

Abstract
The human ether-a-go-go related gene (hERG) encodes two subunits, hERG 1a and hERG 1b, that combine in vivo to conduct the rapid delayed rectifier potassium current (IKr). Reduced IKr slows cardiac action potential (AP) repolarization and is an underlying cause of cardiac arrhythmias associated with long QT syndrome (LQTS). Although the physiological importance of hERG 1b has been elucidated, the effects of hERG 1b disease mutations on cardiac IKr and AP behavior have not been described. To explore the disease mechanism of a 1b-specific mutation associated with a case of intrauterine fetal death, we examined the effects of the 1b-R25W mutation on total protein, trafficking and membrane current levels in HEK293 cells at physiological temperatures. By all measures the 1b-R25W mutation conferred diminished expression, and exerted a temperature-sensitive, dominant-negative effect over the WT hERG 1a protein with which it was co-expressed. Membrane currents were reduced by 60% with no apparent effect on voltage dependence or deactivation. The dominant-negative effects of R25W were demonstrated in iPSC-CMs, where 1b-R25W transfection diminished native IKr compared to controls. R25W also slowed AP repolarization, and increased AP triangulation and variability in iPSC-CMs, reflecting cellular manifestations of pro-arrhythmia. These data demonstrate that R25W is a dominant-negative mutation with significant pathophysiological consequences, and provide the first direct link between hERG 1b mutation and cardiomyocyte dysfunction.
Keywords: hERG, KCNH2, cardiomyocyte, arrhythmia, long QT syndrome
1. Introduction
The first studies of heterologously expressed hERG channels described biophysical and pharmacological properties uniquely characteristic of cardiac IKr (Snyders and Chaudhary, 1996; Trudeau et al., 1995). Later, alternate transcripts of KCNH2 in mouse and human heart were shown to encode two subunits, hERG 1a, the original isolate, and hERG 1b (Lees-Miller et al., 1997; London et al., 1997). The hERG 1a N-terminus contains a Per-Arnt-Sim (PAS) domain, also known as the ether-à-go-go or eag domain (Morais Cabral et al., 1998), which normally modulates channel gating via a concerted interaction between the cytoplasmic linker of the 4th and 5th transmembrane helical segments and the C-terminus (Gianulis et al., 2013; Hull et al., 2014; Ng et al., 2014; Wang et al., 2000; Wang et al., 1998). In the hERG 1b transcript, an alternate 5’ exon replaces 1a exons 1-5, resulting in a shorter and unique N-terminus that lacks a functional PAS domain (Lees-Miller et al., 1997; London et al., 1997). Introduction of the 1b subunit reduces the number of PAS domains in the channel and accelerates kinetics of heteromeric hERG 1a/1b channel compared to 1a homomeric channels (Sale et al., 2008; Trudeau et al., 2011). Overall, the characteristics of hERG 1a/1b channels better resemble those of native IKr (Jones et al., 2014; Larsen and Olesen, 2010; McPate et al., 2009; Sale et al., 2008). Most recently it was shown that disruption of native hERG 1b subunits altered native IKr kinetics and magnitude, as well as prolonged the action potential (AP), thereby demonstrating that hERG 1b is a functional component of human IKr and cardiac repolarization (Jones et al., 2014).
The 1a N-terminus is relatively large (>340 amino acids) with a well-described role in channel trafficking and gating. Numerous 1a-specific mutations have been identified and shown to cause trafficking and functional defects (Anderson et al., 2006; Anderson et al., 2014). Genetic evidence for 1b-specific disease mutations is limited to two clinical cases (Crotti et al., 2013; Sale et al., 2008). And although the 1b subunit’s importance in human IKr and action potential repolarization has been shown (Jones et al., 2014), a direct link between a 1b-specific mutation and cardiomyocyte dysfunction has not been demonstrated.
R25W is a hERG 1b-specific mutation associated with a case of intrauterine fetal death that was initially shown to reduce heteromeric hERG current heterologously expressed in Chinese hamster ovarian cells and recorded at room temperature (Crotti et al., 2013). To further explore the mechanism of disease we recorded membrane currents and APs at physiological temperatures from human embryonic kidney (HEK) cells and human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) exogenously expressing the 1b-R25W mutant. These data identify R25W as a dominant negative mutation that reduces hERG protein levels and native IKr, leading to increased AP duration, triangulation, and variability that represent cellular hallmarks of arrhythmia.
2. Materials and Methods
2.1. Cell Culture
Human iPSC-CMs (iCell® Cardiomyocytes, Cellular Dynamics International) were plated and stored in 12 well dishes as per manufacturer’s instructions. HEK293 cells were cultured in 6-well in DMEM supplemented with 10% fetal bovine serum (FBS).
2.2. Transfection/Infection
The R25W mutation was inserted into a WT hERG 1b pcDNA3.1 vector backbone by quickchange. Mutation was confirmed by automated sequencing (University of Wisconsin-Madison Biotechnology Center). HEK cells stably expressing the hERG 1a subunit were transfected with 2 μg/ml of either wild-type (WT) hERG 1b or the R25W mutant using 1.25 μl/ml Lipofectamine 2000 (Life Technologies). iPSC-CMs were transfected with 1.5 μg/ml WT 1b, R25W, or empty vector DNA using 1.25 μl/ml Lipofectamine 2000. For all voltage clamp experiments we co-transfected hERG 1b constructs with 0.5 μg/ml pcDNA3.1-GFP to identify transfected cells. For AP recordings, iPSC-CM’s were transformed with 1 μl/ml of adenoviral lysate. Adenoviral DNA encoded Kir2.1 in frame with GFP, as described (Jones et al., 2014). We did not transfect GFP for any of the AP recordings because the GFP encoded by the Kir2.1 viral construct masks any transfected GFP. We therefore completed AP recordings without selecting for transfected cells. To minimize recording bias we discarded recordings only if we were unable to elicit an AP with a stimulus current. AP failure occurred equally between wildtype and R25W transfected groups.
2.3. Western Blot
Cells were lysed 48 h post-transfection using buffer containing (in mM): 150 Tris-NaCl, 25 Tris HCl, 10 Na-EGTA, 20 Na-EDTA, 5 glucose, and supplemented with 1% Triton X-100, 50 µg/ml 1,10 phenathroline, 0.7 µg/ml pepstatin A, 1.56 µg/ml benzamidine and 1x Complete Minitab (Roche Applied Science). Solution was sonicated, incubated on ice for 30 min, and centrifuged for 10 min at 13,000 rpm at 4°C. Protein concentration of supernatant was assessed using a BIORAD protein Assay. 30 µg of protein per lane was electrophoresed on a 7.5% SDS-polyacrylamide gel and then transferred onto PVDF membranes. Membranes were blocked and incubated overnight with 1:2000 anti β-actin (ab8226, abCam), and either anti-hERG (CT) (pan) (ALX-215-049-R100, Enzo Life Sciences) or anti-Ribosomal Protein L13A (C-11) (sc-390131, Santa Cruz Biotechnology). Membranes were washed and then incubated with 1:1000 dilutions of secondary antibody Alexa Fluor® 647 Goat Anti-Rabbit IgG A-21245 (Life Technologies) for 1 hour and imaged using a Chemidoc-MP Imaging System (BIORAD). Individual values were calculated from the mean of three parallel experiments completed on the same day and under identical conditions (i.e. n = 1). Protein values were then paired with experiments completed on the same day to increase statistical power.
2.4. Biotinylation
Cell surface proteins were biotinylated with a water soluble sulfo-NHS-SS-biotin (#21331 Thermo Scientific). Cells were washed twice with PBS and labeled with 1 mg/ml sulfo-NHS-SS-biotin in PBS for 30 min at 4ºC. Cells were washed twice and non-reacted biotinylation reagent was quenched with three 5 minute washes using 50 ml of 50 mM glycine in PBS at 4ºC. Cells were lysed in IP buffer containing in mM: 5 EDTA, 5 EGTA, 10 Na-Pyrophosphate, 50 NaF, 1 NaVO3, and supplemented with 1% Triton and 1 protease inhibitor tablet (Roche cat# 1873580). Solution was then centrifuged at 14000 rpm for 10 min at 4ºC. 30 µg of protein is used as lysate input. 100 ul of supernatant containing 500 µg of protein was immunoprecipitated with anti-hERG (See Co-immunoprecipitation methods). Precipitated hERG protein was subjected to 7.5% SDS-polyacrilamide gel electrophoresis and biotin labeled hERG was detected by horseradish peroxidase-conjugated streptavidin (Thermo Scientific). We used the same cell count and plating conditions for each lane.
2.5. Co-immunoprecipitation
Purified protein was co-immunoprecipitated as previously described (Phartiyal et al., 2007). Briefly, transiently HEK293 whole cell lysates (500μg) were precleared with 30µl protein G sepharose beads for 1 hour at 4 °C. Following a 1 minute at 10,000 g, centrifugation at 4 °C to remove beads, 5 µl mouse anti-myc (Abcam) was added to the supernatant and incubated for 16 hours at 4 °C, rotating. For biotinylation preps we replaced anti-myc with the anti-hERG (ALX-215-049-R100, Enzo Life Sciences). Protein G sepharose beads were then added and incubated for an additional 2h. Immunoprecipitates were washed three times in 0.5 ml lysis buffer, and eluted into 30µl Laemmeli sample buffer (25 mM Tris-HCl, pH 6.8, 2% sodium dodecysulfate, 10% glycerol, 0.2 M DL Dithiothreitol) and subjected to a western blot probed with rabbit-pan hERG antibody (Enzo Life Sciences).
2.6. Electrophysiology
iPSC-CM recordings were conducted 5-40 days post-plating. All recordings were completed 48 hrs post-transfection at 36 ± 1°C using whole-cell patch clamp. Recordings were made using an Axon 200A amplifier and Clampex (Molecular Devices). Data were sampled at 10 kHz and low-pass filtered at 1 kHz. Cells were perfused with extracellular solution containing (in mM): 150 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 15 glucose, 10 HEPES, 1 Na-pyruvate, and titrated to pH 7.4 using NaOH. Recording pipettes had resistances of 2-4.5 MΩ when backfilled with intracellular solution containing (in mM): 5 NaCl, 150 KCl, 2 CaCl2, 5 EGTA, 10 HEPES, 5 MgATP and titrated to pH 7.2 using KOH. Intracellular solution aliquots were kept frozen until the day of recording. During recording, the intracellular solution was kept on ice and discarded 2-3 hours post-thaw.
The voltage dependence of hERG 1a/1b currents expressed in HEK cells was assessed by stepping from a −80 mV holding potential to a 3-second pre-pulse between −80 and +50 mV in 10 mV increments. Tail currents were then measured during either a −50 mV, 6-second test pulse. The voltage dependence of IKr in iPSC-CMs was assessed by stepping to from −40 mV holding potential a 3-second pre-pulse between −50 and +30 mV in 10 mV increments. Tail currents were then measured during a −40 mV 6-second test pulse. Cells were held at −50 mV to inactivate voltage-gated sodium channels. Steady-state current in HEK and iPSC-CMs was measured as the 5 ms mean at the end of the pre-pulse. Leak subtraction was performed off-line based on measured current observed at potentials negative to hERG channel activation. To describe the voltage dependence of channel activation, peak tail current was normalized to cellular capacitance, plotted as a function of pre-pulse potential, and fitted with the following Boltzmann equation:
where A1 and A2 represent the maximum and minimums of the fit, respectively, V is the membrane potential, and V0 is the midpoint.
Repolarizing charge was measured from the integral of membrane currents recorded during a voltage protocol designed to mimic a human ventricular action potential (Zhou et al., 1998b). iPSC-CM voltage protocols were completed before and after bath perfusion of 2 μM E-4031, an IKr-specific blocker, and the difference in current was taken to represent IKr (Jones et al., 2014). The time course of deactivation was measured by fitting current decay at −50 mV (HEK) or −40 mV (iPSC-CM) with a single or a double exponential function. iPSC-CM APs were paced at 1 Hz using a 5 ms and 300-1000 pA stimulus. AP variability was measured by recording the AP duration at 90% repolarization (APD90) from 50 sequential APs paced at 1 Hz and reporting the coefficient of variation. The coefficient of variation is defined as the standard deviation of each recording series divided by that series’ mean (Kesteven, 1946).
2.7. Statistical Analysis
Analysis was completed using Clampfit (Molecular Devices) and Origin (OriginLab). All data were reported as mean ± SEM and compared using a Student’s t-tests. When applicable, an ANOVA and Bonferroni post hoc t-tests where used. Statistical significance was taken at p < 0.05.
3. Results
3.1. 1b-R25W reduces membrane current at physiological temperatures
Previous studies demonstrated that heteromeric hERG 1a/1b currents expressed at room temperature in Chinese hamster ovary cells were reduced by 35% when 1b-R25W was substituted for 1b. We repeated these experiments in HEK293 cells at physiological temperatures (36 ± 1°C) (Fig. 1A, B). After transfecting stable hERG 1a cell lines with either 1b or 1b-R25W, we found the mutation reduced maximum peak tail current density by 60% from 64.6 ± 12.6 pA/pF (n = 7) to 25.4 ± 4.4 pA/pF (n = 8, p = 0.01) (Fig. 1C). Similarly, R25W significantly reduced the maximum steady-state current density, measured at the end of a 3-second test pulse, from 71.1 ± 8.3 pA/pF (n = 7) to 40.7 ± 7.7 pA/pF (n = 8, p = 0.01) (Fig. 1D). Thus, the mutant phenotype may be enhanced at physiological temperatures. We found no differences comparing 1a/1b to 1a/1b-R25W channels with respect to voltage dependence (Table 1) or deactivation kinetics (Fig. S1), suggesting the decrease is not obviously attributable to differences in channel gating properties.
Figure 1.
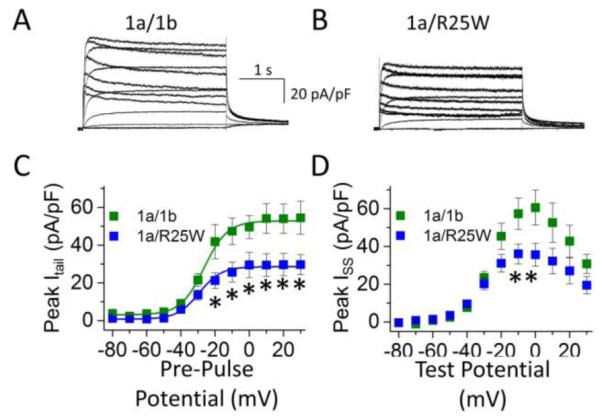
R25W reduces heteromeric peak tail and steady-state current density in human embryonic kidney (HEK) cells at physiological temperatures (36 ± 1°C). Sample current traces, normalized to cellular capacitance, and recorded from HEK cells coexpressing 1a/1b (A) or 1a/1b-R25W (B). (C) Peak tail current density measured at −50 mV, plotted as a function of pre-pulse potential, and fitted with a Boltzmann function for hERG 1a/1b channels (green) and hERG 1a/1b-R25W channels (blue). (D) Steady-state current measured at the end of 3-second test pulse, normalized to cellular capacitance and plotted as a function of test pulse potential for hERG 1a/1b (green) and hERG 1a/1b-R25W (blue). * indicates statistical significance compared to controls at p < 0.05. n = 6-9.
Table 1.
| Table 1 | V1/2
(mV) |
k (mV) |
ITail
(pA/pF) |
ISSmax
(pA/pF) |
Cell Cap. (pF) |
n |
|---|---|---|---|---|---|---|
| 1a/1b | −24.2 ± 1.4 | 11.2 ± 2.1 | 64.6 ± 10.9 | 71.1 ± 8.3 | 23.0 ± 3.7 | 7 |
| 1a/1b-R25W | −26.5 ± 1.2 | 10.1 ± 1.5 | 25.4 ± 4.4* | 40.7 ± 6.8* | 19.3 ± 1.4 | 8 |
indicates statistical significance at p < 0.05 compared to 1a/1b
To predict the potential physiological consequences of 1b-R25W, we measured “repolarizing charge” (Q) during a voltage protocol that mimics a human ventricular action potential (AP) (Fig. 2). Repolarizing charge was calculated as the integral of membrane currents elicited during the AP protocol. The R25W mutation significantly reduced repolarizing charge normalized to cellular capacitance to roughly 45% of controls (1a/1b: 17.9 ± 3.0 pC/pF, n = 7; 1a/1b-R25W: 8.1 ± 1.4 pC/pF, n = 5, p = 0.03) (Fig. 2A, B). To assess if changes in gating kinetics contribute to the reduced repolarizing charge density we normalized repolarizing charge to maximum peak tail current. Normalizing to maximum peak tail current revealed that the repolarizing charge per channel in 1a/1b-R25W channels was not reduced compared to 1a/1b controls, as would have occurred if changes in gating were responsible (1a/1b: 0.30 ± 0.05 pC/pA, n = 7; 1a/R25W: 0.29 ± 0.03 pC/pA, n = 5, p = 0.8) (Fig. 2C, D). This finding reinforces the conclusions from experiments above that the effects of R25W are not caused by alterations in channel gating kinetics.
Figure 2.
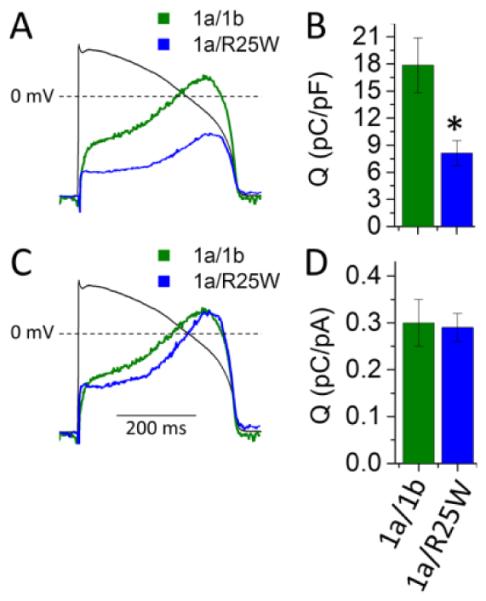
R25W reduces repolarizing charge density in HEK cells at physiological temperature (36 ± 1°C). (A) Representative current profiles recorded during an action potential command (black tracing) and normalized to cellular capacitance from HEK cells expressing hERG 1a/1b (green) or 1a/1b-R25W (blue). (B) Repolarizing charge density (pC/pF) measured from the integral of current profiles as in “A” for hERG 1a/1b (green) and hERG 1a/1b-R25W (blue). Charge density is significantly reduced by 1b-R25W compared to controls. (C) Same traces shown in “A” but normalized to the maximum tail current density (ItailMAX) recorded for that cell. (D) Repolarizing charge normalized to ItailMAX for hERG 1a/1b (green) and hERG 1a/1b-R25W (blue). * indicates statistical significance compared to controls at p < 0.05. n = 6.
3.2. A dominant-negative effect of 1b-R25W on protein levels
To determine if the decrease in current is reflected in protein levels, we carried out quantitative western blot analysis on HEK293 cells stably expressing hERG 1a and transfected with either WT 1b or 1b-R25W (Fig. 3). With values normalized to an actin loading control, we measured 53.8 ± 7.8% less protein for the 1b-R25W mutant compared to WT 1b (Fig. 3A, B). Moreover, hERG 1a protein levels were also reduced when co-expressed with 1b-R25W compared to 1b, exhibiting 46.1 ± 8.9% less protein. Protein levels affected by 1b-R25W were recovered when cells were incubated at 27°C, consistent with a defect in protein folding conferred by the mutant protein (Fig. 3).
Figure 3.

The hERG 1b-specific mutation R25W reduces hERG 1a and 1b protein levels. (A) Sample western blot from HEK cells transfected with either hERG 1a/1b or hERG 1a/R25W and incubated at either 37°C or 27°C. (B) Total hERG 1a protein relative to actin and normalized to control following 24 hour incubation at 37°C and 27°C. (C) Total hERG 1b protein relative to actin and normalized to control following 24 hour incubation at 37°C and 27°C. Experiments completed on the same day were paired to increase statistical power. (D) Western blot showing hERG protein from cell lysate (Lysate) and co-immunoprecipitation (IP). * indicates statistically significance compared to control at p < 0.05. n = 3-7.
The dominant-negative effect of 1b-R25W on hERG 1a protein levels suggests that the mutation does not affect subunit association. To further test this hypothesis, we co-immunoprecipitated myc-tagged 1b and 1b-R25W subunits respectively co-expressed with hERG 1a (Fig. 3D). The 1a subunit effectively co-purified with both WT and mutant subunits. These findings support the idea that the 1b-R25W mutation disrupts protein folding, but not assembly.
To assess defects in channel trafficking commonly associated with hERG mutations (Anderson et al., 2006; Anderson et al., 2014), we measured maturation of protein from the ER-associated glycoform to the mature, Golgi-glycosylated form destined for the plasma membrane (Zhou et al., 1999). Using western blot analysis, we measured the mature glycoform normalized to total protein for hERG 1b and 1b-R25W. (A predominance of the 1a immature glycoform precluded measurements of 1a maturation.) We found that the 1b-R25W mutation significantly reduced mature hERG 1b by roughly 25%, consistent with its effect on protein folding (Fig. 4A, B). Additionally, surface protein labeled with biotin and purified with streptavidin beads showed a corresponding reduction of hERG attributable to 1b-R25W. Under identical cell counts and plating conditions for each lane, biotin labeled hERG protein in the presence of 1b-R25W displayed a 51.1 ± 4.7% and 55.2 ± 3% reduction in 1a and 1b protein, respectively, compared to WT controls (n = 5, p < 0.0001) (Fig 4C, D). The biotin labeled protein did not display protein bands corresponding to the β-actin loading control, indicating that we successfully isolated only the membrane-bound protein.
Figure 4.

1b-R25W impairs hERG 1b trafficking and surface expression. (A) Western blot depicting reduced mature hERG 1a (155 kDa) and hERG 1b (90 kDa) protein in the presence of 1b-R25W. (B) Mature hERG 1b protein reported as a ratio of total protein for hERG 1a (left) and hERG 1b (right). (C) Western blot displaying lysate (left) and biotinylation assay (right) against hERG display reduced surface hERG in the presence of 1b-R25W. (D) Quantification of biotinylation assay displaying surface hERG 1a (left) and hERG 1b (right) protein levels reported relative to control experiments from the same blot. * indicates statistical significance compared to control at p < 0.05. n = 5-8.
3.3. Evaluation of protein instability induced by 1b-R25W
We tested the hypothesis that 1b-R25W destabilizes hERG protein via enhanced lysosomal degradation. We used bafilomycin, a vacuolar H-ATPase inhibitor, to disable lysosomal function (Xu et al., 2003). Western blot analysis revealed that bafilomycin enhanced protein levels in both the control and mutant heteromers, consistent with the role of the lysosome in hERG protein homeostasis (Guo et al., 2009) (Fig. 5A - C). Data displayed in Figure 5B and C were acquired from the three experimental repeats shown in Figure 5A. Similar effects were observed in two additional blots. The enhancement seemed to be greater for the 1a/1b-R25W channels, as expected if they were rendered unstable by the mutation, but this trend did not reach statistical significance (Fig. 5D). Data displayed in Figure 5D are from nine experiments completed on three different days. Experiments completed on the same day were paired to increase statistical power. These experiments suggest 1b-R25W increases degradation through the lysosome but do not rule out the possibility that bafilomycin also acts, in part, as a molecular chaperone.
Figure 5.

Lysosomal inhibition rescues protein levels in the presence of 1b-R25W. (A) Sample western blot from HEK cells coexpressing hERG 1a with either hERG 1b or 1b-R25W. Cells were incubated for 24 hours in control medium (Untreated) or 1 µM of the lysosomal inhibitor, bafilomycin (Bafilo). (B & C) Total hERG 1a (B) and 1b (C) protein levels relative to actin measured from cells cultured in untreated medium and medium supplemented with 1 µM bafilomycin. Data displayed were acquired from the sample blot displayed in “A”. Overnight treatment with bafilomycin restored total hERG 1a (B) and hERG 1b (C) protein levels to WT levels. (D) Percent increase in total hERG 1a (left) and 1b (right) protein following bafilomycin treatment. Data displayed are from nine experiments completed on three different days. Experiments completed on the same day were paired to increase statistical power. * indicates statistical significance compared to control. n = 3-8
3.4. Physiological Consequences of 1b-R25W on Native IKr
The western blot and electrophysiological analyses above suggest a dominant-negative effect of R25W. To further test this hypothesis, we transfected exogenous 1b-R25W into cardiomyocytes derived iPSC-CMs and assessed its effects on native IKr measured as E-4031-sensitive current. The 1b-R25W mutant cDNA significantly reduced peak tail (0.7 ± 0.1 pA/pF, n = 5, p < 0.01) and steady-state (0.8 ± 0.1 pA/pF, p < 0.05) IKr density compared to pcDNA3.1 vector controls or exogenous WT 1b, which expressed in an additive manner (Fig. 6, Table 2, Fig. S2). These data indicate that the dominant-negative effects of R25W observed under conditions of heterologous expression can also be exerted on native IKr.
Figure 6.

1b-R25W displays a dominant negative phenotype on native IKr. Sample E-4031 sensitive current traces, indicative of IKr, recorded from induced pluripotent stem cell-derive cardiomyocytes (iPSC-CMs) transfected with pcDNA3.1 (Vector, A), WT hERG 1b (B), or 1b-R25W (C). (D) Peak tail IKr, measured at −40 mV, normalized to cellular capacitance, plotted as a function of pre-pulse potential, and fitted with a Boltzmann function for vector (black), WT hERG 1b (green), and 1b-R25W (blue) transfected iPSC-CMs. (E) Steady-state IKr, measured at the end of 3-second test pulse, normalized to cellular capacitance, and plotted as a function of test potential for vector (black), hERG 1b (green), and 1b-R25W (blue) transfected iPSC-CMs. * indicates statistical significance compared to Vector at p < 0.05. n = 6–11.
Table 2.
| Table 2 | V1/2
(mV) |
k (mV) |
ITail
(pA/pF) |
ISSmax
(PA/pF) |
Tauw
(ms) |
Cell Cap. (PF) |
n |
|---|---|---|---|---|---|---|---|
| Vector | −30.7 ± 1.1 | 5.1 ± 0.7 | 1.4 ± 0.2 | 1.2 ± 0.2 | 595 ± 33 | 46 ± 11 | 10 |
| hERG 1b | −26.7 ± 1.9 | 6.9 ± 0.8 | 3.0 ± 0.6* | 3.6 ± 0.5* | 74 ± 19* | 95 ± 27 | 7 |
| 1b-R25W | −33.5 ± 4.1 | 7.2 ± 0.6 | 0.7 ± 0.1* | 0.8 ± 0.1* | 547 ± 54 | 68 ± 8 | 5 |
indicates statistical significance compared to Vector at p < 0.05
3.4. Physiological Consequences of 1b-R25W on the Cardiac Action Potential
To determine the effects of 1b-R25W on the cardiomyocyte functional properties we measured APs from Kir2.1-transduced iPSC-CMs (Jones et al., 2014), transfected with either pcDNA3.1 vector or 1b-R25W (Fig. 7). As predicted by its effect on IKr, 1b-R25W significantly slowed repolarization as measured by APD90 (351 ± 25 ms, n = 13, p < 0.01) compared to vector controls (264 ± 19 ms, n = 25) (Fig. 7C), whereas WT 1b transfection significantly shortened the APD90 (Fig. S3). Moreover, compared to controls, 1b-R25W significantly enhanced two important metrics of cellular proarrhyhmia: AP triangulation and AP variability (Altomare et al., 2015; Gallacher et al., 2007; Hondeghem et al., 2001; Jacobson et al., 2011; Ziupa et al., 2014). AP triangulation was assessed using the ratio of Phase 2 (APD40-APD30) divided by Phase 3 (APD80-APD70). We found that 1b-R25W significantly reduced the AP ratio from 5.6 ± 1.0 (n = 25) in controls to 2.2 ± 0.5 (n = 13, p < 0.05) in R25W transfected iPSC-CMs, indicating an increase in triangulation. To determine AP variability, we measured the coefficient of variation of the APD90 recorded from 50 sequential APs paced at 1 Hz (Fig. 7E), and observed a significant increase from 3.2 ± 0.4% in vector controls (n = 22) to 5.1 ± 0.7% in 1b-R25W-transduced cells (n = 12, p < 0.01) (Fig. 7D). The maximum diastolic potential, upstroke velocity, and AP amplitude were unchanged by 1b-R25W (Table 3). These data demonstrate that the 1b-R25W mutant triggers cellular markers of pro-arrhythmia in a dominant-negative manner.
Figure 7.
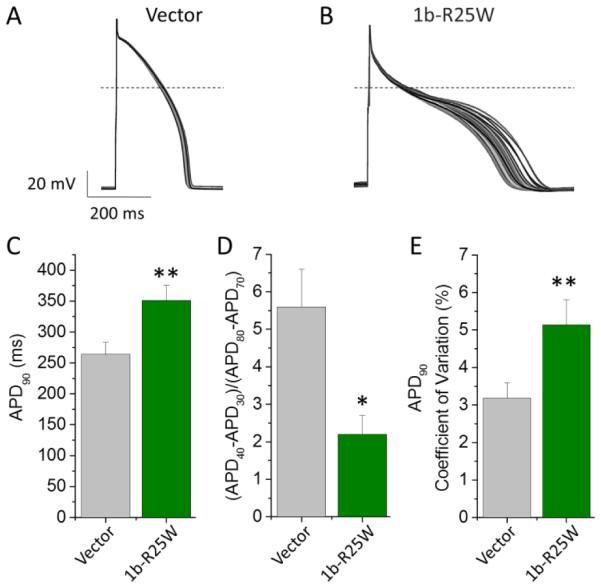
1b-R25W is arrhythmogenic in iPSC-CMs. 50 sequential AP traces paced at 1 Hz recorded from iPSC-CMs transfected with either pcDNA3.1 (Vector, A) or 1b-R25W (B) at 36 ± 1°C. (C) Time to 90% repolarization (APD90) recorded from vector-transfected (gray) or R25W-transfected (green) iPSC-CMs (n = 25 & 13, respectively). (D) Ratio of Phase 2 to Phase 3 repolarization times reported as (APD40-APD30)/(APD80-APD70) for either vector-transfected (gray) or 1b-R25W-transfected (green) iPSC-CMs (n = 25 & 13, respectively). (E) Mean coefficient of variation measured from 50 sequential APs paced at 1 Hz from either vector-transfected (gray) or 1b-R25W-transfected (green) iPSC-CMs (n = 22 & 12, respectively). * indicates statistical significance compared to control at p < 0.05. ** indicates statistical significance compared to control at p < 0.01.
Table 3.
| Table 3 | APD90
(ms) |
|
50 Beat APD Variance (ms2) |
Diastolic Potential (mV) |
dV/dtMax | AP Amplitude (mV) |
n | |
|---|---|---|---|---|---|---|---|---|
| Vector | 264 ± 19 | 5.6 ± 1.0 | 98 ± 28 | −74 ± 5 | 102 ± 13 | 123 ± 6 | 25 | |
| 1b-R25W | 351 ± 25* | 2.2 ± 0.5* | 279 ± 60* | −71 ± 10 | 83 ± 20 | 117 ± 13 | 13 |
indicates statistical significance at p < 0.05 compared to Vector
4. Discussion
Here, we describe a temperature-sensitive, dominant-negative phenotype of the hERG 1b-R25W mutation associated with a case of intrauterine fetal death. Expressed heterologously in HEK293 cells, hERG 1a/1b currents recorded at physiological temperatures and total protein levels are much reduced when the 1b subunit harbors the R25W mutation. The mutation does not disrupt oligomerization with hERG 1a, and association amplifies its effects: both 1b-R25W and WT 1a protein levels are reduced compared to WT controls. Moreover, the mutation retards channel trafficking as measured by maturation. Exogenously expressed 1b-R25W reduces native IKr in iPSC-CMs, consistent with a dominant-negative phenotype. A corresponding increase in APD and variability were also observed. Together these findings provide a mechanistic explanation for the disease phenotype and the first direct link between a hERG 1b-specific mutation and cardiac pro-arrhythmic behavior.
hERG 1b-R25W was first identified in a genetic screen for potential long QT syndrome mutations associated with unexplained intrauterine fetal death (Crotti et al., 2013). That report described a reduction attributable to the mutation in hERG 1a/1b current expressed in Chinese Hamster Ovary (CHO) cells at room temperature. We found a slightly larger reduction in current magnitude but did not see the modest changes observed in gating properties in the previous study (Crotti et al., 2013). Although we have not ruled out differences attributable to different expression systems, our studies in human iPSC-CMs demonstrate physiological differences consistent with our findings using HEK293 cells, a human cell line. The apparently enhanced phenotype in our study could be attributed to elevated temperatures, which are less permissive to protein folding defects associated with many other hERG-associated (LQT2) mutations (Anderson et al., 2006; Zhou et al., 1998a), and can mask or uncover different gating phenotypes (Sale et al., 2008). We conclude that a reduction in membrane density of IKr channels is the predominant determinant of the disease phenotype.
What is the molecular mechanism underlying the 1b-R25W mutant phenotype? Because assembly of hERG 1a and 1b subunits has been reported to involve a co-translational association of the nascent 1a and 1b subunits (Phartiyal et al., 2007), we considered the possibility that assembly of the subunits might be disrupted by the substitution of a charged arginine residue for a bulky tryptophan. This hypothesis was not supported, as the 1b-R25W effectively co-purified with the 1a subunit. Moreover, loss of oligomerization would be expected to result in 1a homomeric-like currents because 1b subunits fail to traffic efficiently without 1a to mask an ER retention signal (Phartiyal et al., 2008). Such changes in current properties were not observed. These observations indicate the arginine at position 25 in the wild type protein is not critical for heteromeric subunit association and place constraints on hypotheses to map the interaction interface. Future experiments will be required to determine at what point in biogenesis or trafficking the mutation exerts its deleterious effects and what the physico-chemical nature of that perturbation might be.
The limitations of our study relate in part to the challenges of studying mutant phenotypes in heterologous expression systems or native-like systems in which exogenous expression of the mutant is used as a proxy for a genomic alteration. For example, it is likely that expression of the cDNA construct in iPSC-CMs results in levels of the mutant 1b protein in excess to what would be expected from allelic expression within the patient’s cardiomyocytes. Our findings provide a strong rationale for generating patient-derived iPSC-CMs to ultimately elucidate the molecular mechanism of this 1b-specific mutation.
Another consideration is the use of the iPSC-CM as a model for the fetal ventricular myocytes. Depolarized diastolic potentials, myogenic AP firing, and a reduced maximum rise rate of the AP upstroke (dV/dtMAX) are several characteristics that iPSC-CMS share with the developing myocardium (Doss et al., 2012; Gennser and Nilsson, 1970; Kong et al., 2010; Liang et al., 2013; Lieu et al., 2013; Mummery et al., 2003; Zhang et al., 2012; Zhang et al., 2009) and that distinguish them from adult ventricular APs (Freud, 1972; Jost et al., 2013). Despite their limitations as proxies for adult cardiomyocytes, these properties of iPSC-CMs are closer to the characteristics of cardiomyocytes isolated from embryonic (Jezek et al., 1982; Tuganowski and Cekanski, 1971) or early fetal heart (Gennser and Nilsson, 1970; Mummery et al., 2003), and therefore may better approximate the native setting for intrauterine cardiac death. Clearly there is a rationale for focused efforts to generate better models not only representing “adult” cardiomyocytes but also the young heart which, at increasingly earlier stages of development, is front and center of studies of life-threatening cardiac arrhythmias (Saul et al., 2014).
R25W was isolated from a fetus whose mother also carried the mutation but was asymptomatic with only borderline QT prolongation (Crotti et al., 2013). Developmentally-dependent disease phenotypes have been described for cardiac sodium channels, where the LQTS mutation L409P/H558R confers a dramatically more deleterious phenotype in the fetal splice variant compared to the adult variant (Murphy et al., 2012). Interestingly, the ratio of hERG 1b/1a transcript is greater in fetal compared to adult heart, suggesting that 1b-specific mutations may be more deleterious early in development (Crotti et al., 2013). The ratios of hERG 1a and 1b protein are also altered in the failing heart (Holzem et al., 2015). How LQTS mutations manifest at different stages of development continues to be active point of research.
5. Conclusions
Evidence supporting the importance of hERG 1b in human cardiac physiology has been growing since it was first isolated in 1997 (Lees-Miller et al., 1997; London et al., 1997). The presence of 1b subunits in cardiac tissue is well documented (Jones et al., 2004a; Jones et al., 2004b; Lees-Miller et al., 2003; Lees-Miller et al., 1997; London et al., 1997) and its relevance in human IKr and AP morphology has been shown using both shRNA knockdown of 1b and overexpression of the 1a PAS domain to mask 1b’s effects on gating (Jones et al., 2014). Here we show that transfection of R25W disrupts hERG protein levels, trafficking and surface expression corresponding to reduced native IKr and increased AP duration, triangulation, and variability. These data strongly support a role for R25W as a proarrhythmic mutation, and provide the first direct link between a naturally occurring hERG 1b mutation and cardiac dysfunction.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank Dr. Greg Starek for critical discussion and acknowledge the following sources of support: NIH Grants 5R01 HL081780 and 5R01 NS081320 (GAR), Training Program in Translational Cardiovascular Science, NIH/NHLBI 5T32HL007936 (DKJ) and the UW Madison Stem Cell and Regenerative Medicine Training Award (DKJ).
Abbreviations
- AP
Action potential
- hERG
human ether-à-go-go related gene
- HEK
human embryonic kidney
- iPSC-CMs
induced pluripotent stem cell-derived cardiomyocytes
- LQTS
long QT syndrome
- PAS
Per-Arnt-Sim
- IKr
rapid delayed rectifier potassium current
- APD90
action potential duration at 90% repolarization
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altomare C, Bartolucci C, Sala L, Bernardi J, Mostacciuolo G, Rocchetti M, Severi S, Zaza A. IKr Impact on Repolarization and Its Variability Assessed by Dynamic-Clamp. Circ Arrhythm Electrophysiol. 2015 doi: 10.1161/CIRCEP.114.002572. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–73. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP, January CT. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun. 2014;5:5535. doi: 10.1038/ncomms6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti L, Tester DJ, White WM, Bartos DC, Insolia R, Besana A, Kunic JD, Will ML, Velasco EJ, Bair JJ, Ghidoni A, Cetin I, Van Dyke DL, Wick MJ, Brost B, Delisle BP, Facchinetti F, George AL, Schwartz PJ, Ackerman MJ. Long QT syndrome-associated mutations in intrauterine fetal death. Jama. 2013;309:1473–82. doi: 10.1001/jama.2013.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss MX, Di Diego JM, Goodrow RJ, Wu Y, Cordeiro JM, Nesterenko VV, Barajas-Martinez H, Hu D, Urrutia J, Desai M, Treat JA, Sachinidis A, Antzelevitch C. Maximum diastolic potential of human induced pluripotent stem cell-derived cardiomyocytes depends critically on I(Kr) PLoS One. 2012;7:e40288. doi: 10.1371/journal.pone.0040288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freud GE. Transmembrane potential in isolated human heart. Cardiovasc Res. 1972;6:75–8. doi: 10.1093/cvr/6.1.75. [DOI] [PubMed] [Google Scholar]
- Gallacher DJ, Van de Water A, van der Linde H, Hermans AN, Lu HR, Towart R, Volders PG. In vivo mechanisms precipitating torsades de pointes in a canine model of drug-induced long-QT1 syndrome. Cardiovasc Res. 2007;76:247–56. doi: 10.1016/j.cardiores.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Gennser G, Nilsson E. Excitation and impulse conduction in the human fetal heart. Acta Physiol Scand. 1970;79:305–20. doi: 10.1111/j.1748-1716.1970.tb04731.x. [DOI] [PubMed] [Google Scholar]
- Gianulis EC, Liu Q, Trudeau MC. Direct interaction of eag domains and cyclic nucleotide-binding homology domains regulate deactivation gating in hERG channels. J Gen Physiol. 2013;142:351–66. doi: 10.1085/jgp.201310995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, Zhang S. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–57. doi: 10.1172/JCI39027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzem KM, Gomez JF, Glukhov AV, Madden EJ, Koppel AC, Ewald GA, Trenor B, Efimov IR. Reduced response to I blockade and altered hERG1a/1b stoichiometry in human heart failure. J Mol Cell Cardiol. 2015 doi: 10.1016/j.yjmcc.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–13. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Hull CM, Sokolov S, Van Slyke AC, Claydon TW. Regional flexibility in the S4-S5 linker regulates hERG channel closed-state stabilization. Pflugers Arch. 2014;466:1911–9. doi: 10.1007/s00424-013-1431-9. [DOI] [PubMed] [Google Scholar]
- Jacobson I, Carlsson L, Duker G. Beat-by-beat QT interval variability, but not QT prolongation per se, predicts drug-induced torsades de pointes in the anaesthetised methoxamine-sensitized rabbit. J Pharmacol Toxicol Methods. 2011;63:40–6. doi: 10.1016/j.vascn.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Jezek K, Pucelik P, Sauer J, Bartak F. Basic electrophysiological parameters and frequency sensitivity of the ventricular myocardium of human embryos. Physiol Bohemoslov. 1982;31:11–9. [PubMed] [Google Scholar]
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC, Robertson GA. hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci U S A. 2014;111:18073–7. doi: 10.1073/pnas.1414945111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EM, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem. 2004a;279:44690–4. doi: 10.1074/jbc.M408344200. [DOI] [PubMed] [Google Scholar]
- Jones EMC, Wang JL, Robertson GA. Expression of HERG1a and HERG1b subunits in the heart. Biophysical Journal. 2004b;86:228A–228A. [Google Scholar]
- Jost N, Virag L, Comtois P, Ordog B, Szuts V, Seprenyi G, Bitay M, Kohajda Z, Koncz I, Nagy N, Szel T, Magyar J, Kovacs M, Puskas LG, Lengyel C, Wettwer E, Ravens U, Nanasi PP, Papp JG, Varro A, Nattel S. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J Physiol. 2013;591:4189–206. doi: 10.1113/jphysiol.2013.261198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesteven GL. The coefficient of variation. Nature. 1946;158:520. doi: 10.1038/158520c0. [DOI] [PubMed] [Google Scholar]
- Kong CW, Akar FG, Li RA. Translational potential of human embryonic and induced pluripotent stem cells for myocardial repair: insights from experimental models. Thromb Haemost. 2010;104:30–8. doi: 10.1160/TH10-03-0189. [DOI] [PubMed] [Google Scholar]
- Larsen AP, Olesen SP. Differential expression of hERG1 channel isoforms reproduces properties of native I(Kr) and modulates cardiac action potential characteristics. PLoS One. 2010;5:e9021. doi: 10.1371/journal.pone.0009021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller JP, Guo J, Somers JR, Roach DE, Sheldon RS, Rancourt DE, Duff HJ. Selective knockout of mouse ERG1 B potassium channel eliminates I(Kr) in adult ventricular myocytes and elicits episodes of abrupt sinus bradycardia. Mol Cell Biol. 2003;23:1856–62. doi: 10.1128/MCB.23.6.1856-1862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller JP, Kondo C, Wang L, Duff HJ. Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res. 1997;81:719–26. doi: 10.1161/01.res.81.5.719. [DOI] [PubMed] [Google Scholar]
- Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC, Wu JC. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong CW, Li RA. Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol. 2013;6:191–201. doi: 10.1161/CIRCEP.111.973420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler CA, Robertson GA. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ Res. 1997;81:870–8. doi: 10.1161/01.res.81.5.870. [DOI] [PubMed] [Google Scholar]
- McPate MJ, Zhang H, Cordeiro JM, Dempsey CE, Witchel HJ, Hancox JC. hERG1a/1b heteromeric currents exhibit amplified attenuation of inactivation in variant 1 short QT syndrome. Biochem Biophys Res Commun. 2009;386:111–7. doi: 10.1016/j.bbrc.2009.05.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998;95:649–55. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, Passier R, Tertoolen L. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–40. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- Murphy LL, Moon-Grady AJ, Cuneo BF, Wakai RT, Yu S, Kunic JD, Benson DW, George AL., Jr. Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm. 2012;9:590–7. doi: 10.1016/j.hrthm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CA, Phan K, Hill AP, Vandenberg JI, Perry MD. Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J Biol Chem. 2014;289:25822–32. doi: 10.1074/jbc.M114.558379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale H, Wang J, O'Hara TJ, Tester DJ, Phartiyal P, He JQ, Rudy Y, Ackerman MJ, Robertson GA. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ Res. 2008;103:e81–95. doi: 10.1161/CIRCRESAHA.108.185249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul JP, Schwartz PJ, Ackerman MJ, Triedman JK. Rationale and objectives for ECG screening in infancy. Heart Rhythm. 2014;11:2316–21. doi: 10.1016/j.hrthm.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyders DJ, Chaudhary A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol Pharmacol. 1996;49:949–55. [PubMed] [Google Scholar]
- Trudeau MC, Leung LM, Roti ER, Robertson GA. hERG1a N-terminal eag domain-containing polypeptides regulate homomeric hERG1b and heteromeric hERG1a/hERG1b channels: a possible mechanism for long QT syndrome. J Gen Physiol. 2011;138:581–92. doi: 10.1085/jgp.201110683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–5. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Tuganowski W, Cekanski A. Electrical activity of a single fibre of the human embryonic heart. Pflugers Arch. 1971;323:21–6. doi: 10.1007/BF00586562. [DOI] [PubMed] [Google Scholar]
- Wang J, Myers CD, Robertson GA. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K(+) channels. J Gen Physiol. 2000;115:749–58. doi: 10.1085/jgp.115.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels [published erratum appears in J Gen Physiol 1999 Feb;113(2):359] J Gen Physiol. 1998;112:637–47. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Feng HT, Wang C, Yip KH, Pavlos N, Papadimitriou JM, Wood D, Zheng MH. Effects of Bafilomycin A1: an inhibitor of vacuolar H (+)-ATPases on endocytosis and apoptosis in RAW cells and RAW cell-derived osteoclasts. J Cell Biochem. 2003;88:1256–64. doi: 10.1002/jcb.10477. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zou B, Yu H, Moretti A, Wang X, Yan W, Babcock JJ, Bellin M, McManus OB, Tomaselli G, Nan F, Laugwitz KL, Li M. Modulation of hERG potassium channel gating normalizes action potential duration prolonged by dysfunctional KCNQ1 potassium channel. Proc Natl Acad Sci U S A. 2012;109:11866–71. doi: 10.1073/pnas.1205266109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem. 1998a;273:21061–6. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–6. doi: 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998b;74:230–41. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziupa D, Beck J, Franke G, Perez Feliz S, Hartmann M, Koren G, Zehender M, Bode C, Brunner M, Odening KE. Pronounced effects of HERG-blockers E-4031 and erythromycin on APD, spatial APD dispersion and triangulation in transgenic long-QT type 1 rabbits. PLoS One. 2014;9:e107210. doi: 10.1371/journal.pone.0107210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.