

Abstract
Objective:
To elucidate the functional consequences of epileptic encephalopathy–causing de novo mutations in DNM1 (A177P, K206N, G359A), which encodes a large mechanochemical GTPase essential for neuronal synaptic vesicle endocytosis.
Methods:
HeLa and COS-7 cells transfected with wild-type and mutant DNM1 constructs were used for transferrin assays, high-content imaging, colocalization studies, Western blotting, and electron microscopy (EM). EM was also conducted on the brain sections of mice harboring a middle-domain Dnm1 mutation (Dnm1Ftfl).
Results:
We demonstrate that the expression of each mutant protein decreased endocytosis activity in a dominant-negative manner. One of the G-domain mutations, K206N, decreased protein levels. The G359A mutation, which occurs in the middle domain, disrupted higher-order DNM1 oligomerization. EM of mutant DNM1-transfected HeLa cells and of the Dnm1Ftfl mouse brain revealed vesicle defects, indicating that the mutations likely interfere with DNM1's vesicle scission activity.
Conclusion:
Together, these data suggest that the dysfunction of vesicle scission during synaptic vesicle endocytosis can lead to serious early-onset epilepsies.
Epileptic encephalopathies are a heterogeneous group of severe childhood neurologic disorders characterized by epileptic activity accompanied by progressive cognitive, behavioral, and sensory impairments.1 In this study, we conduct functional analysis on 3 de novo missense mutations in DNM1 that have been implicated in 2 epileptic encephalopathies: Lennox-Gastaut syndrome (LGS) and infantile spasms.
Dynamin-1 is a 100-kDa mechanochemical GTPase that is required during receptor-mediated endocytosis and synaptic vesicle recycling.2,3 DNM1, expressed predominantly in neurons, localizes to the presynaptic terminal and mediates the uptake of synaptic vesicles.4–7 Its expression is upregulated during postnatal development, accompanying synaptogenesis.4,7,8
DNM1 is organized into 5 domains: a G domain that binds and hydrolyzes GTP, a middle domain that is involved in oligomerization, a GTPase effector domain, a pleckstrin homology domain, and a proline-rich domain.9–12 During receptor-mediated endocytosis, dynamin molecules assemble into tetramers that hydrolyze GTP.12,13 Upon GTP hydrolysis, DNM1 undergoes a conformational change that allows it to pinch vesicles from the membrane.14
A spontaneous mouse mutation in the middle domain of Dnm1 (Dnm1Ftfl) causes seizures and hearing defects.8 The missense mutation affects dynamin oligomerization and decreases endocytosis activity. Patch-clamp recordings demonstrate defects in GABAergic transmission in response to prolonged electrical stimulation.8
In this study, we assess the effects of 3 de novo mutations in DNM1 using cellular assays and in vivo ultrastrucutral studies in mice. Collectively, this work provides a possible mechanistic link between synaptic vesicle trafficking dysfunction and epilepsy.
METHODS
Constructs.
Wild-type (WT) DNM1 cDNA was cloned into the pCMV-AC-GFP (OriGene, Rockville, MD) and pCMV-AC-RFP (OriGene) vectors in frame. Mutagenesis was performed on green fluorescent protein (GFP)-tagged DNM1 by site-directed mutagenesis using the NEB Q5 site-directed mutagenesis kit to create the c.529G>C (A177P), c.618G>C (K206N), and c.1076G>C (G359A) mutations according to the manufacturer's protocol. Plasmid sequences were verified by Sanger sequencing.
Transferrin assay.
COS-7 or HeLa cells were transfected on Mattek glass bottom plates with the DNM1 constructs using Lipofectamine LTX and PLUS reagent (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. Twenty-four hours after transfection, the assay was performed with AlexaFluor-594–conjugated transferrin (Life Technologies), as previously described.8 Cells were mounted in VectaShield with 4',6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) and imaged with a Zeiss Axio Observer with a 63×, 1.4 oil immersion objective. Exposure times were adjusted for the GFP channel to better illustrate localization patterns. The experiment was repeated 3 times with identical conditions.
High-content imaging.
HeLa cells were transfected on Poly-d-Lysine–coated 24-well plates. The transferrin assay was performed as described. Using a Thermo Scientific Arrayscan VTI, cells were filtered for positive DAPI and GFP expression. To quantify transferrin uptake, AlexaFluor-594 (conjugated transferrin fluorophore) intensity was measured for each positively transfected cell. An average of 5,100 cells were analyzed per well. Spot identification was used to quantify the number of puncta per cell. Standardized evaluation was performed by applying the same parameters to each experimental condition. Downstream quantitative analysis was blinded.
Colocalization.
HeLa cells were cotransfected with red fluorescent protein (RFP)-tagged WT DNM1 and each of the GFP-tagged mutants on a 12-well Mattek glass bottom plate using Lipofectamine LTX with PLUS (Life Technologies). Twenty-four hours after transfection, cells were washed 3 times with ice-cold phosphate-buffered saline and fixed in 4% paraformaldehyde for 10 minutes at room temperature. Cells were mounted in VectaShield with DAPI (Vector Laboratories) and imaged with a Zeiss Axio Observer with a 63×, 1.4 oil immersion objective.
Western blot.
Monomer expression assay.
HeLa cells were transfected with the DNM1 constructs. Twenty-four hours after transfection, cells were lysed in 200 μL of radioimmunoprecipitation assay (RIPA) buffer. The lysates were loaded on a 4%–12% NuPAGE Bis-Tris gel and transferred to a PVDF membrane. The blots were blocked in 1% casein solution and then probed with an anti-tGFP antibody (OriGene) at a 1:2,000 dilution overnight at 4°C. The membranes were washed 3 times with 0.1% Tris-buffered saline (TBS)-Tween. They were then probed with IRDye 800CW anti-mouse antibody (LI-COR, Lincoln, NE) at a 1:2,000 dilution for 1 hour at room temperature. The membranes were again washed 3 times in 0.1% TBS-Tween and imaged. Densitometry was performed using ImageJ, and downstream quantification was performed using a blinded analysis.
Cross-linking assay.
HeLa cells were transfected as described above. Twenty-four hours after transfection, the cells were lysed in RIPA buffer and treated with 20 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) for 45 minutes at 4°C. The Western blot was then performed as described for the monomer expression assay.
HeLa cell electron microscopy.
HeLa cells were transfected with WT and mutant constructs using Lipofectamine LTX on a Millipore 4-chamber Millicell EZ slide according to the manufacturer's protocol (Life Technologies). Twenty-four hours after transfection, the cells were fixed in 4% glutaraldehyde in borate buffer, dehydrated, and embedded. Images were acquired on a Phillips CM12.
Mice.
C57BL/6J-Dnm1Ftfl mice arose at The Jackson Laboratory (Bar Harbor, ME) as a spontaneous mutation on the C57BL/6J inbred strain in 2000.8 All mice were housed and procedures performed with approval of Institutional Animal Care and Use Committee. All mice were obtained from The Jackson Laboratory, maintained in a room with a 14-hour light on/10-hour light off cycle, and given free access to LabChow meal and water.
Brain section electron microscopy.
For electron microscopy (EM), littermate C57BL/6J-Dnm1Ftfl/Ftfl and C57BL/6J-Dnm1+/+ mice (aged 2 weeks) were perfused transcardially with 2% paraformaldehyde-2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), and the brains were removed and postfixed overnight at 4°C. Samples were postfixed with 1% OsO4 in 0.1 M cacodylate buffer, dehydrated, and embedded. Grids were scanned on a JEOL 1230 Transmission Electron Microscope, and measurements were performed with ImageJ software. For synaptic vesicle diameter and number analysis, a minimum of 710 vesicles from each sample was measured. Synaptic vesicle number was normalized to the area of each synapse measured.
RESULTS
We examined a total of 3 de novo missense mutations in DNM1 (NM_001005336.1) c.529G>C (p.(Ala177Pro)) (patient 1), c.618G>C (p.(Lys206Asn)) (patient 2), and c.1076G>C (p.(Gly359Ala)) (patient 3). Two amino acid substitutions (A177P and K206N) appear in the G domain of DNM1 and the third substitution (G359A) occurs in the middle domain (figure 1A). Clinical features of the probands harboring variants in DNM1 have been reported.15 All 3 patients had infantile spasms with onset between 2 and 7 months, later evolving into LGS. In addition, each patient presented with severe intellectual disability, pronounced hypotonia, and absence of speech. Patients 1 and 2 are therapy resistant, while patient 3 has been seizure free since beginning a ketogenic diet at age 3. In addition, patient 1 was diagnosed with autism spectrum disorder.
Figure 1. DNM1 mutations and structure.

(A) Structure-based domain architecture of human DNM1. The A177P and K206N mutations occur in the G domain and the G359A mutation occurs in the middle domain. The location of the mouse Dnm1Ftfl (A408T) mutation is indicated above the middle domain. (B) Positions of the amino acid substitutions in the DNM1 crystal structure, shown as a ribbon-type representation.
Available in silico algorithms predict that all 3 missense mutations are likely highly damaging to the encoded DNM1 protein (PolyPhen-2 score = 1.00; SIFT score = 0).16,17 In addition, DNM1 is among the 20% of genes that are the most intolerant of functional genetic variation in the human population.18 The A177P mutation occurs at the C-terminus of β-pleated sheet β4G. The K206N mutation substitutes a charged amide residue for a neutral residue at the C-terminus of β-pleated sheet β5G. The G359A mutation occurs at the N-terminus of α helix α1Ms (figure 1B). The GERP++ scores at these 3 missense positions (5.42, 4.41, and 5.88, respectively) are much higher than the average DNM1 GERP++ score of 3.77, indicating that these sites are highly conserved relative to the overall conservation of the gene.
Dynamin plays a crucial role in the catalysis of clathrin-mediated endocytosis. To determine whether these mutations affect endocytosis, we transfected COS-7 cells with GFP-tagged DNM1 constructs and measured effects on uptake of fluorescently labeled transferrin. Cells containing WT-transfected DNM1 showed a perinuclear accumulation of the fluorescently labeled transferrin (figure 2A). However, overexpression of the G-domain mutants, A177P and K206N, significantly inhibited transferrin uptake (figure 2, B and C). The middle-domain mutation, G359A, showed some transferrin uptake in occasional cells but was largely inhibited (figure 2D). In addition to their effects on endocytosis, the mutant DNM1 proteins exhibited different patterns of localization. WT DNM1 formed distinct, round puncta (figure 2A), while the A177P mutant had a diffuse cytosolic distribution accompanied by puncta (figure 2B). The K206N mutant exhibited abnormally large protein aggregates with a nonuniform distribution throughout the cell (figure 2C). Finally, the G359A mutant dynamin lacked puncta and exhibited a reticular distribution throughout the cell (figure 2D).
Figure 2. DNM1 mutations inhibit transferrin uptake.

Inhibition of transferrin internalization in mammalian cell lines. COS-7 cells were transfected with green fluorescent protein (GFP)-tagged DNM1 constructs and then treated with fluorescently labeled transferrin. Scale bar, 20 μm. (A) Cells expressing wild type (WT) DNM1 exhibit transferrin uptake with a perinuclear accumulation. WT DNM1 forms round puncta that are evenly dispersed throughout the perimeter of the cell. (B) The A177P mutant inhibits transferrin uptake. DNM1 shows some diffuse GFP signal throughout the cytosol accompanied by puncta. (C) The K206N mutant also inhibits transferrin uptake and shows abnormal aggregation of DNM1. (D) The G359A mutant shows some transferrin uptake in certain cells. There is a distinct lack of puncta, and DNM1 shows a reticular GFP signal throughout the cytosol.
To quantify differences in endocytosis levels and localization of DNM1, we performed high-content imaging analyses on transfected HeLa cells. First, intensity of the transferrin fluorophore was measured in positively transfected cells. Compared to cells transfected with the WT DNM1 construct, overexpression of each mutant protein conferred nearly a 60% reduction in transferrin uptake (figure 3A). Spot identification analysis was used to quantify differences in puncta per cell and revealed a 50% reduction in the number of puncta in the G359A mutant (figure 3B).
Figure 3. Quantification of transferrin uptake and cellular localization patterns.
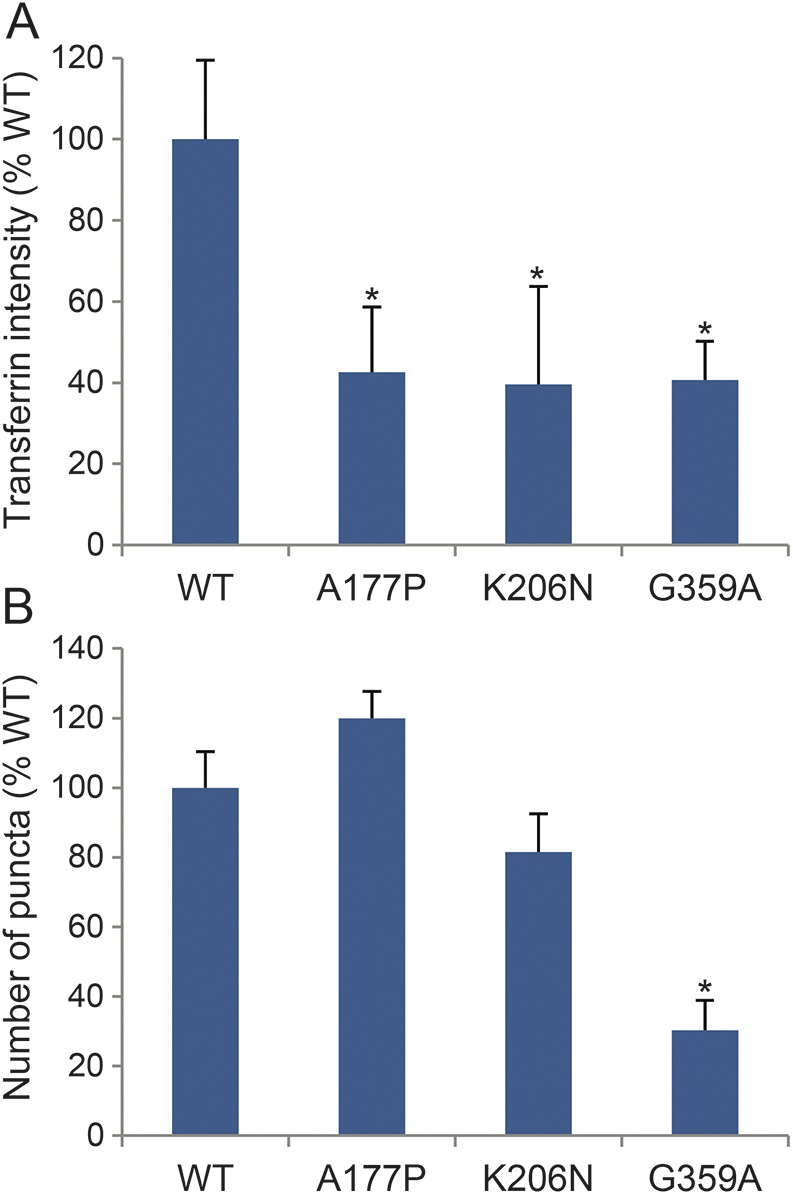
(A) High-content imaging analysis of transferrin fluorophore intensity in positively transfected HeLa cells. All 3 mutations confer nearly a 60% reduction in transferrin uptake compared to wild type (WT). Error bars represent SD between 5 replicate wells. p < 0.05 by Student t test. (B) High-content imaging spot identification revealed a 50% reduction in the number of puncta in cells expressing the G359A mutation. Error bars represent SD among 5 replicate wells. *p < 0.05 by Student t test.
Because DNM1 is the predominant dynamin in neurons,19 we were interested to see whether the mutant forms of the protein could colocalize with WT DNM1. To test this hypothesis, RFP-tagged WT DNM1 was cotransfected with each of the GFP-tagged mutants. Each mutant protein significantly colocalized with WT DNM1, suggesting likely interaction between the WT and mutant proteins (figure e-1 at Neurology.org/ng). Additionally, in contrast to the aberrant localization patterns when transfected alone, all 3 mutant proteins formed puncta in the presence of WT DNM1.
Because these mutations were predicted to be highly damaging to functional DNM1, we performed a Western blot on transfected HeLa cells to assay possible differences in expression levels (figure 4A). Average normalized WT and mutant DNM1 levels were determined and are shown in figure 4B. The K206N mutant protein showed a 75% decrease in steady-state levels compared to WT. It is interesting that the G359A transfected cells showed nearly a 2-fold increase in expression.
Figure 4. DNM1 mutations affect protein levels and self-dimerization.
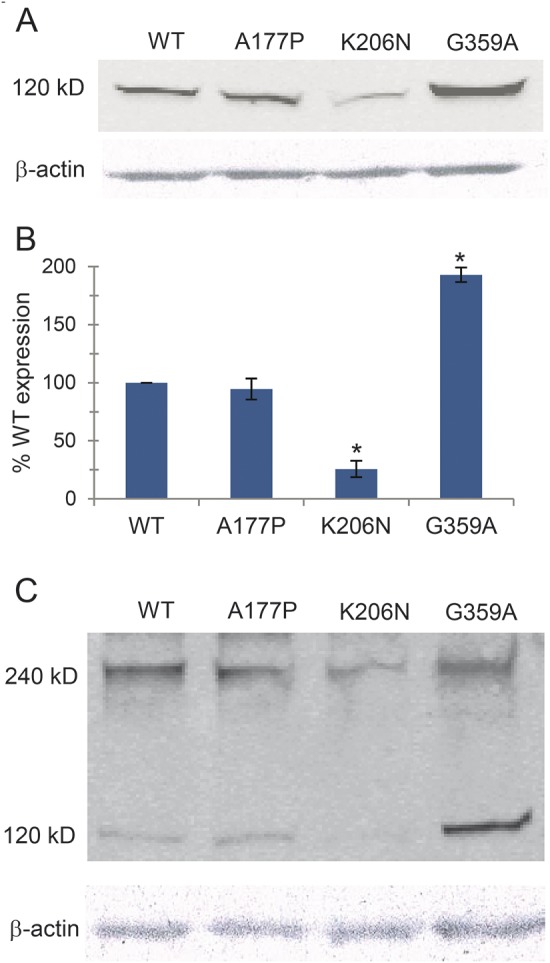
(A) HeLa cells were transfected with green fluorescent protein (GFP)-tagged mutant constructs. The blots were probed with anti-GFP antibodies. A representative blot is shown. (B) Quantification of protein expression levels from 3 independent Western blot experiments is shown. Actin levels were used for normalization and error bars indicate SD values. *p < 0.05 by Student t test. (C) HeLa cell lysates from cells transfected with GFP-tagged mutant constructs were treated with 20 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) cross-linking agent and analyzed by Western blot. The monomeric and dimeric forms are indicated. WT = wild type.
We next conducted a cross-linking assay to test whether the mutations disrupt DNM1 protein dimerization. We suspected that mutant G359A would disrupt dimerization, as the middle domain is involved in dynamin self-assembly, as shown previously for the mouse Dnm1Ftfl mutation.8 Forty-eight hours after transfecting HeLa cells with mutant constructs, cell lysates were treated with 20 mM EDC, a zero-length cross-linking agent, and analyzed by Western blot with anti-GFP antibody. The relative levels of monomer and dimer are shown in figure 4C. The G-domain mutants showed similar dimerization patterns to WT, but the G359A middle-domain mutant was associated with nearly a 50% decrease in dimerization, consistent with the Dnm1Ftfl result.
During endocytosis, DNM1 pinches forming vesicles from the plasma membrane. We conducted EM on transfected HeLa cells to determine effects of the mutants on vesicle scission. In cells transfected with the A177P construct, we observed extremely large abnormally shaped vesicles and smaller vesicles that clustered at the edge of the plasma membrane (figure 5A). This finding supports our hypothesis that G-domain DNM1 mutations significantly affect vesicle formation. Previously reported dominant-negative G-domain DNM1 mutants exhibited deep clathrin-coated invaginations in the membrane by EM,14 but while we observed this phenotype in the G359A construct by immunofluorescence, we were not able to detect it by EM for these mutations. In contrast, in cells transfected with the G359A mutant construct, there were no obvious vesicle abnormalities (figure 5A).
Figure 5. Electron microscopy of transfected HeLa cells and Dnm1Ftfl neurons.

(A) Electron microscopy (EM) of HeLa cells transfected with wild type (WT) and mutant DNM1 constructs. There are larger and abnormal vesicles in the A177P mutant. Cells transfected with the G359A show no obvious vesicle defects. Scale bars, 1 μm. (B) EM of WT mouse brain sections. (C) EM of Dnm1Ftfl mouse neurons reveals increased vesicle size. (D) A box plot depicting the number of vesicles in both WT and Dnm1Ftfl neurons. Outliers are excluded from the plot. Significance was determined by the Mann-Whitney-Wilcoxon test (**p < 0.005, ***p < 0.0005). (E) A box plot depicting the size of vesicles in both WT and Dnm1Ftfl neurons. There were significantly larger vesicles in both hippocampal and cortical neurons. Outliers are excluded from the plot. Significance was determined by the Mann-Whitney-Wilcoxon test.
Because the previously reported Dnm1Ftfl mutation (figure 1A) results in an early-onset seizure disorder in the mouse, decreases endocytosis activity in heterologous expression assays, and decreases oligomerization of DNM1,8 we reasoned that this mutation acts as a good model of the 3 human mutations. To understand the effects of DNM1 mutations on vesicle trafficking in vivo, we conducted EM on brain sections of the Dnm1Ftfl mouse. Overall, we observed a significant general decrease in vesicle number and an increase in vesicle size in synapses from both hippocampal and cortical regions when compared with WT (figure 5, B–D). Together, these findings suggest a stall in the overall fission activity of mutant DNM1, resulting in larger and fewer vesicles indicative of defects in endocytosis.
DISCUSSION
In this study, we analyzed the functional consequences of 3 de novo DNM1 missense mutations that were identified among patients with LGS, a severe epileptic encephalopathy. When expressed alone in COS-7 and HeLa cells, all 3 mutant constructs significantly inhibited endocytosis. Cells that overexpress WT DNM1 do not show an increase in overall transferrin uptake compared to cells transfected with a control GFP construct. Thus, the significant decrease in transferrin uptake suggests a dominant-negative effect of all 3 mutations, consistent with several other reported DNM1 mutations.14 Furthermore, mammalian genomes contain a total of 3 dynamin genes: DNM1, DNM2, and DNM3.20 DNM2 is expressed ubiquitously,21 while DNM3 is expressed in the brain and the testes.4 Given the ubiquitous expression of DNM2, the profound inhibitory effect on endocytosis suggests that the mutant DNM1 proteins bind to endogenous DNM2, thus interrupting DNM2 function and causing an accumulation of endocytotic intermediates.4,8
Both G-domain mutant proteins, A177P and K206N, showed different localization patterns from WT when expressed alone. EM revealed dramatic vesicle abnormalities, and our Western blot results indicate that neither mutation affects dimerization, but there is a significant decrease in steady-state levels of the K206N mutant, possibly suggesting decreased stability of this protein. Our co-transfection studies indicate that both mutant proteins colocalize—and presumably dimerize—with WT DNM1. We hypothesize that these mutations decrease GTP hydrolysis in a fashion similar to that reported for other dominant-negative G-domain mutants.2,14,22 Because GTP hydrolysis is necessary for vesicle scission,14 disruptions to this process likely lead to the observed decreased endocytosis activity.
The middle-domain mutation, G359A, also affected DNM1 localization. It is interesting that this mutant protein exhibited a reticular, or tube-like, distribution correlating with the transferrin signal. This is highly similar to what is observed when the Dnm1-Ftfl protein is overexpressed in COS-7 cells.8 Other studies of dominant-negative DNM1-expressing cells have reported reticular localization of some mutants, proposing that mutant dynamin can assemble on and tubulate the membrane but cannot function to fission the membrane, thus resulting in a longer lifetime on the membrane than WT dynamin.14
The middle-domain mutation interfered with DNM1 dimerization and showed nearly a 2-fold increase in monomer expression compared to WT. Since each of these proteins is expressed from cDNA constructs with constitutive cytomegalovirus promoters, these observations suggest different stabilities of the WT and mutant proteins. However, it is unclear why the monomeric form is more stable. Membrane invagination and fission are mediated by 2 separate dynamin mechanisms that require both assembly-independent and assembly-dependent GTPase activity.23,24 Thus, we hypothesize that an increase in the monomeric form of this mutant protein may allow it to bind to and sequester certain interacting vesicle trafficking partners to achieve its dominant-negative effect. It is interesting that cells expressing the G359A mutant protein did not show the same vesicle defects as cells transfected with the G-domain mutant constructs in EM. As previously hypothesized for the mouse fitful mutation, which also occurs in the middle domain, we believe that the G359A mutant would not participate in assembly-stimulated activity required for vesicle scission but would retain the assembly-independent activity required for membrane invagination.8
The G359A and Dnm1Ftfl constructs produce the same cellular phenotype when expressed in COS-7 cells: decreased transferrin endocytosis, a reticular DNM1 distribution, and decreased dimerization. Thus, the Dnm1Ftfl mutation is a very good model for this mutation. Our EM data from Dnm1Ftfl neurons reveal an overall increase in vesicle size coupled with a decrease in vesicle number. Because dynamin-mediated scission is the rate-limiting step of endocytosis,25 these results suggest a stall in the endocytosis process due to decreased dynamin-mediated membrane fission. Together, these data illustrate that dominant-negative DNM1 mutations are also likely to disrupt vesicle trafficking in vivo.
Although they may achieve a dominant-negative interaction through different mechanisms, each mutant protein causes endocytosis defects, as suggested by our transferrin assay. One possible consequence is that decreased endocytosis activity results in a depleted pool of synaptic vesicles, as supported by EM of fitful neurons. It has been previously shown that inhibitory neurons are more sensitive to the lack of DNM1, presumably due to their tonic activity.26 Thus, a lack of synaptic vesicles in inhibitory neurons could cause decreased GABA transmission, leading to network-level hyperactivity and an epileptic phenotype. Alternatively, these mutations may affect neurite formation, as recent evidence illustrates that inhibiting dynamin-dependent endocytosis disrupts neurite outgrowth.27 Additional neuronal-based studies are required to fully elucidate the downstream effects of inhibited neuronal endocytosis.
Our study adds to the increasing evidence that dysfunctional synaptic vesicle cycling is a mechanism for neurologic disorders. Mutations in other vesicle trafficking genes, such as STXBP1, SYN1, and LRRK2, have been implicated in autism spectrum disorder, epileptic encephalopathies, and Parkinson disease.27–29 Together, these studies support the emerging paradigm in neurology that disruptions in the same pathway can increase risk for a broad range of neuropsychiatric disorders.30
Finally, our results emphasize that functional analysis driven by genomic data can play an important role in discovering novel disease mechanisms. A wide spectrum of proteins, including channel proteins, protein modifiers, RNA binding proteins, and others, have been implicated in LGS alone.31 Thus, due to their genetic heterogeneity, treatment considerations for epileptic encephalopathies will eventually be guided by the underlying pathogenic mechanism in any given patient. Further understanding of the relation between synaptic vesicle dysfunction and epilepsy will facilitate the discovery of treatment opportunities.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to Epi4K, EPGP, the patients, their families, and referring physicians for their participation and provision of phenotypic and genetic data used in this study. The authors also thank Neil Medvitz and Sara Miller at Duke Electron Microscopy Services and So Young Kim at the Duke RNAi Screening Facility.
GLOSSARY
- DAPI
4',6-diamidino-2-phenylindole
- EDC
1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide
- EM
electron microscopy
- GFP
green fluorescent protein
- LGS
Lennox-Gastaut syndrome
- RFP
red fluorescent protein
- RIPA
radioimmunoprecipitation assay
- TBS
Tris-buffered saline
- WT
wild type
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Ryan S. Dhindsa, Shelton S. Bradrick, Brian J. Krueger, Steven Petrou, Rebecca M. Boumil, and David B. Goldstein designed and conceptualized the study. Ryan S. Dhindsa, Shelton S. Bradrick, Xiaodi Yao, Erin L. Heinzen, Slave Petrovski, Brian J. Krueger, Wayne N. Frankel, Steven Petrou, Rebecca M. Boumil, and David B. Goldstein analyzed and interpreted the data. Ryan S. Dhindsa and David B. Goldstein drafted the manuscript. Shelton S. Bradrick, Xiaodi Yao, Erin L. Heinzen, Slave Petrovski, Brian J. Krueger, Michael R. Johnson, Wayne N. Frankel, Steven Petrou, and Rebecca M. Boumil revised the manuscript for intellectual content.
STUDY FUNDING
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (Epi4K—Administrative Core NS077274; Epi4K Sequencing, Biostatistics and Bioinformatics Core NS077303).
DISCLOSURE
Mr. Dhindsa, Dr. Bradrick, and Dr. Yao report no disclosures. Dr. Heinzen has received research support from the NIH, CURE AHC, and Epilepsy Genome Initiative. Dr. Petrovski reports no disclosures. Dr. Krueger has acted as a consultant for Genomics Technology Consulting. Dr. Johnson has a patent pending for a potential new drug target in epilepsy. Dr. Frankel has served on the editorial boards of PLOS Genetics and Genes, Brain and Behavior and has received research support from the NIH. Dr. Petrou has received research support from CSL, Therapeutic Innovation Australia (NCRIS), ARC Centre of Excellence for Integrative Brain Function (CIBF), NHMRC, Australian Research Council (ARC), UoM, DHB Foundation, and Macquarie Bank. Dr. Boumil has received research support from the NIH. Dr. Goldstein reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Dulac O. Epileptic encephalopathy. Epilepsia 2001;42(suppl 3):23–26. [DOI] [PubMed] [Google Scholar]
- 2.van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol 1993;122:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 2012;13:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray NW, Fourgeaud L, Huang B, et al. Dynamin 3 is a component of the postsynapse, where it interacts with mGluR5 and Homer. Curr Biol 2003;13:510–515. [DOI] [PubMed] [Google Scholar]
- 5.Powell KA, Robinson PJ. Dephosphin/dynamin is a neuronal phosphoprotein concentrated in nerve terminals: evidence from rat cerebellum. Neuroscience 1995;64:821–833. [DOI] [PubMed] [Google Scholar]
- 6.van der Bliek AM, Meyerowitz EM. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature 1991;351:411–414. [DOI] [PubMed] [Google Scholar]
- 7.Ferguson SM, Brasnjo G, Hayashi M, et al. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science 2007;316:570–574. [DOI] [PubMed] [Google Scholar]
- 8.Boumil RM, Letts VA, Roberts MC, et al. A missense mutation in a highly conserved alternate exon of dynamin-1 causes epilepsy in fitful mice. PLoS Genet 2010;6:e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Achiriloaie M, Barylko B, Albanesi JP. Essential role of the dynamin pleckstrin homology domain in receptor-mediated endocytosis. Mol Cell Biol 1999;19:1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ford MG, Jenni S, Nunnari J. The crystal structure of dynamin. Nature 2011;477:561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faelber K, Posor Y, Gao S, et al. Crystal structure of nucleotide-free dynamin. Nature 2011;477:556–560. [DOI] [PubMed] [Google Scholar]
- 13.Ramachandran R, Surka M, Chappie JS, et al. The dynamin middle domain is critical for tetramerization and higher-order self-assembly. EMBO J 2007;26:559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marks B, Stowell MH, Vallis Y, et al. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 2001;410:231–235. [DOI] [PubMed] [Google Scholar]
- 15.EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013;7.20.1–7.20.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu YW, Neumann S, Ramachandran R, Ferguson SM, Pucadyil TJ, Schmid SL. Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. Proc Natl Acad Sci USA 2011;108:E234–E242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao H, Garcia F, McNiven MA. Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell 1998;9:2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook TA, Urrutia R, McNiven MA. Identification of dynamin 2, an isoform ubiquitously expressed in rat tissues. Proc Natl Acad Sci USA 1994;91:644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol 1994;127:915–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song BD, Yarar D, Schmid SL. An assembly-incompetent mutant establishes a requirement for dynamin self-assembly in clathrin-mediated endocytosis in vivo. Mol Biol Cell 2004;15:2243–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayanan R, Leonard M, Song BD, Schmid SL, Ramaswami M. An internal GAP domain negatively regulates presynaptic dynamin in vivo: a two-step model for dynamin function. J Cell Biol 2005;169:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sever S, Damke H, Schmid SL. Dynamin:GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J Cell Biol 2000;150:1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi M, Raimondi A, O'Toole E, et al. Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. Proc Natl Acad Sci USA 2008;105:2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stafa K, Tsika E, Moser R, et al. Functional interaction of Parkinson's disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet 2014;23:2055–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788. [DOI] [PubMed] [Google Scholar]
- 29.Garcia CC, Blair HJ, Seager M, et al. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet 2004;41:183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 2014;17:773–781. [DOI] [PubMed] [Google Scholar]
- 31.Epi4K Consortium. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.