

Abstract
Adenosine deaminase (ADA) is a ubiquitous enzyme that catabolizes adenosine and deoxyadenosine. During cerebral ischemia, extracellular adenosine levels increase acutely and adenosine deaminase catabolizes the increased levels of adenosine. Since adenosine is a known neuroprotective agent, adenosine deaminase was thought to have a negative effect during ischemia. In this study, however, we demonstrate that adenosine deaminase has substantial neuroprotective effects in the striatum, which is especially vulnerable during cerebral ischemia. We used temporary oxygen/glucose deprivation (OGD) to simulate ischemia in rat corticostriatal brain slices. We used field potentials as the primary measure of neuronal damage. For stable and efficient electrophysiological assessment, we used transgenic rats expressing channelrhodopsin-2, which depolarizes neurons in response to blue light. Time courses of electrically evoked striatal field potential (eFP) and optogenetically evoked striatal field potential (optFP) were recorded during and after oxygen/glucose deprivation. The levels of both eFP and optFP decreased after 10 min of oxygen/glucose deprivation. Bath-application of 10 µg/ml adenosine deaminase during oxygen/glucose deprivation significantly attenuated the oxygen/glucose deprivation-induced reduction in levels of eFP and optFP. The number of injured cells decreased significantly, and western blot analysis indicated a significant decrease of autophagic signaling in the adenosine deaminase-treated oxygen/glucose deprivation slices. These results indicate that adenosine deaminase has protective effects in the striatum.
Keywords: Adenosine, neuroprotection, optogenetics, brain ischemia, brain slice
Introduction
Adenosine deaminase (ADA; EC 3.5.4.4) is broadly distributed in the brain.1,2 It deaminates adenosine and 2′-deoxyadenosine to inosine and deoxyinosine.2,3 During an ischemic brain event, the extracellular adenosine concentration increases over 10-fold4,5 and adenosine is deaminated to inosine by ADA.2,6,7 Although the neuroprotective and adverse effects of adenosine receptor ligands have been well studied,2,8,9 the role of ADA in ischemia is unclear.8
Selective neuronal loss10 has been reported in the ischemic brain. The striatal medium spiny neurons are some of the most vulnerable cells in the human brain, similar to hippocampus CA1 and cortex layer 5 pyramidal neurons.10 In humans, age-related small vessel disease (prevalence >80% among the elderly) is frequently observed in the striatum and is accompanied by vascular Parkinsonism.11 The vulnerability of the striatum is also observed in the developing human brain. Neonatal ischemic brain injuries such as periventricular leukomalacia often leave striatal damage with neurological abnormalities and movement disorders.12
Congenital ADA deficiency2 results in severe combined immunodeficiency (SCID). The cause of SCID is an increase in deoxy-adenosine levels in the serum and tissues. This increases deoxy-ATP levels in the T-cells and causes T-cell apoptosis.13 Although hematopoietic stem cell transplantation (HSCT) alleviates SCID, HSCT-treated patients continue to have increased adenosine/deoxy-adenosine levels and suffer from cognitive and neurological developmental disabilities.14–16 Neurologic abnormalities with volume loss in the basal ganglia including the striatum have been observed in non-infected ADA deficient children.15,17 The average IQ for HSCT-treated patients is 85, significantly lower than the population average.16 In contrast to the immunodeficiency, the causes of neurological developmental abnormalities have not yet been well studied.16 Since ADA-deficient patients suffer from non-infectious respiratory distress and severe hypoxia,18,19 it is possible that they have a higher risk of cerebral hypoxia, which could result in brain lesions including the basal ganglia. Assuming that this increased risk of hypoxia is seen in ADA deficiency, there are four candidates to explain the abnormalities in the central nervous system (CNS): (1) the increased adenosine levels impair the CNS via A2A receptors,20,21 (2) the increased deoxy-adenosine levels are toxic,13 (3) the decreased inosine levels impair the CNS neuron survivability,22–24 or (4) ADA affects adenosine receptors directly.2
Since adenosine A1 receptor agonists decrease brain damage induced by ischemic stress,8 adenosine is a known neuroprotective agent. Therefore, ADA catabolism of adenosine was expected to have negative effects.8,25,26 However, the protective role of adenosine A2A receptor antagonists has recently received increasing attention.2,9,21 Assuming that a reduction of the A2A receptor dependent signaling cascade counteracts ischemic stress, the ADA-induced decrease of adenosine receptor ligands could also have neuroprotective effects, especially in regions with abundant A2A receptors such as the striatum.
Based on the above, we were interested in determining whether ADA is protective or harmful in the striatum. In order to determine the effects of ADA on transient ischemic stress of the striatum, we used acute rat corticostriatal slices. An acute slice preserves the neuro-glial connections and enables pharmacological testing with artificially controlled oxygen/glucose deprivation (OGD) conditioning. We used field potentials as the primary measure of neuronal damage. The striatal field potentials were evoked in two ways: (1) electrical stimulation of the white matter between the cortex and the striatum and (2) optogenetic stimulation27,28 of the striatum. The latter is an original method to enable reproducible evaluation of the OGD-induced neuronal damage. Since the striatum lacks the layered dendritic structure seen in the hippocampus, it is difficult to observe robust post-synaptic field responses with conventional electrical white matter stimulation. Therefore, we concomitantly used the optogenetic method, which enables the measurement of the striatal neuronal responses as extracellular DC potential. In this study, we tested two hypotheses: (1) ADA modulates neuronal damage during and after OGD, and (2) optogenetic stimulation is as useful as conventional electric stimulation for the detection of OGD-induced neuronal damage in striatum.
Materials and methods
Animals
The care and use of all rats in this study was carried out in accordance with the institutional ethical guidelines for animal experiments and the safety guidelines for gene manipulation experiments of the National Defense Medical College (Tokorozawa, Saitama, Japan). All experimental procedures were approved by the Animal Research Committee of the National Defense Medical College. Reporting of the results conforms to the ARRIVE (Animal Research Reporting in In Vivo Experiments) guidelines. We used a transgenic Wistar W-TChR2V428 rat line, which expresses channelrhodopsin-2 (ChR2) with the Venus fluorescence marker under the regulation of the Thy-1.2 promoter which drives neuron-specific expression of transgenes.28,29 ChR2 acts as a directly light-gated cation-sensitive ion channel30 and ChR2-transfected neurons become photosensitive. Most of the neurons, including striatal medium spiny neurons, express ChR2 (Figure S1) and show robust depolarization under blue light stimulation.28,31 Cell-type specific ChR2 expression localization has not yet been identified.
Corticostriatal slice
Male and female rats 8–12 weeks of age (160–250 g body weight) were anesthetized with isoflurane and sodium pentobarbital. Ice-cold oxygenated (95% O2/5% CO2) cutting solution containing (in mM) 93 N-Methyl-D-glucamine (NMDG)-Cl, 30 NaHCO3, 20 HEPES, 2.5 KCl, 10 MgSO4, 0.5 CaCl2, 1.2 NaH2PO4, 25 glucose, 2 thiourea, 5 sodium ascorbate, and 3 sodium pyruvate32,33 (pH 7.4) was perfused transcardially (hereinafter NMDG solution). The animals were decapitated quickly and 6 to 8 500-µm-thick parasagittal brain slices were cut using a microslicer (Linear Slice Pro 7, Dosaka, Kyoto, Japan) in ice-cold NMDG solution. Prior to recording, individual slices were pre-incubated for 10–15 min in the NMDG solution at 32 ± 1℃. Then they were transferred into oxygenated artificial cerebrospinal fluid (aCSF) at 23 ± 1℃, which consists of (in mM) 120 NaCl, 26 NaHCO3, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4 and 11 glucose (pH 7.4). The slices were mounted in the stage chamber of an upright microscope equipped with infrared differential interference contrast (IR-DIC) optics (AxioSkop 2FS, Zeiss, Jena, Germany). The slices were constantly perfused with aCSF at 1.5–2 ml/min at 32 ± 1℃ throughout the recordings. The slices derived from each animal were randomized into the experimental (with ADA) and the control (without ADA) groups.
Electrophysiology: DC field potential
Extracellular recordings from the slice were performed under direct visual control using the microscope equipped with a 5x objective lens. Extracellular field potentials were recorded with glass microelectrodes (4–7 MΩ) filled with 3 M NaCl and placed on the striatum near the white matter. Data from the electrode were stored on a PC using an A-D/D-A converter (30 kHz/channel, 16-bit; USB-6259BNC, National Instruments, Austin, USA) and Matlab (Mathworks Japan, Tokyo, Japan).
Electrical stimulation was delivered through a bipolar electrode positioned in the white matter between the cortex and the striatum.34 Constant current stimuli (0.2–0.8 mA, 100 µs duration) were generated every 15 s by a constant current stimulus isolator (SS-102J, Nihon Koden, Tokyo, Japan).
Photo-stimulation was delivered by a blue LED light source (Luxeon Rebel LXML-PB01-0023, Phillips Lumileds Lighting Company, San Jose, USA) through a camera double port with a standard GFP filter cube. The focused light spot was a 90 × 90 µm square and had a light power density of 2 mW/mm.2 The pulse intensity and the pulse timing of the LED light were electronically controlled by a computer through a custom-designed current regulator.
To emulate ischemic stress, the slice was temporarily (10 min)35 perfused with oxygen/glucose deprived34 aCSF (OGD-aCSF) under electrical recording. Compared with the composition of standard aCSF, the OGD-aCSF had an equimolar substitution of sucrose for glucose and was deoxygenated using 5% CO2 and 95% N2 for at least 1 h.36 ADA was added only to the OGD-aCSF solution (OGD-ADA). After the OGD period, to emulate reperfusion stress, slices were quickly reperfused with standard aCSF oxygenated with 5% CO2 and 95% O2. The temperature of the slice bath-solutions for both normal aCSF and OGD-aCSF was controlled at 32 ± 1℃.21,35–37 In this experiment, two slices (one slice for OGD and one slice for OGD-ADA) were obtained from a single rat. Seven rats were used in total, meaning that seven slices were used in each experimental group. One experiment in eFP for OGD-ADA and one experiment in optFP for OGD were excluded because of mechanical recording trouble.
The successful detection rate after pre-incubation with and without ADA was calculated by dividing the number of successful eFP trials (in which the responses to stimulation were detected) by the number of total trials. In this experiment, four slices (two slices for control and two slices for pre-incubation with ADA) were obtained from each rat. Two rats were used in total, with a total of four slices used in each experimental group. Twenty-five trials were carried out on a single slice, and thus, 100 trials were performed in each experimental group.
Electrophysiology: multiunit recording
A wire tetrode was constructed with an 18 µm-diameter nickel-chrome wire (761000, A-M Systems, Washington, USA) for multiunit extracellular recordings. Signal from the tetrode was amplified with a 4-channel voltage follower pre-amplifier and an amplifier (AB-611J, Nihon Koden, Tokyo, Japan). Data from the tetrode were stored on a PC using an A-D/D-A converter and Matlab. Multiunit recording data were processed to isolate spike events using the semi-automatic spike-sorting method.38 Four animals, with two to three slices derived from each animal, were assigned to the multiunit recording experiment.
Propidium iodide (PI) staining
After pre-incubation, the slices were transferred onto a 30 mm Millicell-CM insert membrane (with 0.4 µm pore size, Millipore, MA, USA) in six-well plates (Corning Costar, Tokyo, Japan), with two slices on one insert. Twenty-two sister corticostriatal slices were assigned to the PI staining experiment; 2 animals were assigned, with 10–12 slices derived from each animal. Slices were subjected to ischemic treatment by submersion in OGD-aCSF. OGD treatments were performed at 32 ± 1℃.21,35–37 ADA (10 µg/ml) was applied only during the period of submersion. After 30 min of OGD treatment, slices were returned to wells containing 1.5 ml of standard aCSF oxygenated with 5% CO2 and 95% O2. For cell viability evaluation, each slice (OGD or OGD-ADA) was maintained in a separate chamber in oxygenated aCSF at room temperature (23 ± 1℃) for 5.5 h starting from the end of OGD.
PI dye was added directly to the aCSF in the last 15 min of post-OGD incubation to yield a 2 µM concentration. After PI staining, each slice was washed for >15 min with oxygenated aCSF and was mounted onto a glass bottom dish for microscopic examination 6–7 h after OGD termination (modified from Pugliese et al., 2009).21
Slices were mounted on the stage of a laser confocal scanning microscope (LSM510, Carl Zeiss, Tokyo, Japan) with the imaging software ZEN (Carl Zeiss) to estimate z-depth. PI fluorescence was elicited by excitation at 543 nm and was collected by a 10x dry objective. We obtained 3D images of 20-µm-thick sections located between 30 and 50 µm below the slice surface.33 Stored images were then counted manually with NIS elements D 4.2 image analyzing software (Nikon Instruments Inc., Tokyo, Japan). The number of PI-positive cells in an area of 250,000 µm2 was used as the dead cell count of the respective slice. Cells with length of over 5 µm along the major axis were counted. The dead cell counts of OGD-ADA groups were compared with those of sister slices derived from the same animals that were only exposed to OGD.39 No image data were excluded from this analysis.
Immunoblotting
Autophagic death signaling was immunologically evaluated by examining the expression of microtubule-associated protein light chain 3-II (LC3-II).40 The presence of LC3-II in the slices in the 6-well plate (see above PI staining section) was measured. In the control group, OGD treatment was replaced with standard oxygenated aCSF at 32 ± 1℃. Each slice was transferred to a 1.5 ml tube 1 h after OGD/OGD-ADA, quickly frozen using liquid N2, and stored at −80℃ (Figure 4(a)). Three animals were used and nine sister slices were derived from each animal. In total, 27 sister corticostriatal slices were assigned to the immunoblotting experiment. The frozen samples were sonicated in boiling 1% sodium dodecyl sulfate (SDS) containing 50 mM sodium fluoride, boiled for an additional 10 min, and centrifuged at 15,000 g for 20 min at 4℃ in order to precipitate debris. The supernatant was collected, and the protein concentration was determined using a Protein Assay Reagent Kit (Thermo Fischer Scientific Inc., Illinoi, USA). The samples were run using electrophoresis on a TGX precast gel (Bio-Rad Laboratories Inc., Hercules, CA, USA), and transferred electrically to nitrocellulose membranes (pore size, 0.2 µm; Thermo Fischer Scientific Inc.) by loading at 20 V for 45 min with a semidry blot transfer system (Trans-blot, Bio-Rad Laboratories). After blocking the membrane with 5% skim milk in 1x Tris buffered saline (TBS) and 0.1% Tween-20 for 1 h at RT, the membranes were exposed to specific primary antibodies (rabbit anti-LC3-II monoclonal antibody #4599, diluted 1:5000; Cell Signaling Technology Inc., Danvers, MA, USA) in 1x TBS with 10% bovine serum albumin and 0.1% Tween 20. The blots were washed and incubated for 1 h at RT with HRP-conjugated anti-rabbit IgG antibody (#7074, diluted 1:4000; Cell Signaling Technology Inc.). For the immunoblotting of β-actin, the membranes were exposed to a rabbit anti-β-actin antibody (#5125, diluted 1:10,000; CST). For the detection of chemiluminescent signals, an image analyzer (LAS-4000, GE Healthcare, Buckinghamshire, England) and ECL Prime (RPN2232, GE Healthcare) were used. The levels of each protein were determined by measuring the area of each band by outlining the band on blots and measuring the average optical density by subtracting background signals. For densitometry, NIH ImageJ software was used.
Figure 4.

Adenosine deaminase (ADA) supplementation during oxygen-glucose deprivation (OGD) attenuated LC3-II expression in the corticostriatal slice. (a) The slices were incubated with and without ADA (10 µg/ml) during 30-min OGD. (b) Western blotting was carried out on corticostriatal slice homogenate obtained 60 min after the end of the OGD period. (c) LC3-II levels when treated with ADA were significantly lower than control (***p < 0.0001) and without ADA (***p < 0.0001).
In individual experiments, samples from control, OGD and OGD-ADA-treated slices were analyzed on the same immunoblot. For each experiment, values obtained for slices were calculated relative to values for the OGD or OGD-ADA-treated slices. Normalized data from multiple experiments were averaged and statistical analysis was carried out as described in the figure legends. Protein extraction and densitometory analysis were performed by the authors who were blinded to the ADA-treatment.
IR morphological evaluation
To quantify the morphological changes of living cells without staining, the slices were mounted on the stage of an infrared (IR) differential interference contrast microscope. Two animals were used and nine slices were obtained from each animal. In total, 18 sister corticostriatal slices were assigned to the IR morphological evaluation experiment. A series of manually incremented images of each slice from the surface to 50 µm depth were obtained. The focused cell bodies were manually identified. The major and minor axis lengths of cells in an area of 26,900 µm2 of each slices were manually measured. Cell diameter was determined as the average of the lengths of the major axis and the minor axis. No image data were excluded from this analysis.
Drugs
Calf ADA, the ADA isoenzyme (ADA 1, 200 IU/mg of protein; Roche, Basel, Switzerland), was used; 7-nitro-2,3-dioxo-1,4-dihydroquinoxaline-6-carbonitrile (CNQX) and D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5) were obtained from Wako (Osaka, Japan). PI was obtained from Dojindo (Osaka, Japan).
Results
Reproducible electrical viability test
Evoked field potential measurements are required to determine the effects of ADA on the OGD-induced reduction of striatal electrical activity. Although the electrical stimulation to white matter has been used for the measurement of evoked striatal responses,36 the low reproducibility disturbs the recording of OGD-induced changes, possibly due to the lack of layered dendritic structure and robust compound post-synaptic potentials in the striatum. Therefore, we used an optogenetically induced extracellular DC potential to accumulate dendritic and somatic current in striatal neurons. We used a transgenic W-TChR2V4 rat line, which expresses ChR2 (see Materials and methods section). Large extracellular DC potential changes during striatal photo-stimulation can be observed in the transgenic rat striatum. We utilized them to determine the viability of striatal neurons.
Figure 1 shows the electrically and optogenetically evoked field potentials (eFP and optFP respectively) of the dorsal striatum near the white matter. For eFP, the wire stimulation electrode was placed on the white matter, and the field potential was recorded at the dorsal striatum (Figure 1(a)). The eFP was defined as the amplitude between the mean of the two positive peaks and the minimum of the negative peak, based on previously proposed methods.34 For optFP, we applied 20-ms blue light pulse stimulation to the striatum and measured the DC field responses at the striatum (Figure 1(b) and S2). The recording electrode tip was placed at the center of the light spot. Two types of responses were obtained. One was a DC potential with an action potential, which has a strong sharp potential change and was only observed at the onset of stimulation (Figure 1(b), right). The other response obtained was without the action potential (Figure 1(b), left). The optFP indicator was defined as the mean of a 10-ms-long DC negative field response, 10-ms after the onset of photo-stimulation, to exclude the action potential. The DC response was considered to be mainly the result of the continuous inward current in striatal cells near the recording electrode, because the optFP was not affected by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA-R) blocker CNQX and N-Methyl-D-aspartate receptor (NMDA-R) blocker AP5 bath-applications (Figure S3).
Figure 1.

Field responses to electrical and optogenetic stimulation. (a) Electrically evoked striatal field response (top; eFP). Electrical stimulation was applied through a twisted bipolar wire electrode placed on the white matter between the cortex (CTX) and the dorsal striatum (STR). (b) Optogenetically evoked striatal field responses (top; optFP). Optical stimulation was applied to the striatum. DC potentials with (left) and without (right) the action potential were recorded. (c) The success rate of eFP and optFP detection. OptFPs were detected in all trials.
We compared the reproducibilities of both optFP and eFP by counting the ratio of successfully detected evoked potentials. A negative peak which was isolated from the stimulation artifact34 was defined as a detection of eFP. A negative DC potential, which was over 100 µV in amplitude was defined as a detection of optFP. One hundred trials at different sites showed that the detected eFP to optFP ratio was 18 to 100. OptFP was detected in all recording trials (Figure 1(c)). Therefore, optFP can enable consistent measurements of striatal electrical changes during OGD.
ADA rescues OGD-induced electrical attenuation
To obtain the time courses of eFP and optFP during and after OGD, both electrical stimulation and photo-stimulation were applied 4 times for 1 min with 15 s stimulation onset intervals. The 10-min long OGD inhibited eFP (Figure 2(a), black line, CTRL, 65.1 ± 5.0% 35 min after the end of OGD, n = 7). The 10-min long OGD with the application of 10 µg/ml ADA significantly reduced the inhibition 35 min after the end of OGD (Figure 2(a), red line, ADA, 103.9 ± 11.8%, n = 6, p < 0.01, t = 3.20, df = 11, two-sided Student’s t test). The OGD also inhibited optFP (Figure 2(b), black line, CTRL, 82.4 ± 4.5%, n = 6), and the inhibition was significantly attenuated by the application of ADA during OGD (Figure 2(b), red line, ADA, 101.3 ± 5.5%, n = 7, p < 0.01, t = 3.45, df = 11, two-sided Student’s t test).
Figure 2.

Neuroprotective effects of adenosine deaminase (ADA) in the rat striatum. (a) and (c) White matter electrical stimulation evoked striatal field potential (eFP). The oxygen-glucose deprivation (OGD) reduced eFP (a: CTRL-black line, n = 7, 65.1 ± 5.0% at 50 min). ADA supplementation during the OGD significantly enhanced the post-OGD eFP recovery (a: ADA-red line, n = 6, 103.9 ± 11.8% at 50 min, p < 0.01). In (c), the stimulus artifact has been erased. (b) and (d) Optogenetic striatal stimulation evoked striatal field potential (optFP). OGD reduced optFP (b: CTRL-black line, n = 6, 82.4 ± 4.5 % at 50 min). ADA significantly enhanced the post-OGD optFP recovery (b: ADA-red line, n = 7, 101.3 ± 5.5 % at 50 min, p < 0.01).
ADA reduces OGD-induced late onset cell death
In order to determine the effects of ADA on cell viability, we measured late-onset cell death after OGD by counting the number of PI positive cells.33 Instead of using OGD conditioning on slices in the stage chamber of the microscope, the slices were incubated and conditioned on the membrane filter in a 6-cell incubation chamber to reduce slice damage. Our preliminary test suggested that a 6-h incubation after OGD was required to detect obvious cell death. In 10 slices, a 30-min long OGD induced evident cell death in the entire area of the striatum after 6-h (Figure 3(a), CTRL). This late onset cell death is consistent with other investigations.33 ADA application during OGD induction significantly decreased the number of PI positive cells (Figure 3(b) and (c), p < 0.05, t = 3.18, df = 20, two-sided Student’s t test).
Figure 3.
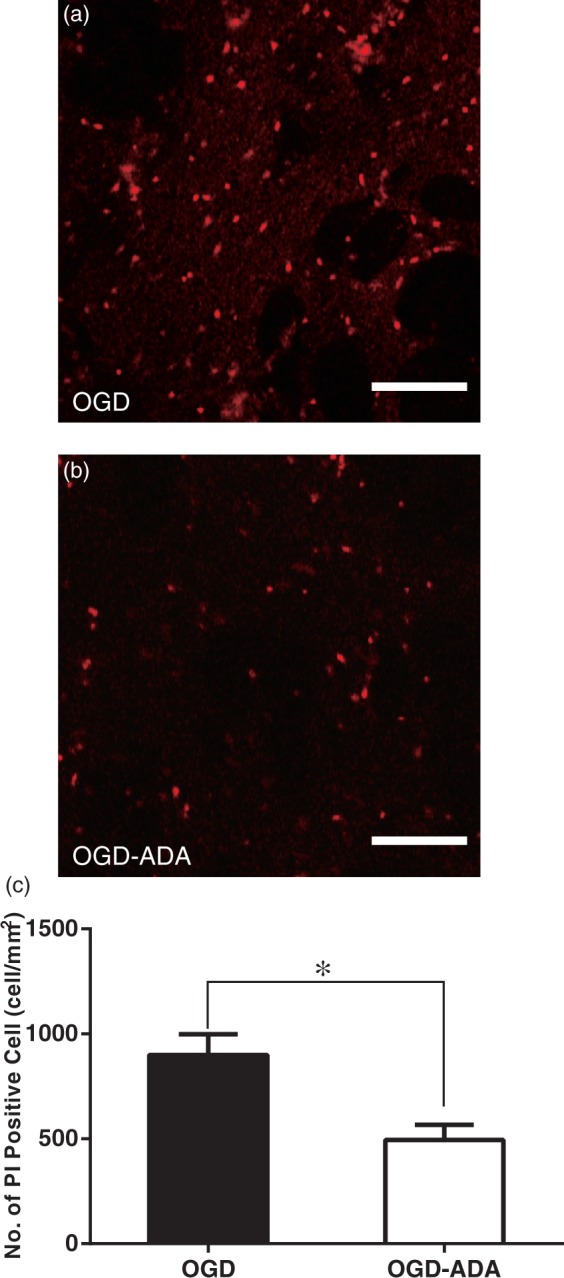
Adenosine deaminase (ADA) decreased propidium iodide (PI)-positive injured cells in the striatum. PI staining was carried out 5.25–5.5 h after the end of 30-min oxygen-glucose deprivation (OGD) with (b) or without (a) 10 µg/ml ADA. Scale bars represent 100 µm. (c) The number of PI positive cells in the OGD-slices with ADA (n = 10) was significantly smaller than in those without ADA (n = 12, p < 0.05).
ADA alters autophagy
We sought to elucidate the effects of ADA on autophagic cell death signaling by examining the expression of microtubule-associated protein light chain 3-II (LC3-II) as a biochemical footprint.40 With the stress of starvation40,41 or ischemia,42 cytosolic LC3-I is known to convert to phosphatidylethanolamine-conjugated active LC3-II. LC3-II promotes the formation of autophagosomes that ultimately lead to lysosomal autodigestion and autophagic cell death.41 Western blot analyses (Figure 4) revealed significant differences of LC3-II expression among the control, OGD and OGD-ADA groups (df = 9, F = 41.4, one-way ANOVA). We observed significantly increased LC3-II expression in the OGD treated group compared to the control group (p < 0.0001, Tukey’s post-hoc test) and significantly reduced LC3-II expression in the OGD-ADA group compared to the OGD group (p < 0.0001, Tukey’s post-hoc test).
ADA preserves striatal cell morphology and electrical activity
We examined the effects of ADA on the initial starvation and high-concentration adenosine exposure during the pre-incubation process. Traditionally, the acute slice-OGD method has been used in order to investigate ischemic stress related changes.6,34,36 However, during pre-incubation the acute slices suffer from OGD-like starvation and exposure to high concentrations of adenosine before the standard OGD induction. Therefore, the traditional OGD is a second conditioning. In the slice preparation process,33 the slice experiences ice-cold induced dormancy on the cutting stage followed by a 32℃ pre-incubation (Figures 4(a) and 5(a)) induced recovery challenge from the dormancy. During the recovery process, slices are thought to be exposed to increased extracellular adenosine,6 in addition to suffering from ATP depletion due to the resumption of internal cell metabolism.43 Takahashi et al.6 reported that extracellular adenosine release during OGD in the isolated spinal cord is highly dependent on temperature: the OGD-induced increase in adenosine levels at 35℃ is about 10-fold higher than that at 25℃. In the acute slice tissue, it is probable that the levels of ADA1,2 decrease due to a lack of ADA supply from blood circulation2 and diffusive loss of ect-ADA.2
Figure 5.

Long-term effects of ADA 3–6 h after washout. (a) ADA was added to 32℃ pre-incubation solution (NMDG solution). (b) ADA pre-incubation increased the eFP detection rate (% of total trials). The eFP counts were measured 3–6 h after the pre-incubation and the washout of NMDG solution. (c) Raw waveform of striatal firing response to a 300-ms photo-stimulation. The spikes were observed mainly late in the photo-stimulation. Top bar indicates the photo-stimulation period. The right inset illustrates an expanded single spike (at the arrow). (d) Averaged peri-stimulus time histogram (PSTH) of isolated spike responses (n = 48). (e) Raw waveform obtained from ADA pre-incubated slice. The onset of spike responses to the photo-stimulation was accelerated. (f) Averaged PSTH from the ADA pre-incubated slice (n = 28). (g) ADA pre-incubation significantly increased the mean rate of optogenetically evoked firings during the 150 ms after the onset of the photo-stimulation (p < 0.05).
Pre-incubation with ADA (Figure 5(a)) increased the eFP detection ratio 3–6 h after the incubation (Figure 5(b), CTRL: n = 100, 18%; ADA: n = 100, 40%. Here, four slices were used in each group, and 25 trials were carried out on each slice). The eFP detection ratio was calculated in the same way as described above (see reproducible electrical viability test section). To confirm the effects of ADA on the striatal neurons, we also analyzed the evoked striatal firing pattern by 300 ms striatal photo-stimulation. The isolated spikes (Figure 5(c)) were obtained by using the multiunit recording and spike sorting method. Pre-incubation with ADA accelerated the initial firing response during the 300-ms length photo-stimulation (Figure 5(e)). The number of evoked spikes from multiple neurons was counted in each 50-ms length bin, averaged for the whole sample, and displayed as a peri-stimulus time histogram (PSTH)44 (Figure 5(d) and (f)). The spike counts during the first 150-ms after the onset of photo-stimulation significantly increased after the addition of ADA to the pre-incubation medium (Figure 5(g); CTRL: 7.91 ± 1.53 spikes/s, n = 48 neurons; ADA: 19.2 ± 4.40 spikes/s, n = 28, p < 0.05, t = 3.56, df = 224 by two-way ANOVA with Bonferroni post-hoc test).
Finally, we compared the morphological conditions of the slices 3–6 h after pre-incubation. Although the control slices that were pre-incubated without ADA showed increased cell body diameter (Figure 6(a)), the slices with ADA pre-incubation preserved the cell shapes keeping the cell body size small (Figure 6(b)). To clarify the preservative effect of ADA pre-incubation on cell shapes, we measured the body size of all cells in the ROI (164 µm × 164 µm area) 3–5 h after pre-incubation with and without ADA. With ADA, the number of small cells with 8 ± 0.5, 9 ± 0.5, 10 ± 0.5 µm diameters were significantly greater than the control (Figure 6(c), p < 0.0001 by two-way ANOVA with Bonferroni post-hoc test, CTRL: n = 9 slices; ADA: n = 9), and the median body diameters were smaller than the control.
Figure 6.

ADA pre-incubation preserved the morphology of striatal cells. The IR images (40x objective lens) were obtained from 78-day-old rat striatal slices 6 h after the 32℃ pre-incubation with (b) and without ADA (a). Scale bars represent 15 µm. (c) Cell size histograms. ADA pre-incubation increased the small cell population. The cell axis lengths were manually measured in 26,900 µm2 area of each slice. Cell diameter was determined as the average of the lengths of the major axis and the minor axis. Asterisk indicates significantly different from control in each bin (*p < 0.0001; CTRL n = 9, ADA n = 9). Small circle with vertical line indicates the median cell diameter.
Discussion
Although a global extracellular distribution of ADA has been reported in the CNS,1 the physiological function of ADA has not yet been determined. Our present study indicates that ADA has a neuroprotective role under ischemic conditions in the striatum. We demonstrated that ADA inhibits the OGD-induced electrophysiological depression (Figure 2), cell death (Figure 3) and autophagy processes (Figure 4). The 10-min OGD decreased the striatal field responses to the white matter electrical stimulation and the optogenetic photo-stimulation during and after the OGD. The ADA supplementation to OGD-aCSF reduced the OGD-induced decrease of field responses 35 min after OGD (Figure 2). Excessive autophagy processes induced by severe ischemia are related to cell death. Activation of LC3-I by covalent conjugation with phosphatidylethanolamine occurs in the first step of autophagy, and is induced by starvation or ischemic stress in the nervous system.42 Our results of ADA-induced active LC3-II reduction indicate that ADA attenuates the autophagy process. We also confirmed that ADA application during the pre-incubation process with its associated ATP starvation and exposure to high-concentration adenosine results in an increase of electrophysiological responses (Figure 5) and the reduction of morphological cell damage (Figure 6) 3–5 h after the washout of ADA. All of these results consistently show that ADA plays a neuroprotective role in the striatum.
We hope these findings will help re-evaluate the bovine ADA supplementation treatment15,18 for ADA deficient patients. In the 2000s many epidemiological reports from various countries indicated strong relationships between ADA-SCIDs and neurological deficits.14–16 At present, hematopoietic stem cell transplantation (HSCT) is the first-line of treatment for congenital immunodeficiency, while ADA supplementation is a second-line therapy.13 HSCT does not sufficiently decrease excessive adenosine and deoxy-adnosine,15 and HSCT leaves serious neurological problems including mental retardation, seizures, and basal ganglia volume loss in some recipients.14–17 On the other hand, ADA supplementation results in higher systemic ADA concentrations than HSCT therapy16 and patients do not show progressive neuronal abnormalities.15 Our results suggest that conventional ADA supplementation should be reevaluated from the combined viewpoint of neurological pathophysiology and CNS stress. ADA-deficient patients and ADA lacking mice repeatedly suffer from non-infectious respiratory distress and severe hypoxia.18,19 Thus, there is a higher probability of cerebral hypoxia, which could result in striatal lesions. Therefore, it is possible that ADA supplementations introduced at earlier stages could protect the CNS during the perinatal and developmental periods. Although the neurological problems might originate from complex interactions and the fundamental assessment is difficult, the investigation of the functional role of ADA in the CNS should be the next target of ADA deficiency research.
One of the main limitations of our study is that our observation is performed only in corticostriatal acute slices from adult rats. In vivo and in culture studies would indicate a more comprehensive result. It should be noted that age, time intervals from ischemia and doses interfere with adenosine-CNS related issues strongly.9,10,36 It should also be noted that the effects of systemic ADA administration for other brain regions have not been determined.
During and after ischemia, CNS cells show a cascade of cellular level catastrophes and intracellular interferences. Brisson et al. revealed that the striatal neurons show anoxic depolarization, losing the resting membrane potential during OGD and returning to an abnormal potential after 10-min of OGD.35 Since the potential loss was mimicked by the application of a Na+/K+-ATPase inhibitor ouabain, they hypothesized that ischemia shuts down the ATP-dependent Na+/K+ pump, resulting in the potential loss. Anoxic depolarization is most probably the major cause of the reduction of the electrically/optogenetically evoked field responses (Figure 2). The Na+/K+ pumping fault breaks down the cell osmolality, resulting in cellular morphological changes and damage35 (Figures 6 and 7). While this is the most common main process of ischemia induced cell death, a more detailed discussion of how ADA additionally modulates this process and the 4 possible mechanisms of ADA neuroprotection can be found below.
Figure 7.

Possible ADA neuroprotection mechanisms. OGD and ischemia induce intracellular ATP loss resulting in cell damage. In addition to this damage process, excessive glutamate release potentiates cell damage. Although adenosine inhibits the excessive glutamate release via A1-R activation and reduces the additional excitotoxicity, adenosine has a harmful effect by enhancing the glutamate release via A2A-R activation. ADA considerably reduces the A2A-R based excitotoxicity (1). In the striatum, expression of A2A-R is much higher than compared with other regions (see review, Cortes et al. 20152)(*). In addition, deoxy-adenosine and deoxy-ATP are also known causes of apoptosis. ADA could inhibit this cell damaging process (2).
During ischemia, CNS cells effuse a high concentration of adenosine.4,5,8 The concentrated intracellular adenosine is well-known to protect neurons through the A1 receptor.2,8 However, it is also known that the A2A antagonist plays a protective role during OGD/ischemia and leads to greater neuronal survival.9,21 These controversial effects are explained by the opposing actions on glutamatergic excitotoxicity: the A1 receptor activation inhibits excessive glutamate release and the A2A receptor conversely promotes it.2,8,9,25 In the striatum, expression of A2A receptor is much more abundant compared with the other brain regions,2 and thus, adenosine deamination with ADA might reduce the A2A receptor activation and the associated excitotoxicity (Figure 7).
However, from the viewpoint of the ADA deficiency mechanisms and the adverse effects of ADA inhibitor treatment in chemotherapy,2,45,46 there could be an alternative explanation of the effects of ADA, which could derive its neuroprotective role through the deamination of deoxyadenosine (Figure 7). ADA deficient patients suffer from immunodeficiency based on T-cell extinction. ADA deficiency increases not only adenosine but also deoxyadenosine.47,48 Adenosine is deoxidized into deoxyadenosine and deoxyadenosine is converted to deoxy-ATP by the ubiquitous enzyme ribonucleotide-diphosphate reductase (EC 1.17.4.1).13 Deoxyadenosine is cytotoxic to T-cells.13,49 In the treatment of lymphoid malignancy, an ADA inhibitor deoxycoformycin is used2,45 and is well-known to have severe neurotoxicity resulting in fatal coma, encephalopathy with seizures and coma, generalized seizures, long-lasting ataxia and dementia.45 It is reported that the neurotoxicity of the ADA inhibitor is associated with high levels of CSF deoxyadenosine and high erythrocyte deoxy-ATP/ATP ratios.46 Although at this time it is unclear how deoxyadenosine and deoxy-ATP relate to neurotoxicity, the possibility of their negative effects on the CNS should not be ignored.
There remain two other possible explanations of the protective effect of ADA. On one hand, highly concentrated inosine derived from adenosine by ADA could be anaerobically metabolized into intracellular ATP and utilized by oxygen deprived and starved cells.23 The neuroprotective effects of inosine during OGD have been reported in cultured cells, although the culture systems do not perfectly simulate the real brain during ischemia.22,24 On the other hand, ADA itself may also modify the adenosine receptors directly. A1, A2A, and A2B receptors are ADA-anchoring membrane proteins and the anchored ADA could alter the properties of the adenosine receptors.2
In the present study, we introduced optFP as an alternative to eFP as an indicator of neuronal viability. It is worth noting that the optFP procedure is highly reproducible (Figure 1(c)). Although it is only possible to stimulate a limited number of neurons in the examined region by the eFP procedure, the optFP procedure can stimulate most neurons in the photo-stimulated region and enable extracellular recordings of the strong DC responses. Just by setting the recording electrode and starting photo-stimulation, experimenters can start the examination and maintain responses stably for over an hour. In addition, the 100% efficacy rate of optFP can dramatically reduce the number of animals in a given experiment.
In conclusion, this report shows the neuroprotective effects of the ADA enzyme in the striatum. The possible therapeutic usages of ADA in ischemia and the treatment of neurological problems in ADA-deficiency should be explored in future studies.
Acknowledgements
We are grateful to S. Kanda for great technical assistance, to D. A. Tyurmin for language assistance, and to K. Takishima and Y. Watanabe for advising.
Funding
This work was supported by MEXT/JSPS KAKENHI Grant Number 24700200, a grant for Specific Research from National Defense Medical College, Defense Medicine Propulsion Research of the Ministry of Defense, Japan, the Kawano Memorial Foundation for the Promotion of Pediatrics of Japan, and the Research Foundation for Opto-Science and Technology, Hamamatsu, Japan.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
RT, HO, YS, MN, and SN designed the experiments and interpreted the data. RT and HO collected the data. HO, YS, MN, and YN analyzed the data. RT, HO, YS, MN, and SN prepared the manuscript. HO is the guarantor of the work and, as such, had full access to all the data in the study and takes full responsibility for the integrity of the data and accuracy of the data analysis.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data.
References
- 1.Nagy J, LaBella L, Buss M, et al. Immunohistochemistry of adenosine deaminase: implications for adenosine neurotransmission. Science 1984; 224: 166–168. [DOI] [PubMed] [Google Scholar]
- 2.Cortes A, Gracia E, Moreno E, et al. Moonlighting adenosine deaminase: a target protein for drug development. Med Res Rev 2015; 35: 85–125. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Corriden R, Inoue Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006; 314: 1792–1795. [DOI] [PubMed] [Google Scholar]
- 4.Melani A, Pantoni L, Corsi C, et al. Striatal outflow of adenosine, excitatory amino acids, gamma-aminobutyric acid, and taurine in awake freely moving rats after middle cerebral artery occlusion: correlations with neurological deficit and histopathological damage. Stroke 1999; 30: 2448–2454. discussion 2455. [DOI] [PubMed] [Google Scholar]
- 5.Hillered L, Hallström A, Segersvärd S, et al. Dynamics of extracellular metabolites in the striatum after middle cerebral artery occlusion in the rat monitored by intracerebral microdialysis. J Cereb Blood Flow Metab 1989; 9: 607–616. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi T, Otsuguro K, Ohta T, et al. Adenosine and inosine release during hypoxia in the isolated spinal cord of neonatal rats. Br J Pharmacol 2010; 161: 1806–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballarin M, Fredholm B, Ambrosio S, et al. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand 1991; 142: 97–103. [DOI] [PubMed] [Google Scholar]
- 8.De Mendonça A, Sebastião AM, Ribeiro JA. Adenosine: does it have a neuroprotective role after all? Brain Res Rev 2000; 33: 258–274. [DOI] [PubMed] [Google Scholar]
- 9.Melani A, Pantoni L, Bordoni F, et al. The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res 2003; 959: 243–250. [DOI] [PubMed] [Google Scholar]
- 10.Baron J-C, Yamauchi H, Fujioka M, et al. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab 2014; 34: 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Laat KF, Van Norden AGW, Gons RAR, et al. Cerebral white matter lesions and lacunar infarcts contribute to the presence of mild parkinsonian signs. Stroke 2012; 43: 2574–2579. [DOI] [PubMed] [Google Scholar]
- 12.Turner CP, Seli M, Ment L, et al. A1 adenosine receptors mediate hypoxia-induced ventriculomegaly. Proc Natl Acad Sci U S A 2003; 100: 11718–11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaspar HB, Aiuti A, Porta F, et al. How I treat ADA deficiency. Blood 2009; 114: 3524–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers MH, Lwin R, Fairbanks L, et al. Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J Pediatr 2001; 139: 44–50. [DOI] [PubMed] [Google Scholar]
- 15.Hoenig M, Albert M, Schütz C, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood 2006; 109: 3595–3602. [DOI] [PubMed] [Google Scholar]
- 16.Titman P, Pink E, Skucek E, et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood 2008; 112: 3907–3913. [DOI] [PubMed] [Google Scholar]
- 17.Nofech-Mozes Y, Blaser SI, Kobayashi J, et al. Neurologic abnormalities in patients with adenosine deaminase deficiency. Pediatr Neurol 2007; 37: 218–221. [DOI] [PubMed] [Google Scholar]
- 18.Booth C, Algar VE, Xu-Bayford J, et al. Non-infectious lung disease in patients with adenosine deaminase deficient severe combined immunodeficiency. J Clin Immunol 2012; 32: 449–453. [DOI] [PubMed] [Google Scholar]
- 19.Dhanju R, Min W, Ackerley C, et al. Pulmonary alveolar proteinosis in adenosine deaminase-deficient mice. J Allergy Clin Immunol 2014; 133: 1467–1471. [DOI] [PubMed] [Google Scholar]
- 20.Chen JF, Huang Z, Ma J, et al. A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci 1999; 19: 9192–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pugliese AM, Traini C, Cipriani S, et al. The adenosine A2A receptor antagonist ZM241385 enhances neuronal survival after oxygen-glucose deprivation in rat CA1 hippocampal slices. Br J Pharmacol 2009; 157: 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haun SE, Segeleon JE, Trapp VL, et al. Inosine mediates the protective effect of adenosine in rat astrocyte cultures subjected to combined glucose-oxygen deprivation. J Neurochem 1996; 67: 2051–2059. [DOI] [PubMed] [Google Scholar]
- 23.Litsky ML, Hohl CM, Lucas JH, et al. Inosine and guanosine preserve neuronal and glial cell viability in mouse spinal cord cultures during chemical hypoxia. Brain Res 1999; 821: 426–432. [DOI] [PubMed] [Google Scholar]
- 24.Jurkowitz MS, Litsky ML, Browning MJ, et al. Adenosine, inosine, and guanosine protect glial cells during glucose deprivation and mitochondrial inhibition: correlation between protection and ATP preservation. J Neurochem 1998; 71: 535–548. [DOI] [PubMed] [Google Scholar]
- 25.Donaghy KM, Schofield CN. Concentration dependence of adenosine and the protection of rat cortical neurones during anoxia. Brain Res 1994; 656: 174–176. [DOI] [PubMed] [Google Scholar]
- 26.Phillis JW, O’Regan MH. Deoxycoformycin antagonizes ischemia-induced neuronal degeneration. Brain Res Bull 1989; 22: 537–540. [DOI] [PubMed] [Google Scholar]
- 27.Tomita H, Sugano E, Fukazawa Y, et al. Visual properties of transgenic rats harboring the channelrhodopsin-2 gene regulated by the thy-1.2 promoter. PLoS One 2009; 4: e7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohta H, Sakai S, Ito S, et al. Paired stimulation between CA3 and CA1 alters excitability of CA3 in the rat hippocampus. Neurosci Lett 2013; 534: 182–187. [DOI] [PubMed] [Google Scholar]
- 29.Aigner L, Arber S, Kapfhammer JP, et al. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 1995; 83: 269–278. [DOI] [PubMed] [Google Scholar]
- 30.Nagel G, Szellas T, Huhn W, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A 2003; 100: 13940–13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji Z-G, Ito S, Honjoh T, et al. Light-evoked somatosensory perception of transgenic rats that express channelrhodopsin-2 in dorsal root ganglion cells. PLoS One 2012; 7: e32699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao S, Ting JT, Atallah HE, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods 2011; 8: 745–752. Light-evoked somatosensory perception of transgenic rats that express channelrhodopsin-2 in dorsal root ganglion cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka Y, Tanaka Y, Furuta T, et al. The effects of cutting solutions on the viability of GABAergic interneurons in cerebral cortical slices of adult mice. J Neurosci Methods 2008; 171: 118–125. [DOI] [PubMed] [Google Scholar]
- 34.Calabresi P, Pisani A, Mercuri NB, et al. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci 1992; 4: 929–935. [DOI] [PubMed] [Google Scholar]
- 35.Brisson CD, Hsieh Y-T, Kim D, et al. Brainstem neurons survive the identical ischemic stress that kills higher neurons: insight to the persistent vegetative state. PLoS One 2014; 9: e96585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klapstein GJ, Levine MS. Age-dependent biphasic changes in ischemic sensitivity in the striatum of Huntington’s disease R6/2 transgenic mice. J Neurophysiol 2005; 93: 758–765. [DOI] [PubMed] [Google Scholar]
- 37.Joshi I, Andrew RD. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J Neurophysiol 2001; 85: 414–424. [DOI] [PubMed] [Google Scholar]
- 38.Takekawa T, Isomura Y, Fukai T. Accurate spike sorting for multi-unit recordings. Eur J Neurosci 2010; 31: 263–272. [DOI] [PubMed] [Google Scholar]
- 39.Frantseva M, Carlen PL, El-Beheiry H. A submersion method to induce hypoxic damage in organotypic hippocampal cultures. J Neurosci Methods 1999; 89: 25–31. [DOI] [PubMed] [Google Scholar]
- 40.Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117: 2805–2812. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 2007; 581: 2156–2161. [DOI] [PubMed] [Google Scholar]
- 42.Weis SN, Toniazzo AP, Ander BP, et al. Autophagy in the brain of neonates following hypoxia-ischemia shows sex- and region-specific effects. Neuroscience 2014; 256: 201–209. [DOI] [PubMed] [Google Scholar]
- 43.Aitken PG, Breese GR, Dudek FF, et al. Preparative methods for brain slices: a discussion. J Neurosci Methods 1995; 59: 139–149. [DOI] [PubMed] [Google Scholar]
- 44.Shinomoto S. Analysis of parallel spike trains, Boston, MA: Springer US, 2010. [Google Scholar]
- 45.Cheson B, Vena D, Foss F, et al. Neurotoxicity of purine analogs: a review. J Clin Oncol 1994; 12: 2216–2228. [DOI] [PubMed] [Google Scholar]
- 46.Grever M, Siaw M, Jacob W, et al. The biochemical and clinical consequences of 2′-deoxycoformycin in refractory lymphoproliferative malignancy. Blood 1981; 57: 406–417. [PubMed] [Google Scholar]
- 47.Wakade AR, Przywara DA, Palmer KC, et al. Deoxynucleoside induces neuronal apoptosis independent of neurotrophic factors. J Biol Chem 1995; 270: 17986–17992. [DOI] [PubMed] [Google Scholar]
- 48.Phan TG, Wright PM, Markus R, et al. Salvaging the ischaemic penumbra: more than just reperfusion? Clin Exp Pharmacol Physiol 2002; 29: 1–10. [DOI] [PubMed] [Google Scholar]
- 49.Wakamiya M, Blackburn MR, Jurecic R, et al. Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc Natl Acad Sci U S A 1995; 92: 3673–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]