

Abstract
A stereoselective aza-Henry reaction between an arylnitromethane and Boc-protected aryl aldimine using a homogeneous Brønsted acid–base catalyst was translated from batch format to an automated intermittent-flow process. This work demonstrates the advantages of a novel intermittent-flow setup with product crystallization and slow reagent addition which is not amenable to the standard continuous equipment: plug flow tube reactor (PFR) or continuous stirred tank reactor (CSTR). A significant benefit of this strategy was the integration of an organocatalytic enantioselective reaction with straightforward product separation, including recycle of the catalyst, resulting in increased intensity of the process by maintaining high catalyst concentration in the reactor. A continuous campaign confirmed that these conditions could effectively provide high throughput of material using an automated system while maintaining high selectivity, thereby addressing nitroalkane safety and minimizing catalyst usage.
Keywords: organocatalysis, asymmetric catalysis or synthesis, sustainable or green chemistry, continuous processes, flow chemistry, aza-Henry reaction
Graphical Abstract
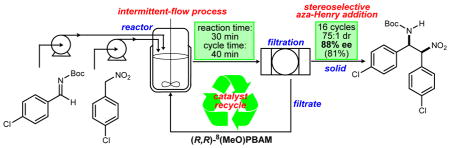
INTRODUCTION
Versatile methods to prepare nitrogen-containing small molecules remain in high demand in the pharmaceutical industry, driving the discovery of new reactions and the refinement of existing methods.1 Although heterocycles devoid of chiral centers can be effective therapeutics (e.g., tyrosine kinase inhibitors), the selective disruption of more complex interactions (e.g., protein–protein interactions) have increased attention on the expansion and tailoring of molecular space, leading to drug candidates with complex structures containing several chiral centers.2 Chiral auxiliaries, kinetic resolution, and hydrogenation methods are well-established in industrial settings and continue to be successfully applied to the large scale preparations of APIs.3,4,5 However, ever more complex targets drive the invention and development of approaches outside of the conventional paradigm, and novel methodologies must answer emerging challenges as they appear. Contemporary enzymatic approaches have risen to meet this demand in recent years.6 These tools offer the potential to address the bulk production of material but still find developmental hurdles while engineering both high specificity and reactivity.
In the past decade alone, the field of organocatalysis has achieved major advances, particularly in reactions where metal-catalyzed methods have traditionally dominated. As a result, an array of transformative technologies have provided functionally dense intermediates with high levels of diastereo- and enantioselectivity.7 These methods also allow straightforward access to previously unavailable chiral molecules using robust and easy to handle catalysts, themselves readily prepared inexpensively and without metal contamination. Adaptation of these methods, however, to a process chemistry environment has been slower, and only a few applications of organocatalysts are reported in the literature (predominantly Cinchona alkaloids, proline, and chiral amine catalysts).8 Analysis of this situation leads to a conclusion that successful implementation of organocatalytic procedures on industrial scale would require finding an appropriate answer to several major challenges. Generally high catalyst loadings (5–10%) are considered unattractive, specifically in comparison with metal–mediated hydrogenation methods where conditions with minute catalyst amounts are quite effective (S/C ≥ 1000–4000). With high catalyst loadings, economical constraints play an important role in the selection process. Slow reaction rates (typically ~24 h) followed by the necessity to maintain cryogenic conditions during the reaction cycle significantly decreases the intensity of the process and increases the cost of operation, which is further magnified when specialized equipment is required. Finally, an organocatalyst needs to be effectively separated from the organic reaction product without the use of chromatography. In this first report using Bis(AMidine) [BAM] organocatalysis, an aza-Henry reaction is scaled by translation of the original batch reaction into a continuous process. This effort leveraged the advantages of this format to maintain high selectivity and yield while intensifying the process.
Continuous processing is a powerful alternative to classic batch chemistry and is often applied to procedures where safety (high temperatures, pressures, large exotherms and aggressive reagents) and fast heat or mass transfer (bi- and triphasic systems, reactions with gases or light) have a major impact.9 Pharmaceutical companies have embraced continuous technologies to benefit from fully automatic processes and a continuous analytical data stream while using dedicated, inexpensive equipment where scale–up often involves a longer running time.10 These methodologies are also easily aligned with the majority of the highest standards of quality and green chemistry principles recommended by the FDA and EPA.11 By relying less on process-tailored infrastructure and more on flexible continuous process equipment, chemists can benefit from shorter optimization campaigns, improved and simplified synthetic strategies, timely delivery of the desired quantities of APIs, and reduced risk and cost associated with the development and production of APIs.
The use of nitroalkanes has blossomed in recent years due to an appreciation for, and better understanding of, the chemistry of the nitro functional group12 and its ability to behave as a masked amine. To highlight just one case, the aza-Henry reaction provides a carbon–carbon bond-forming pathway to the stereoselective construction of vic-diamines in protected form.13 Furthermore, methods to address the stereocontrol issues associated with this reaction have proliferated since the early contributions of Shibasaki and Jørgenson.14 Our interest has been driven, in part, by the need for stereocontrolled aza-Henry reactions extending beyond simple aliphatic nitroalkanes.15 Among these is the aryl nitromethane addition to imines, which had limited success16 until 2011.17 These discoveries have been leveraged to create molecular complexity in key intermediates that have also been shown to be competent precursors to heterocyclic pharmacophores. The focus on protic acid salts of chiral bis(amidine) ligands has produced a collection of organocatalytic methods capable of generating highly functionalized and enantioenriched diamine building blocks, all comparing favorably to traditional metal–based methods.18
Our initial goal was to translate the enantioselective aza-Henry reaction from batch format to continuous conditions in order to accelerate the large scale production of diamine building blocks. The formation of a less soluble product is often the case during heterocycle formation. This provides a unique opportunity to couple reaction development with separation/purification. However, a consequence of the insoluble product is that a typical plug flow reactor (PFR) is not suitable because of plugging and clogging due to the formation of solids. Heterogeneous reactions with solids precipitation can be run in continuous stirred tank reactors (CSTR) which are more suitable for handling the solids in flow. For example reactive crystallization in mixed suspension/mixed product removal (MSMPR) crystallizers is a legitimate option for handling this type of process physically. MSMPRs have been used for antisolvent,19 cooling,20 or reactive21 crystallizations. The intent is to use a continuous reactive crystallization platform for an aza-Henry reaction. However, because of the slow reaction kinetics, the need for full conversion of imine, and the desire for controlled addition of imine to nitroalkane, a truly continuous MSMPR is not the best equipment choice for this reaction. Therefore, an automated intermittent flow stirred tank reactor was selected instead. Automated intermittent flow is an equivalent process to continuous flow. Under these operating conditions, the aza-Henry product continuously crystallizes from the reaction mixture and is intermittently filtered while the mother liquor with the solubilized catalyst is returned to the reactor. Intermittent separation and catalyst recycling should achieve formally low catalyst loadings and achieve a high catalyst concentration in the reactor. Thus, a single operating unit effectively fulfills a triple goal (function): promoting a faster organocatalytic reaction, crystallizing the product and also separating the catalyst for the recycle stage.
Unlike nitroarenes, nitroalkanes are relatively underutilized due to the perception that they are hazardous and unsafe materials. This proposal to merge organocatalysis and continuous processing also provided an opportunity to create a system which utilizes nitroalkanes in a safe and controlled (reliable) way.
Batch Aza-Henry Chemistry
The initial scale up conditions for the organocatalytic aza-Henry were first developed to secure a significant throughput of material necessary for biological testing (Scheme 1).22 The original procedure17 was refined23 to provide the desired adduct 4 in 90% yield and with high stereoselectivity. During these studies we have discovered that to achieve consistent enantioselectivites on a larger scale, it was advantageous to add imine 1 slowly to the mixture of nitroalkane 2 and catalyst 3 in toluene at −20 °C. This protocol was both effective and reproducible, generating the β-amino nitroalkane 4 on decagram scale (23.1 g in a single batch). However, after thoughtful analysis we concluded that for further development and scale up, several issues required additional attention:
Scheme 1.

Initial Parameters for a Batch aza-Henry Reaction, Targeting a Viable Intermediate to (−)-Nutlin 3
Safety: Low molecular weight nitroalkanes can be high-energy compounds, but the hazards of functionalized derivatives are largely unstudied.24,25
Preparation of aliphatic nitroalkanes: general methods to synthesize nitroalkanes on large scale are limited.
Productivity (moles product/hour): Batch organocatalytic aza-Henry reactions can require long reaction times (~24 h).23 Reduced cycle times on the order of 30–40 min would provide an opportunity for translation of these processes into an automated intermittent flow format.
Economy: Highest selectivity is achieved under cryogenic conditions, leading to lower output of the product and greater expense. Batch catalyst loadings of 0.5–1% are acceptable but additional catalyst economy and recycle are desirable.
Reactor System Design and Major Drivers for a Translation of Aza-Henry Process to Intermittent-Flow Format with Recycle
The two main types of continuous reactors are plug flow reactors (PFRs)26 and continuous stirred tanks (CSTRs).27 Neither one of these truly continuous reactor types is ideally suited for the aza-Henry reaction targeted here. The product precipitates during the course of the reaction and PFRs generally cannot handle solids in flow due to plugging and clogging in the tube, pipe, or microchannel. Second PFRs normally have all-at-once stoichiometric addition of reagents at the PFR inlet, while the aza-Henry chemistry performs best with controlled addition of the imine to the nitroalkane. It is possible to mimic controlled addition by injecting the imine at multiple points along the length of the PFR,28 but this requires additional feed pumps and flow meters. CSTRs are more suited to heterogeneous reactions with solids because the agitation system keeps the solids suspended in the stirred tank. The main limitations of a simple CSTR for this application are (1) very long residence times are needed to achieve full conversion of the imine in a single CSTR, and (2) controlled addition of imine to nitroalkane is not possible in a CSTR, since both reagents are added together (coaddition) and the reactor runs at end-of-reaction conditions. Although the limitations can be overcome by using a train of CSTRs in series, and feeding the imine portion-wise into the individual CSTRs, this adds complexity and still requires a significantly longer overall residence time compared to batch or PFR to achieve full conversion. A mathematical comparison of residence times in batch, PFR, CSTR, and CSTRs in series is provided in the Supporting Information.
The answer to this reactor design challenge was an intermittent-flow CSTR. It is an automated repeating batch, with a 40 min total turnover time. The feed pumping, product slurry pumping to the filter, filtration, and recycle of a desired fraction of the filtrate back to the reactor are all fully automated and repeating. The reaction design allows full conversion of the imine and the product separated from the product slurry with the solids forward-processed into the next step.
Compared to a PFR, the intermittent flow reactor is better suited for solids in flow. Compared to a CSTR, the intermittent flow reactor achieves the same conversion at much shorter reaction time for positive order reactions, and thus smaller reactor volumes. Furthermore, it can accomplish all-at-once addition, controlled addition of one or more feeds, any order of addition of multiple feeds, or coaddition, depending on which gives a higher yield and/or minimizes key impurities.
This type of reaction process is analogous to a truly continuous process. Continuous processing enables smaller reactor volumes and also facilitates the integrated recycle. The higher the recycle ratio, the lower the overall catalyst usage. The reactor is about 36 times smaller than a batch reactor for the same kg/week throughput, assuming 1-day start-to-start cycle time for a typical batch process. The overall logic of the design, as well as other benefits of automatic intermittent-flow CSTR, are summarized in Figure 1.
Figure 1.

Major drivers for using the intermittent-flow system
The intermittent flow approach has been taken by other researchers. In the work of Adams, intermittent flow is termed semicontinuous operation.29 Adams describes it as a forced cyclic process, in which there are no steady states. He explains that it is possible to achieve multiple separation steps and high reaction conversion using fewer vessels than would be required in a truly continuous operation.
RESULTS AND DISCUSSION
1. Synthesis of Starting Materials and Catalyst
Imine 1 was prepared on decagram scale using previously described procedures (Scheme 2).23 In a typical experiment chloro-benzaldehyde 6, tert-butylcarbamate 7, and the sodium salt of sulfinic acid were stirred in a mixture of methanol and water in the presence of formic acid at room temperature for 9 days. Filtration and trituration with diethyl ether to remove residual aldehyde provided the desired amido-sulfone 8 in 80–82% yield overall. This method is based on slow formation and precipitation of the insoluble α-amido sulfone. In this case, the ease of preparation and straightforward product collection contrasted the impractical reaction time and use of an ether solvent. However, purification of these sulfones is generally a challenge due to low solubility in a majority of nonpolar organic solvents, and significant decomposition is often observed upon crystallization from protic solvents (water, alcohols). This protocol was utilized to prepare the sulfone on 100 g scale; however, improvements to this are actively investigated.
Scheme 2.

Large Scale Synthesis of Imine 1.
Elimination was performed in dry THF by refluxing sulfone 8 for 3 h with excess potassium carbonate and sodium sulfate. The salts were filtered at the end of the reaction, and the removal of the solvent furnished imine 1 which was utilized in the next step without further purification. Aldehyde levels were consistently observed at 1–2% in the crude material. Attempts to apply more rigorous conditions with extensively dried solvents and an inert atmosphere did not further lower the amount of aldehyde. Since we have found that residual aldehyde does not affect yield or stereoselectivity of the aza-Henry reaction in a recycling sequence, the initial conditions were employed for the scale-up of the material for the automated intermittent flow runs.
We have previously utilized two different methods to construct the 1-chloro-4-(nitromethyl)benzene. The first method entailed a radical bromination of para-chlorotoluene in order to prepare the corresponding benzyl bromide followed by the subsequent substitution of the bromide with a nitro group, an approach often referred to as Kornblum’s procedure.23 Workup with phloroglucinol improved the yield by converting the nitrite (formed by O-alkylation) to alcohol. The resulting crude mixture could be purified by column chromatography to provide nitroalkane in 33% yield over 2 steps containing up to 2% of the corresponding aldehyde. In another method, aldehyde 6 was easily converted to the oxime 9 in nearly quantitave yield and then oxidized with MCPBA to generate the desired nitroalkane 2 (Scheme 3). Recently we have shown that this procedure is highly versatile and could be applied to a variety of the substrates on a small scale.22 However, upon scale-up of this process the yields plummeted, extensive chromatographic purification was required, and excessive amounts of meta-chloroperbenzoic acid raised safety concerns. An alternative oxidant was considered in order to produce the nitroalkane on larger scale. Based on literature precedent30 we attempted the same reaction using peracetic acid solution. In comparison with MCPBA oxidation these conditions have shown a slightly better reaction profile with decreased reaction times, diminished side product formation and a simple purification protocol. However, direct crystallization of the product with desired purity from the crude reaction mixture was unattainable, and an exothermic profile of the reaction was a concern. The crude reaction mixture was initially subjected to chromatographic purification followed by crystallization from the mixture of heptanes/EtOAc to furnish nitroalkane 2 as white low-melting needles in 46–49% yield. This route was amenable to 50 g scale and provided an appropriate throughput of material for the automated intermittent flow experiments. This method is suboptimal, and further development of a safe and reliable protocol continues.
Scheme 3.

Nitroalkane Synthesis
A preliminary safety evaluation of nitroalkane 2, oxime 9 and aza-Henry product 4 was conducted. All compounds exhibit complex decomposition behavior and decomposition energies >350 J/g. Analysis of differential scanning calorimetry (DSC) data using Yoshida’s correlation did not indicate that these compounds were shock-sensitive or explosion-propagating (see Supporting Information for further details). However, this analysis has also shown that nitroalkane 2 characteristics are borderline; therefore, more detailed evaluation of this compound would be beneficial and particularly critical upon further scale-up.
Catalyst 3 was prepared according to the previously reported procedure.23,31 Several minor modifications were introduced in order to improve yields and overall throughput of the material (Scheme 4). In the Combes synthesis of 2,4-dichloro-8-methoxyquinoline 12, dilution of the reaction mixture improved the yield of the quinoline 3-fold. Buchwald–Hartwig amination using potassium carbonate instead of sodium tert-butoxide resulted in a 10% increase in the yield of ClQuinBAM. Finally, nucleophilic aromatic substitution with pyrrolidine was executed under milder microwave conditions. The original procedure required 180 °C for 30 min and provided the product in 48% yield. Moreover, extensive chromatographic purification was needed to isolate the pure material. By reducing the reaction temperature to 110 °C the formation of the key byproducts was suppressed, and after 3.5 h 8(MeO)PBAM catalyst 3 was generated as the major product. Trituration from benzene/hexanes provided the catalyst in 81% yield as a light brown solid.
Scheme 4.

Catalyst Synthesis
2. Automated Intermittent-Flow Process with Recycle
Batch Approach to Develop Parameters for the Process
To find acceptable starting conditions for automated intermittent flow process, we used series of batch reactions. After each batch reaction, the solids were filtered off, and the filtrate was returned to the reaction flask. This created a simplified model for a run in automated mode. Although improving selectivity was not the primary goal of the project, maintaining the levels of selectivity equal to the batch reaction was critical. The initial choice of parameters (concentration, temperature, catalyst loading, mode of imine addition) was based on prior work.23 The major goal of the optimization at this level was to achieve intensification of the process.
For our optimization we have selected the 8(MeO)PBAM catalyst 3 based on two major reasons:
Access: the catalyst can be easily prepared on a large scale in 3 steps.
Process intensification: 8(MeO)PBAM has exhibited the highest reaction rate and maintained reasonable stereoselectivity, achieving intensification of the process.
Since direct kinetic studies of this aza-Henry system were complicated by the precipitation of the solid product during the course of the reaction, a brief screen of different reaction times and catalyst loadings was conducted. It was found that reaction time can be shortened from 24 to 4 h, and the catalyst loading was reduced to 0.5% without loss of yield or selectivity. An attempt at 0.1% catalyst and 4 h reaction time furnished a very low yield of the aza-Henry product and unreacted imine was observed in the reaction mixture.
Excesses of nitroalkane and concentration of reaction solution were evaluated. Experiments were usually performed with a slight excess of nitroalkane (1.2–1.5 equiv at 0.10–0.15M). High stoichiometric amounts of nitroalkane proved to be detrimental to selectivity since larger quantity of the acidic nitroalkane relative to Brønsted basic catalyst leads to either catalyst modulation or inactivation as a result of a nitroalkane solvation phenomenon by hydrogen bond donation. To further support this data, an attempt to increase concentration further resulted in lower selectivity.
In order to identify key variables necessary for translation from batch to intermittent flow with recycle, several iterative series of batch reactions were conducted. In general, representative sequences consisted of several (5–8) consecutive aza-Henry reactions performed at −20 °C. The product was removed by filtration and washed with ice-cold toluene, and the residual filtrate was concentrated and used as a starting point for the next step. Each step was initiated with dissolution of residual solid, cooling to the corresponding temperature followed by a recharge with fresh reactants. The mass of the filtrate was registered after each iteration, and the composition of the filtrate was carefully determined by HPLC and NMR analysis. The final product isolated from each iteration was washed further with cold hexanes, dried, and similarly analyzed.
Initial batch series were performed using 100% recycle, and the catalyst was introduced only once in the first iteration. However, it was shown that in this case good enantioselectivity could be maintained only during the first 4–5 iterations and usually dropped (10–15%) after the fifth or sixth iteration, and declined thereafter.32
Batch Experiment with Recycle
Since limited success was achieved when recycling 100% of the catalyst, introduction of fresh catalyst at the beginning of each iteration might translate into a more effective batch sequence without loss of selectivity. This also aligned well with the overall plan to transform current batch recycling into a completely automated intermittent flow process.33 We conducted a sequence with 1.25% of the catalyst in the flask and an 80% recycling ratio (). The recycling number 80% indicates that only 80% of the catalyst was recycled, but 20% of fresh catalyst was introduced at the beginning of each cycle. The removal of 20% of the reaction mixture at the end of each cycle was another important difference of this recycling sequence in comparison with those using a 100% recycle ratio. The enantioselectivity and diastereoselectivity of the process remained high throughout the entire sequence, and after the eighth iteration the product was isolated in good yield and close to 90% ee (Figure 2).34 For this protocol, nitroalkane (1.4 equiv) was introduced at each step to maintain a proper stoichiometric ratio of the reagent in the sequence. At the end of the eighth iteration, the overall catalyst loading was 0.38%. Overall catalyst loading was low because 1.25% catalyst was added for the first iteration but only 0.25% fresh catalyst was added in each subsequent iteration. The recycle kept catalyst loading at 1.25% in the reactor for all iterations. Significant intensification of the process was achieved by reduction of the reaction time from 24 to 4 h while maintaining high concentrations of the catalyst in the flask due to 80% recycling of the catalyst.
Figure 2.

Enantioselectivity trends in the sequence with 80% recycle and 1.25% catalyst loading.
Manual Proof of Concept
With initial success the next goal of development was to further demonstrate the viability of an automated process with semibatch filtration of product away from the solution with catalyst and remaining starting materials. The batch sequence was designed to closely imitate the planned sequence. In order to achieve this goal several alterations to the previously described sequence (vide supra) were introduced: solvent removal, drying, and redissolution of the recycled material were omitted between iterations. Thus, upon completion of each cycle the reaction mixture was filtered while the collecting vessel for the filtrate was placed in the cooling bath and kept at temperatures between −15 and −25 °C during the entire process. Upon conclusion of the filtration, the starting materials were charged directly to the same vessel and the next iteration was initiated. Additional toluene and catalyst were added in order to keep concentration of the reactants and catalyst constant. Under the described conditions using manual filtration and house vacuum, full transfer of the recycle was unattainable and on average 10–20% of the recycle was retained in the filtered solid product. It is also important to mention that these sequences allowed catalyst examination (stability and sustainability) under conditions similar to the actual automated intermittent flow runs which should provide improved longevity of the catalyst and superior selectivities.35
Our goal was to achieve a reduction of cycle time to 40 min or less in order to improve the overall intensity of the process. The sequence was conducted with 1.0% catalyst. At the beginning of each cycle, 7% of the initial amount (0.5 mg) of fresh catalyst was introduced (Scheme 6). Initial iterations have shown lower stereoselectivity. But from one batch to another, enantioselectivity progressively increased and plateaued for the last three runs at 88% (reaching a steady state, Figure 3).36 Diastereoselectivty of the solid decreases from cycle to cycle but remains high (25:1). This trend is an expected result since overall diastereoselectivity of the reaction measured for the product isolated after chromatographic purification is ~10:1.23,37 After 8 cycles product was isolated in 78% yield overall. Based on the mass of material collected in the washes the recycling ratio for this sequence was 80%. This combination of robust results and significantly improved material throughput (30 min vs. 4 h reaction cycle) provided support for the basic concept of our design. This series also revealed the stability of the catalyst under these reaction conditions, including encouraging levels of selectivity at 1% loading. The potential advantages of reaching a steady state operation where a higher enantioselectivity is achieved in comparison with a single batch reaction were clearly demonstrated.
Scheme 6.
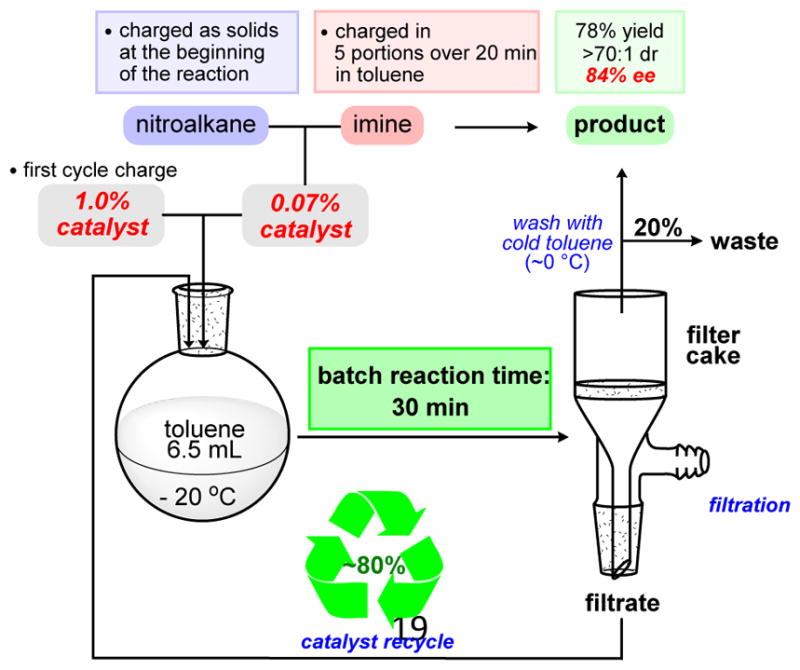
Sequence with 1.0% Catalyst and 80% Recycle Ratio
Figure 3.

Enantioselectivity and diastereoselectivity trends in 1.0% catalyst loading sequence.
Automated Intermittent Flow Development
The automated intermittent flow unit was designed to conduct an organocatalytic aza-Henry reaction in a repeating batch fashion with semibatch filtration of the precipitated product followed by recycling of the filtrate to the reactor. Recycling catalyst kept catalyst loading high in the reactor, thereby allowing a reduction in the cycle time and intensification of the process. The technical schematic for the process is shown in Figure 5.
Figure 5.

Technological scheme of the process and mass streams for the 3-day intermittent flow campaign.
In order to ensure homogeneous solutions for the feeds for long time durations, a solubility screen was conducted. The imine exhibited good solubility under the reaction conditions. While cooling nitroalkane to −20 °C, in concentrations between 0.5 and 0.7 g per 1 mL, slow crystallization of the starting material was observed.
After brief optimization of automation and reaction parameters, a test 10-cycle sequence was conducted with 40 min cycle time, 4.0 % 8(MeO)PBAM catalyst loading and 80% recycle ratio. The mode of reagent addition was designed to keep concentration of the imine low relative to nitroalkane, while simultaneously maintaining excess nitroalkane acid in a range that does not affect activity of the Brønsted basic catalyst. Enantioselectivity of the crystallized aza-Henry product after the first cycle was 85%; it reached 87% in the second cycle, and thereafter remained unchanged for the rest of the first day campaign (Figure 4). Enantioselectivity in the recycle was slightly higher: in the first two cycles 91.3% ee was observed, whereas the next 8 cycles gave lower but very consistent results in a range between 90.2 and 90.8 % ee. The isolated yield of the filtered product was 15.5 g (80.9% yield, 87% ee, with 96.2% area by HPLC, and 2.1% area of diastereomer). In this experiment 80% of the filtrate was recycled back to the reactor, and 20% flowed to a waste container. Unfortunately, we did not account for the amount of filtrate sequestered with the solids on the filter; therefore, the amount of catalyst and excess nitroalkane in the reactor gradually decreased over the 8 iterations for this automated experiment. Even so, the results were good for all 10 iterations. Due to the short cycle time and multiple volume turnovers per day, this setup is significantly smaller in size compared to a required single batch reactor that achieves similar throughput. In batch processing without catalyst recycle, lower catalyst loadings lead to longer reaction times and require larger reaction vessels.
Figure 4.

Enantioselectivity and diastereoselectivity trends in the automated intermittent flow run with 80% recycle ratio.
With successful results from the first automated intermittent flow campaign, a longer 24-cycle sequence was completed using a lower catalyst loading (2.64%) and higher recycle ratio. 97% of the filtrate was recycled to the reactor, while 3% of the filtrate was pumped to waste. Taking into account loss of solution to the filter cake, 89% of the liquid that flowed out of the reactor was recycled back to the reactor each cycle. Therefore, 89% of the catalyst was returned back to the reactor after each cycle. The overall structure and timing of the cycle remained unchanged (23.5 min addition of the imine and 30 min reaction cycle with 5 min filtration and 5 min precooling of the recycled filtrate). The average temperature of the process was sustained at −13 to −15 °C. Initially, three cycles were required to achieve a steady-state condition, and enantioselectivity of the solid product steadily increased during these cycles (82.6, 86.0, 88.3% ee, Figure 6). During the following three cycles selectivity stabilized and remained on the same level (89.4, 89.4, 89.2% ee). This mirrored the situation observed in the recycle where selectivity decreased during the first three cycles (91.0, 90.3, 89.5% ee) and again remained essentially unchanged for the next three (87.9, 88.3, 88.2% ee). After the sixth cycle, the process was interrupted by stopping for the night and reinitiated the next morning. During this overnight period, the arrested reactor with recycle inside was maintained at the standard temperature. However, after this 24 h period under consistent operating conditions the reactor slowly warmed due to circulator problems. During the second day of the campaign the average temperature increased and was maintained at −10 to −12 °C. Overall 10 cycles were conducted during the second day of the campaign. Expectedly, the first three were necessary to achieve a steady state, and these cycles delivered slightly lower selectivity (88.0, 87.5, 87.4% ee). Selectivity then restabilized, and during the last five cycles minimal fluctuations were observed (89.2, 89.1, 89.3, 89.2, 89.2% ee). After a two day campaign, 25.1 g of the product was isolated (81.3% yield, 88.5% ee and purity of the product was 98.6% HPLC area). The resulting aza-Henry product could be further recrystallized from toluene to provide a single stereoisomer (>200:1 dr, 99% ee).23
Figure 6.

Enantioselectivity and diastereoselectivity trends in the continuous run with 97% recycle ratio.
Careful analysis of the filter cake from the second day of the campaign led to an interesting observation. The sample from the top of the cake measured 91.2% ee, while the sample from the bottom of the cake measured 87.1% ee, and the average selectivity of the dry product was 88.8% ee. This phenomenon can be explained by an increased solubility of a single enantiomer in comparison with the racemate. Thus, after multiple filtrations, the bottom of the cake is enriched with racemate, and the top section contains product with the highest enantioenrichment. In order to eliminate this heterogeneity in the future, solid product will need to be washed and removed from the filters more frequently to minimize the influence of the filtrate solution on the cake. There are many options for doing this. A centrifugal filter with automated solids peeling or inverting basket could be used. Dual filters with automated washing dissolving of solids from the off-line filter are another option.
After 16 cycles the process was interrupted again. But this time the cooling system was turned off, and the recycle solution was warmed to room temperature for a prolonged period of time. Eight additional cycles were executed during the third day of the campaign. In comparison with the first reinitiation, the selectivity of the process dropped more, and ~85% ee was observed in the first three cycles. Slow improvement in selectivity was observed in the subsequent cycles; however, the maximum selectivity of 87.5% was observed in this sequence (1.5–1.8% lower than during the second day of campaign). Surprisingly, the selectivities of the product in the recycle solution remained mostly unchanged and consistently high (87–90%). A degree of catalyst deactivation as it is recycled, the result of prolonged exposure to room temperature, could be an explanation for the observation.
CONCLUSIONS
This platform both preserves the advantages commonly ascribed to the continuous systems (reduced waste and production footprint, atom economy and improved safety) and incorporates the effectiveness of an organocatalyst to provide access to a privileged (structurally and stereochemically) class of diamines. An automated intermittent flow reactor was used instead of a truly continuous setup due to solids precipitation, reaction kinetics, and the selectivity advantages of controlled addition of the imine to the nitroalkane. Automated intermittent flow is equivalent in this context to a continuous process. It enabled high throughput for a small reactor size, a low level of material hazard at any one time, fast cooling by flowing through heat exchangers leading to the reactor, and integrated recycle of catalyst and unreacted nitroalkane. The following improvements of the developed protocol over the standard batch were accomplished:
Safety: Operating an automated intermittent flow reactor affords the safety benefit of only having small amounts of nitroalkane present at any given time, as opposed to a batch reaction where the full amount of nitroalkane is subjected to the reaction from the start.
Productivity (moles product/hour/reactor volume): Reaction can be safely run with a higher output and a single cycle reduced to 40 min from 22 h for the batch process.
Recycle of nitroalkane and catalyst: higher ratio of the catalyst and nitroalkane to imine results in process intensification.
Reduced production footprint: atom economy, waste minimization, decreased amount of solvents and catalyst.
-
Fully automated process:
Full control of variables and high reproducibility (constant enantioselectivity)
Flexibility of production volume and scale up with minimum optimization
Reduced cost: Reaction performed at a higher temperature, with a smaller reactor size and diminished single cycle time provides significant cost benefit. The recycle allows a lower catalyst loading and reduced solvent amounts.
This process was applied to the multigram synthesis of chiral, densely functionalized precursors to differentially protected diamines which are useful building blocks with a broad range of applications.
EXPERIMENTAL SECTION
Materials
Starting materials (nitroalkane, imine and catalyst) were prepared according to the description provided in section “Synthesis of Starting Materials and Catalyst” and following previously published procedures.22–23 Toluene was purchased commercially and used without further purification.
Equipment specification
| Description | Part Number | Supplier |
|---|---|---|
| Peristaltic Pump | 7523-80 | Cole-Palmer |
| Peristaltic tubing size 35 | EW9619-35 | Cole-Palmer |
| 250 mL Jacketed Reactor | CG-1930-22 | Chemglass |
| 600 mL Pressure Filter Complete | 6384-231 | Ace Glass |
| Pressure Transmitter, SS, 0-4000 psig | 3051S1TG4A2E11A1AQ4 | Rosemount |
| 1/4″ Actuated valve, 60 series | SS-62TS4-31C | Indiana Fluid |
Reagent Solutions
The process is not sensitive to brief exposure to residual moisture from the air as can be seen from the recycling sequences described above (Error! Reference source not found. & Scheme 6) where the product was filtered and manual iterations were reinitiated under open-air conditions. Despite the tolerance to adventitious moisture, some precautions are necessary since imine hydrolysis may eventually be observed. To avoid potential complications during long term campaigns, automated intermittent flow runs were conducted under nitrogen atmosphere, rigorously dried conditions, and with all feeding solutions prepared in a glovebox environment at room temperature in a separate feed bottle using dried degassed toluene and volumetric flasks.
Imine (18.0 g, 75.2 mmol) was dissolved in toluene to provide 65.3 mL of the solution. Nitroalkane (13.44 g, 78.4 mmol) and catalyst (122 mg, 0.214 mmol) were dissolved in toluene to give total 38.2 mL total volume of the desired solution. The initial charge (38.4 mL total volume) was prepared by dissolving nitroalkane (287 mg, 1.67 mmol) and catalyst (62.7 mg, 0.111 mmol). Reagent feed solutions were transferred to feed vessels.
Intermittent flow experiment
The following steps were performed to start the cycle:
The circulator was turned on and set to temperature.
Utilities were turned on (nitrogen, vacuum, valve to humidifier was opened).
Premeasured material was charged into the reactor.
Once the material was at temperature (−15 °C), DeltaV sequence was initiated
One Automated Cycle of the Intermittent Flow Reactor Is As Follows
The catalyst/nitroalkane solution (2.39 mL total volume, 0.84 g nitroalkane, 7.6 mg catalyst) was pumped into the stirred tank reactor (250 mL volume, with overhead stirring, Figure 7) through a cooling heat exchanger over a 53 s period and entered the reactor at −20 °C temperature (Figure 7). Once finished the imine solution (4.08 mL total volume, 1.125 g imine) was introduced separately and pumped slowly into the reactor with a predetermined flow rate during the cycle (over a period of 23.5 min). The solution mixed in the reactor for 5.5 min after the addition of the imine was complete to ensure full conversion of imine into the aza-Henry product. The overall reaction time amounted to 30 min. Upon conclusion of each reaction cycle, 90% of the slurry in the reactor was immediately pulled out of the reactor from a dip tube and into a transfer zone (wide diameter tubing) using trapped vacuum. 10% of the slurry was left behind in the reactor as a heel because the dip tube did not go completely to the bottom. Slurry moves at high velocity through 1/4″ o.d. 1/8″ i.d. PFA tubing. The “transfer zone” for intermittent pumping of slurry at small scale was introduced earlier.19b The transfer zone was pressurized with nitrogen and the slurry was pushed to the filter. The filtration was performed in a 5 min time period. There was a sample port located between the first transfer zone and the filter. The slurry sample (~1.0 mL) was taken from the port and filtered/washed manually each cycle (samples were taken using a Whatman Autovial Syringeless Filter). The composition of solid product and filtrate, enantioselectivity, diastereoselectivity of the product, and levels of impurities were determined by HPLC. The filtrate (38.4 mL total volume, 0.287 g of nitroalkane, 63 mg of catalyst) immediately flowed out of the filter and directly back to the reactor. Depending on the established recycle ratio, only the desired amount of filtrate is recycled back to the reactor, in this case 97% of the filtrate. The ratio between the recycled and discarded filtrate was adjustable. The recycled filtrate was passed through a cooling loop before reentering the reactor. The desired amount of filtrate waste (1.041 g total, 9 mg nitroalkane, 2 mg catalyst) was then pushed into a waste container. Five minutes was designated for the cool down of the recycled filtrate before initiation of the next cycle. This completes one cycle. The automation repeats these cycles for the number designated by the operator. The overall cycle time in the reactor was extremely consistent because of the automated sequence. When the experiment was complete, DeltaV was stopped and circulator was turned off. Material from the filtrate waste bottle, filter, and reactor were collected and measured.
Figure 7.

Reactor with a cooling jacket for the continuous crystallization process and peristaltic pumps used for the continuous feeding of solutions.
After 16 cycles 25.1 g of the product was isolated (81.3% yield, 88.5% ee, and purity of the product was 98.6% HPLC area). Analytical data of the isolated product is in accordance with the previous report.
Supplementary Material
Scheme 5.
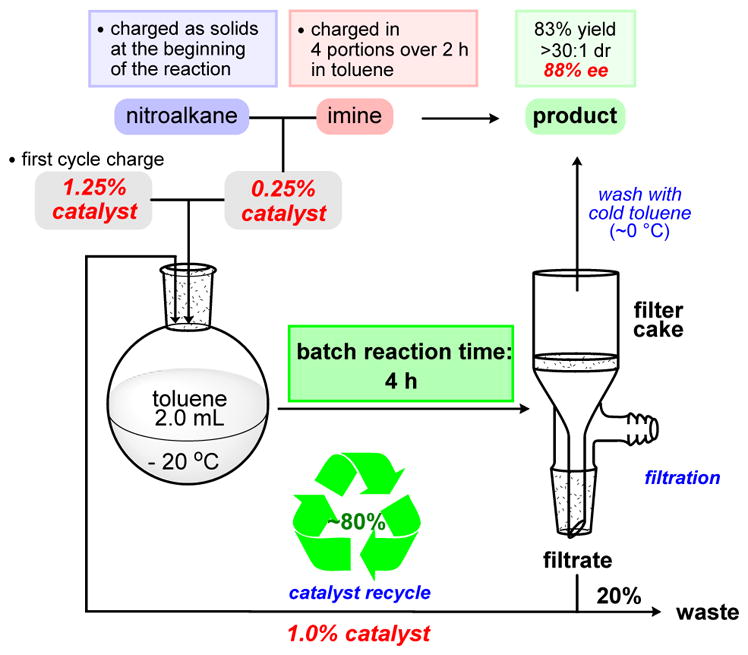
Sequence with 1.25% of the Catalyst and 80% Recycle
Acknowledgments
Support from the Lilly Innovation Fellowship Award (LIFA) for SVT is gratefully acknowledged. The authors also thank James R. Stout and Megan M. Wong for their extensive help with the continuous campaign. Catalyst development was supported by funding from the National Institutes of Health (GM 084333).
Footnotes
Additional engineering drawings, calculations, HPLC analytical data for the continuous campaign and preliminary safety evaluation.
Notes
The authors declare no competing financial interest
This Supporting Information is available free of charge on the ACS Publications website DOI: 10.1021/acs.oprd.5b00245.
References
- 1.Alvarez-Builla J, Vaquero JJ, Barluenga J, editors. Modern Heterocyclic chemistry. Wiley; 2011. Heterocyclic reviews. [Google Scholar]; Katritzky AR, Ramsden CA, Joule JA, Zhdankin VV, editors. Handbook of heterocyclic chemistry. 3. Oxford; New York: 2011. [Google Scholar]
- 2.(a) Lovering F, Bikker J, Humblet C. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; (b) Marson CM. Chem Soc Rev. 2011;40:5514–5533. doi: 10.1039/c1cs15119c. [DOI] [PubMed] [Google Scholar]
- 3.Recent examples from the literature using chiral auxiliaries: Maddess ML, Scott JP, Alorati A, Baxter C, Bremeyer N, Brewer S, Campos K, Cleator E, Dieguez-Vazquez A, Gibb A, Gibson A, Howard M, Keen S, Klapars A, Lee J, Li J, Lynch J, Mullens P, Wallace D, Wilson R. Org Process Res Dev. 2014;18:528–538.Singer RA, Ragan JA, Bowles P, Chisowa E, Conway BG, Cordi EM, Leeman KR, Letendre LJ, Sieser JE, Sluggett GW, Stanchina CL, Strohmeyer H, Blunt J, Taylor S, Byrne C, Lynch D, Mullane S, O’Sullivan MM, Whelan M. Org Process Res Dev. 2014;18:26–35.Bellizzi ME, Bhatia AV, Cullen SC, Gandarilla J, Kruger AW, Welch DS. Org Process Res Dev. 2014;18:303–309.
- 4.Recent examples from the literature using classic and metal mediated dynamic kinetic resolutions: Prashad M, Liu Y, Shieh WC, Schaefer F, Hu B, Girgis M. Org Process Res Dev. 2015;19:290–295.Xu F, Zacuto MJ, Kohmura Y, Rosen J, Gibb A, Alam M, Scott J, Tschaen D. Org Lett. 2014;16:5422–5425. doi: 10.1021/ol502661g.Lucas BS, Fisher B, McGee LR, Olson SH, Medina JC, Cheung E. J Am Chem Soc. 2012;134:12855–12860. doi: 10.1021/ja305123v.
- 5.Recent examples from the literature using metal mediated hydrogenations: Mangion IK, Chen C, Li H, Maligres P, Chen Y, Christensen M, Cohen R, Jeon I, Klapars A, Krska S, Nguyen H, Reamer RA, Sherry BD, Zavialov I. Org Lett. 2014;16:2310–2313. doi: 10.1021/ol500971c.Fandrick KR, Mulder JA, Patel ND, Gao J, Konrad M, Archer E, Buono FG, Duran A, Schmid R, Daeubler J, Desrosiers JN, Zeng X, Rodriguez S, Ma S, Qu B, Li Z, Fandrick DR, Grinberg N, Lee H, Bosanac T, Takahashi H, Chen Z, Bartolozzi A, Nemoto P, Busacca CA, Song JJ, Yee NK, Mahaney PE, Senanayake CE. J Org Chem. 2015;80:1651–1660. doi: 10.1021/jo502550h.DeBaillie AC, Magnus NA, Laurila ME, Wepsiec JP, Ruble JC, Petkus JJ, Vaid RK, Niemeier JK, Mick JM, Gunter TZ. Org Process Res Dev. 2012;16:1538–1543.
- 6.(a) Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. Science. 2010;329:305–309. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]; (b) Chung JYL, Zhong YL, Maloney KM, Reamer RA, Moore JC, Strotman H, Kalinin A, Feng R, Strotman NA, Xiang B, Yasuda N. Org Lett. 2014;16:5890–5893. doi: 10.1021/ol5028249. [DOI] [PubMed] [Google Scholar]; (c) Frodsham L, Golden M, Hard S, Kenworthy MN, Klauber DJ, Leslie K, Macleod C, Meadows RE, Mulholland KR, Reilly J, Squire C, Tomasi S, Watt D, Wells AS. Org Process Res Dev. 2013;17:1123–1130. [Google Scholar]; (d) Chung CK, Bulger PG, Kosjek B, Belyk KM, Rivera N, Scott ME, Humphrey GR, Limanto J, Bachert DC, Emerson KM. Org Process Res Dev. 2014;18:215–227. [Google Scholar]; (e) Peng Z, Wong JW, Hansen EC, Puchlopek-Dermenci ALA, Clarke HJ. Org Lett. 2014;16:860–863. doi: 10.1021/ol403630g. [DOI] [PubMed] [Google Scholar]; (f) Mangion IK, Sherry BD, Yin J, Fleitz FJ. Org Lett. 2012;14:3458–3461. doi: 10.1021/ol3014123. [DOI] [PubMed] [Google Scholar]
- 7.For reviews, see: Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r.Enders D, Niemeier O, Henseler H. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016.Erkkilä A, Majander I, Pihko PM. Chem Rev. 2007;107:5416–5470. doi: 10.1021/cr068388p.Davie EAC, Mennen SM, Xu Y, Miller SJ. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w.Volla CMR, Atodiresei I, Rueping M. Chem Rev. 2014;114:2390–2431. doi: 10.1021/cr400215u.Moyano A, Rios R. Chem Rev. 2011;111:4703–4832. doi: 10.1021/cr100348t.
- 8.(a) Wan ZK, Chenail E, Li HQ, Kendall C, Wang Y, Gingras S, Xiang J, Massefski WW, Mansour TS, Saiah E. J Org Chem. 2011;76:7048–7055. doi: 10.1021/jo200958a. [DOI] [PubMed] [Google Scholar]; (b) Huang X, Broadbent S, Dvorak C, Zhao SH. Org Process Res Dev. 2010;14:612–616. [Google Scholar]; (c) Belyk KM, Xiang B, Bulger PG, Leonard WR, Jr, Balsells J, Yin J, Chen C. Org Process Res Dev. 2010;14:692–700. [Google Scholar]; (d) Scott JP, Ashwood MS, Brands KMJ, Brewer SE, Cowden CJ, Dolling UH, Emerson KM, Gibb AD, Goodyear A, Oliver SF, Stewart GW, Wallace DJ. Org Process Res Dev. 2008;12:723–730. [Google Scholar]; (e) Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2008;130:16358–16365. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For reviews see: Malet-Sanz L, Susanne F. J Med Chem. 2012;55:4062–4098. doi: 10.1021/jm2006029.Hartman RL, McMullen JP, Jensen KF. Angew Chem, Int Ed. 2011;51:7502–7519. doi: 10.1002/anie.201004637.Tsubogo T, Ishiwata T, Kobayashi S. Angew Chem Int Ed. 2013;52:6590–6604. doi: 10.1002/anie.201210066.Mak XY, Laurino P, Seeberger PH. Beilstein J Org Chem. 2009;5:19. doi: 10.3762/bjoc.5.19.Wegner J, Ceylan S, Kirschning A. Adv Synth Catal. 2012;354:17–57.Mason BP, Price KE, Steinbacher JL, Bogdan AR, McQuade DT. Chem Rev. 2007;107:2300–2318. doi: 10.1021/cr050944c.Ley SV. Chem Record. 2012;12:378–390. doi: 10.1002/tcr.201100041.Gutmann B, Cantillo D, Kappe CO. Angew Chem, Int Ed. 2015;54:6688–6728. doi: 10.1002/anie.201409318.
- 10.Recent examples from industry: Mascia S, Heider PL, Zhang H, Lakerveld R, Benyahia B, Barton PI, Braatz RD, Cooney CL, Evans JMB, Jamison TF, Jensen KF, Myerson AS, Trout BL. Angew Chem Int Ed. 2013;52:12359–12362. doi: 10.1002/anie.201305429.White TD, Alt CA, Cole KP, Groh JM, Johnson MD, Miller RD. Org Process Res Dev. 2014;18:1482–1491.LaPorte TL, Spangler L, Hamedi M, Lobben P, Chan SH, Muslehiddinoglu J, Wang SSY. Org Process Res Dev. 2014;18:1492–1502.
- 11.(a) Lee SL, O’Connor TF, Yang X, Cruz CN, Chatterjee S, Madurawe RD, Moore CMV, Yu LX, Woodcock J. J Pharm Innov Chem. 2015;10:191. [Google Scholar]; (b) Poechlauer P, Colberg J, Fisher E, Jansen M, Johnson MD, Koenig SG, Lawler M, Laporte T, Manley J, Martin B, O’Kearney-McMullan A. Org Process Res Dev. 2013;17:1472–1478. [Google Scholar]
- 12.Ono N. The Nitro Group in Organic Synthesis; John Wiley & Sons, Inc; New York, USA: 2001. [Google Scholar]
- 13.Anderson JC, Blake AJ, Howell GP, Wilson C. J Org Chem. 2005;70:549–555. doi: 10.1021/jo048304h. [DOI] [PubMed] [Google Scholar]
- 14.(a) Yamada K, Harwood SJ, Groger H, Shibasaki M. Angew Chem Int Ed. 1999;38:3504–3506. doi: 10.1002/(sici)1521-3773(19991203)38:23<3504::aid-anie3504>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (b) Knudsen KR, Risgaard T, Nishiwaki N, Gothelf KV, Jorgensen KA. J Am Chem Soc. 2001;123:5843–5844. doi: 10.1021/ja010588p. [DOI] [PubMed] [Google Scholar]
- 15.(a) Davis T, Wilt JC, Johnston JN. J Am Chem Soc. 2010;132:2880–2882. doi: 10.1021/ja908814h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wilt JC, Pink M, Johnston JN. Chem Commun. 2008;35:4177–4179. doi: 10.1039/b808393b. [DOI] [PubMed] [Google Scholar]; (c) Singh A, Johnston JN. J Am Chem Soc. 2008;130:5866–5867. doi: 10.1021/ja8011808. [DOI] [PubMed] [Google Scholar]; (d) Singh A, Yoder RA, Shen B, Johnston JN. J Am Chem Soc. 2007;129:3466–3467. doi: 10.1021/ja068073r. [DOI] [PubMed] [Google Scholar]; (e) Nugent BM, Yoder RA, Johnston JN. J Am Chem Soc. 2004;126:3418–3419. doi: 10.1021/ja031906i. [DOI] [PubMed] [Google Scholar]; (f) Shen B, Johnston JN. Org Lett. 2008;10:4397–4400. doi: 10.1021/ol801797h. [DOI] [PubMed] [Google Scholar]
- 16.(a) Rueping M, Antonchick AP. Org Lett. 2008;10:1731–1734. doi: 10.1021/ol8003589. [DOI] [PubMed] [Google Scholar]; (b) Nishiwaki N, Knudsen KR, Gothelf KV, Jorgensen KA. Angew Chem, Int Ed. 2001;40:2992–2995. doi: 10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Davis T, Johnston JN. Chem Sci. 2011;2:1076–1079. doi: 10.1039/C1SC00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.See also: Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. Org Lett. 2012;14:6322–6325. doi: 10.1021/ol303092v.Davis TA, Danneman MW, Johnston JN. Chem Commun. 2012;48:5578–5580. doi: 10.1039/c2cc32225k.Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125.Dobish MC, Johnston JN. Org Lett. 2010;12:5744–5747. doi: 10.1021/ol1025712.Hess AS, Yoder RA, Johnston JN. Synlett. 2006:147–149.
- 19.(a) White TD, Berglund KD, Groh JM, Johnson MD, Miller RD, Yates MH. Org Process Res Dev. 2012;16:939–957. [Google Scholar]; (b) Johnson MD, May SA, Calvin JR, Remacle J, Stout JR, Diseroad WD, Zaborenko N, Haeberle BH, Sun WM, Miller MT, Brennan J. Org Process Res Dev. 2012;16:1017–1038. [Google Scholar]
- 20.(a) Quon JL, Zhang H, Alvarez A, Evans J, Myerson AS, Trout BL. Cryst Growth Des. 2012;12:3036–3044. [Google Scholar]; (b) Hou G, Power G, Barrett M, Glennon B, Morris G, Zhao Y. Cryst Growth Des. 2014;14:1782–1793. [Google Scholar]
- 21.Zhang H, Lakerveld R, Heider PL, Tao M, Su M, Testa CJ, D’Antonio AN, Barton PI, Braatz RD, Trout BL, Myerson AS, Jensen KF, Evans JMB. Cryst Growth Des. 2014;14:2148–2157. [Google Scholar]
- 22.Vara B, Mayasundari A, Tellis J, Danneman M, Arredondo V, Davis T, Min J, Finch K, Guy RK, Johnston JN. J Org Chem. 2014;79:6913–6938. doi: 10.1021/jo501003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis T, Vilgelm AE, Richmond A, Johnston JN. J Org Chem. 2013;78:10605–10616. doi: 10.1021/jo401321a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Technical data (product safety assessment) for several nitroalkanes (e.g. nitromethane, nitroethane) could be found at www.dow.com (accessed Dec 10, 2015): http://msdssearch.dow.com/PublishedLiteratureDOWCOM/dh_0914/0901b80380914379.pdf?filepath=productsafety/pdfs/noreg/233-00287.pd...&fromPage=GetDoc http://msdssearch.dow.com/PublishedLiteratureDOWCOM/dh_091d/0901b8038091d474.pdf?filepath=productsafety/pdfs/noreg/233-00579.pdf&fromPage=GetDoc
- 25.Markofsky SB. Nitro Compounds, Aliphatic Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KgaA; 2000. [Google Scholar]
- 26.(a) Anderson NG. Org Process Res Dev. 2012;16:852–869. [Google Scholar]; (b) Hessel V, Kralisch D, Kockmann N, Noel T, Wang Q. ChemSusChem. 2013;6:746–789. doi: 10.1002/cssc.201200766. [DOI] [PubMed] [Google Scholar]; (c) Baxendale IR. J Chem Technol Biotechnol. 2013;88:519–552. [Google Scholar]; (d) Baxendale IR, Braatz RD, Hodnett BK, Jensen KF, Johnson MD, Sharratt P, Sherlock JP, Florence AJ. J Pharm Sci. 2015;104:781–791. doi: 10.1002/jps.24252. [DOI] [PubMed] [Google Scholar]
- 27.(a) Li B, Guinness S. Managing Hazardous Reactions and Compounds in Process Chemistry. In: Pesti JA, Abdel-Magid AF, editors. ACS Symposium Series. American Chemical Society; Washington, DC: 2014. pp. 383–402. [Google Scholar]; (b) Proctor L, Dunn PJ, Hawkins JM, Wells AS, Williams MT. In: Green Chemistry in the Pharmaceutical Industry. Dunn PJ, Wells AS, Williams MT, editors. Wiley-VCH; Weinheim: 2010. pp. 221–242. [Google Scholar]; (c) Kopach ME, Roberts DJ, Johnson MD, McClary Groh J, Adler JJ, Schafer JP, Kobierski ME, Trankle WG. Green Chem. 2012;14:1524–1536. [Google Scholar]; (d) Pedersen MJ, Holm TL, Rahbek JP, Skovby T, Mealy MJ, Dam-Johansen K, Kiil S. Org Process Res Dev. 2013;17:1142–1148. [Google Scholar]
- 28.Barthe P, Guermeur C, Lobet O, Moreno M, Woehl P, Roberge DM, Bieler N, Zimmermann B. Chem Eng Technol. 2008;31:1146–1154. [Google Scholar]
- 29.Adams TA, II, Pascall A. Chem Eng Technol. 2012;35(7):1153–1170. [Google Scholar]
- 30.Stiefel FJ. Synthesis of 2-phenyl-1,3-propanediol. 5,072,056. US Patent. 1991 Dec 10;
- 31.Davis T, Dobish M, Schwieter KE, Chun AC, Johnston JN. Org Synth. 2014;89:380–393. doi: 10.15227/orgsyn.089.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.100% recycle only designates that all collected filtrate was reused in the following step, since some catalyst can be retained with the solid product after filtration leading to loss of the catalyst after every iteration.The potential deactivation pathways of the catalyst were examined in this case. For example, upon recycling, some of the catalyst can be either doubly protonated by a strong acid or increasingly solvated by a weaker hydrogen bond donor. In this form, the catalyst would exit the catalytic cycle, leading to a diminished ratio of the active catalyst and reduced stereoselectivity. To test this hypothesis stirring the residual recycle with inorganic base (i.e., potassium carbonate) or with mildly basic Amberlyst 21 resin (dimethylamine functionality) before resubmitting it to the next iteration was attempted. Mixed and inconclusive results were observed during these experiments and did not provide direct evidence that double protonation of the catalyst affects the yield or stereoselectivity of the process.
- 33.In automated intermittent flow process necessity to maintain a constant volume of the solvent in the reactor leads to a recycling percentage not equal to 100%. A percentage of the liquid phase remains wetted on the solid product, and wash with solvent would result in dilution of the recycle stream. Therefore the automated intermittent flow process would need to pump a small amount of fresh catalyst into the reactor along with the reagent feeds to maintain constant catalyst loading over time.
- 34.Observed fluctuations in enantioselectivity of the product could be associated with manual handling, and in case of automated intermittent flow processes this variability is eliminated.
- 35.The catalyst in the previous sequences underwent a series of manipulations: concentration, drying, and dissolution. While there is no direct evidence that these iterative operations were detrimental to the catalyst activity, the new sequence gives an opportunity to constantly maintain the catalyst in solution under invariable reaction conditions (~ −20 °C).
- 36.The overall phenomenon of increasing ee upon recycle at the beginning of the process could be attributed to the difference in solubility of the product racemate and product single enantiomer. The racemate is less soluble than a single enantiomer, and the ee eventually equilibrates upon achieving a steady state. Similar results observed in intermittent-flow continuous runs (For example in 24 cycle run: after the first cycle solid product is 82% ee, but selectivity of the product in the filtrate is 91%. However, moving from cycle to cycle the solid product becomes more enantiomerically enriched: after the second cycle it is 86% for the solid and 90% in filtrate, third cycle is 88% for solid, and 89% for the filtrate).
- 37.The analysis and comparison of diastereoselectivity data in the solid product and in the filtrate (full numbers for the continuous runs presented in the supporting information) shows that the ratio in the filtrate is much lower than in the solid product from the beginning of the process, meaning the overall diastereoselectivity of the reaction is lower than observed in the solid product during the first several cycle. Since filtrate gets recycled, diastereomer gets also recycled, and its concentration in the reactor increases and results in crystallization of the larger amounts of the diastereomer with the solid product. By fourth cycle in continuous system this ratio between solid product and filtrate equilibrates, and relative stabilization of the diastereomeric ratio is observed. Processes with recycle usually require several turnovers to reach steady state.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.