

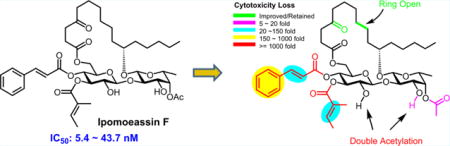
Abstract
Ipomoeassin F, the flagship congener of a resin glycoside family exhibited single-digit nanomolar IC50 values against several cancer cell lines. To facilitate drug discovery based on this unique yet underexplored natural product, we performed the most sophisticated SAR studies of ipomoeassin F to date, which not only greatly bettered our understanding of its pharmacophore but also led to the discovery of two new derivatives (3 and 27) with similar potency but improved synthetic profile. The work presented here opens new avenues toward harnessing the medicinal potential of the ipomoeassin family of glycolipids in the future.
Graphical abstract
In the ipomoeassin family of resin glycosides, ipomoeassin F showed exceptionally potent activity against cancer cell growth (Table 1).1,2 Its IC50 values are usually in the single-digit nanomolar range. More excitingly, its structural homologue, ipomoeassin A, showed a distinct activity profile compared to other anticancer agents in the NCI 60-cell line screen.3 This result indicates that the ipomoeassins likely hit some novel biological targets. Since the discovery of ipomoeassin F, three total syntheses have been reported;4–6 however, the information on the structure–activity relationship (SAR) of ipomoeassin F is very scarce. To extend ipomoeassin research into the fields of chemical biology and drug discovery, it is crucial to understand how structural modifications would influence the biological activity of the ipomoeassins.
Table 1.
Structures and IC50 Values of Ipomoeassin A–F
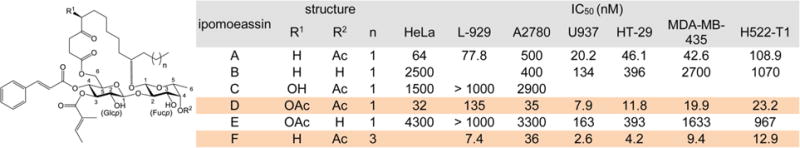
|
Based on the cytotoxicity data of ipomoeassins A–F (Table 1), the overall lipophilicity is essential to the molecule’s potency. When the R1 group is OAc vs OH (D vs C), the R2 group is OAc vs OH (A vs B, or D vs E), or n is equal to 3 vs 1 (F vs A), the activity increases significantly. In the process of the total syntheses of the ipomoeassins, Fürstner and co-workers found two other pieces of information on SAR.5 First, in the structure of ipomoeassin B, the removal of the C-4 ketone functionality in the fatty acid chain damaged the biological activity. Second, simultaneous migration of tiglate from 3-O-Glcp (glucosylpyranoside) to 2-O-Glcp and cinnamate from 4-O-Glcp to 3-O-Glcp was detrimental to the antitumor activity of ipomoeassin F. In our very recent studies, we demonstrated the critical role of the stereochemistry at C-11 of the fatty acid for the first time.6 To facilitate future biomedical evaluation, we continued to conduct more systematic medicinal chemistry explorations of ipomoeassin F and its congeners by using the scalable and flexible synthetic route we recently developed, and the results are presented in this report.
The ipomoeassins are distinguished from other resin glycosides7 by two pronounced structural features. First, the carbohydrate core is densely decorated with ester functionalities, especially the two α,β-unsaturated (tiglate and cinnamate) esters. Second, there is a ketone functional group at the C-4 position of the fatty acid chain. To check the connection between the biological activity and these structural units in ipomoeassin F, we removed them individually to generate a minilibrary of four analogues 1–4 (Figure 1).
Figure 1.
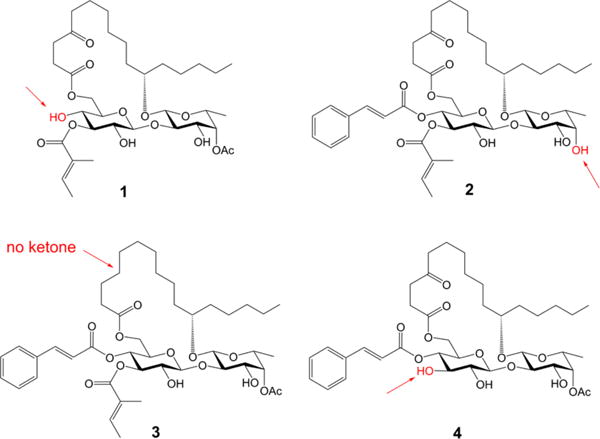
Ipomoeassin F analogues 1–4.
The syntheses of analogues 1, 2, and 3 were straightforward by adopting the same strategy of our earlier total synthesis of ipomoeassin F (starting from intermediates 28, 29, and 30, respectively; see the SI for details). In this route, we introduced the tigloyl group at a rather early stage. Since analogue 4 is missing a tigloyl moiety, we designed a new glucosyl donor to achieve its preparation (Scheme 1). Esterification of the diol 58 with levulinic acid (Lev) and then replacement of the anomeric protecting group p-methoxyphenyl (PMP) with C(NH)CCl3 provided glucosyl donor 7 in high yield (72% over three steps). Next, regioselective glycosylation of 7 with the fucoside acceptor 8 gave β-(1 → 2)-linked disaccharide 9 in 91% yield. Being aware that the final removal of Lev with hydrazine acetate will likely affect the ketone on the lipophilic tether, instead of carrying Lev in disaccharide 10 any further, we replaced them with the chloroacetyl (CA) groups that are orthogonal to the other protecting groups and can be easily removed by 1,4-diazabicyclo[2.2.2]octane (DABCO).9 The cleavage of the isopropylidene group using CSA smoothly afforded diol 13. To our surprise, the subsequent DCC-mediated acylation with acid 1410 did not give the desired ester 15 (22% yield) as the major product. In contrast, a significant amount of 4-O-Glcp acylated isomer 16 (32% yield) was afforded (more details are available in the SI). The diene 15 was then subjected to the RCM reaction using Hoveyda–Grubbs catalyst (II) (20 mol %), followed by hydrogenation over Pd/C, to give 17 in 70% yield over two steps. Esterification of 17 to give 18 went smoothly. Finally, as expected, the selective dechloroacetylation of 18 using DABCO, followed by the final TBS-cleavage with TBAF, delivered the analogue 4 in very good yield for the last two steps and in 4.3% overall yield from intermediates 5 and 8.
Scheme 1.
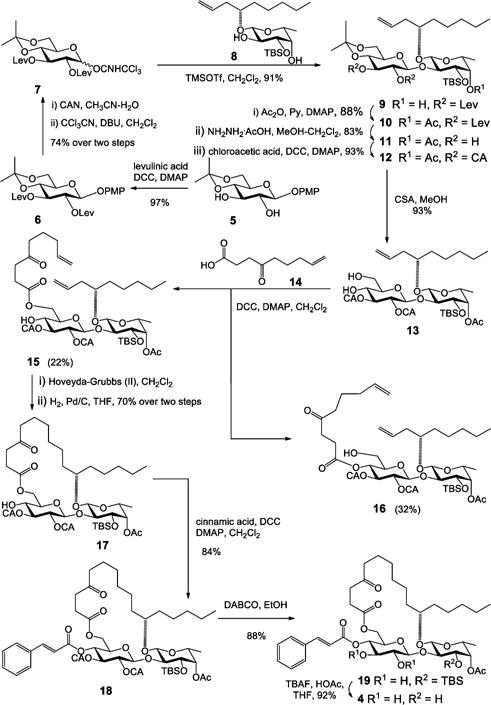
Synthesis of Analogue 4 of Ipomoeassin F
Analogues 1–4 were first screened against two breast cancer cell lines (MDA-MB-231 and MCF7), and their cytotoxicity data are shown in Table 2. Not surprisingly, the two unsaturated ester functional groups on the sugar ring, namely cinnamate and tiglate, are crucial for the cytotoxicity of ipomoeassin F. The cytotoxicity dropped dramatically for analogue 1 without cinnamate (200–1000-fold) and analogue 4 without tiglate (100–1000-fold). On the other hand, the analogue 2 without acetate almost retained the cytotoxicity against MCF7 (dropped 2-fold) but is much less potent than ipomoeassin F against MDA-MB-231 (dropped 20-fold). Interestingly, analogue 3 without the carbonyl group in the lipophilic tether almost retained the cytotoxicity against all five tested cancer cell lines compared to ipomoeassin F, suggesting that the carbonyl group is not as essential as the three esters for the cytotoxicity. Although the conclusion seems contradictory to the previous finding,5 the activity change (~3 fold) is quite similar. Experimentally, we have proven for the first time that the order of influence on the cytotoxicity of the ipomoeassins is cinnamoyl > tigloyl ≫ acetyl > ketone.
Table 2.
| MDA-MB-231 | MCF7 | HeLa | U937 | Jurkat | MCF-10A | NIH/3T3 | |
|---|---|---|---|---|---|---|---|
| ipomoeassin F | 6.5 | 43.7 | 16.4 | 5.4 | 6.1 | 5.4 | 16.8 |
| 1 | 7129 | 9090 | –c | – | – | – | – |
| 2 | 131 | 86.7 | 133 | 72.5 | 139 | 297 | 281 |
| 3 | 16.1 | 33.0 | 14.9 | 11.1 | 9.7 | 30.7 | 24.7 |
| 4 | 6848 | 5929 | – | – | – | – | – |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
MCF-10A and NIH/3T3 are two nontumor cell lines.
–, not tested.
The selectivity index (SI) of a compound is derived from its IC50 value against nontumor cells divided by its IC50 value against tumor cells. The higher the SI, the more selective the compound against tumor cells. Using MCF-10A cells as the reference nontumor cell line, the SI values of ipomoeassin F, its analogue 3, and paclitaxel are listed in Table 3. While paclitaxel showed good selectivity in general, unfortunately, ipomoeassin F did not exhibit any selectivity (SI < 1). Somewhat surprisingly, analogue 3 without the carbonyl group in the fatty acid tether showed better selectivity (ca. 2.3, 7.8, 6.2, 2.8, and 3.6 times, respectively) than ipomoeassin F for the five tested cancer cell lines over MCF-10A. This finding sends the encouraging message that the overall biological activity profile of ipomoeassin F may be improved through appropriate structural modifications.
Table 3.
Selectivity Index (SI) of Ipomoeassin F, Its Analogue 3, and Paclitaxel over MCF-10A Cellsa
| MDA-MB-231 | MCF7 | HeLa | U937 | Jurkat | |
|---|---|---|---|---|---|
| ipomoeassin F | 0.83 | 0.12 | 0.33 | 1 | 0.89 |
| 3 | 1.91 | 0.93 | 2.06 | 2.77 | 3.16 |
| paclitaxel | 8.04 | 3.73 | 25.6 | 15.2 | 14.6 |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
After confirming the great importance of both cinnamate and tiglate moieties (1 and 4 in Table 2), we turned our attention to the structural elements within them, namely the double bonds and the phenyl group. The double-bond reduced analogues 20 and 21 and the double-bond missing analogue 22 were designed (Figure 2). In addition, to investigate the role of the phenyl group within cinnamate, analogue 23 was also designed (Figure 2). Syntheses of these four analogues went well without difficulty by adopting the same strategy we developed earlier (for the synthetic details, see the SI).
Figure 2.
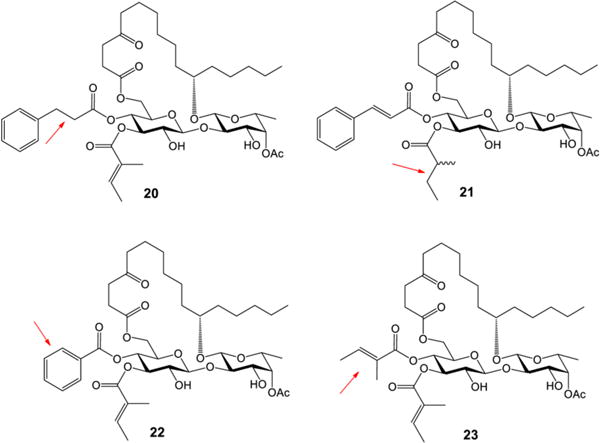
Ipomoeassin F analogues 20–23
The cytotoxicity data of the ipomoeassin F analogues 20–23 are shown in Table 4, providing clear evidence for the first time that the double bond within either cinnamate or tiglate is a subtle but critical parameter, variations of which would remarkably spoil cytotoxicity. To elaborate, the reduction or removal of the cinnamate double bond (analogue 20 or 22) caused a huge loss in cytotoxicity. On the other hand, the activity change caused by the reduction of the tiglate double bond (analogue 21) is a little less pronounced. Although 21 is not stereochemically pure, the data still lead to a conclusion that the cinnamate double bond is more crucial than the tiglate double bond. Furthermore, the replacement of cinnamate with tiglate made the cytotoxicity of analogue 23 drop even more dramatically. Further investigation is needed to clarify the role of the α-methyl group in tiglate; however, the data strongly suggest that the sole aromatic group, i.e., the phenyl group, is of great importance to the biological activity of ipomoeassin F likely through π–π stacking interaction. For the first time, we demonstrated that the phenyl group in cinnamate and the double bonds in both cinnamate and tiglate are far more important structural features than the acetate group at 4-C-Fucp (fucosylpyranoside).
Table 4.
Cell Cytotoxicity (IC50, nM) of Ipomoeassin F Analogues 20–23a
| MDA-MB-231 | MCF7 | |
|---|---|---|
| ipomoeassin F | 6.5 | 43.7 |
| 20 | 980 | 2560 |
| 21 | 429 | 477 |
| 22 | 930 | 2890 |
| 23 | 4500 | 3930 |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
Michael acceptor systems are present in many electrophilic natural products to form covalent adducts with biomacromolecules, which are responsible for the biological activities of those natural products.11 Because of the significant loss in the cytotoxicity of 20–22 compared to ipomoeassin F (Table 4), we hypothesized that α,β-unsaturated cinnamate and/or tiglate may enable irreversible binding between ipomoeassin F and its biological targets. Further experiments are being conducted to test this hypothesis and will be reported in due course.
In ipomoeassin F, two nearby free hydroxyl groups (3-OH-Fucp and 2-OH-Glcp) constitute the only hydrophilic region of the molecule, which may serve as an exit tunnel to the aqueous environment outside the binding pocket. Therefore, we speculated that one of those two hydroxyl groups, if not both, may be modified without a significant activity loss. To test this hypothesis and also in consideration of the great importance of overall lipophilicity, two additional analogues 24 and 25 (Figure 3) were designed. These two analogues were synthesized in one pot by treating ipomoeassin F with an excess amount of acetic anhydride (see the SI for synthetic details). Regioselectivity of acylation is the same as that reported in the literature.1 Whereas the fully protected analogue 25 was completely inactive (Table 5) presumably due to poor aqueous solubility, 24 with free 2-OH-Glcp remained significantly cytotoxic. In the future, the nature of a substituent at 3-OH-Fucp deserves further studies. Currently, we are developing new synthetic strategies to investigate how modifications of 2-OH-Glcp would affect biological activity.
Figure 3.
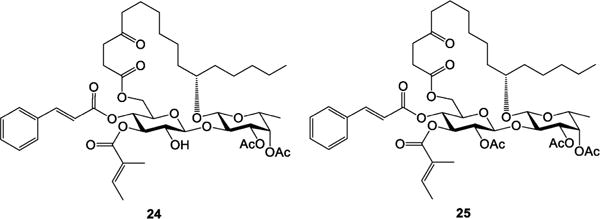
Ipomoeassin F analogues 24 and 25.
Table 5.
| MDA-MB-231 | MCF7 | HeLa | U937 | Jurkat | MCF-10A | NIH/3T3 | |
|---|---|---|---|---|---|---|---|
| ipomoeassin F | 6.5 | 43.7 | 16.4 | 5.4 | 6.1 | 5.4 | 16.8 |
| 24 | 16.5 | 216 | 138 | 79.8 | 82.6 | 24.0 | 55.9 |
| 25 | IAc | IA | –d | – | – | – | – |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
MCF-10A and NIH/3T3 are two nontumor cell lines.
IA means IC50 > 10 μM.
–, not tested.
Finally, it is a well-accepted assumption that the cyclic structural framework is essential for the biological activity of a macrocycle.12,13 However, a cyclization step usually limits the scale of production because of competition from intermolecular reactions, which made us very curious about the impact of the macrocyclic skeleton of ipomoeassin F on its inhibition activity against cancer cell growth. We reasoned that open-chain analogues may be folded in a binding pocket without much entropy penalty due to the great flexibility of both the fatty acid chain and the large ring system. Considering the most convenient preparation, we designed an open-chain analogue 26 (Figure 4), which could be obtained from a key intermediate (36, synthesized at gram scale in our previous total synthesis) over only two steps. Surprisingly, 26 retained good activities against tested cancer cell lines with even improved selectivity between tumor and nontumor cells (Table 6; selectivity index data are available in the SI; see Table S1). To check the role of the two terminal alkenes, we designed the alkene-reduced analogue 27. Biological assays showed increased activity for 27 up to 13-fold when compared to 26 (Table 6). In the future, it would be of great interest to know whether the open-chain analogues are a new chemotype or just mimics of the ipomoeassins.
Figure 4.
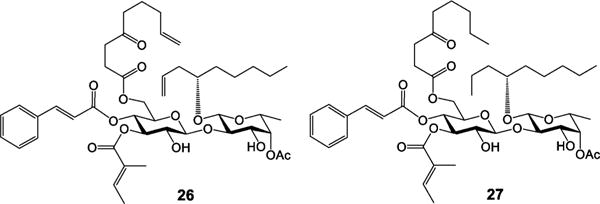
Ipomoeassin F analogues 26 and 27.
Table 6.
| MDA-MB-231 | MCF7 | HeLa | U937 | Jurkat | MCF-10A | NIH/3T3 | |
|---|---|---|---|---|---|---|---|
| ipomoeassin F | 6.5 | 43.7 | 16.4 | 5.4 | 6.1 | 5.4 | 16.8 |
| 26 | 59.1 | 124 | 87.2 | 27.4 | 242 | 151 | 137 |
| 27 | 18.8 | 78.2 | 30.0 | 17.1 | 22.5 | 11.8 | 20.7 |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
MCF-10A and NIH/3T3 are two nontumor cell lines.
In conclusion, deliberate digression from our newly developed synthesis path brought a set of analogues into reach that allowed the most systematic SAR studies of ipomoeassin F to date. The biological data clearly demonstrated the importance of different structural features in the rank of cinnamate > tiglate > phenyl > alkene in cinnamate > alkene in tiglate > acetate >3-OH-Fucp > cyclic skeleton ≈ ketone by greater than 3 orders of magnitude compared to ipomoeassin F. Together with the known information about the significant role of the fatty acid tail, our work lays a solid foundation for ensuing medicinal chemistry studies of the ipomoeassins. Furthermore, the critical role of two α,β-unsaturated esters, especially cinnamate, suggests that they may covalently anchor the natural product to its molecular targets, which will benefit our ongoing effort to identify cellular targets of the ipomoeassin family of glycoresins for drug development. In addition, the open-chain analogues of ipomoeassin F with potent cytotoxicity but enhanced selectivity and relatively easy production show great promise for future anticancer drug development. How to improve the therapeutic window between tumor and nontumor cells is a great challenge that needs to be tackled for future ipomoeassin research.
Supplementary Material
Acknowledgments
This work was primarily supported by startup funds from the University of Arkansas and also in part by Grant Nos. P30GM103450 and R15GM116032 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) and by seed money from the Arkansas Biosciences Institute (ABI). We thank Dr. Jianjun Zhang at CAU (China Agricultural University) for providing per-acetylated D-fucopyranose (NKT R&D Program of China, 2015BAK45B01). We also acknowledge Dr. Charles Kwangyul Moon in the synthetic core facility for supplying some building blocks.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b00555.
Experimental procedures and analytical data for all of the new compounds; cytotoxicity assays (PDF)
1H and 13C NMR spectra (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Cao S, Guza RC, Wisse JH, Miller JS, Evans R, Kingston DGI. J Nat Prod. 2005;68:487. doi: 10.1021/np049629w. [DOI] [PubMed] [Google Scholar]
- 2.Cao S, Norris A, Wisse JH, Miller JS, Evans R, Kingston DGI. Nat Prod Res. 2007;21:872. doi: 10.1080/14786410600929576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kingston DGI. J Org Chem. 2008;73:3975. doi: 10.1021/jo800239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Postema MHD, TenDyke K, Cutter J, Kuznetsov G, Xu Q. Org Lett. 2009;11:1417. doi: 10.1021/ol900086b. [DOI] [PubMed] [Google Scholar]
- 5.Nagano T, Pospisil J, Chollet G, Schulthoff S, Hickmann V, Moulin E, Herrmann J, Mueller R, Fürstner A. Chem – Eur J. 2009;15:9697. doi: 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]
- 6.Zong G, Barber E, Aljewari H, Zhou J, Hu Z, Du Y, Shi WQ. J Org Chem. 2015;80:9279. doi: 10.1021/acs.joc.5b01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereda-Miranda R, Rosas-Ramirez D, Castaneda-Gomez J. Prog Chem Org Nat Prod. 2010;92:77. doi: 10.1007/978-3-211-99661-4_2. [DOI] [PubMed] [Google Scholar]
- 8.Tamura JI, Neumann KW, Ogawa T. Liebigs Ann. 1996;1996:1239. [Google Scholar]
- 9.Lefeber DJ, Kamerling JP, Vliegenthart JFG. Org Lett. 2000;2:701. doi: 10.1021/ol005567z. [DOI] [PubMed] [Google Scholar]
- 10.Killen JC, Leonard J, Aggarwal VK. Synlett. 2010;2010:579. [Google Scholar]
- 11.Gersch M, Kreuzer J, Sieber SA. Nat Prod Rep. 2012;29:659. doi: 10.1039/c2np20012k. [DOI] [PubMed] [Google Scholar]
- 12.Driggers EM, Hale SP, Lee J, Terrett NK. Nat Rev Drug Discovery. 2008;7:608. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 13.Marsault E, Peterson ML. J Med Chem. 2011;54:1961. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.