

Abstract
Objective:
To identify novel genes implicated in epilepsy with auditory features (EAF) in phenotypically heterogeneous families with unknown molecular basis.
Methods:
We identified 15 probands with EAF in whom an LGI1 mutation had been excluded. We performed electroclinical phenotyping on all probands and available affected relatives. We used whole-exome sequencing (WES) in 20 individuals with EAF (including all the probands and 5 relatives) to identify single nucleotide variants, small insertions/deletions, and copy number variants.
Results:
WES revealed likely pathogenic variants in genes that had not been previously associated with EAF: a CNTNAP2 intragenic deletion, 2 truncating mutations of DEPDC5, and a missense SCN1A change.
Conclusions:
EAF is a clinically and molecularly heterogeneous disease. The association of EAF with CNTNAP2, DEPDC5, and SCN1A mutations widens the phenotypic spectrum related to these genes. CNTNAP2 encodes CASPR2, a member of the voltage-gated potassium channel complex in which LGI1 plays a role. The finding of a CNTNAP2 deletion emphasizes the importance of this complex in EAF and shows biological convergence.
Epilepsy with auditory features (EAF) is an uncommon presentation of focal epilepsy characterized by focal seizures with auditory auras and symptoms suggesting lateral temporal onset.1 EAF is the hallmark of autosomal dominant EAF (ADEAF),1–3 a familial epilepsy syndrome due to mutations of the leucine-rich, glioma-inactivated 1 (LGI1) gene (Online Mendelian Inheritance in Man [OMIM] *604619)4 in 50% of families.5 In ADEAF, affected members typically have EAF, although to make the diagnosis only 2 family members need to have EAF, irrespective of their degree of relatedness and the total number of individuals with epilepsy in the family.5,6
EAF also occurs in sporadic cases or in pedigrees with no LGI1 mutation. In these pedigrees, EAF is indistinguishable from LGI1-positive ADEAF. However, the pattern of inheritance may be unclear, there may be phenotypic heterogeneity and EAF may occur in only a minority of cases. Such families do not usually fulfill the criteria for any specific familial epilepsy syndrome.
The small size, clinical heterogeneity, and incomplete penetrance displayed by such families with EAF may have hampered gene mapping through traditional approaches, such as linkage analysis. No mutation in genes other than LGI1 has been identified so far in EAF.7–10 The advent of next-generation sequencing has made the identification of causal variants possible even in small families. In this study, we describe a comprehensive whole-exome sequencing (WES) approach that enabled us to identify likely pathogenic variants in EAF probands and their affected relatives, demonstrating genetic heterogeneity underlying this epilepsy phenotype.
METHODS
Families.
From 2001 to 2014, we recruited consecutive probands with EAF and family history of epilepsy (at least 1 relative within 3 degrees of kinship affected with EAF or other epilepsy).
All affected family members underwent phenotyping, including medical and seizure history, neurologic examination, EEG recordings, and brain MRI. Previous medical records were obtained.
Standard protocol approvals, registrations, and patient consents.
We received approval from institutional ethical standards committee on human experimentation, and written informed consent was obtained from all patients participating in the study or their parents in the case of minors.
Molecular genetic analysis.
WES was performed on 20 individuals: 15 probands and 5 relatives with EAF. DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Venlo, the Netherlands) following the manufacturer's protocol. Whole-exome DNA was captured using the NimbleGenSeqCap EZ Exome v3 array (Nimblegen Inc., Madison, WI) and sequenced as 91-bp paired-end reads on the Illumina HiSeq2000 platform (Illumina Inc., Santa Clara, CA) at Beijing Genomic Institute (Shenzhen, China). Reads were processed following a general analysis pipeline described elsewhere.11
Single nucleotide variants (SNVs) and small insertions and deletions (InDels) were called with the Genome Analysis ToolKit,12 and copy number variants (CNVs) were called from WES data using EXCAVATOR.13 Among SNVs and InDels, nonsynonymous SNVs, splice-site substitutions, and coding small insertions/deletions with a population allele frequency <1‰ in public databases (1000 Genomes, http://www.1000genomes.org; Exome Variant Server, http://evs.gs.washington.edu/EVS/; ExAC, http://exac.broadinstitute.org) and absent from approximately 200 patients without epilepsy in our internal exome database were considered further.
The priority assigned to variants was higher in those with a lower Residual Variation Intolerance Score (RVIS) percentile (indicating greater intolerance to functional variation)14 and in those predicted to have a likely functional effect by Combined Annotation-Dependent Depletion (CADD) score15 (higher CADD scores indicate that the variant is more likely to be functionally relevant), or predicted to alter splicing or to be protein-truncating (frame-shift InDels or nonsense SNVs). Among CNVs, a higher priority was assigned to events that were not observed to occur in regions known to be involved in segmental duplications or were rare (allele frequency <1‰) in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) by manual inspection. When WES of 2 affected members of the family (table 1) had been performed, only the variants shared by the 2 were retained.
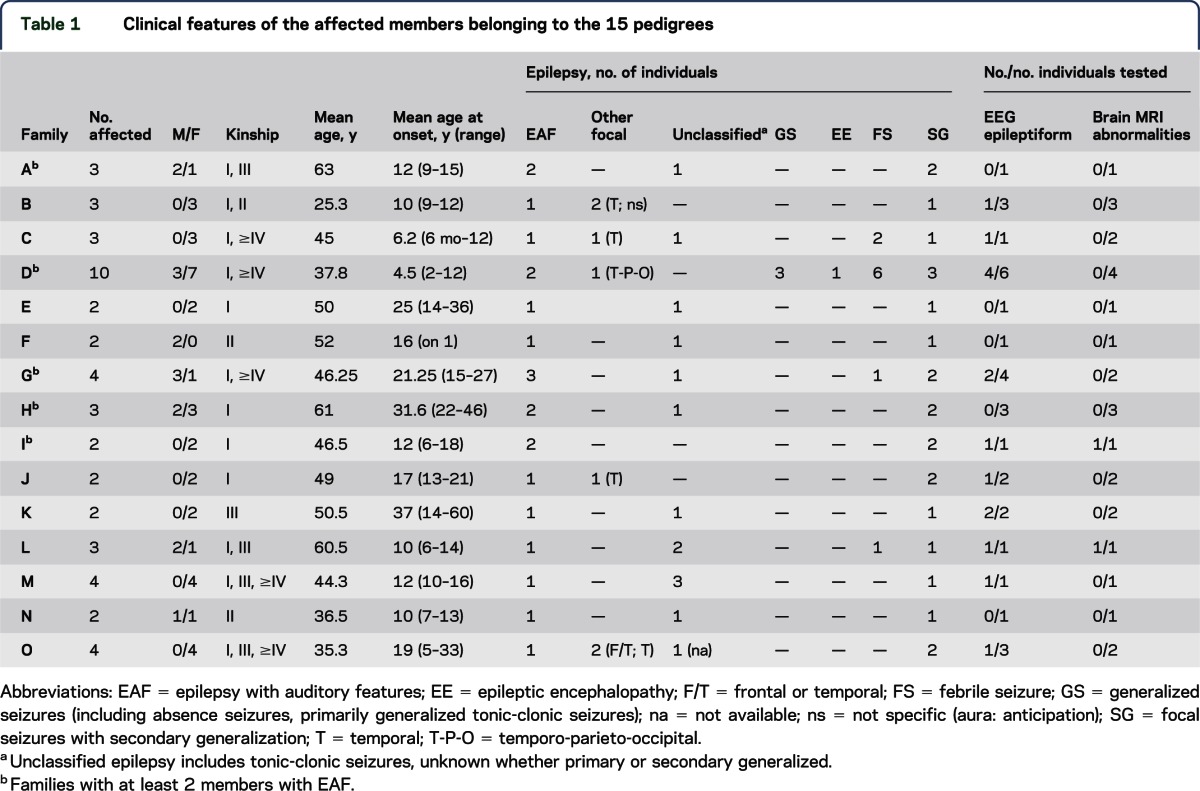
Table 1.
Clinical features of the affected members belonging to the 15 pedigrees
Validation and segregation analysis of candidate variants and CNVs were performed using Sanger sequencing and quantitative PCR analyses, respectively, following standard protocols.
RESULTS
We included 12 consecutive probands of Italian ancestry attending the Epilepsy Center of our Institute in Bologna. Three additional cases were referred by 2 other epilepsy centers in Italy. A mutation or CNV of LGI116 was excluded in all 15 probands with EAF. Pedigree analysis revealed a total of 34 affected relatives: 6 with EAF (in the 5 families A, D, G, H, and I in table 1) and 28 with other epilepsy (table 1). Two families have been previously reported,6,17 while the remaining 13 pedigrees are described here for the first time.
Molecular genetic findings in 344 “epilepsy genes.”
To analyze WES data, we initially focused on a set of 344 genes (table e-1 at Neurology.org/ng) implicated in monogenic disorders including epilepsy as a phenotypic feature based on previously published work18 and the OMIM database (www.omim.org). We identified 7 candidate variants of 5 genes, each in one of 7 families. These variants were validated in every individual who underwent exome testing. We then tested segregation in all the available family members, both unaffected and affected by EAF or other epilepsies, who had no WES performed. After testing segregation, 6 variants remained: 1 CNV in CNTNAP2 (contactin-associated protein-like 2; OMIM *604569), 1 SNV and 1 InDel in DEPDC5 (DEP domain-containing protein 5; OMIM *614191), 1 SNV in SCN1A (sodium channel, voltage-gated, type 1, alpha subunit; OMIM *182389), 1 SNV in CACNA1H (calcium channel, voltage-dependent, T type, alpha-1H subunit; OMIM *607904), and 1 SNV in RELN (REELIN, OMIM *600514).
Likely pathogenic variants.
CNTNAP2.
An intragenic deletion of CNTNAP2 (p.Gln33Argfs*7) was identified in both the affected members of family A (figure 1). The proband (A:IV:1), aged 47 years, presented at 9 years with episodes characterized by confusion, distortion of voices, and inability to speak, occasionally followed by a bilateral tonic-clonic seizure. From 37 years of age, she had an aura of the noise of a train followed by an aphasic phase and sometimes twitching of her mouth and left arm, drooling, and head deviation to the right. Interictal EEG showed left temporal theta activity, and brain MRI was unremarkable. Her father (A:III:1) experienced seizures with auditory phenomena and rare bilateral tonic-clonic seizures from 15 to 18 years. The CNV of CNTNAP2 identified spans about 270 kb across exons 2-3 and is predicted to produce an out-of-frame transcript p.Gln33Argfs*7 (NM_014141:c.98_402del) and to result in loss of function.
Figure 1. Segregation and localization of the identified variants.

Pedigrees of the mutated families with mutations and schematic representation of the genes and the identified variants are presented. Phenotype labels: EAF = epilepsy with auditory features; GTCS = generalized tonic-clonic seizures; GS = generalized seizures; FS = febrile seizures; unclassified: it is undetermined whether the seizure type is focal or generalized. Gene diagrams and variants: blue lines represent the genomic regions spanned by the gene, while the blue bars represent the exons. Colored blocks surrounding gene regions correspond to the protein domains encoded by the embedded exons. DEP = Dishevelled, Egl-10 and Pleckstrin domain; D1-4 = domains 1–4; F5/8 = discoidin domain; LGL = laminin G-like domain; EL = EGF-like domain; FCT = fibrinogen C-terminal domain; TM = transmembrane domain. The probands' Sanger sequencing electropherogram surrounding the variant site is reported. The histogram plot represents the result of the real-time PCR. Log2ratios of ΔΔCt from patient A:III:1, patient A:IV:1, and a normal individual each against a control sample are plotted. The control sample is one that was negative for CNTNAP2 rearrangements by array comparative genomic hybridization analyses. Four probes were tested. Probe 1 and probe 2 are located in intron 1, while probe 3 and probe 4 are located in intron 3.
DEPDC5.
Two different truncating mutations of DEPDC5 were found in 2 probands. Both DEPDC5 variants were predicted to be loss-of-function alleles.
A single base out-of-frame deletion p.Leu1371Argfs*14 (NM_001136029:c.4112delT) was detected in 3 affected members of family B (figure 1). The proband (B:III:1) with EAF had onset at 12 years of episodes comprising “a sensation of a whistle in the right ear,” sometimes associated with a rising epigastric sensation and followed by impairment of awareness and gestural automatisms. Interictal EEG showed bitemporal theta activity. The other 2 affected members have other types of focal epilepsy. Her mother (B:II:1) developed focal seizures at 9 years characterized by a rising epigastric sensation and autonomic phenomena followed by impaired awareness and head deviation. Her maternal aunt (B:II:2) experienced focal seizures from 9 years that began with a nonspecific aura and sometimes evolved to a bilateral tonic-clonic seizure. Her EEG showed right temporal sharp wave complexes.
In family C (figure 1), we identified a stop-gain substitution p.Tyr306*(NM_001136029:c.C918G). The proband (C:IV:1) had a single febrile seizure (FS) at 6 months of age and then had rare right tonic seizures, occasionally followed by a bilateral tonic-clonic seizure, predominantly during sleep until 2 years of age. Since childhood, she has also had seizures preceded by auditory phenomena and/or déjà vécu, sometimes followed by impaired consciousness. By 29 years, seizures occurred weekly, despite trials of several antiepileptic drugs. Interictal EEG showed bitemporal theta activity. Her mother (C:III:2) had FSs at 2 years and focal seizures beginning with visual phenomena, déjà vu, and an epigastric aura between 12 and 35 years. Brain MRI was normal in all patients from both families.
SCN1A.
In family D (figure 1), a missense change p.Met956Thr (NM_001165963:c.T2867C) of SCN1A was identified in the proband (D:III:1) and her daughter (D:IV:1) with EAF. The family comprised 10 affected members (3 male, 7 female; mean age at onset: 4.5 years, range: 2–12). Seven had epilepsy (D:III:1, D:III:7, D:III:8, D:IV:1, D:IV:6, D:IV:7, and D:IV:8), while 3 had FSs alone (D:III:6, D:III:11, and D:IV:4). In addition to individuals D:III:1 and D:IV:1 with EAF, 1 patient had focal epilepsy (D:III:8), 3 had generalized seizures in childhood (D:III:7, D:IV:6, and D:IV:8), and 1 had infantile epileptic encephalopathy (D:IV:7). Three of the family members with epilepsy had a history of prolonged FSs (D:III:8, D:IV:1, and D:IV:8). The SCN1A change segregated with affected status in this large pedigree. There was incomplete penetrance (61.5%) since 2 unaffected obligate carriers (D:II:4 and D:III:4) and 3 healthy individuals (D:III:3, D:III:5, and D:III:9) were heterozygous for the mutation. The same mutation has been recently reported in a family with intractable childhood epilepsy with generalized tonic-clonic seizures where it was described as a loss-of-function, fully penetrant allele that led to incorrect folding of the alpha 1 subunit of the voltage-gated sodium channel type 1 (Nav1.1) encoded by SCN1A.19
Variants of unclear significance.
CACNA1H.
A missense change p.Arg1669Gln (NM_001005407:c.G5006A) of CACNA1H was identified in the 38-year-old proband of family E (table 1).
The CACNA1H SNV identified was inherited by her mother (E:I:1), who experienced sleep-related bilateral tonic-clonic seizures at 36 years of age. Rare amino acid substitutions altering function of the channel encoded by CACNA1H have been reported in families with predominantly generalized epilepsies, as well as temporal lobe epilepsy.20 The present change was potentially deleterious (table e-2). Functional assessment of the pathogenic potential was not performed as part of this study but is needed to confirm a causal role.
RELN.
A missense change p.Asp3323Glu (NM_005045:c.T9969G) of RELN was identified in the 52-year-old proband of family F (table 1). Segregation of the variant could not be tested since no relative was available for this study. The father's grandfather, who was the only other affected member of the family (table 1), was already deceased at the time of enrollment. RELN encodes reelin, which is essential for cerebral cortex development and synaptic function in the adult brain.21 Homozygous recessive loss-of-function RELN alleles have been shown to cause lissencephaly22 (OMIM #257320), while heterozygous variants have been purported to play a role in neuropsychiatric disorders.21 Reelin is a huge protein (3460 aa, Uniprot ID P78509). Although RELN is highly intolerant to functional variation and the present variant is potentially deleterious (table e-2), many rare missense variants accumulate in RELN (ExAC database). Due to the lack of segregation testing and uncertainty of its functional effect, we cannot assess a causal role of this heterozygous variant.
Possible novel candidate genes.
We further analyzed WES data looking at variants in genes beyond the initial set. In order to identify possible candidates for EAF, we examined variants within genes shared by at least 2 unrelated probands. We validated and tested the segregation of variants in 4 genes, selected based on the lowest RVIS scores and the highest pathogenic potential. After testing the segregation, only the echinoderm microtubule associated protein-like 6 gene (EML6) remained as a possible candidate gene. EML6 showed a novel p.Ala186fs frameshift event (c.558delA) in family M and a rare p.Thr918Ala missense variant (c.A2752G) in family F (table e-2). The function of EML6 is largely unknown, and apart from its reported expression in cerebral temporal cortex (http://www.braineac.org/), no data support its possible dysfunction being implicated in epilepsy. Moreover, a major role in disease for any of the variants in EML6 and RELN, which have both been found in family F, is difficult to ascertain. Study of larger cohorts will be needed to assess whether this gene can contribute to EAF.
DISCUSSION
In this study, we describe defects in novel genes contributing to the genetic architecture of EAF. We identified likely pathogenic genetic variants in 4 of 15 unrelated EAF probands: an intragenic deletion of CNTNAP2, 2 truncating mutations of DEPDC5, and 1 missense mutation in SCN1A. All genes have been previously associated with other forms of focal epilepsy but not EAF.23–26 Several factors support the pathogenicity of the identified mutations, including their rarity, their predicted deleterious nature, their segregation with affected status within kindreds, and the recognized role of mutations of these genes in the etiology of the associated epilepsies. In these families, EAF is a phenotype that occurs in few (often the minority) of the affected members. Such phenotypically heterogeneous families are generally of small size and have usually been considered poorly informative for genetic studies, as recurrence of the disease tends to be low, expressivity of the phenotype widely variable, and penetrance reduced. These families likely reflect the more common scenario in clinical practice compared with the rare familial epilepsy syndrome of ADEAF. LGI1 mutations are associated with ADEAF in about half of the large multigenerational families displaying autosomal dominant inheritance of EAF. However, LGI1 mutations have been rarely detected in pedigrees with EAF and a less clear-cut pattern of inheritance or phenotypic heterogeneity.5
The discovery of DEPDC5 mutations in 2 of 15 probands is in line with the frequency of DEPDC5 mutations observed in other focal epilepsies (around 10%).23,27 The 2 identified DEPDC5 mutations (c.C918G and c.4112delT) are loss-of-function events, both predicted to generate a prematurely truncated product. These mutated transcripts likely undergo nonsense-mediated decay leading to haploinsufficiency, which is the main mechanism believed to underlie pathogenesis.23,28 These results enlarge the DEPDC5-related phenotypic spectrum to include EAF with lateral temporal lobe semiology, further strengthening the emerging association of defects in this gene with a broad range of focal epilepsies. It could be anticipated that different epilepsy phenotypes, including EAF, will be associated with DEPDC5 mutations in other small families.
Similarly, the discovery of an SCN1A c.T2867C change in family D widens the already large compendium of epilepsy phenotypes associated with SCN1A mutations. In this interesting pedigree, the EAF phenotype is limited to the proband and her daughter, but expanding the clinical and genetic study of this family allowed us to define the diagnosis of the familial epilepsy syndrome of generalized epilepsy with febrile seizures plus (GEFS+). SCN1A has been implicated in different epilepsy syndromes ranging from GEFS+29 to Dravet syndrome (OMIM #607208)30 and occasionally includes focal seizures.24,25 Lateral temporal semiology with auditory symptoms due to SCN1A mutation has not been previously reported, and our findings indicate EAF as a novel phenotype within the SCN1A phenotypic spectrum.
Mutations in SCN1A and DEPDC5 result in specific epilepsy syndromes, whereas CNTNAP2 is disrupted in a broad range of neurologic conditions31–33 in which seizures play a part.26 Diallelic loss of normal CNTNAP2 has been linked to complex phenotypes, including cortical dysplasia and focal epilepsy,34,35 but most of the disruptive events involving CNTNAP2 are heterozygous and show incomplete penetrance. These include heterogeneous alterations ranging from intronic/exonic deletions to splice-site mutations and truncating and missense changes. Family A consists of a nuclear pedigree with only 2 affected members, father and daughter, who both have EAF. The exon 2-3 deletion we report, predicted to code for an abnormal product (p.Gln33Argfs*7), lies within the CNV hotspot spanning the CNTNAP2 exon 2-9 genomic region implicated in complex neuropsychiatric disorders (figure 2). Epilepsy or seizures are reported in many of the patients with this deletion, but they are not commonly the primary diagnosis and are therefore not described in detail. Accurate epilepsy phenotyping in these patients may reveal other EAF cases associated with deletions of this gene. Neither affected member of family A showed features of intellectual disability, speech delay, or psychiatric disturbances. Notably, CASPR2, the protein product of CNTNAP2, and LGI1 are closely associated with the voltage-gated potassium channel (VGKC) complex in the human brain (figure 2),36 and LGI1 has been found to be part of the CASPR2 interactome.37 This functional relationship highlights the possible molecular link between CNTNAP2 disruption and the EAF phenotype. Haploinsufficient Lgi1 in adult rat brains has been demonstrated to cause changes in the inactivation gating of presynaptic A-type channels, of which Kv1.1 is a major constituent.38 This has been proposed to be the molecular mechanism promoting epileptic activity in patients with defective LGI1 protein. In addition, the predicted haploinsufficiency of CASPR2, another protein complexed with Kv1.1, may alter channel kinetics. Recently, consistent evidence has shown that mutations in the same gene can increase the risk of a broad range of complex neuropsychiatric disorders.39 This suggests the involvement of genetic or environmental modifiers. Notwithstanding the challenge in interpreting the complexity of CNTNAP2 genotype-phenotype correlations, the present data together with the accumulated knowledge about CNTNAP2 mutations and CASPR2 function strongly suggest that disrupted CNTNAP2 contributes to EAF in family A. This perspective is fascinating because other pre- or postsynaptic membrane-bound proteins (ADAM22, ADAM23, and Kv1 channel genes) have been explored before in search for mutations in EAF, but no mutations were detected.8–10
Figure 2. The CNTNAP2 locus in disease and localization of its encoded product, CASPR2, at the synapse.

(A) Schematic indicating the functional relationship linking LGI1 and CASPR2 proteins through the voltage-gated potassium channel (VGKC). (B) Schematic indicating the previously reported microdeletions (horizontal bars) of CNTNAP2 in neuropsychiatric conditions (modified from Rodenas-Cuadrado, 2013). The deletion reported in this study, spanning CNTNAP2 exons 2-3, overlaps most of the CNTNAP2 deletions implicated in disorder before. It is represented in red with a halo surrounding it. ADHD = attention-deficit/hyperactivity disorder; ASD = autism spectrum disorder.
Additional variants were identified in different genes, either previously implicated in epilepsy (CACNA1H, RELN) or not (EML6), but their actual contribution to EAF cannot be assessed based on our present observations.
Overall, defects in 3 novel genes were associated with EAF. Of the pedigrees in which these mutations were found, 1 with a CNTNAP2 deletion may have ADEAF, as both affected individuals have the EAF phenotype and the genetic inheritance model is autosomal dominant. The association of ADEAF with a CNTNAP2 deletion shows biological convergence and opens new perspectives on the contribution of non-LGI1 VGKC-complex proteins to the EAF phenotype. The identification of mutations in DEPDC5 and SCN1A in families showing heterogeneous phenotypic patterns enlarges the phenotypic spectrum of the epilepsies associated with these genes. Overall, our results suggest that EAF is an epilepsy phenotype originating from alterations in a heterogeneous group of genes implicated in a broader range of epileptic syndromes or neurologic diseases. This overlap, frequently observed in other forms of epilepsy (e.g., autosomal dominant nocturnal frontal lobe epilepsy),27,28 reflects the complexity of the involved molecular mechanisms and reveals the challenge of incorporating the underlying biology into the current classification of epilepsies. This study emphasizes the use of a comprehensive WES strategy, tackling both mutations and CNVs, in an effort to solve challenging diagnostic puzzles in epilepsy.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to the families for participation in the study, especially to the probands for their valuable assistance during in loco assessment of the family. The authors acknowledge Mrs. Annalia Cesare for secretarial help in keeping in contact with patients, Ms. Elena Zoni for help in manuscript editing, and Dr. Lara Alvisi and the other EEG technicians at our institute for recording the patients. The authors acknowledge the genetic commission of the Italian League Against Epilepsy for discussion of the pedigrees and valuable suggestions for the study design.
GLOSSARY
- ADEAF
autosomal dominant EAF
- CADD
Combined Annotation-Dependent Depletion
- CNV
copy number variant
- EAF
epilepsy with auditory features
- FS
febrile seizure
- GEFS+
generalized epilepsy with febrile seizures plus
- InDels
insertions and deletions
- OMIM
Online Mendelian Inheritance in Man
- RVIS
Residual Variation Intolerance Score
- SNV
single nucleotide variant
- VGKC
voltage-gated potassium channel
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Dr. Pippucci, Dr. Licchetta, S. Baldassari, and Dr. Bisulli conceptualized and designed the study and drafted the manuscript. Dr. Menghi, Dr. Leta, Dr. Stipa, Dr. Boero, Dr. d'Orsi, and Dr. Bisulli phenotyped the patients. Dr. Pippucci, Dr. Licchetta, S. Baldassari, F. Palombo, Dr. D'Aurizio, Dr. Magi, and Dr. Bisulli collected, analyzed, and interpreted the data. Dr. Scheffer, Dr. Seri, and Dr. Tinuper critically revised the manuscript for important intellectual content.
STUDY FUNDING
Italian Ministry of Health Young Investigators project GR-2009-1574072 to T.P. and F.B., Telethon Foundation Project GGP13200 to P.T. and T.P.
DISCLOSURE
Dr. Pippucci has worked for Azienda Ospedaliero-Universitaria Careggi and has received research support from the Italian Ministry of Health and Telethon Foundation. Dr. Licchetta reports no disclosures. S. Baldassari has received research support from the Italian Ministry of Health and Telethon Foundation. F. Palombo has received research support from the Italian Ministry of Health and Telethon Foundation. Dr. Menghi, Dr. D'Aurizio, Dr. Leta, Dr. Stipa, Dr. Boero, Dr. d'Orsi, and Dr. Magi report no disclosures. Dr. Scheffer has received funding for travel and/or speaker honoraria from Sanofi, UCB, and Transgenomics; has served on editorial boards for Neurology, Epilepsy Currents, Annals of Neurology, Epileptic Disorders, Progress in Epileptic Disorders series, and Virtual Neuro Centre; holds patents for methods of treatment and diagnosis of epilepsy by detecting mutations in the SCN1A gene, A Diagnostic Method for Epilepsy (also published as Methods for the Diagnosis and Treatment of Epilepsy), Mutations in Ion Channels, Diagnostic and Treatment Methods Relating to Autosomal Dominant Nocturnal Frontal Lobe Epilepsy and pending patents for gene and mutations thereof associated with seizure disorders, gene and mutations thereof associated with seizure and movement disorders, and diagnostic and therapeutic methods for EFMR; and has received research support from NHMRC, NIH, The University of Melbourne, Austin Health Medical Research Foundation, Melbourne Neurosciences Institute, Weizmann Institute, CURE SUDEP, and Perpetual Philanthropic Services. Dr. Seri reports no disclosures. Dr. Tinuper has served on scientific advisory boards of UCB Pharma and Cyberonics; has received funding for travel and/or speaker honoraria from Cyberonics; and has served on the editorial board of Seizures. Dr. Bisulli has received research support from the Italian Ministry of Health Young Investigators and Telethon. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Ottman R, Risch N, Hauser WA, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet 1995;10:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 3.Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet 2002;11:1119–1128. [DOI] [PubMed] [Google Scholar]
- 4.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ottman R, Winawer MR, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology 2004;62:1120–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michelucci R, Pasini E, Malacrida S, et al. Low penetrance of autosomal dominant lateral temporal epilepsy in Italian families without LGI1 mutations. Epilepsia 2013;54:1288–1297. [DOI] [PubMed] [Google Scholar]
- 7.Bisulli F, Naldi I, Baldassari S, et al. Autosomal dominant partial epilepsy with auditory features: a new locus on chromosome 19q13.11-q13.31. Epilepsia 2014;55:841–848. [DOI] [PubMed] [Google Scholar]
- 8.Chabrol E, Gourfinkel-An I, Scheffer IE, et al. Absence of mutations in the LGI1 receptor ADAM22 gene in autosomal dominant lateral temporal epilepsy. Epilepsy Res 2007;76:41–48. [DOI] [PubMed] [Google Scholar]
- 9.Diani E, Di Bonaventura C, Mecarelli O, et al. Autosomal dominant lateral temporal epilepsy: absence of mutations in ADAM22 and Kv1 channel genes encoding LGI1-associated proteins. Epilepsy Res 2008;80:1–8. [DOI] [PubMed] [Google Scholar]
- 10.Rigon L, Vettori A, Busolin G, et al. ADAM23, a gene related to LGI1, is not linked to autosomal dominant lateral temporal epilepsy. Epilepsy Res Treat 2011;2011:258365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magini P, Pippucci T, Tsai IC, et al. A mutation in PAK3 with a dual molecular effect deregulates the RAS/MAPK pathway and drives an X-linked syndromic phenotype. Hum Mol Genet 2014;23:3607–3617. [DOI] [PubMed] [Google Scholar]
- 12.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magi A, Tattini L, Cifola I, et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol 2013;14:R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magini P, Bisulli F, Baldassari S, et al. LGI1 microdeletions are not a frequent cause of partial epilepsy with auditory features (PEAF). Epilepsy Res 2014;108:972–977. [DOI] [PubMed] [Google Scholar]
- 17.Bisulli F, Tinuper P, Avoni P, et al. Idiopathic partial epilepsy with auditory features (IPEAF): a clinical and genetic study of 53 sporadic cases. Brain 2004;127:1343–1352. [DOI] [PubMed] [Google Scholar]
- 18.Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398. [DOI] [PubMed] [Google Scholar]
- 19.Bechi G, Rusconi R, Cestèle S, Striano P, Franceschetti S, Mantegazza M. Rescuable folding defective Na(V)1.1 (SCN1A) mutants in epilepsy: properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol Dis 2015;6:100–114. [DOI] [PubMed] [Google Scholar]
- 20.Heron SE, Khosravani H, Varela D, et al. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol 2007;62:560–568. [DOI] [PubMed] [Google Scholar]
- 21.Folsom TD, Fatemi SH. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013;68:122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 2000;26:93–96. [DOI] [PubMed] [Google Scholar]
- 23.Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551. [DOI] [PubMed] [Google Scholar]
- 24.Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology 2001;57:2265–2272. [DOI] [PubMed] [Google Scholar]
- 25.Colosimo E, Gambardella A, Mantegazza M, et al. Electroclinical features of a family with simple febrile seizures and temporal lobe epilepsy associated with SCN1A loss-of-function mutation. Epilepsia 2007;48:1691–1696. [DOI] [PubMed] [Google Scholar]
- 26.Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet 2014;22:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 2014;82:2101–2106. [DOI] [PubMed] [Google Scholar]
- 28.Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24:343–345. [DOI] [PubMed] [Google Scholar]
- 30.Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lesca G, Rudolf G, Labalme A, et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia 2012;53:1526–1538. [DOI] [PubMed] [Google Scholar]
- 32.Gregor A, Albrecht B, Bader I, et al. Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med Genet 2011;12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman JI, Vrijenhoek T, Markx S, et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry 2008;13:261–266. [DOI] [PubMed] [Google Scholar]
- 34.Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med 2006;354:1370–1377. [DOI] [PubMed] [Google Scholar]
- 35.Zweier C, de Jong EK, Zweier M, et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet 2009;85:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen N, Koopmans F, Gordon A, et al. Interaction proteomics of canonical Caspr2 (CNTNAP2) reveals the presence of two Caspr2 isoforms with overlapping interactomes. Biochim Biophys Acta Epub 2015 Feb 21. [DOI] [PubMed]
- 38.Schulte U, Thumfart JO, Klöcker N, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron 2006;49:697–706. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 2014;17:773–781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.