

Abstract
Background
The N100 is a negative deflection in the surface EEG approximately 100ms after an auditory signal. It has been shown to be reduced in individuals with schizophrenia and those at clinical high risk (CHR). N100 blunting may index neural network dysfunction underlying psychotic symptoms. This phenomenon has received little attention in pediatric populations.
Method
This cross-sectional study compared the N100 response measured via the average EEG response at the left medial frontal position FC1 to 150 sinusoidal tones in participants ages 5 to 17 years with a CHR syndrome (n = 29), a psychotic disorder (n = 22), or healthy controls (n=17).
Results
Linear regression analyses that considered potential covariates (age, gender, handedness, family mental health history, medication usage) revealed decreasing N100 amplitude with increasing severity of psychotic symptomatology from healthy to CHR to psychotic level.
Conclusions
Longitudinal assessment of the N100 in CHR children who do and do not develop psychosis will inform whether it predicts transition to psychosis and if its response to treatment predicts symptom change.
1. Introduction
Research suggests that psychotic disorders involve a neurodevelopmental vulnerability and an active process of brain dysfunction beginning before full psychosis (Asami et al., 2014; Cannon et al., 2015; Kahn and Sommer, 2015; Thermenos et al., 2013). As this process unfolds, cognitive decline and social withdrawal increase (Giuliano et al., 2012; Johnstone et al., 2005; Mesholam-Gately et al., 2009; Piskulic et al., 2012), and the attenuated positive symptoms of the clinical high risk (CHR) state emerge (Cornblatt et al., 2012). Approximately one third of adolescents and young adults with CHR transition to full psychosis (Cornblatt et al., 2003; Fusar-Poli et al., 2012). Many nonconverters continue to experience functional impairment (Addington et al., 2011). Prevention of the disability of CHR and psychosis will require understanding the underlying biological processes and biomarkers sensitive to these processes.
The auditory N100 is an evoked response potential (ERP) associated with neurons in the primary and association auditory cortices in the superior temporal gyrus, Heschl’s gyrus (Zouridakis et al., 1998), and planum temporale (Godey et al., 2001; Milner et al., 2014). In animals, the N100 originates from activation of superficial supragranular layers of the cortex (Moore, 2002; Stroganova et al., 2013), the primary origin and termination of intracortical connections (Swenson, 2006). Thus, pathology affecting these layers and projections may reduce N100 responses. Notably, in primarily adult studies, N100 amplitude is reduced in first episode and chronic schizophrenia (SZ) patients, both medicated and unmedicated, and in their relatives (Brockhaus-Dumke et al., 2008; Brown et al., 2002; Hsieh et al., 2012; Mukundan, 1986; Neuhaus et al., 2013; Rosburg et al., 2008; Turetsky et al., 2008). Studies have differed as to whether the N100 is decreased in CHR and whether it decreases further with progression to psychosis (Brockhaus-Dumke et al., 2008; Hsieh et al., 2012; van Tricht et al., 2015).
To our knowledge, N100 abnormalities have not been studied in fully pediatric samples with CHR or psychosis (PS). Importantly, the N100 can be measured with minimal risk beginning in early childhood (Pang and Taylor, 2000) and thus may serve as an early index of psychosis liability. This study compared N100 responses in pediatric patients with PS or CHR and healthy controls. We hypothesized that decreasing N100 amplitude would be observed from healthy controls to CHR to PS.
2. Method
2.1 Participants
Patients with PS (n=22), patients with CHR (n=29), and healthy controls (HC; n=17) between 5 and 17 years of age were recruited. PS and CHR participants were drawn from the psychiatry service at Boston Children’s Hospital (BCH); the Commonwealth Research Center (CRC, PI L. J. Seidman); and the Social Neuroscience and Psychopathology Laboratory (SNAP Lab, PI C. Hooker) at Harvard University. The Structured Interview for Psychosis-risk Syndromes (SIPS; described in 2.2.1.) was utilized to determine whether PS or CHR syndrome criteria were met (McGlashan et al., 2010). PS participants met DSM-IV criteria for Schizophrenia (n = 7), Schizoaffective Disorder (n = 8), Schizophreni form Disorder (n = 3), Bipolar Disorder with Psychotic Features (n=2), or Major Depression with Psychotic Features (n=2). HCs were identified through advertisements and word of mouth. HCs could not meet CHR criteria or have a current or past Axis I diagnosis or any first-, second-, or third-degree biological relative with a psychotic disorder. Exclusion criteria for all participants included a lifetime diagnosis of substance abuse or dependence, neurological disease (e.g., epilepsy) or head injury, medical illness with cognitive sequelae, sensory impairments, or intellectual disability. Fig. 1 displays the results of the screening process (described in 2.2.1.).
Figure 1.

Flow charts depicting participants screened, reasons for exclusion, and number of participants retained for each clinical group.
2.2 Procedures and measures
Eligible participants were invited to complete demographic and clinical interviews/questionnaires and an auditory ERP paradigm. BCH’s Institutional Review Board approved all procedures. Participants provided assent, and a parent or legal guardian provided written informed consent.
2.2.1 Screening assessment
Participants and their parents/guardians were administered the Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime Version (K-SADS-PL), a validated semistructured interview used to diagnose DSM-IV psychiatric disorders in youth under age 18 years (Kaufman et al., 1996). Participants were administered the SIPS and the Scale of Prodromal Symptoms (SOPS) (McGlashan et al., 2010) to assess current and past psychosis and CHR status and to rate positive and negative symptom severity. If the ERP paradigm visit occurred more than one month after the screening assessment, the SIPS/SOPS were re-administered to confirm clinical group assignment. No participant was reclassified based upon reassessment. SIPS/SOPS raters were trained and certified by Yale University’s PRIME Research Clinic, and several attended North American Prodromal Longitudinal Study (NAPLS-2) SIPS interview reviews for 9 months. Sixty-six participants were administered the SIPS/SOPS by study staff and two by their referral source (CRC or SNAP Lab).
2.2.2 Demographic and clinical assessment
Race, ethnicity, date of birth, medical and psychiatric history, medication usage, and school functioning were determined from parent/guardian interview and record review. Intellectual disability was ruled out if (a) previous IQ testing results were N70 for full scale or verbal or performance IQ, (b) school functioning was at grade level without special education services, or (c) administration of the Scales of Independent Behavior—Revised (SIB-R; Bruininks et al., 1996), a comprehensive, norm-referenced assessment of functional level, indicated normal functioning.
2.2.3 Auditory ERP (AER) paradigm to form the AER N100 component
Following a 10-min baseline, EEG recordings were collected with an EGI™ 128-channel Geodesic Net System (Electrical Geodesics Inc., Eugene, OR) while the participant was seated in a quiet, electrically shielded room. Auditory stimuli were presented with TDH-49P headphones. To facilitate state stabilization, all participants viewed an age appropriate video with the sound muted during the auditory stimuli.
The individual auditory stimuli were 1000 Hz digitally constructed sinusoidal tone pulses; each tone pulse was 50 ms in duration bounded by 0.005-second onset and offset ramps. Sinusoidal tone pulses were adjusted to a Sound Pressure Level (SPL) of 75 dB and routed to participants via binaural earphones. Tones were presented binaurally on a randomly determined interstimulus interval (Noesis software), varying 2000–3000ms, to avoid rhythm artifact. A trial marker corresponding to stimulus onset was recorded along with the EEG data.
EEG data were recorded using FCZ as the physical reference electrode at a 500 Hz sampling rate with bandpass filtering from 0.53 to 100 Hz and with 60 Hz mains filters turned on. Prior to signal averaging, trained staff visually edited EEG data recordings for movement and electrode artifact, eyeblink storms, state changes, muscle bursts, and timeout intervals; such epochs were marked for omission from subsequent signal averaging. Automated eyeblink and eye movement artifact removal procedures were then implemented using BESA 6.0 (Berg and Scherg, 1994). When a signal averaging segment of EEG contained amplitudes greater than±200 μV, they were visually inspected; if likely artifact was noted, the identified segment was excluded from signal averaging.
Photogrammetry™ was performed on 11 photographic images of the original 128-channel EGI net placement to determine true scalp electrode location. Utilizing three-dimensional spline interpolation (BESA), the 128 channels were converted to a full 81 channel 10–10 electrode standard data set. Analyses were based upon common average reference for all channels. Data analysis was primarily limited to EEG channel FC1, which lies immediately to the left of the scalp vertex region at the center of the rectangle formed by traditional electrodes FZ, CZ, F3, and C3. Evoked potentials were formed and analyzed with band pass set to 1–50 Hz to form the tone-burst auditory evoked response (AER). These contained epochs 500 ms before and after tone presentation, with the entire prestimulus epoch used for baseline amplitude correction. For each participant, tone burst AERs were calculated by signal averaging 150 stimulus presentations, with the N100 component measured at FC1-common average reference as the amplitude of and latency to the maximal negativity in the interval 80–125 ms post stimulus presentation.
2.2.3.1. Justification of analysis limited to FC1 and the common average reference
All scalp manifestations of evoked potential data constitute projections of the primary electrical source dipole within the brain to the scalp, where they may be measured with surface electrodes. In healthy participants, the click AER source dipole localizes within the brain to the posterior superior temporal gyrus of both temporal lobes. All such sources have a physical location(s), a source orientation, and a source polarity (positive–negative sides/ends of the source vector). The N100 component of the AER typically points superiorly towards the medial frontal–central cortex bilaterally. Its frontally projected negativity is seen over a broad scalp region, usually involving standard electrodes FZ, CZ, F3, C3, FC1, F4, C4, and FC2. The negative aspect of the source dipole from both the left and right temporal regions, which mostly project vertically, typically show stronger ipsilateral projections with some degree of projection across the midline, contralaterally. In some cases, the bihemispheric frontal overlap is minimal, and the N100may appear maximal at a non-midline electrode, often FC1 or FC2. In a noise-free environment in healthy samples, CZ, and sometimes FZ, typically provide the strongest signal. As the signal distribution at the above frontal electrode coverage regions are assumed to be passive projections from the temporal source dipole, any of the above electrodes should contain the same information. In healthy participants, the positive component of the N100 AER dipole typically points inferiorly, posteriorly, and laterally and is seen exclusively ipsilaterally, almost always maximal at A1 (left ear) or TP9 (left mastoid) or homologously at electrodes A2 and TP10 on the right. In healthy participants, the largest N100 signal can typically be observed by recording between CZ and either linked ears [(A1 + A2) / 2] or linked mastoids [(TP9 + TP10) / 2].
Notably, we have found among SZ patients that the largest negative N100 component is often medial to the midline, typically more on the left, at FC1 (Morstyn et al., 1983a; Morstyn et al., 1983b; Faux, Shenton, et al., 1987, Faux et al., 1988; Faux, Torello et al., 1987a; Faux, Torello et al., 1987b). Although results for the P200 and P300 were reported in these articles, an N100 asymmetry was also observed. Prior to measuring N100 amplitudes for the current study, we screened the data and confirmed that the optimal electrode for N100 measurements in our sample was the FC1. Furthermore, prior work has demonstrated that in SZ, the scalp manifestation of the positive pole may be found to be maximal anterior to the ears and mastoids, even occasionally to the frontal regions, although these findings have not yet been formally reported. Accordingly we decided, a priori, to avoid the advantage ears and/or mastoid use would provide in healthy participants, but which may not be true for SZ, by measuring from FC1 to the common average reference, which would be a “neutral” reference across groups. Thus, by using the common average reference, we avoided potential “noise” related to a shifting positive N100 dipole scalp presentation, and by taking measurements at FC1 for all groups, we avoided issues related to negative N100 scalp vertex presentation asymmetry in the PS group.
2.3 Data analysis
Differences among clinical groups on demographic and clinical characteristics were tested using the Freeman–Halton extension of Fisher’s exact tests for categorical variables and ANOVAs for continuous variables. Significant differences were followed by 2 × 2 Fisher’s exact tests with Bonferroni corrected p threshold for categorical variables and Student–Newman–Keuls (SNK) tests for continuous variables. Consistent with prior studies (del Re et al., 2015; Salisbury et al., 2010), a one-way ANOVA revealed no significant association between clinical group and N100 mean latency [F(2, 65) = 0.020, p = 0.980]; thus, N100 latency was not considered further.
Linear regression models tested the effect of clinical group on the N100 mean amplitude, consistent with methods of similar analyses (Hsieh et al., 2012). Potential covariates, including age, gender, handedness, first-degree family history of mental illness (psychosis, nonpsychotic major depression, nonpsychotic bipolar disorder) and current medication usage (antipsychotics, antidepressants, mood stabilizers, benzodiazepines, stimulants), were regressed against the N100 mean amplitude individually. Each variable that reached a significance level of p < .10 in its individual regression was included in the linear regression model testing the effect of clinical group on N100 amplitude. Variables were then removed using backward elimination with a threshold of p ≥ .10 to produce the final linear regression model.
For all linear regression models in which clinical group emerged as a significant predictor, we assessed the appropriateness of using a linear model by running ANOVAs to test deviation from linearity and by examining the residuals to ensure homoscedasticity and normal distributions. Follow-up pair-wise comparisons specified group differences. For all analyses, p < .05 was considered statistically significant except where Bonferroni correction was used as indicated below.
3. Results
3.1 Descriptive analysis
Table 1 depicts the distributions of study variables across clinical groups. ANOVAs revealed significant differences between groups on age [F(2, 65) = 4.493, p < .015] and SIB-R scaled scores [F(2, 54) = 8.366, p=.001]. Fisher’s exact tests revealed group differences on gender (p = .007) and usage of antipsychotics (p < .001) and antidepressants (p = .003). Table 1 specifies the significant pairwise group differences. Clinical groups did not differ on the remaining variables (ps ≥.12).
Table 1.
Demographic and Clinical Characteristics of Clinical Groups.
| Clinical group | ||||||||
|---|---|---|---|---|---|---|---|---|
| Variable | PS (n = 22) | CHR (n = 29) | HC (n = 17) | |||||
| % | M | SD | % | M | SD | % | M | SD |
| Demographics | ||||||||
| 86.4a | 44.8b | 52.9 | ||||||
| 11.4a | 2.8 | 13.5b | 2.7 | 11.0a | 4.2 | |||
| 95.2 | 79.3 | 94.1 | ||||||
| 13.6 | 6.9 | 5.9 | ||||||
| 75.6a | 27.9 | 89.6a | 13 | 107.1b | 20.9 | |||
| First-degree family mental health history d | ||||||||
| 14.3 | 4.4 | 0 | ||||||
| 14.3 | 21.7 | 0 | ||||||
| 4.8 | 17.4 | 11.8 | ||||||
| Medication use at assessment | ||||||||
| 50.0a | 260.3 | 382.8 | 27.6 | 149.1 | 107.6 | 0b | 0 | 0 |
| 45.5a | 31.1 | 0b | ||||||
| 18.2 | 6.9 | 0 | ||||||
| 4.5 | 3.5 | 0 | ||||||
| 4.5 | 0 | 0 | ||||||
Note: PS, psychosis; CHR, clinical high risk; HC, healthy control; SIB-R SS, Scales of Independent Behavior—Revised scaled scores.
Groups noted by different superscripted letters were significantly different in post hoc pairwise Student–Newman–Keuls (SNK) tests for continuous variables and Bonferroni corrected (p < .017) Fisher’s exact tests for categorical variables.
The SIB-R was administered to 20 PS participants, 21 CHR participants, and 16 HC participants.
One PS participant and six CHR participants were unable to provide information about family history of mental illness because they had limited contact with their biological parents or their adoptive parents were unsure of mental illness history among first-degree biological relatives.
Means and standard deviations are for chlorpromazine (CPZ) equivalence. Antipsychotic medications used (not mutually exclusive)included Risperidone (n = 10), Aripiprazole (n = 6), Clozapine (n = 2), Haloperidol(n = 1), Lurasidone (n = 1), Olanzapine (n = 1), and Ziprasidone (n = 1).
3.2 clinical group differences in N100 amplitute
In analyses regressing the study variables individually against N100 amplitude, age, gender, antipsychotic medication usage, and clinical group met criterion for inclusion in the next stage of analysis. In the subsequent linear regression model including these variables, backward elimination removed gender and antipsychotic medication usage, leaving clinical group (β= .465, t= 4.413, p < .001) and age (β =.258, t=2.447, p = .017) as significant predictors of N100 amplitude [R2 = .279, F(2, 65)=12.603, p < .001]. As shown in Fig. 2, there were smaller N100 amplitudes from HC (M = 5.43, SD = 3.29) to CHR (M = 4.10, SD = 1.85) to PS (M = 2.21, SD = 2.25). N100 mean amplitude increased with age. The interaction between age and clinical group on N100mean amplitude was not significant (β= .043, t=.095, p = .924).
Figure 2.
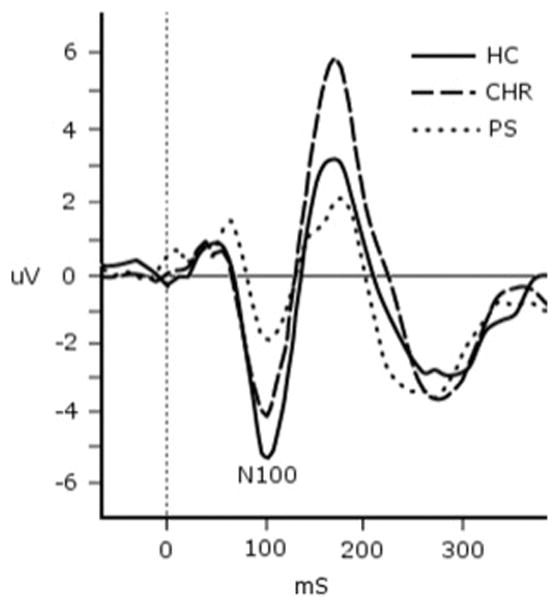
Average auditory N100 response by clinical group. HC = healthy control; CHR = clinical high risk; PS = psychosis. All groups differed significantly from each other in pairwise regression analyses that controlled for age: HC N CHR N PS.
In pairwise analyses, N100 amplitude was predicted by group (β = .527, t = 3.881, p < .001) and age (β = .276, t =2.003, p = .049) when comparing HC and PS [R2 = .338, F(2, 36) = 9.202, p = .001]; by group (β = .353, t = 2.323, p = .025) and marginally by age (β = .273, t = 1.799, p = .079) when comparing HC and CHR [R2 = .131, F(2, 43) = 3.250, p = .048]; and by group (β = .325, t = 2.414, p = .020) and age (β = .272, t = 2.021, p = .049) when comparing CHR and PS [R2 = .245, F(2, 48) = 7.776, p = .001].
4. Discussion
We believe that this is the first study in a pediatric sample to find smaller N100 amplitudes with increasing severity of psychosis spectrum from HC to CHR to PS. Studies have been inconsistent as to whether CHR individuals differ from HCs or PS patients inN100 amplitude. van Tricht et al. (2015) found increasing blunting of the N100 in CHR individuals transitioning to psychosis, Hsieh et al. (2012) found a decrease in N100 amplitude from HC to CHR to first-episode PS patients, and Salisbury et al. (2010) found that the N100 blunted from first episode to chronic SZ. However, del Re et al. (2015) found similar diminished N100 amplitudes in CHR and PS patients, and others did not find N100 blunting in CHR patients (Bramon et al., 2008; Brockhaus-Dumke et al., 2008; Hsieh et al., 2012). Notably, these were studies of adults.
Our results argue for decreasing N100 amplitude when comparing HC to CHR to PS in children and adolescents. These findings need to be replicated in larger samples and with longitudinal assessments to determine whether the N100 amplitude predicts which CHR youth will progress to psychosis and whether the N100 decreases when progressing from CHR to PS. Future studies should also test whether N100 blunting is evident in individuals at high genetic risk for psychosis (e.g., 22q11.2 deletion syndrome; Salisbury et al., 2010; Turetsky et al., 2008). If observed, then the N100 may serve as a useful biomarker when testing whether new treatments that target these genetic mechanisms perform as expected.
In the current study, groups were compared on the average N100 amplitude assessed via the average EEG response at the left medial frontal position FC1 to 150 tones. To date, many studies examining N100 responses in CHR and PS patients have utilized channel CZ. As described in the Method section, in healthy samples, CZ, and sometimes FZ, typically provide the strongest signal. However, in our own work, we have repeatedly found that, among SZ patients, the largest negative N100 component is often medial to the midline, typically more on the left, at FC1 (Morstyn et al., 1983a; Morstyn et al., 1983b; Faux, Shenton, et al., 1987, Faux et al., 1988; Faux, Torello et al., 1987a; Faux, Torello et al., 1987b). For that reason, we screened our data prior to conducting statistical analyses and confirmed that the optimal electrode for N100 measurements in our sample was the FC1. We conducted supplemental analyses (not reported above) to test for clinical group differences in the N100 response utilizing CZ, FZ, and FC2 to linked ears. The results demonstrated a similar decrease in N100 amplitude from HC to CHR to PS as reported above using FC1; however, the former channels produced greater inter-individual variation, and the group differences were statistically weaker than those found using FC1. These findings support our decision to use FC1 in this sample. Notably, our sample was a fully pediatric sample, unlike the majority of studies in this area, which have been conducted largely with adults. Future studies should explore the optimal channels for assessing N100 and other ERP responses in CHR and PS patients of varying ages.
This study has limitations. Its small sample size restricted statistical power. The sample was almost exclusively non-Hispanic White, and the PS subsample was largely male. We attempted to minimize the role of intelligence in contributing to group differences on N100 measures by excluding individuals with intellectual disability. However, future studies should match participants on IQ and/or control for IQ in analyses, as intellectual capabilities are associated with N100 responses (Lijffijt et al., 2009). N100 amplitude increased with age, and the clinical groups differed in age. However, the CHR group was the oldest group but its N100 amplitude fell between the HC and PS groups, and group predicted N 100 amplitude after controlling for age. Although we considered medication usage as a covariate in analyses, we cannot rule out medication effects given that no HC participants were medicated. Exclusion criteria for all participants included a lifetime diagnosis of substance abuse or dependence based on participant and parent/guardian responses to the K-SADS-PL. Urine toxicology testing was not performed to confirm lack of substance use. Another limitation relates to the assessment of CHR in pediatric populations. Although childhood onset of prodromal symptoms is not rare (Woodberry et al., 2014), identification of CHR in pediatric populations is less reliable than in adults (Schultze-Lutter et al., 2015). Furthermore, the predictive validity of the SIPS, particularly in children under age 10 years, is not established. Finally, the PS group was not limited to DSM-IV SZ due to (a) difficulty in determining if patients with early psychosis would settle into a categorical diagnosis of SZ or an affective psychosis (Consoli et al., 2014; Remberk et al., 2014) and (b) increasing biological evidence for heterogeneity among patients with SZ and for pleiotropy in the phenotypic expressions of SZ risk alleles (Kavanagh et al., 2015). Consequently, we employed a Research Domain Criteria (RDoC) approach, as recently advocated by the National Institute of Mental Health (Insel, 2014).
4.1 Conclusions
The N100 response appears smaller in pediatric CHR and PS patients compared to HCs and thus may be a biomarker of brain pathology underlying CHR and PS emerging early in life. Longitudinal studies in children at high genetic risk are required to determine if N100 abnormalities can identify those who will develop psychotic disorders prior to the emergence of CHR symptoms. With additional study, the N100 may serve as a useful biomarker of network pathology and treatment efficacy in pediatric psychosis and CHR treatment trials (Turetsky et al., 2008).
Acknowledgments
Role of Funding Sources
This work was supported by the Tommy Fuss Fund (Joseph Gonzalez-Heydrich, Michelle Bosquet Enlow, Eugene D’Angelo, Sarah Gumlak, April Kim, Ashley Rober, Sahil Tembulkar, Kelsey Graber, Kyle O’Donnell, Kara Kimball, Frank H. Duffy). During preparation of this manuscript, the authors were supported by the National Institute of Mental Health (1R01MH100186, Lindsay Oberman), the Simons Foundation (Lindsay Oberman), the Nancy Lurie Marks Family Foundation (Lindsay Oberman), a Faculty Development Fellowship from Boston Children’s Hospital (Hesham M. Hamoda), and the Program for Behavioral Science in the Department of Psychiatry at Boston Children’s Hospital (Michelle Bosquet Enlow). Some participants were referred by the NAPLS-2 grant (U01 MH08198-06A1, Larry J. Seidman, Kristen A. Woodberry). None of the funding agencies had any role in the study design, the collection, analysis or interpretation of data, the writing of the manuscript, or the decision to submit the manuscript for publication. The content is solely the responsibility of the authors and does not represent the official views of any funding entity.
We thank the patients and families whose generous donation of time made this project possible.
Abbreviations
- CHR
clinical high risk
- ERP
evoked response potential
- SZ
schizophrenia
- PS
psychosis
- HC
healthy control
- BCH
Boston Children’s Hospital
- CRC
Commonwealth Research Center
- SNAP Lab
Social Neuroscience and Psychopathology Laboratory
- K-SADS-PL
Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime Version
- SIPS
Structured Interview for Psychosis risk Syndromes
- SOPS
Scale of Prodromal Symptoms
- NAPLS-2
second phase of the North American Prodromal Longitudinal Study
- SIB-R
Scales of Independent Behavior—Revised
Footnotes
Contributors
Joseph Gonzalez-Heydrich and Frank H. Duffy designed the study. Joseph Gonzalez-Heydrich, Eugene D’Angelo, Larry J. Seidman, and Kristen A. Woodberry participated in the recruitment, assessment, and selection of study participants. Joseph Gonzalez-Heydrich, Eugene D’Angelo, Sarah Gumlak, April Kim, Ashley Rober, Sahil Tembulkar, Kyle O’Donnell, Kara Kimball, and Hesham M. Hamoda collected the clinical data. Joseph Gonzalez-Heydrich and Eugene D’Angelo interpreted the clinical data. Sarah Gumlak, April Kim, Ashley Rober, Sahil Tembulkar, Kyle O’Donnell, Kara Kimball, and Frank H. Duffy collected and managed the neurophysiologic data. Kelsey Graber managed the clinical and neurophysiologic data. Joseph Gonzalez-Heydrich, Michelle Bosquet Enlow, and Kyle O’Donnell performed the statistical analyses. Joseph Gonzalez-Heydrich, Michelle Bosquet Enlow, Larry J. Seidman, Alexander Rotenberg, Lindsay M. Oberman, Alvaro Pascual-Leone, Matcheri S. Keshavan, and Frank H. Duffy interpreted the results. Michelle Bosquet Enlow wrote the manuscript drafts, with assistance from Joseph Gonzalez-Heydrich, Kyle O’Donnell, April Kim, and Sahil Tembulkar. Joseph Gonzalez-Heydrich directed the conduct of the study, had full access to the clinical data, and takes full responsibility for the accuracy of participant selection. Frank H. Duffy had full access to the neurophysiologic data and takes responsibility for all aspects of these data, including collection, processing, and analyses. All authors collaborated in writing and editing the manuscript and approved the final manuscript.
Conflict of Interest
In the past 3 years, Joseph Gonzalez-Heydrich has received grant support from the Tommy Fuss Fund, the Al Rashed Family, and Glaxo-SmithKline. In previous years, he served as a consultant to Abbott Laboratories, Pfizer Inc., Johnson & Johnson (Janssen, McNeil Consumer Health), Novartis, Parke-Davis, Glaxo-SmithKline, AstraZeneca, and Seaside Therapeutics; was a speaker for Abbott Laboratories, Pfizer Inc., Novartis, Bristol-Meyers Squibb; and received grant support from Abbott Laboratories, Pfizer Inc., Johnson & Johnson (Janssen, McNeil Consumer Health), and Akzo-Nobel/Organon. He is a founder, equity holder, and consultant to Neuro’motion Labs and is an inventor on a patent pending for technologies to enhance the development of emotional regulation. Alvaro Pascual-Leone serves on the scientific advisory boards for Nexstim, Neuronix, Starlab Neuroscience, Neuroelectrics, and Neosync and is listed as an inventor on several issued and pending patents on the real-time integration of transcranial magnetic stimulation (TMS) with electroencephalography (EEG) and magnetic resonance imaging (MRI). Alexander Rotenberg has served as an advisor for NexstimInc., Sage Therapeutics Inc., and NeuroRex Inc. and has joint grants with Brainsway Inc. and Vivonics Inc. He is a founder, equity holder, and consultant to Neuro’motion Labs and is an inventor on a patent pending for technologies to enhance the development of emotional regulation.
References
- Addington J, Cornblatt BA, Cadenhead KS, Cannon TD, McGlashan TH, Perkins DO, Seidman LJ, Tsuang MT, Walker EF, Woods SW, Heinssen R. At clinical high risk for psychosis: outcome for nonconverters. Am J Psychiatry. 2011;168(8):800–805. doi: 10.1176/appi.ajp.2011.10081191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asami T, Hyuk Lee S, Bouix S, Rathi Y, Whitford TJ, Niznikiewicz M, Nestor P, McCarley RW, Shenton ME, Kubicki M. Cerebral whitematter abnormalities and their associations with negative but not positive symptoms of schizophrenia. Psychiatry Res. 2014;222(1–2):52–59. doi: 10.1016/j.pscychresns.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg P, Scherg M. A multiple source approach to the correction of eye artifacts. Electroencephalogr Clin Neurophysiol. 1994;90(3):229–241. doi: 10.1016/0013-4694(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Bramon E, Shaikh M, Broome M, Lappin J, Bergé D, Day F, Woolley J, Tabraham P, Madre M, Johns L, Howes O, Valmaggia L, Pérez V, Sham P, Murray RM, McGuire P. Abnormal P300 in people with high risk of developing psychosis. NeuroImage. 2008;41(2):553–560. doi: 10.1016/j.neuroimage.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Brockhaus-Dumke A, Schultze-Lutter F, Mueller R, Tendolkar I, Bechdolf A, Pukrop R, Klosterkoetter J, Ruhrmann S. Sensory gating in schizophrenia: P50 and N100 gating in antipsychotic-free subjects at risk, first-episode, and chronic patients. Biol Psychiatry. 2008;64(5):376–384. doi: 10.1016/j.biopsych.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Brown KJ, Gonsalvez CJ, Harris AWF, Williams LM, Gordon E. Target and non-target ERP disturbances in first episode vs. chronic schizophrenia. Clin Neurophysiol. 2002;113(11):1754–1763. doi: 10.1016/s1388-2457(02)00290-0. [DOI] [PubMed] [Google Scholar]
- Bruininks RH, Woodcock RW, Weatherman RF, Hill BK. Scales of Independent Behavior—Revised. Riverside Publishing; Itasca IL: 1996. [Google Scholar]
- Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG, McEwen S, Addington J, Bearden CE, Cadenhead K, Cornblatt B, Mathalon DH, McGlashan T, Perkins D, Jeffries C, Seidman LJ, Tsuang M, Walker E, Woods SW, Heinssen R North American Prodrome Longitudinal Study Consortium. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 2015;77(2):147–157. doi: 10.1016/j.biopsych.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consoli A, Brunelle J, Bodeau N, Louët E, Deniau E, Perisse D, Laurent C, Cohen D. Diagnostic transition towards schizophrenia in adolescents with severe bipolar disorder type I: an 8-year follow-up study. Schizophr Res. 2014;159(2–3):284–291. doi: 10.1016/j.schres.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Cornblatt BA, Carrion RE, Addington J, Seidman L, Walker EF, Cannon TD, Cadenhead KS, McGlashan TH, Perkins DO, Tsuang MT, Woods SW, Heinssen R, Lencz T. Risk factors for psychosis: impaired social and role functioning. Schizophr Bull. 2012;38(6):1247–1257. doi: 10.1093/schbul/sbr136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornblatt BA, Lencz T, Smith CW, Correll CU, Auther AM, Nakayama E. The schizophrenia prodrome revisited: a neurodevelopmental perspective. Schizophr Bull. 2003;29(4):633–651. doi: 10.1093/oxfordjournals.schbul.a007036. [DOI] [PubMed] [Google Scholar]
- Faux SF, Shenton M, McCarley RW, Torello M, Duffy FH. P200 topographic alterations in schizophrenia: evidence for left temporal centroparietal amplitude deficits. Electroencephalogr Clin Neurophysiol Suppl. 1987;40:681–687. [PubMed] [Google Scholar]
- Faux SF, Shenton M, McCarley RW, Torello M, Duffy FH. Differentiation of schizophrenics and normal controls is enhanced by the Good in subtraction procedure. Int J Neurosci. 1988;39(1–2):117–135. doi: 10.3109/00207458808985697. [DOI] [PubMed] [Google Scholar]
- Faux SF, Torello M, McCarley RW, Shenton M, Duffy FH. P300 in schizophrenia: confirmation and statistical validation of temporal region deficit in P300 topography. Biol Psychiatry. 1987a;23(8):776–790. doi: 10.1016/0006-3223(88)90066-2. [DOI] [PubMed] [Google Scholar]
- Faux SF, Torello M, McCarley RW, Shenton M, Duffy FH. P300 topograpy alterations in schizophrenia: a replication study. Electroencephalogr Clin Neurophysiol Suppl. 1987b;40:688–694. [PubMed] [Google Scholar]
- Fusar-Poli P, Bonoldi I, Yung AR, Borgwardt S, Kempton MJ, Valmaggia L, Barale F, Caverzasi E, McGuire P. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Arch Gen Psychiatry. 2012;69(3):220–229. doi: 10.1001/archgenpsychiatry.2011.1472. [DOI] [PubMed] [Google Scholar]
- Giuliano AJ, Li H, Mesholam-Gately RI, Sorenson SM, Woodberry KA, Seidman LJ. Neurocognition in the psychosis risk syndrome: a quantitative and qualitative review. Curr Pharm Des. 2012;18(4):399–415. doi: 10.2174/138161212799316019. [DOI] [PubMed] [Google Scholar]
- Godey B, Schwartz D, de Graaf JB, Chauvel P, Liégeois-Chauvel C. Neuromagnetic source localization of auditory evoked fields and intracerebral evoked potentials: a comparison of data in the same patients. Clin Neurophysiol. 2001;112(10):1850–1859. doi: 10.1016/s1388-2457(01)00636-8. [DOI] [PubMed] [Google Scholar]
- Hsieh MH, Shan JC, Huang WL, Cheng WC, Chiu MJ, Jaw FS, Hwu HG, Liu CC. Auditory event-related potential of subjects with suspected pre-psychotic state and first episode psychosis. Schizophr Res. 2012;140(1–3):243–249. doi: 10.1016/j.schres.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Insel TR. The NIMH research domain criteria (RDoC) project: precision medicine for psychiatry. Am J Psychiatry. 2014;171(4):395–397. doi: 10.1176/appi.ajp.2014.14020138. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Ebmeier KP, Miller P, Owens DG, Lawrie SM. Predicting schizophrenia: findings from the Edinburgh high risk study. Br J Psychiatry. 2005;186(1):18–25. doi: 10.1192/bjp.186.1.18. [DOI] [PubMed] [Google Scholar]
- Kahn RS, Sommer IE. The neurobiology and treatment of first-episode schizophrenia. Mol Psychiatry. 2015;20(1):84–97. doi: 10.1038/mp.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman J, Birmaher B, Brent D, Rao U, Ryan N. Diagnostic Interview: Kiddie-Sads—Present and Lifetime Version (K-SADS-PL) (Retrieved April. 1996;19:2012. from http://www.psychiatry.pitt.edu/node/8233) [Google Scholar]
- Kavanagh DH, Tansey KE, O’Donovan MC, Owen MJ. Schizophrenia genetics: emerging themes for a complex disorder. Mol Psychiatry. 2015;20(1):72–76. doi: 10.1038/mp.2014.148. [DOI] [PubMed] [Google Scholar]
- Lijffijt M, Moeller G, Boutros NN, Burroughs S, Lane SD, Steinberg JL, Swann AC. The role of age, gender, education, and intelligence in P50, N100, and P200 auditory sensory gating. J Psychophysiol. 2009;23(2):52–62. doi: 10.1027/0269-8803.23.2.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlashan TH, Walsh BC, Woods SW. Structured Interview for Psychosis-Risk Syndromes, Version 5.0. PRIME Research Clinic, Yale School of Medicine; New Haven, CT: 2010. [Google Scholar]
- Mesholam-Gately R, Giuliano AJ, Goff KP, Faraone SV, Seidman LJ. Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology. 2009;23(3):315–336. doi: 10.1037/a0014708. [DOI] [PubMed] [Google Scholar]
- Milner R, Rusiniak M, Lewandowska M, Wolak T, Ganc M, Piatkowska-Janko E, Bogorodzki P, Skarżyński H. Towards neural correlates of auditory stimulus processing: a simultaneous auditory evoked potentials and functional magnetic resonance study using an odd-ball paradigm. Med Sci Monit. 2014;20:35–46. doi: 10.12659/MSM.889712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK. Maturation of human auditory cortex: implications for speech perception. Ann Otol Rhinol Laryngol Suppl. 2002;189:7–10. doi: 10.1177/00034894021110s502. [DOI] [PubMed] [Google Scholar]
- Morstyn R, Duffy FH, McCarley RW. Altered P300 topography in schizophrenia. Arch Gen Pyschiatry. 1983a;40(7):729–734. doi: 10.1001/archpsyc.1983.01790060027003. [DOI] [PubMed] [Google Scholar]
- Morstyn R, Duffy FH, McCarley RW. Altered topography of EEG spectral content in schizophrenia. Electroencephalogr Clin Neurophysiol. 1983b;56(4):263–271. doi: 10.1016/0013-4694(83)90251-1. [DOI] [PubMed] [Google Scholar]
- Mukundan CR. Middle latency components of evoked potential responses in schizophrenics. Biol Psychiatry. 1986;21(11):1097–1100. doi: 10.1016/0006-3223(86)90295-7. [DOI] [PubMed] [Google Scholar]
- Neuhaus AH, Popescu FC, Rentzsch J, Gallinat J. Critical evaluation of auditory event-related potential deficits in schizophrenia: evidence from large-scale single subject pattern classification. Schizophr Bull. 2013;40(5):1062–1071. doi: 10.1093/schbul/sbt151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang EW, Taylor MJ. Tracking the development of the N1 from age 3 to adulthood: an examination of speech and non-speech stimuli. Clin Neurophysiol. 2000;111(3):388–397. doi: 10.1016/s1388-2457(99)00259-x. [DOI] [PubMed] [Google Scholar]
- Piskulic D, Addington J, Cadenhead K, Cannon TD, Cornblatt BA, Heinssen R, Perkins DO, Seidman LJ, Tsuang MT, Walker EF, Woods SW, McGlashan TH. Negative symptoms in individuals at clinical high risk of psychosis. Psychiatry Res. 2012;196(2–3):220–224. doi: 10.1016/j.psychres.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Re EC, Spencer KM, Oribe N, Mesholam-Gately RI, Goldstein J, Shenton ME, Petryshen T, Seidman LJ, McCarley RW, Niznikiewicz MA. Clinical high risk and first episode schizophrenia: auditory event-related potentials. Psychiatry Res. 2015;231(2):126–133. doi: 10.1016/j.pscychresns.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remberk B, Bażyńska AK, Krempa-Kowalewska A, Rybakowski F. Adolescent insanity revisited: course and outcome in early-onset schizophrenia spectrum psychoses in an 8-year follow-up study. Compr Psychiatry. 2014;55(5):1174–1181. doi: 10.1016/j.comppsych.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Rosburg T, Boutros NN, Ford JM. Reduced auditory evoked potential component N100 in schizophrenia—a critical review. Psychiatry Res. 2008;161(3):259–274. doi: 10.1016/j.psychres.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Salisbury DF, Collins KC, McCarley W. Reductions in the N1 and P2 auditory event related potentials in first-hospitalized and chronic schizophrenia. Schizophr Bull. 2010;36(5):991–1000. doi: 10.1093/schbul/sbp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze-Lutter F, Michel C, Schmidt SJ, Schimmelmann BG, Maric NP, Salokangas RK, Riecher-Rössler A, van der Gaag M, Nordentoft M, Raballo A, Meneghelli A, Marshall M, Morrison A, Ruhrmann S, Klosterkötter J. EPA guidance on the early detection of clinical high risk states of psychoses. Eur Psychiatry. 2015;30(3):405–416. doi: 10.1016/j.eurpsy.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Stroganova TA, Kozunov VV, Posikera IN, Galuta IA, Gratchev VV, Orekhova EV. Abnormal pre-attentive arousal in young children with autism spectrum disorder contributes to their atypical auditory behavior: an ERP study. PLoS One. 2013;8(7):e69100. doi: 10.1371/journal.pone.0069100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson RS. The Cerebral Cortex, in: Review of Clinical and Functional Neuroscience. (Retrieved February. 2006;20:2015. from http://www.dartmouth.edu/~rswenson/NeuroSci/chapter_11.html) [Google Scholar]
- Thermenos HW, Keshavan MS, Juelich RJ, Molokotos E, Whitfield-Gabrieli S, Brent BK, Makris N, Seidman LJ. A review of neuroimaging studies of young relatives of individuals with schizophrenia: a developmental perspective from schizotaxia to schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(7):604–635. doi: 10.1002/ajmg.b.32170. [DOI] [PubMed] [Google Scholar]
- van Tricht MJ, Nieman DH, Koelman JT, Mensink AJ, Bour LJ, van derMeer JN, van Amelsvoort TA, Linszen DH, de Haan L. Sensory gating in subjects at ultrahigh risk for developing a psychosis before and after a first psychotic episode. World J Biol Psychiatry. 2015;16(1):12–21. doi: 10.3109/15622975.2012.680911. [DOI] [PubMed] [Google Scholar]
- Turetsky BI, Greenwood TA, Olincy A, Radant AD, Braff DL, Cadenhead KS, Dobie DJ, Freeman R, Green MF, Gur RE, Gur RC, Light GA, Mintz J, Nuechterlein KH, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Calkins ME. Abnormal auditory N100 amplitude: a heritable endophenotype in first-degree relatives of schizophrenia probands. Biol Psychiatry. 2008;64(12):1051–1059. doi: 10.1016/j.biopsych.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodberry KA, Serur RA, Hallinan SB, Mesholam-Gately RI, Giuliano AJ, Wojcik JD, Keshavan MS, Frazier JA, Goldstein JM, Shenton ME, McCarley RW, Seidman LJ. Frequency and pattern of childhood symptom onset reported by first episode schizophrenia and clinical high risk youth. Schizophr Res. 2014;158(1):45–51. doi: 10.1016/j.schres.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouridakis G, Simos PG, Papanicolaou AC. Multiple bilaterally asymmetric cortical sources account for the auditory N1m component. Brain Topogr. 1998;10(3):183–189. doi: 10.1023/a:1022246825461. [DOI] [PubMed] [Google Scholar]