Bacteriol. 1998;180:6415–18. doi: 10.1128/jb.180.23.6415-6418.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartosik D, Szymanik M, Wysocka E. Identification of the partitioning site within the repABC-type replicon of the composite Paracoccus versutus plasmid pTAV1. J Bacteriol. 2001;183:6234–43. doi: 10.1128/JB.183.21.6234-6243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abeles AL, Friedman SA, Austin SJ. Partition of unit-copy miniplasmids to daughter cells: III. The DNA sequence and functional organization of the P1 partition region. J Mol Biol. 1985;185:261–72. doi: 10.1016/0022-2836(85)90402-4. [DOI] [PubMed] [Google Scholar]
- 10.Macartney DP, Williams DR, Stafford T, Thomas CM. Divergence and conservation of the partitioning and global regulation functions in the central control region of the IncP plasmids RK2 and R751. Microbiology. 1997;143:2167–77. doi: 10.1099/00221287-143-7-2167. [DOI] [PubMed] [Google Scholar]
- 11.Gerdes K, Larsen JE, Molin S. Stable inheritance of plasmid R1 requires two different loci. J Bacteriol. 1985; 161:292-298.12. Nordstrom K. Plasmid R1-replication and its control. Plasmid. 2006;55:1–26. doi: 10.1016/j.plasmid.2005.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nordstrom K. Plasmid R1-replication and its control. Plasmid. 2006;55:1–26. doi: 10.1016/j.plasmid.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Pansegrau W, Lanka E, Barth PT, Figurski DH, Guiney DG, Haas D, et al. Complete nucleotide sequence of Birmingham IncPα plasmids: compilation and comparative analysis. J Mol Biol. 1994;239:623–63. doi: 10.1006/jmbi.1994.1404. [DOI] [PubMed] [Google Scholar]
- 14.Gerdes K, Moller-Jensen J, Bugge JR. Plasmid and chromosome partitioning: surprises from phylogeny. Mol Microbiol. 2000;37:455–66. doi: 10.1046/j.1365-2958.2000.01975.x. [DOI] [PubMed] [Google Scholar]
- 15.Gerdes K, Howard M, Szardenings F. Pushing and pulling in prokaryotic DNA segregation. Cell. 2010;141:927–42. doi: 10.1016/j.cell.2010.05.033. [DOI] [PubMed] [Google Scholar]
- 16.Keen NT, Tamaki S, Kobayashi D, Trollinger D. Improved broad-host-range plasmids for DNA cloning in Gram-negative bacteria. Gene. 1988;70:191–97. doi: 10.1016/0378-1119(88)90117-5. [DOI] [PubMed] [Google Scholar]
- 17.Jagura-Burdzy G, Khanim F, Smith CA, Thomas CM. Crosstalk between plasmid vegetative replication and conjugative transfer: repression of the trfA operon by trbA of broad host range plasmid RK2. Nucleic Acids Res. 1992;20:3939–44. doi: 10.1093/nar/20.15.3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah DS, Cross MA, Porter D, Thomas CM. Dissection of the core and auxiliary sequences in the vegetative replication origin of promiscuous plasmid RK2. J Mol Biol. 1995;254:608–22. doi: 10.1006/jmbi.1995.0642. [DOI] [PubMed] [Google Scholar]
- 19.Thomas CM, Cross MA, Hussain AA, Smith CA. Analysis of copy number control elements in the region of the vegetative replication origin of the broad host range plasmid RK2. EMBO J. 1984;3:57–63. doi: 10.1002/j.1460-2075.1984.tb01761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shingler V, Thomas CM. Analysis of the trfA region of broad host-range plasmid RK2 by transposon mutagenesis and identification of polypeptide products. J Mol Biol. 1984;175:229–49. doi: 10.1016/0022-2836(84)90346-2. [DOI] [PubMed] [Google Scholar]
- 21.Ayres EK, Saadi S, Schreiner HC, Thomson VJ, Figurski DH. Differentiation of lethal and nonlethal, kor-regulated functions in the kilB region of broad host-range plasmid RK2. Plasmid. 1991;25:53–63. doi: 10.1016/0147-619X(91)90006-I. [DOI] [PubMed] [Google Scholar]
- 22.Ziegelin G, Pansegrau W, Strack B, Balzer D, Kroger M, Kruft V, et al. Nucleotide sequence and organization of genes flanking the transfer origin of promiscuous plasmid RP4. DNA Seq. 1991;1:303–27. doi: 10.3109/10425179109020786. [DOI] [PubMed] [Google Scholar]
- 23.Mather MW, McReynolds LM, Yu CA. An enhanced broad-host-range vector for gram-negative bacteria: avoiding tetracycline phototoxicity during the growth of photosynthetic bacteria. Gene. 1995;156:85–88. doi: 10.1016/0378-1119(95)00074-G. [DOI] [PubMed] [Google Scholar]
- 24.Thomas CM, Hussain AA. The korB gene of broad host range plasmid RK2 is a major copy number control element which may act together with trfB by limiting trfA expression. EMBO J. 1984;3:1513–19. doi: 10.1002/j.1460-2075.1984.tb02004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motallebi-Veshareh M, Rouch DA, Thomas CM. A family of ATPases involved in active partitioning of diverse bacterial plasmids. Mol Microbiol. 1990;4:1455–63. doi: 10.1111/j.1365-2958.1990.tb02056.x. [DOI] [PubMed] [Google Scholar]
- 26.Williams DR, Macartney DP, Thomas CM. The partitioning activity of the RK2 central control region requires only incC, korB and KorB-binding site OB3 but other KorB-binding sites form destabilizing complexes in the absence of OB3. Microbiology. 1998;144:3369–78. doi: 10.1099/00221287-144-12-3369. [DOI] [PubMed] [Google Scholar]
- 27.Zukowski MM, Gaffney DF, Speck D, Kauffmann M, Findeli A, Wisecup A, et al. Chromogenic identification of genetic regulatory signals in Bacillus subtilis based on expression of a cloned Pseudomonas gene. Proc Natl Acad Sci U S A. 1983;80:1101–5. doi: 10.1073/pnas.80.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falcon CM, Matthews KS. Operator DNA sequence variation enhances high affinity binding by hinge helix mutants of lactose repressor protein. Biochemistry. 2000;39:11074–83. doi: 10.1021/bi000924z. [DOI] [PubMed] [Google Scholar]
- 29.Bartosik AA, Lasocki K, Mierzejewska J, Thomas CM, Jagura-Burdzy G. ParB of Pseudomonas aeruginosa: interactions with its partner ParA and its target parS and specific effects on bacterial growth. J Bacteriol. 2004;186:6983–98. doi: 10.1128/JB.186.20.6983-6998.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lobocka M, Yarmolinsky M. P1 plasmid partition: a mutational analysis of ParB. J Mol Biol. 1996;259:366–82. doi: 10.1006/jmbi.1996.0326. [DOI] [PubMed] [Google Scholar]
- 31.Lynch AS, Wang JC. SopB protein-mediated silencing of genes linked to the sopC locus of Escherichia coli F plasmid. Proc Natl Acad Sci U S A. 1995;92:1896–900. doi: 10.1073/pnas.92.6.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodionov O, Lobocka M, Yarmolinky M. Silencing of genes flanking the P1 plasmid centromere. Science. 1999;283:546–49. doi: 10.1126/science.283.5401.546. [DOI] [PubMed] [Google Scholar]
- 33.Kusiak M, Gapczynska A, Plochocka D, Thomas CM, Jagura-Burdzy G. Binding and spreading of ParB on DNA determine its biological function in Pseudomonas aeruginosa. J Bacteriol. 2011;193:3342–55. doi: 10.1128/JB.00328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jagura-Burdzy G, Ibbotson JP, Thomas CM. The korF region of broad-host-range plasmid RK2 encodes two polypeptides with transcriptional repressor activity. J Bacteriol. 1991;173:826–33. doi: 10.1128/jb.173.2.826-833.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furste JP, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, et al. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 1986;48:119–31. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 36.Calos MP. DNA sequence for a low-level promoter of the lac repressor gene and an “up” promoter mutation. Nature. 1978;274:762–65. doi: 10.1038/274762a0. [DOI] [PubMed] [Google Scholar]
- 37.Lee C, Kim J, Shin SG, Hwang S. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 2006;123:273–80. doi: 10.1016/j.jbiotec.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 38.Guzman L-M, Belin D, Crason MJ, Beckwith J. Tight Regulation, Modulation, and High-Level Expression by Vectors Containing the Arabinose PBAD Promoter. J. Bacteriol. 1995;177:4121–30. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cronan JE. A family of arabinose-inducible Escherichia coli expression vectors having pBR322 copy control. Plasmid. 2006;55:152–57. doi: 10.1016/j.plasmid.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Kulinska A, Czeredys M, Hayes F, Jagura-Burdzy G. Genomic and functional characterization of the modular broad-host-range RA3 plasmid, the archetype of the IncU group. Appl Environ Microbiol. 2008;74:4119–32. doi: 10.1128/AEM.00229-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kulinska A, Cao Y, Macioszek M, Hayes F, Jagura-Burdzy G. The centromere site of the segregation cassette of broad-host-range plasmid RA3 is located at the border of the maintenance and conjugative transfer modules. Appl Environ Microbiol. 2011;77:2414–27. doi: 10.1128/AEM.02338-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ingram C, Brawner M, Youngman P, Westpheling J. xylE functions as an efficient reporter gene in Streptomyces spp.: use for the study of galP1, a catabolite-controlled promoter. J Bacteriol. 1989;171:6617–24. doi: 10.1128/jb.171.12.6617-6624.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon R, O’Connell M, Labes M, Puhler A. Plasmid vectors for the genetic analysis and manipulation of rhizobia and other gram-negative bacteria. Methods Enzymol. 1986;118:640–59. doi: 10.1016/0076-6879(86)18106-7. [DOI] [PubMed] [Google Scholar]
- 44.Czarnecki J, Dziewit L, Kowalski L, Ochnio M, Bartosik D. Maintenance and genetic load of plasmid pKON1 of Paracoccus kondratievae, containing a highly efficient toxin-antitoxin module of the hipAB family. Plasmid. 2015;80:45–53. doi: 10.1016/j.plasmid.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 45.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–80. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 46.Kahn M, Kolter R, Thomas C, Figurski D, Meyer R, Remaut E, et al. Plasmid cloning vehicles derived from plasmids ColE1, F, R6K, and RK2. Methods Enzymol. 1979;68:268–80. doi: 10.1016/0076-6879(79)68019-9. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, a laboratory manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989. [Google Scholar]
- 48.Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;1:263–73. doi: 10.1101/SQB.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 49.Bartosik AA, Markowska A, Szarlak J, Kulinska A, Jagura-Burdzy G. Novel broad-host-range vehicles for cloning and shuffling of gene cassettes. J Microbiol Methods. 2012;88:53–62. doi: 10.1016/j.mimet.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 50.Reece KS, Phillips GJ. New plasmids carrying antibiotic-resistance cassettes. Gene. 1995;165:141–42. doi: 10.1016/0378-1119(95)00529-F. [DOI] [PubMed] [Google Scholar]
- 51.Thomas CM, Smith CA. The trfB region of broad host range plasmid RK2: the nucleotide sequence reveals incC and key regulatory gene trfB/korA/korD as overlapping genes. Nucleic Acids Res. 1986;14:4453–69. doi: 10.1093/nar/14.11.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marx CJ, Lidstrom ME. Development of improved versatile broad-host-range vectors for use in methylotrophs and other Gram-negative bacteria. Microbiology. 2001;147:2065–75. doi: 10.1099/00221287-147-8-2065. [DOI] [PubMed] [Google Scholar]
- 53.Irani VR, Rowe JJ. Enhancement of transformation in Pseudomonas aeruginosa PAO1 by Mg2+ and heat. Biotechniques. 1997;22:54–56. doi: 10.2144/97221bm09. [DOI] [PubMed] [Google Scholar]
- 54.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 55.Spitzer M, Wildenhain J, Rappsilber J, Tyers M. BoxPlotR: a web tool for generation of box plots. Nat Methods. 2014;11:121–22. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spratt BG, Hedge PJ, te Heesen S, Edelman A, Broome-Smith JK. Kanamycin-resistant vectors that are analogues of plasmids pUC8, pUC9, pEMBL8 and pEMBL9. Gene. 1986;41:337–42. doi: 10.1016/0378-1119(86)90117-4. [DOI] [PubMed] [Google Scholar]