

Abstract
Experiments were carried out to detect cysteine residues on human Keap1 protein that may be sensors of oxidative stress that gives rise to changes in the GSH/GSSG redox couple. Human Keap1 protein, at a final concentration of 6 μM, was incubated for two hours in aqueous buffer containing 0.010 M GSH, pH 8, in an argon atmosphere. Subsequently, excess iodoacetamide and trypsin were added to generate a peptide map effected by LCMS analysis. Peptides containing all 27 carboxamidomethylated cysteines were identified. Replacement of GSH by 0.010 M GSSG yielded a map in which 13 of the original carboxamidomethylated peptides were unperturbed, while other caboxamidomethylated cysteine-containing peptides were undetected, and a number of new cysteine-containing peptide peaks were observed. By mass analysis, and in some cases, by isolation, reduction, carboxamidomethylation, and reanalysis, these were identified as S-glutathionylated (Type 1) or Cys-Cys (Type 2) disulfides. Such peptides derived from the N-terminal, dimerization, central linker, Kelch repeat and C-terminal domains of Keap1. Experiments were carried out in which Keap1 was incubated similarly but in the presence of various GSH/GSSG ratios between 100 and 1 ([GSH + GSSG] = 0.010 M), with subsequent caraboxamidomethylation and trypsinolysis to determine differences in sensitivities of the different cysteines to the type 1 and type 2 modifications. Cysteines most sensitive to S-glutathionylation include Cys77, Cys297, Cys319, Cys368, and Cys434, while cysteine disulfides most readily formed are Cys23-Cys38 and Cys257-Cys297. The most reducing conditions at which these modifications are at GSH/GSSG = 10, which computes to an oxidation potential of Eh = −268.5 mV, a physiologically relevant value. Under somewhat more oxidizing, but still physiologically relevant, conditions, GSH/GSSG = 1 (Eh = −231.1 mV), a Cys319-Cys319 disulfide is detected far from the dimerization domain of the Keap1 homodimer. The potential impact on protein structure of the glutathionylation of Cys434 and Cys368, the two modified residues in the Kelch repeat domain, was analyzed by docking and energy minimizations of glutathione residues attached to the Kelch repeat domain, whose coordinates are known. The energy minimizations indicated marked alterations in structure with a substantial constriction of Neh2 binding domain of the Keap1 Kelch repeat domain. This alteration appears to be enforced by an extended hydrogen-bonding network between residues on the glutathione moiety attached to Cys434 and amino acid side chains that have been shown to be essential for repression of Nrf2 by Keap1. The modifications of Keap1 detected in the present study are discussed in the context of previous work of others who have examined the sensitivity of cysteines on Keap1 to electrophile assault.
Introduction
The protein Keap1 plays a central role in the compartmentalization and trafficking of Nrf2, a transcription factor that mediates upregulation of the expression of a large array of cytoprotective enzymes in response to electrophilic and oxidative assault (1–3). Keap1 is an ∼70 kD protein that is proposed to function as a homodimer (4–7), binds an equivalent of Zn2+ per subunit, and contains a number of functional domains, as indicated by the green linear schematic at the bottom of Figure 1 (5, 8–10). These domains include the BTB dimerization region, toward the N-terminus; the Kelch repeat domain that has been demonstrated to bind a peptide fragment from the Neh2 (Kelch binding) domain of Nrf2 (7) and is also an Actin binding domain; and the central linker region (CLR) that conjoins the former two domains. Keap1 facilitates ubiquitination of Nrf2, targeting it for proteasomal degradation by serving as an adapter protein for Cul3, an E3 ubiquitin ligase (11–13). The Cul3 binding domain of Keap1 spans adjacent regions of the BTB and CLR domains. Keap1 contains a nuclear export signal, originating in the C-terminal end of the CLR, that is hypothesized to assist in the escort of Nrf2 from the nucleus (14). Keap1 is proposed to tether Nrf2 to the Actin cytoskeleton at the Kelch repeat domain thereby preventing its nuclear translocation (15, 16).
Figure 1.
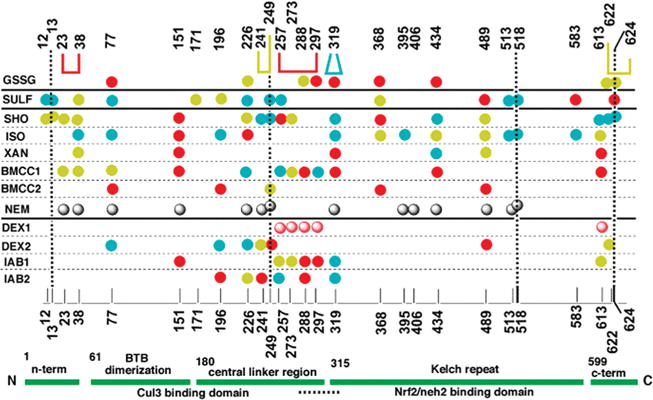
Domain schematic and map of the most reactive cysteine residues of human and mouse Keap1 protein toward electrophiles. The structure and domains of Keap1 are indicated by the labeled green ribbon at the bottom of the figure, numbers indicate the amino acid residue at which the top domains nominally begin, and the horizontal dashed line below indicates the nuclear export sequence. Numbers of the cysteine residues are indicated at the bottom and top of the (main) central portion, and the vertical dashed lines are for clarity. Electrophiles that have been studied previously and the oxidant described in the present work are listed in the left column. Abbreviated name, chemical name and reference: GSSG, glutathione disulfide, present work; SULF, sulforaphane (R-1-isothiocyanato-4-methylsulfinylbutane), ref 20; SHO, ISO, XAN, and BMCC1, 10-shogaol, isoliquiritigenin, xanthohumol, and 1-biotinamido-4-(4′-[maleimidiethylcyclohexane]-carboxamido)butane, respectively, ref 22; BMCC2, 1-biotinamido-4-(4′-[maleimidiethylcyclohexane]-carboxamido)butane, ref 17; NEM, N-ethylmaleimide, ref 18; DEX1, dexamethasonemesylate, ref 18, DEX2, dexamethasonemesylate, ref 19; IAB1, N-iodoacetyl-N-biotinylhexylenediamine, ref 17; IAB2, N-iodoacetyl-N-biotinylhexylenediamine, ref 21. Solid circles indicate cysteine residues of human Keap1 that react with the electrophile in the same row, and the shaded circles are for experiments with mouse Keap1. Descending order of reactivity, based on detection at various (except for GSSG, see text) concentrations, is indicated as red > azure > canary > no symbol at the given position (see Supporting Information). Gray symbols indicate detectable reactivity. For GSSG, solid circles represent type 1 disulfides, while connectors indicate type 2 disulfides formed from the connected residues. The bold horizontal lines separate the electrophiles/oxidant into chemical classes (see Discussion).
Human Keap1 contains 27 cysteines (17), some of which are purported to be the targets of electrophiles and oxidants that, when modified, facilitate the derepression of Nrf2 and give rise to enzyme induction; thus, there has been considerable interest in identifying the subset of cysteines that might be the key sensors of electrophilic/oxidative assault. Data concerning the reactivity of the various cysteines of human and mouse Keap1 toward a variety of electrophiles is summarized in Figure 1 (17–22). The left-most column contains the identities of various electrophiles. Above the green schematic of the domains of Keap1 in Figure 1 are the sequence numbers of the various cysteines of human Keap1, and the numbering is repeated, for clarity, at the top of the figure. Along the rows corresponding to each electrophile entered in the leftmost column are indicated, by means of a circle, the cysteine residues that were observed to react with the electrophile in a given experiment. Where possible (detailed in Supporting Information), the relative reactivity found in a given experiment is indicated by color differences, with red, azure, and canary signifying the descending order of reactivity. Except for the reaction of glutathione disulfide (top row), the subject of the present article, all of the data in Figure 1 have been previously published.
The previously published data included in Figure 1 illustrate that no single cysteine or group of cysteines exhibits exalted reactivity toward all of the electrophiles. The germinal work involving the reaction of mouse Keap1 and the electrophile dexamethasone mesylate (DEX1, Figure 1) reported five most reactive cysteines, four grouped in the CLR (18). These latter four were purported to engage in disulfide formation upon reaction with 2,2′- and 4,4′-bispyridyldisulfides, though direct evidence of this was not obtained. The conclusion was based on the implicit assumption that the reactivity order of Keap1 cysteines in the reaction of DEX1 is identical to that in the reaction of other electrophiles or oxidants; an assumption that is contradicted by the facts summarized in Figure 1. Further, the results described in the study implicating the most reactive cysteines in disulfide formation are difficult to reconcile with the experimental protocols (18). To wit, three sequential additions of one equivalent of disulfide were described to give three reactions in “excellent” agreement with pseudofirst order kinetics, and the derived rate constants varied by less than a factor of 3. How equimolar constituents in a bimolecular reaction could give rise to pseudofirst order kinetics was not rationalized. Further, simple inspection of the data (Figure 6 in ref 18) indicates that the initial two reactions, at least, are of mixed kinetic order. Additionally, it is physically impossible to separately titrate three reactive cysteines whose reactivities differ by less than a factor of 3. Finally, the claim that reactions at these CLR cysteines lead to dissociation of Nrf2 (18) appears to be in error (17). There is other evidence, discussed later, that implicates some of the CLR cysteines, as well as others outside the CLR, as important residues in the maintenance of Keap1 repression of Nrf2. Beyond this, the data summarized in Figure 1 indicate that many powerful cytoprotective enzyme inducers target cysteine residues outside the CLR (17, 20, 22); indeed others have demonstrated that DEX itself, targets a distinctly different set of cysteines of Human Keap1 (DEX2, third row from the bottom of Figure 1) (21). Thus, there appears to be little basis for the original model and more recent ones (6, 15, 23, 24), which focus on the original four most reactive CLR cysteines as principal or exclusive mediators of electrophile/oxidative stress. Others have reached similar conclusions (19–22, 25).
Among all of the cysteines in human Keap1, Cys151 is most consistently detected as being of high reactivity. In one case, the failure to detect its reactivity, IAB1 Figure 1 (21), has been demonstrated to have been artifactual, IAB2 Figure 1 (25). Cys151 has been found to be required for Nrf2 activation by a number of inducers (26–28), including tBHQ in mice (29), and recently, it has been elegantly demonstrated in HEK293 cells that IAB targets Cys151, among others, initiating the dissociation of Keap1 from Cul3 resulting in derepression of Nrf2 (30).
As indicated, Keap1 appears to mediate the upregulation of cytoprotective enzymes in response to oxidants, as well as electrophiles, but there has been no attempt to determine which, if any, of the cysteines are modified by such physiologically relevant agents or cellular oxidants derived therefrom; this is the subject of the present article. Oxidative stress is known to at least temporally alter the cellular redox couple defined by glutathione (GSH) and its disulfide (GSSG) (31–41). Increased relative concentrations of GSSG can result in the glutathionylation of cellular proteins, as in eq 1, and the closure of protein disulfide bonds, as in eq 2. Schafer and Buettner termed the product of the former reaction a Type 1 disulfide and the product of the latter reaction a Type 2 disulfide (31). Indeed, a recent
| (1) |
| (2) |
report deduced that Keap1 undergoes type 1 disulfide formation in cells treated with the redox cycling agent tert-butylhydroquinone (33). We report here experiments designed to detect type 1 and type 2 disulfides in Keap1 challenged with different oxidation potentials, by varying GSH/GSSG ratios that span the physiologically relevant range.
Experimental Procedures
Protein Sample
Keap1 was prepared as described previously (17). A 100 μM stock was prepared in 25 mM Tris-HCl buffer (pH 8.0) containing 2 mM Tris-HCl 2-carboxyethyl phosphine hydrochloride (TCEP) and 20% glycerol.
Redox Reactions
Except where noted, all manipulations below were carried out in argon bags, at 21 ±s 2 or 4 °C, that were frequently vented and flushed with fresh argon. All stock solutions were vigorously bubbled with argon for 10 min prior to being transferred to the argon bags.
At 4 °C, 3 μL of the 100 μM stock solution of Keap1 were suspended in 50 μL of 0.025 M Tris-HCl buffer, pH 8.0, resulting in a final concentration of 6 μM Keap1. This solution was transferred to a YM-10 Microcon (Millipore) nitrocellulose membrane centrifugation filter, and the sample was centrifuged at 12,000 × g for 5 min, after which ∼25 μL of buffered protein solution remained above the membrane. Spinning the solution to dryness was avoided to obviate peptide loss (25). This solution was then diluted with 400 μL of 0.025 M Tris-HCl, pH 8.0, and again, the sample was centrifuged at 12,000 × g for ∼30 min, leaving 25 μL of buffered protein solution above the membrane. The protein was then eluted with an additional ∼25 μL of Tris, pH 8.0, resulting in a final volume of 50 μL (2 μL. The sample was then moved to an argon purged glovebag at room temperature. Here, 0.01 M total glutathione was added to the protein. In successive experiments, the GSH/GSSG ratios were altered in the following series: 100% GSH, 100% GSSG, and GSH/GSSG) 100, 50, 10, 5, and 1. The reactions were allowed to equilibrate for 2 h. The sample was then moved to the argon bag at 4 °C, and the glutathione was washed from the solution with the Microcon centrifugation filter as stated above. After removing the glutathione, the protein was eluted from the filter with 50 μL of 25 mM Tris-HCl buffer (pH 8), and iodoacetamide, at a final concentration of 20 mM, and 25 μg of trypsin were added. This reaction was transported back to the room temperature argon bag. After a 2 h digestion, the reaction mixture was acidified with a dilute stock solution of formic acid to a final concentration of 0.1% formic acid by volume and analyzed by LC/MS.
LC/MS Conditions
Peptides were analyzed on an Agilent 1100 series high-performance liquid chromatography (HPLC) system coupled with a Bruker Daltonics Esquire 3000plus ion-trap mass spectrometer. Separations were performed using a Phenomenex Jupiter reverse phase C18 column (5 μm, 2.0 × 150 mm, 300 Å). The mobile phase consisted of water and acetonitrile containing 0.1% formic acid at a flow rate of 200 μL/min. The first 2 min of the run were isocratic 97:3 (water/acetonitrile). A linear gradient was applied over the next 73 min increasing the percent of acetonitrile to 42%. Over the next 8 min, the acetonitrile increased to 95%, where it remained for an additional 20 min. The eluate was analyzed by electrospray ionization in positive ion mode. Selected precursor ions were isolated in the gas phase and submitted to collision-induced dissociation (CID) for sequence characterization.
Molecular Docking
Docking of GSH onto Keap1 was completed using AutoDock version 4.0.1 (42). This program allows the identification of possible binding sites for small, flexible ligands on a fixed macromolecular target by using algorithms designed to perform docking of the former onto affinity grid maps defined on the surface of the latter. In the present case, the receptor molecule consisted of the structure of the Kelch repeat domain of Keap 1, which was obtained from the corresponding high-resolution coordinates deposited in the Protein Data Bank (PDB ID: 1u6d) (43). Before any docking calculation was initiated, the auxiliary program AutoDockTools (ADT) was employed to setup the docking experiments. Imput charges were preserved on the receptor Kelch repeat domain, and ADT was used to merge nonpolar hydrogens and add salvation parameters. The ligand GSH was treated as a separate entity and was constructed using Pymol (http://pymol.sourceforge.net/). ADT was used to merge nonpolar hydrogens and add Gasteiger charges to GSH. With the exception of amide bonds, all other bonds were defined as rotatable during docking, which resulted in a total of 13 active torsion angles in the GSH molecule. In contrast, the receptor coordinates were kept fixed throughout the process. The receptor surface sampled by the docking algorithm was based on the proximity to Cys434, a residue with demonstrated susceptibility to formation of a type 1 disulfide (vide infra). The AutoGrid program included in the AutoDock suite was employed to prepare a grid map containing 58 × 52 × 64 grid points, which was centered on Cys434 and whose diagonal length exceeded that of a fully extended GSH molecule by more than a factor of 4. One corner and parts of three edges of the cube were in close contact with the protein surface that binds the Neh2 domain of Nrf2. The remaining, larger, portion of the cube projected into space, away from the Kelch repeat domain. A default value of 0.375 Å was selected for grid spacing. The Lamarckian genetic algorithm was employed for the actual docking calculations, which were completed according to the following parameters: 150 individuals as initial population; 27,000 generations corresponding to 2,500,000 energy evaluations; 0.02 mutation rate; 0.8 crossover rate. Each docking experiment was repeated 50 times, and the solutions were ranked according to the respective binding energy.
Energy Minimization. Type 1 Disulfide at Cys434
The coordinates of the lowest energy Kelch repeat domain-GSH complex identified by the docking calculations were submitted to CNS version 1.1 for further model refinement (44). First, an appropriate input file was generated by enforcing a disulfide bond between GSH and Cys434 (vide infra) and by applying a patch that made the atypical gamma-glutamyl linkage present in GSH compatible with CNS. Subsequently, conjugate-gradient energy minimization was performed using the model_minimize.inp module with no experimental energy terms. In particular, 1,000 cycles of energy minimization with 200 minimization steps were completed using a 13 Å nonbonded cutoff and implicit solvation.
Type 1 Disulfides at Cys434 and Cys368
Starting with the original PDB coordinates, input files were generated, which enforced disulfide bonds between GSH and Cys434 and between GSH and Cys368. The starting conformation of the type 1 disulfide at Cys434 was identical to that in the preceding section, while that for the type 1 disulfide at Cys368 was chosen with GSH parallel to the inside of the barrel structure with the gamma-glutamate residue projecting away from the Neh2 binding domain. The resulting structure was subjected to conjugate-gradient minimization as indicated.
Results
100% GSH Reactions
After incubation in an argon tent for 2 h in the presence of 0.010 M GSH and subsequent addition of iodoacetamide and trypsinolysis, peptides were separated and analyzed by LC/MS. As has been reported under other reducing conditions (17), tryptic peptides containing all 27 carboxamidomethylated cysteines of human Keap1 could be identified in each of three replicate experiments. Peptide identities were confirmed by mass mapping and sequencing in the ion trap mass spectrometer. For example, Figure 2 shows a typical electrospray ionization mass spectrum obtained from a representative chromatographic peak (panel A) and the corresponding product ion spectrum provided by collision-induced dissociation (panel B). Singly and doubly charged ions were detected for a species with a 1401.5 Da molecular mass, which corresponds to the carboamidomethylated peptide LNSAECYYPER (1401.5 Da monoisotopic mass calculated from the sequence). As expected from ion trap experiments, gas-phase activation of the doubly charged precursor ion provided mainly y-type fragments that confirmed the identity of the carboxamidomethylated peptide.
Figure 2.
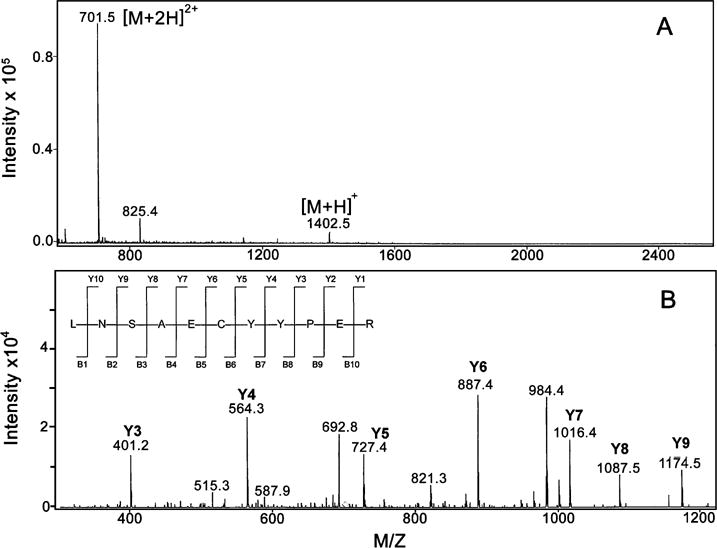
Identification of a human Keap1 peptide by LC-MS/MS analysis. Panel A depicts the [M + H]+ and [M + 2H]2+ ions for a carboxyamidomethylated peptide from a Keap1 tryptic digest. The observed monoisotopic mass matches well with the theoretical mass calculated from the sequence (1401.51 Da). Panel B is the product ion spectrum obtained by gas-phase activation of of the [M + 2H]2+ precursor observed above. The peptide sequence with theoretical fragmentation ions are illustrated in the upper left corner of panel B.
100% GSSG Reactions
In a similar fashion, a tryptic map was obtained from human Keap1 equilibrated for 2 h with 0.010 M GSSG in an argon tent, followed by the addition of iodoacetamide and trypsinolysis. LC/MS analysis yielded the chromatogram depicted in Figure 3. As indicated by solid arrows in Figure 3, 13 of the 27 cysteines were unperturbed by the presence of GSSG, as demonstrated by the fact that the corresponding peptides were efficiently carboxamidomethylated and exhibited retention times identical to those observed after preincubation with 100% GSH. In contrast, a number of other cysteine-containing carboxamidomethylated peptides were not observed, including those comprising Cys257, Cys297, Cys319, and two peptides containing two cysteines, Cys23/38 and Cys241/249. In addition, a larger number of new peptides were observed, as indicated by the dashed arrows in Figure 3. These were identified by mass mapping and sequencing as either S-glutathionylated peptides (Type 1 disulfides) or peptide cysteine (Type 2) disulfides. A peptide containing both Cys 23 and Cys 38 was identified as a new peak with a 2542.3 Da molecular mass, consistent with the presence of an intrapeptide disulfide bond. Its identity was confirmed by the detection of a species with 2658.3 Da molecular mass obtained after isolation, reduction with NaBH4, neutralization, and labeling with iodoacetamide. The peptide containing Cys297 was found to be involved in both type 1 and type 2 disulfides, in the latter case conjugated with the peptide containing Cys257. Similarly, Cys319 was found as both a type 1 and type 2 disulfide, in the latter case with the identical peptide containing Cys319, indicating an intersubunit cross-link between Keap1 monomers. New peptides containing type 1 disulfides were detected for cysteine residues 77, 226, 288, 368, and 434, and the peptide containing both Cys622 and Cys 624 (with a single one of the cysteines modified), although the corresponding carboxamidomethylated peptides were also detectable in all these cases. A type 2 disulfide involving Cys622 and Cys624 was also detected and confirmed by reisolation, reduction, and carboxamidomethylation, as above. These modifications are summarized in the first column of Table 1.
Figure 3.
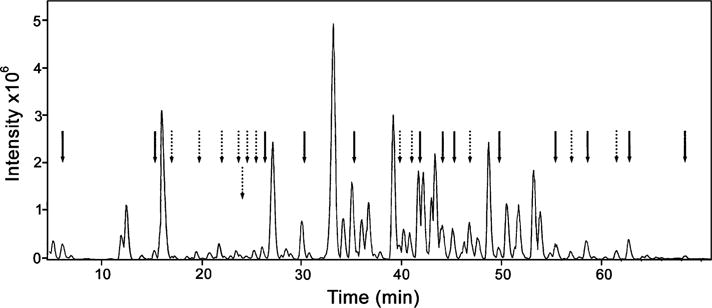
LCMS chromatogram of a tryptic digest of human Keap1. The protein was incubated with 10 mM GSSG, followed by the addition of iodoacetamide and trypsin (see Experimental Procedures). Carboxyamidomethylated cysteine residues are indicated by a solid arrow. Type 1 and type 2 disulfides are indicated by dotted arrows.
Table 1.
| cysteine modifications observed at GSH/GSSG < 0.003 | GSH/GSSG ratio at which the disulfide is first observed |
|---|---|
| Type 1 Disulfides | |
| Cys77a | 5 |
| Cys226a | <0.003 |
| Cys288a | <0.003 |
| Cys297 | 10 |
| Cys319 | 10 |
| Cys368a | 10 |
| Cys434a | 10 |
| Cys622/624a,b | <0.003 |
| Type 2 Disulfides | |
| Cys23-Cys38 | 10 |
| Cys241-Cys249 | <0.003 |
| Cys257-Cys297 | 10 |
| Cys319-Cys319 | 1 |
| Cys622-Cys624a | <0.003 |
Carboxamidomethylated cysteine was also detected at GSH/GSSG < 0.003.
Tryptic peptide contains both cysteines, one was carboxamidomethylated, and the other was glutathionylated, the identities of which, if either, cysteine was uniquely modified was undetermined.
Redox Titration
In order to determine whether the modifications observed upon exposure to GSSG could be anticipated at more moderate oxidation potentials, Keap1 was incubated, in an argon atmosphere, in solutions containing various ratios of GSH/GSSG (100, 50, 10, 5, and 1), at a constant total concentration of glutathione (0.010 M). Each sample was treated with iodoacetamide and trypsin before carrying out LC/MS analysis. In this case, the fate of specific residues of interest was monitored by plotting the current recorded for the corresponding ions as a function of retention time, producing an extracted ion chromatogram (EIC). Representative data in Figure 4 show the EIC’s of ions corresponding to tryptic peptides containing Cys257 and Cys297 and their disulfide products generated by GSH/GSSG treatment. Figure 4A shows that only two species were detected from the sample treated with a GSH/GSSG ratio of 100, consisting of the carboxamidomethylated peptides containing Cys257 and Cys297 (∼10 and ∼25 min retention time, respectively). As the GSH/GSSG ratio was decreased to 10 (Figure 4B), the peak corresponding to the product with a disulfide bond bridging the two peptides was observed with ∼23.5 min retention time. This product increased further by decreasing the GSH/GSSG ratio to 1 (Figure 4C) and became the sole detectable species at GSH/GSSG < 0.003 (Figure 4D). As a control, the original peptides were regenerated as separate species by isolating the fraction with ∼23.5 min retention time, reducing with NaBH4, treating with iodoacetamide, and resubmitting the products to the usual analytical protocol (Figure 4E). The same process was completed for all the type 2 disulfides. The highest GSH/GSSG ratio at which each of the disulfides was detectable is recorded in the second column of Table 1.
Figure 4.
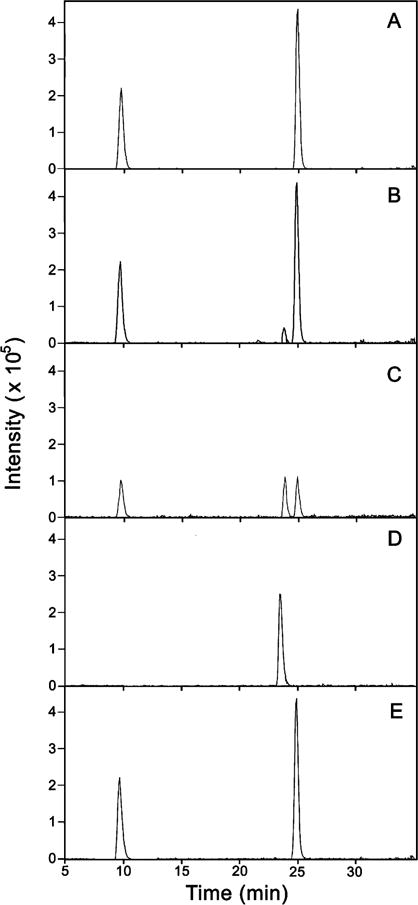
Redox titration of Cys257 and Cys297 of human Keap1. The protein was incubated with varying ratios of GSH/GSSG, followed by the addition of iodoacetamide and trypsin (see Experimental Procedures). The reaction mixture was analyzed by LCMS. Each panel is an extracted ion chromatogram containing the following ions: m/z = 839.9, 870.4, and 925.5. Panel A is the protein in a GSH/GSSG = 100 redox buffer, panel B is GSH/GSSG = 100 and GSH/GSSG = 10, panel C is GSH/GSSG = 1, and panel D is Keap1 in “100% GSSG”. Panel E is the isolation, reduction by NaBH4, and carboxyamidomethylation of the peak seen in panel D.
Molecular Docking
It was of interest to understand the potential impact on the known structure of the Kelch repeat domain of the type 1 disulfide formation observed at Cys 434 and Cys368, which reside in the Kelch repeat domain. Residue Cys368 is inside the inner barrel of the propeller of the Kelch domain, whereas Cys434 lies on the propeller face. To obtain an initial structure with which to begin minimization with Cys434, docking calculations of GSH to the face of the propeller were undertaken. The results of 50 independent docking calculations between GSH and Keap1’s Kelch domain were inspected using the ADT module of AutoDock version 4.0.1 (42). Despite the large volume of the cube of space sampled by GSH (see Experimental Procedures), all 50 of the lowest energy structures placed GSH in close proximity, within hydrogen bonding distance, of residues on the surface of the Kelch repeat domain (data not shown). Indeed, the lowest energy configuration placed the S atom of GSH within 4 Å of the S atom of Cys434, with remaining parts of the GSH in close proximity to the edges of the Kelch repeat domain. The fact that the all of the 50 lowest energy structures placed GSH near the protein validated initiating an energy minimization study of the glutathionylated Cys434 with the glutathionyl moiety proximate to the protein.
Energy Minimization
The lowest energy configuration, alluded to above, was employed for further refinement in CNS by performing conjugate-gradient minimization with no experimental energy terms (see Experimental Procedures). As a control, the original protein coordinates were also submitted to conjugate-gradient minimization to evaluate the effects of this procedure on the crystal structure in the absence of modification by GSH. The results of this control are depicted in Figure 5A. Overlaying the coordinates before and after minimization indicated close agreement and only minor variations of the crystal structure due to minimization (not shown). In contrast, minimization of the type 1 disulfide involving Cys434 engendered substantial alteration of structure, depicted in Figure 5B. The alteration involves constriction of the Neh2 binding domain, apparently enforced by a hydrogen bonding network: the amino group of the Glu residue of GSH is within hydrogen bonding distance of –OH group of T525; the γ-glutamyl amide hydrogen is in similar proximity to one of the imidazole ring nitrogen atoms of His436; the carboxylate of the Gly residue of GSH makes hydrogen bond contact with both the guanidino groups of Arg380 and Arg415, while Tyr334 contacts both the guanidino group of Arg380 and the carbonyl oxygen of Asn382 (see Supporting Information). Docking and minimization of GSH to a second Cys, Cys368, suggests the possibility of a somewhat further contracted Neh2 binding domain, with a similar hydrogen bonding network, as depicted in Figure 5C.
Figure 5.

(Panel A) Structures of the Kelch repeat domain after energy minimization. Minimized structure of unmodified Keap1 starting from the coordinates of the crystal structure in the PDB. (Panel B) Minimized structure containing a type 1 disulfide at Cys434, starting from the lowest energy structure of Keap1 docked to noncovalently bound GSH, as described in Experimental Procedures. (Panel C) Minimized structure containing type 1 disulfides at Cys434 and Cys368. In orange are Cys434 and Cys368, the GSH fragment (B and C) is in red, green residues are those that have been shown to contact a peptide fragment of the Neh2 domain of Nrf2 in Keap1-peptide cocrystals, blue is His436, and in mauve are Gly364 and Gly430 (see Discussion).
Discussion
Protein Modifications
Even under relatively highly oxidizing, nonphysiological, conditions nearly half of the cysteines on Keap1 are unreactive to a large excess of GSSG; thus, these cysteines are unlikely candidates as the ultimate barometers of, or ultimate switches for detection of, oxidative stress that gives rise to changes in the GSH/GSSG ratio. Experiments in which 6 μM protein was incubated with 0.010 M GSSG involve a 60-fold molar excess of oxidant over each cysteine of human Keap1. The stock solution of GSSG was assayed for contaminating GSH, and it was determined that the final concentration of GSH in the reaction solution was less than 0.00003 M, giving an upper limit of GSH/GSSG < 0.003. This equates, by use of eq 3, to an oxidation potential of Eh > −93.4 mV. In healthy dividing and confluent mammalian cultured cells, typical values of Eh range from −280 mV < Eh < −180 mV, while those stressed to the point of apoptosis or necrosis have values of Eh > −170 (31, 34, 38, 41).
| (3) |
Inaccessibility of any of the cysteines is an unlikely explanation for unreactivity. Inspection of Figure 1, all rows but the top, shows that all cysteines except Cys171 and Cys406 are reactive with more than one electrophile, some of them of considerable steric bulk. In addition, it has been demonstrated that disulfide oxidant, 2,2′-bis-dithiopyridine, a more powerful oxidant than GSSG, reacts with nearly all of the 25 cysteine residues in murine Keap1 (18). Thus, failure to detect the formation of either type 1 or type 2 disulfide formation from candidate cysteines under these highly oxidizing conditions, while the carboxamidomethylated peptides are still detected, makes it unlikely that these cysteines act as oxidation sinks that could serve as ultimate redox switches.
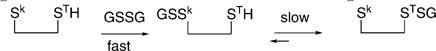
Employment of the caveat ultimate on the detection of prospective switches is based on the following considerations. The incubation time of 2 h is sufficient to obtain equilibrium, on the basis of rate constants known for reactions of GSSG and related bis-disulfides to form mixed disulfides (45–50). However, the absence of a detectable type 1 or type 2 disulfide product at a given cysteine does not rule out its involvement in some redox sensitive dithiol–disulfide switch; it simply suggests that the particular cysteine is not a thermodynamic, or ultimate, product. A situation in which a kinetically most highly reactive cysteine is in fact not a thermodynamic sink can be readily conceived, for either type 1 or type 2 disulfides, as in eqs 4 and 5, respectively. The superscripts k and T in eqs 4 and 5 denote
 |
(4) |
| (5) |
kinetic and thermodynamic, respectively, target cysteines, employing a distinction in the common parlance of organic chemistry. In the situations depicted in eqs 4 and 5, the kinetically most reactive cysteines, labeled Sk, would ultimately be detected as carboxamidomethylated cysteines under the experimental conditions employed, and thus, their involvement in the redox shuttles of eqs 4 and 5 would not be detected.
On the basis of further experiments, a subset of the cysteine modifications observed at nonphysiological ratios of GSH/GSSG were observed to occur at more physiological conditions and were thus identified as candidate cysteines that may be sensors of oxidative stress. The ratio of GSH/GSSG measured in normal cultured cells is typically reported as being on the order of 100–200, but under varying conditions of oxidative stress, it has been measured to decrease to levels approaching GSH/GSSG) 1 (31, 34, 36–38, 41). Experiments carried out here at varying GSH/GSSG) 100, 10, 5, and 1 equate to oxidation potentials of Eh = −298.8, −268.5, −258.3, and −231.1 mV, respectively, overlapping the physiological ranges alluded to earlier (−280 to −180 mV). The second column of Table 1 indicates the highest ratio of GSH/GSSG at which the various disulfides were observed. From inspection of Table 1, it can be seen that type 1 disulfides at Cys77, Cys297, Cys319, Cys368, and Cys434, and type 2 disulfides involving Cys23-Cys38 and Cys257-Cys297 form at relatively moderate conditions of oxidative stress. The intersubunit type 2 disulfide Cys319-Cys319 forms under somewhat more oxidizing conditions. Given that the other modifications observed (Table 1) only appear at the lowest level of GSH/GSSG < 0.003, equal to an oxidation potential of −93.4 mV, these are unlikely candidates for sensors of physiological oxidative stress. Of note, formation of type 1 disulfides at Cys77, Cys368, and Cys434 was observed to commence at modest levels of GSH/GSSG, second column of Table 1, but the modification was incomplete even when the GSH/GSSG ratio was reduced by a factor of >103, (see column 1 of Table 1, footnote a). Perhaps this is due to conformational changes of the dimer that are imbued by some oxidation reactions, at these or other residues, that subsequently limit accessibility or alter redox potential of the remaining unreacted cysteine residues at these positions.
This is the first report of Keap1 cysteine modification by a physiologically relevant oxidant. It seems likely that such type 1 modifications occur in vivo with oxidative stress in which the GSH/GSSG redox couple is altered because glutathionylation of proteins in general is elevated under such conditions (37, 39, 40, 51). There is a suggestion, but as yet no direct detection, of glutathionylation of Keap1 in vivo (33). There is no report that Keap1 undergoes cysteine disulfide closure in response to oxidative stress, but it seems likely on the basis of the present work. As indicated above, the oxidation potentials in these experiments are well within the physiological range; indeed, it is possible that some of the type 2 disulfides identified are normally intact under conditions of little or no oxidative stress.
The present work has not detected Cys273 and Cys288 as involved in type 2 disulfide switches, though these have been suggested to be the key oxidant sensors that release the Zn2+ ion in the process of forming a type 2 disulfide (5, 6). It cannot be ruled out from the present work that they in fact form a disulfide to some extent, but this may have escaped detection (vide supra). However, it should be recognized that all evidence that Cys273 and Cys288 are prime targets that react with oxidants/electrophiles, release a Zn2+ ion, form a disulfide, and release Nrf2 is indirect. Indeed, a mutant Keap1 in which four CLR cysteines, Cys257, Cys273, Cys288, and Cys297, are replaced by alanine residues is kinetically only ∼50% less reactive with electrophiles and has only a ∼50% reduced affinity for Neh2. Covalent modification of these cysteines was later shown not to reduce the affinity of Keap1 for Neh2 (17). In addition, the double mutant C273A/C288A retains a binding affinity for Zn2+ that is just 1.6 kcal/mol lower than the binding affinity of native Keap1 of 15.1 kcal/mol! Despite this, there is evidence that Cys273 and Cys288 are critical in Keap1 repression of Nrf2. Homodimeric mutant Keap1 proteins harboring either C273A or C288A mutations fail to repress Nrf2, while heterodimeric mutant Keap1 formed from mixing mutant monomers C273A and C288A shows significant recovery of the ability to repress Nrf2 mediated expression in an extranuclear reporter gene assay (6). The Keap1 mutants C273S and C288S fail to repress Nrf2 and fail to upregulate cytoprotective enzymes on electrophile assault, and fail to both mediate ubiquitination of Nrf2 and repress its transcription (26, 52).
Notably, the present study did not detect Cys151 as the site of type 1 or type 2 disulfide formation, though there may be a number of reasons for this. Cys151 has been demonstrated to be a sensitive target for a range of electrophiles and a few oxidants (see Introduction). As alluded to earlier, it is possible that Cys151 in fact is a site of disulfide formation but is not a thermodynamically stable site, instead passing the oxidizing equivalents to a proximal cysteine residue. It is also possible that Cys151 remains a sentinel for other types of assault during flux of the GSH/GSSG redox couple. In this regard, it is interesting that it has been recently shown that arsenite activation of Nrf2 is not dependent on Cys151 (53).
Biological Correlates
Information relevant to some of the cysteines that have been here determined to engage in type 1 and type 2 disulfides is of interest regarding the possible biological significance of these modifications. The type 2 disulfide formed by Cys23 and Cys38 is remarkable in that cysteine residues in the N-terminal domain of Keap1 are significantly targeted only by sulforaphane among all the electrophiles examined to date (Figure 1). Interestingly though, a C23Y Keap1 mutant has recently been identified in breast cancer cells (54). The mutant binds Nrf2 but fails to mediate its ubiquitination and therefore does not fully repress Nrf2-stimulated transcription. Thus, formation of a type 2 disulfide involving Cys23 could indeed alter Keap1 repression of Nrf2. The Cys257/Cys297 disulfide is in the CRL, a region widely believed to communicate covalent modification to the adjacent Kelch repeat domain that is the main binding site for Nrf2. It should be noted though that it has been shown that multiply mutated Keap1 containing C257A and C297A mutations successfully represses Nrf2 in a reporter assay containing an extra nuclear construct (6). In addition, C257S and C297S mutants also repress Nrf2 and derepress Nrf2 in response to lipid oxidation products in a similar assay (52). In the latter case though, the inducing agents presumably act as Michael acceptors not oxidants. Further, the Cys257/Cys297 cross-link, if intramolecular, traverses the nuclear export sequence (horizontal dashed line on the Keap1 domain schematic in Figure 1) (14), and the consequence of this to normal Keap1 function, as opposed to function in an extranuclear reporter assay, is unknown.
Two of the observed modifications potentially affect Keap1 dimerization. First, type 1 disulfide formation at Cys77 lies in the BTB dimerization domain (green Keap1 schematic, Figure 1) and could potentially disrupt either dimerization or dimer geometry that affects Cul3 and/or Nrf2 interactions. Site-directed mutagenesis of three residues in this region, both individually and in combination, led to loss of Nrf2 repression in some cases but in no case to inability to dimerize (7). Second, the curious observation here of the type 2 disulfide Cys319/Cys319, far from the dimerization domain, suggests the possibility that oxidation can in effect temporarily lock in substantial conformational alterations in close proximity to the Kelch repeat, the Nrf2 binding domain.
Type 1 disulfides detected within the Kelch repeat domain involving Cys368 and Cys434 are proximate to residues that have recently been demonstrated to be critical for Keap1 binding of Nrf2. Two Keap1 mutants isolated from human lung cancer patients carry single mutations in nearby residues, G364C and G430C, that result in a weakened affinity for NrF2, as shown by coimmunoprecipitation, and upregulation of antioxidant genes engendering a cytoprotective advantage to the transformed cells (55). In addition, Keap1 mutant H436A demonstrates dramatically decreased affinity for an Neh2-derived peptide and shows no detectable ability to repress Nrf2 in a reporter assay (56). Clearly, fairly small changes in structure in these regions can significantly alter Keap1 function; therefore, presumably glutathionylation here might engender similar alterations in function.
Structural Consequences of Type 1 Modifications in the Kelch Repeat Domain
Docking and energy minimization of GSH to the Kelch repeat domain suggests that formation of the observed type 1 disulfides of Cys434 and Cys368 in this region may dramatically alter the protein surface that is critical to Nrf2 binding. In the structures of Figure 5, residues in green make direct contact with an Neh2 fragment peptide in the cocrystallized protein–peptide complex (56). Half of these, as well as the blue His436, which is not in contact with the peptide in the cocrystal, have been shown to be critical for repression of Nrf2 dependent transcription on the basis of site-directed mutagenesis studies in which the individual residues are separately converted to Ala (56). The mauve Gly residues are those mutated to Cys in the lung cancer mutant Keap1 proteins mentioned in the immediately previous paragraph, which manifest weakened Nrf2 binding and upregulation of phase 2 proteins (55). The two orange Cys residues, Cys434 and Cys368, unadorned in Figure 5A, were modified by simulated formation of a type 1 disulfide at Cys434 (Figure 5B) and at both residues (Figure 5C). The structure in Figure 5A is a control experiment in which the unadorned protein structure taken from the coordinates in the PDB underwent minimization. The good agreement between the original coordinates (43) and the minimized structure in 5A suggests that the minimization does not arbitrarily induce dramatic alterations in structure. The minimized structure in Figure 5B indicates that there is a dramatic constriction of the propeller structure at the surface that has been shown to bind the peptide fragment of the Neh2 domain. This contraction appears to be enforced by an extended GSH–protein and protein–protein hydrogen bonded network that recruits a substantial number of the residues that are either known to contact the Neh2 peptide fragment–Kelch repeat domain cocrystal or are otherwise demonstrably critical for Nrf2 repression, as in the case of the shielded His436 (Figure 5B) (56). Minimization of Keap1 after attachment of GSH to both Cys368 and Cys434 generates the structure in Figure 5C in which the contact distances in the proposed hydrogen bonding network of 5B are, in most cases, somewhat contracted, as is the barrel structure as a whole. These minimizations suggest that the formation of type 1 disulfides in the Kelch repeat domain that have been observed experimentally in the present study could indeed influence the association of Keap1 with Nrf2 or perhaps sufficiently alter the structure of Keap1 in a way that leads to its ubiquitination and proteasomal degradation.
Comparison with Other Chemical Modifications
The observed type 1 and type 2 modifications of Keap1 are placed in the context of the reactivities of the cysteines of Keap1 with electrophiles in the top row of Figure 1. In this row, circles represent type 1 modifications, while connectors indicate type 2 modifications. Figure 1 is divided into four sections by bold horizontal lines. The bottom section contains SN2 electrophiles. The next section above, the largest section, includes Michael acceptors. The next section above contains a single entry, sulforaphane, acting as a thiocarbamoylating agent. Some technical sources of the diversity, among electrophiles, of the most reactive (Figure 1, red and azure) target cysteines have been discussed previously and may explain some disagreement evident in studies of the same electrophile by different groups (19–21, 25). As pointed out, the diverse nature of the electrophiles can certainly represent a major factor in these differences. But Figure 1 offers some additional insight. Of all of the agents in Figure 1, only the SN2 agents certainly target the kinetically most reactive cysteines, and these do appear to be clustered, but not exclusively, in the CLR. Of all of the agents in Figure 1, only GSSG certainly targets the thermodynamically most reactive cysteines, and these are scattered across all the domains of Keap1 except the C-terminal region. In this regard, GSSG is most similar to the Michael acceptors, for some of which it has been suggested that the reactions are sufficiently reversible such as to skew the product distribution to more thermodynamic products (22). With sulforaphane, it was argued, on the basis of the relatively small number of most easily detected cysteines and on the behavior of a model peptide, that those cysteines detected were kinetic products, but this is not certain (20). Additionally, there is uncertainty about whether the thiocarbamoylation reaction detected is in fact the reaction that leads to Nrf2 derepression. Sulforaphane administration has been shown, in some cases, to be accompanied by a flux of reactive oxygen species that could be the active agents in Nrf2-mediated enzyme upregulation (57–60).
It is clear from an inspection of Figure 1, as has been pointed out (19–22, 25), that a range of cysteines react with the variety of electrophiles/oxidant, and a rationale for this is not presently clear. Of those most relevant in vivo, the natural products SHO, ISO, XAN, and SULF (to the extent its effects stem from thiocarbamoylation), and GSSG, most reactive cysteines (red and azure symbols), spanning all domains are targeted. It was suggested that this arrangement allows Keap1 to be a most efficient detector of the array of electrophile/oxidant assaults to which a cell responds with a multipronged defense initiated by the single event: Nrf2 nuclear translocation and consequent transcriptional activation (19). This hypothesis is reasonable, and currently not refutable. Alternative hypotheses are tenable. As currently understood, the Keap1 dimer complex in vivo is attached to Actin, to one or two molecules of Nrf2, and also binds the Cul3–Rbx1 complex, a ubiquitin ligase whose propinquity to the Keap1 subunits is presently unknown. It is possible that the multiple reactive cysteines of Keap1 act to disassemble the multiple attachment points in this multiprotein complex, thus allowing for different mechanisms or degrees of Nrf2 derepression. Alternatively, certain proposals maintain Keap1-bound Nrf2 is irretrievably destined for the proteasome and that the unadorned Keap1 dimer is the important victim of electrophile/oxidant assault, allowing newly synthesized Nrf2 to translocate and stimulate transcription (15, 24). If this is indeed true, the existence of multiple reactive cysteines similarly may ensure effective deactivation of Keap1, inhibiting the formation of nonfunctional multiprotein complexes and presumably effecting Keap1 ubiquitination and disposal.
Supplementary Material
Acknowledgments
This work was supported in part by RO1CA91032 from the National Cancer Institute of the National Institutes of Health. The authors wish to acknowledge helpful discussions with Professor Colin Garvie, University of Maryland, Baltimore County, and Dr. Elsa Garcin, Scripps Research Institute, La Jolla, California.
Footnotes
Supporting Information Available: A description of the basis for the relative reactivities arrived at in Figure 1, a Table S1 of tryptic peptide mass and product ions for the type 1 cysteine-disulfide containing peptides identified, and a close up of Figure 5B and C, depicting the hydrogen-bonding network deduced from the energy minimizations of the modified Kelch repeat domain. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–91. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 2.Kensler TW. Chemoprevention by inducers of carcinogen detoxication enzymes. Environ Health Perspect. 1997;105(Suppl 4):965–70. doi: 10.1289/ehp.97105s4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talalay P, Dinkova-Kostova AT, Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul. 2003;43:121–34. doi: 10.1016/s0065-2571(02)00038-9. [DOI] [PubMed] [Google Scholar]
- 4.Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–52. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
- 5.Dinkova-Kostova AT, Holtzclaw WD, Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44:6889–99. doi: 10.1021/bi047434h. [DOI] [PubMed] [Google Scholar]
- 6.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–5. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–68. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 8.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2001;20:3906–17. doi: 10.1038/sj.onc.1204506. [DOI] [PubMed] [Google Scholar]
- 10.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–91. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–71. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–49. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, Itoh K, Yamamoto M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells. 2007;12:1163–78. doi: 10.1111/j.1365-2443.2007.01118.x. [DOI] [PubMed] [Google Scholar]
- 16.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the Actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci USA. 2004;101:2046–51. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophilesensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci USA. 2005;102:10070–5. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–13. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liebler DC, Hong F, Sekhar KR, Freeman ML. Site-Specific Modification of the Electrophile Sensor Protein Keap1 and Activation of Nrf2-Dependent Gene Expression. Adv Mol Toxicol. 2006;1:55–71. [Google Scholar]
- 20.Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol. 2005;18:1917–26. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 21.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–75. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 22.Luo Y, Eggler AL, Liu D, Liu G, Mesecar AD, van Breemen RB. Sites of alkylation of human Keap1 by natural chemoprevention agents. J Am Soc Mass Spectrom. 2007;18:2226–32. doi: 10.1016/j.jasms.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–9. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–20. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 25.Eggler AL, Luo Y, van Breemen RB, Mesecar AD. Identification of the highly reactive cysteine 151 in the chemopreventive agent-sensor Keap1 protein is method-dependent. Chem Res Toxicol. 2007;20:1878–84. doi: 10.1021/tx700217c. [DOI] [PubMed] [Google Scholar]
- 26.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc Natl Acad Sci USA. 2006;103:768–73. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakurai T, Kanayama M, Shibata T, Itoh K, Kobayashi A, Yamamoto M, Uchida K. Ebselen, a seleno-organic antioxidant, as an electrophile. Chem Res Toxicol. 2006;19:1196–204. doi: 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–70. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21:705–10. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 31.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol Med. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 32.Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–9. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci. 2004;82:308–17. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 34.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radical Biol Med. 1999;27:1208–18. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 35.Watson WH, Jones DP. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003;543:144–7. doi: 10.1016/s0014-5793(03)00430-7. [DOI] [PubMed] [Google Scholar]
- 36.Han D, Hanawa N, Saberi B, Kaplowitz N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic Biol Med. 2006;41:627–39. doi: 10.1016/j.freeradbiomed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Zou J, Salminen WF, Roberts SM, Voellmy R. Correlation between glutathione oxidation and trimerization of heat shock factor 1, an early step in stress induction of the Hsp response. Cell Stress Chaperones. 1998;3:130–41. doi: 10.1379/1466-1268(1998)003<0130:cbgoat>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radical Biol Med. 2000;28:625–35. doi: 10.1016/s0891-5849(99)00275-0. [DOI] [PubMed] [Google Scholar]
- 39.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481–90. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 40.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–44. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 41.Jones DP. Extracellular redox state: refining the definition of oxidative stress in aging. Rejuvenation Res. 2006;9:169–81. doi: 10.1089/rej.2006.9.169. [DOI] [PubMed] [Google Scholar]
- 42.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 43.Li X, Zhang D, Hannink M, Beamer LJ. Crystal structure of the Kelch domain of human Keap1. J Biol Chem. 2004;279:54750–8. doi: 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- 44.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr Sect D. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 45.Szajewski RP, Whitesides GM. Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J Am Chem Soc. 1980;102:2011–2025. [Google Scholar]
- 46.Whitesides GM, Lilburn JE, Szajewski RP. Rates of thiol disulfide interchange reactions between mono- and di-thiols and Ellman’s reagent. J Org Chem. 1977;42:332–338. [Google Scholar]
- 47.Guo W, Pleasants J, Rabenstein DL. Nuclear magnetic resonance studies of thiol/disulfide chemistry. 2. Kinetics of symmetrical thiol/disulfide interchange reactions. J Org Chem. 1990;55:373–376. [Google Scholar]
- 48.Keire DA, Strauss E, Guo W, Noszal B, Rabenstein DL. Kinetics and equilibria of thiol/disulfide interchange reactions of selected biological tiols and related molecules with oxidized glutathione. J Org Chem. 1992;57:123–127. [Google Scholar]
- 49.Houk J, Singh R, Whitesides GM. Measurement of Thiol-Disulfide Interchange Reactions and Thiol pKa Values. Vol. 143. Academic Press; Orlando, FL: 1987. [DOI] [PubMed] [Google Scholar]
- 50.Shaked Z, Szajewski RP, Whitesides GM. Rates of thiol-disulfide interchange reactions involving proteins and kinetic measurements of thiol pKa values. Biochemistry. 1980;19:4156–66. doi: 10.1021/bi00559a004. [DOI] [PubMed] [Google Scholar]
- 51.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–42. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 52.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–82. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang XJ, Sun Z, Chen W, Li Y, Villeneuve NF, Zhang DD. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: enhanced Keap1-Cul3 interaction. Toxicol Appl Pharmacol. 2008;230:383–389. doi: 10.1016/j.taap.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun. 2007;362:816–21. doi: 10.1016/j.bbrc.2007.08.051. [DOI] [PubMed] [Google Scholar]
- 55.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 56.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–17. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L, Baskaran R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J Biol Chem. 2004;279:25813–22. doi: 10.1074/jbc.M313538200. [DOI] [PubMed] [Google Scholar]
- 58.Jakubikova J, Sedlak J, Bod’o J, Bao Y. Effect of isothiocyanates on nuclear accumulation of NF-kappaB, Nrf2, and thioredoxin in caco-2 cells. J Agric Food Chem. 2006;54:1656–62. doi: 10.1021/jf052717h. [DOI] [PubMed] [Google Scholar]
- 59.Kim H, Kim EH, Eom YW, Kim WH, Kwon TK, Lee SJ, Choi KS. Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma cells to TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of DR5. Cancer Res. 2006;66:1740–50. doi: 10.1158/0008-5472.CAN-05-1568. [DOI] [PubMed] [Google Scholar]
- 60.Payen L, Courtois A, Loewert M, Guillouzo A, Fardel O. Reactive oxygen species-related induction of multidrug resistance-associated protein 2 expression in primary hepatocytes exposed to sulforaphane. Biochem Biophys Res Commun. 2001;282:257–63. doi: 10.1006/bbrc.2001.4531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.