

Abstract
Methamphetamine (METH) is a neurotoxic drug of abuse that damages the dopamine (DA) neuronal system in a highly delimited manner. The brain structure most affected by METH is the caudate–putamen (CPu) where long-term DA depletion and microglial activation are most evident. Even damage within the CPu is remarkably heterogenous with lateral and ventral aspects showing the greatest deficits. The nucleus accumbens (NAc) is largely spared of the damage that accompanies binge METH intoxication. Increases in cytoplasmic DA produced by reserpine, L-DOPA or clorgyline prior to METH uncover damage in the NAc as evidenced by microglial activation and depletion of DA, tyrosine hydroxylase (TH), and the DA transporter. These effects do not occur in the NAc after treatment with METH alone. In contrast to the CPu where DA, TH, and DA transporter levels remain depleted chronically, DA nerve ending alterations in the NAc show a partial recovery over time. None of the treatments that enhance METH toxicity in the NAc and CPu lead to losses of TH protein or DA cell bodies in the substantia nigra or the ventral tegmentum. These data show that increases in cytoplasmic DA dramatically broaden the neurotoxic profile of METH to include brain structures not normally targeted for damage by METH alone. The resistance of the NAc to METH-induced neurotoxicity and its ability to recover reveal a fundamentally different neuroplasticity by comparison to the CPu. Recruitment of the NAc as a target of METH neurotoxicity by alterations in DA homeostasis is significant in light of the important roles played by this brain structure.
Keywords: dopamine, methamphetamine, neurotoxicity, nucleus accumbens, tyrosine hydroxylase, ventral tegmentum
Methamphetamine (METH) causes persistent neurochemical deficits within the dopamine (DA) neuronal system. The neurotoxicity of METH is remarkable in that lateral aspects of the caudate–putamen (CPu) show greater damage (Fukui et al. 1986; Hirata et al. 1996; Harvey et al. 2000a) and microglial activation (Thomas et al. 2004b) than more medial aspects. Other elements of the striatum, including the nucleus accumbens (NAc), are quite resistant to METH neurotoxicity (Eisch et al. 1992; Cass 1997; Haughey et al. 2000) although some evidence of METH-induced DA deficits within the NAc core has been presented (Broening et al. 1997; Wallace et al. 1999). DA-containing neurons of the substantia nigra (SNc) show limited damage after METH (Sonsalla et al. 1996; Brown et al. 2006), effects that recover without neuronal losses (Harvey et al. 2000b). The mechanisms determining the remarkably heterogenous pattern of METH-induced neurotoxicity are not understood, but brain-regional differences in expression of the DA transporter (DAT) and the vesicular monoamine transporter (VMAT), and the manner in which they interact with METH to cause DA release may play a role (Volz et al. 2007).
Dopamine has long been implicated as a key factor in METH neurotoxicity. Wagner et al. (1983) first showed that inhibition of tyrosine hydroxylase (TH) with α-methyl-p-tyrosine prevented METH-induced neurotoxicity. Reserpine, which disrupts vesicle storage of DA while leaving the METH-releasable pool intact, enhances its neurotoxicity (Wagner et al. 1983; Albers and Sonsalla 1995; Thomas et al. 2008). The combined effects of METH to cause DA release and block its reuptake expose extracellular DA to non-enzymatic degradation by reactive oxygen species (Yamamoto and Bankson 2005; Cadet et al. 2007). Oxidant attack on DA leads to the formation of DA quinone (Graham 1978; Nappi and Vass 2001), a redox-active species implicated in METH-induced toxicity (LaVoie and Hastings 1999) and which alters several of the same critical proteins inhibited by METH to include TH (Kuhn et al. 1999) and the DAT (Whitehead et al. 2001; Park et al. 2002). DA quinone also causes microglial activation (Le et al. 2001; Kuhn et al. 2006; Thomas et al. 2006), an effect that is emerging as an important element of METH toxicity. The ability of reserpine, which reduces steady state DA to near zero, to increase METH toxicity points to the fact that METH need only mobilize a very small pool of cytoplasmic DA to exert damaging effects.
We recently observed that increases in cytoplasmic DA with L-DOPA, clorgyline, or reserpine led to significant enhancement of METH toxicity within the CPu (Thomas et al. 2008). In this study, we confirm the resistance of DA nerve endings of the NAc to METH and reveal novel elements of its neurotoxic cascade by showing that increases in cytoplasmic DA uncover a METH neurotoxic profile in NAc. DA nerve ending damage in the NAc also shows partial recovery after METH, in sharp contrast to the persistence of damage in the CPu.
Materials and methods
Materials
(+)Methamphetamine hydrochloride, pentobarbital, horseradish peroxidase (HRP)-conjugated isolectin B4 (from Griffonia simplicifolia), 3,3′-diaminobenzidine, L-DOPA, carbidopa, clorgyline, and p-formaldehyde, Triton X-100, Tween 20, DA, methanol, EDTA, all buffers, and HPLC reagents were purchased from Sigma-Aldrich (St Louis, MO, USA). CitriSolv and Permount were products of Fisher Scientific (Pittsburgh, PA, USA). Bicinchoninic acid protein assay kits were obtained from Pierce (Rockford, IL, USA). Polyclonal antibodies against rat TH were produced as previously described (Kuhn and Billingsley 1987). Monoclonal antibodies against rat DAT were generously provided by Dr Roxanne A. Vaughan (University of North Dakota, Grand Forks, ND, USA). HRP-conjugated secondary antibodies were obtained from Amersham (Piscataway, NJ, USA). Alexa Fluor 488-conjugated secondary antibodies were obtained from Invitrogen (Carlsbad, CA, USA).
Animals
Female C57BL/6 mice (Harlan, Indianapolis, IN, USA) weighing 20–25 g at the time of experimentation were housed five per cage in large shoebox cages in a light- and temperature-controlled room. Mice had free access to food and water. The Institutional Care and Use Committee of Wayne State University approved the animal care and experimental procedures. All procedures were also in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Pharmacological and physiological procedures
Mice were exposed to a neurotoxic regimen of METH comprised of four injections of 5 mg/kg i.p. with a 2 h interval between each injection. This METH regimen is known to cause microglial activation and DA nerve ending damage (Thomas et al. 2004b). To assess the effect of alterations in the newly synthesized pool of DA on METH toxicity, mice were treated with (i) L-DOPA, the immediate precursor to DA, 1 h before the first and third METH injections in a dose of 50 mg/kg along with carbidopa (25 mg/kg) to inhibit peripheral decarboxylase enzymes; (ii) clorgyline (10 mg/kg), an inhibitor of monoamine oxidase A, 1 h before the METH regimen; or (iii) reserpine (2.5 mg/kg) 24 h prior to METH. Controls for drug pre-treatments and METH received i.p. injections of physiological saline on the same schedule used for each respective compound. All injections were given via the i.p. route. Mice were killed at various times after METH treatment (specified below). Body temperature was monitored by telemetry using IPTT-300 implantable temperature transponders from Bio Medic Data Systems, Inc. (Seaford, DE, USA). Temperatures were recorded every 20 min non-invasively using the DAS-5001 console system from Bio Medic.
Determination of nucleus accumbens dopamine content
Depletion of DA after METH treatment is widely used as an index of METH-induced toxicity to DA nerve endings and faithfully reflects other measures of DA nerve ending damage caused by METH, such as reduced TH immunoreactivity or reduced ligand binding to the DAT. The NAc was dissected bilaterally from 2 mm thick slices of brain by micropunch (1.5 mm diameter) 2, 7, or 14 days after treatments and stored at −80°C. Frozen tissues were weighed and sonicated in five volumes of 0.16 N perchloric acid at 4°C. Insoluble protein was removed by centrifugation (18 000 × g) and DA was determined by HPLC with electrochemical detection. Values for DA are reported as ng/mg tissue (wet weight).
Determination of tyrosine hydroxylase and dopamine transporter protein levels by immunoblotting
The effects of the pharmacological treatments on expression of TH and DAT proteins were determined by immunoblotting. Mice were killed by decapitation 2, 7, or 14 days after treatments and the CPu, NAc, and SNc/ventral tegmentum (VTA) were dissected bilaterally from 3 mm thick tissue slices of brain by micropunch (1.5 mm diameter). Tissue was stored at −80°C. Frozen tissue was disrupted by sonication in 1% sodium dodecyl sulfate at 95°C and insoluble material was sedimented by centrifugation (18,000 × g). Protein was determined by the bicinchoninic acid method and equal amounts of protein (20 μg/lane for NAc and CPu and 10 μg/lane for SNc) were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then electroblotted to nitrocellulose. Blots were blocked in Tris-buffered saline containing Tween 20 (0.1% v/v) and 5% non-fat dry milk for 1 h at 23°C. Anti-TH (1 : 2000) or anti-DAT (1 : 1000) was added to blots and allowed to incubate for 16 h at 4°C. Blots were washed 3x in Tris-buffered saline to remove unreacted primary antibodies and then incubated with species-appropriate HRP-conjugated anti-IgG secondary antibody (1 : 4000) for 1 h at 23°C. Immunoreactive bands were visualized by enhanced chemiluminescence and the relative densities of TH- and DAT-reactive bands were determined by scanning films on an Alpha Innotech (San Leandro, CA, USA) AlphaImager HP using automatic spot detection.
Lectin histochemical staining of microglia
Microglial activation was assessed by staining fixed brain sections with HRP-conjugated ILB4 as developed by Streit (1990) and as previously described in our studies with METH (Thomas et al. 2004b, 2008). At the time of killing, mice were deeply anesthetized with pentobarbital (120 mg/kg) and perfused transcardially with ice-cold 4% p-formaldehyde in phosphate-buffered saline (PBS). Brains were removed and stored overnight in fixative at 4°C. Sections of 50-μm thickness were cut through the NAc (approximately +1.7 through +0.9 mm with respect to Bregma) as well as through the SNc/VTA. Sections were floated into PBS containing 0.3% H2O2 for 30 min, washed once in PBS + 0.1% Triton X-100, and then incubated in fresh PBS + 0.1% Triton X-100 for an additional 30 min. Microglia were labeled with HRP-conjugated ILB4 (10 μg/mL in PBS + 0.1% Triton X-100) overnight at 4°C. Excess ILB4 was removed by three washes with PBS + 0.1% Triton X-100 (5 min each) followed by a single wash in PBS before exposure to 3,3′-diaminobenzidine substrate (0.1 mg/mL) in PBS for 25 min. After three washes with PBS, all sections were transferred to glass slides, air dried, and dehydrated through a series of graded ethanol washes. Sections were incubated in Citrisolv for 5 min then coverslipped under Permount. For all pharmacological studies presented below, brain sections from drug-treated mice were processed simultaneously with controls to normalize staining among treatment groups. Microglial reactivity was viewed under the light microscope and the number of stained cells observed after various treatments was quantified using NIH (Bethesda, MD, USA) Image. To facilitate counting, a circle with a 1 mm diameter was drawn with its center over the anterior commissure. ILB4-positive microglia were counted in the lower half of this circle (area = 0.39 mm2) to include both shell and core regions of the NAc. Cell counts were made by a person blinded to the treatment conditions. Counts were taken from three independent sections from all like-treated mice, bilaterally, generating an average count for each treated subject.
Immunohistochemistry of tyrosine hydroxylase-containing neurons of the SNc and VTA
Dopamine cell bodies of the SNc and VTA, in the area of the parabrachial pigmented nucleus, were examined for treatment effects on cell number because these structures contain the cell bodies of origin of the DA nerve endings of the CPu and NAc, respectively. TH-positive neurons were identified by incubating fixed sections with polyclonal antibodies against TH (diluted 1 : 2000) overnight at 4°C in PBS + 0.1% Triton X-100. Unbound anti-TH was removed by three washes in PBS + 0.1% Triton X-100 and then sections were incubated with Alexa Fluor 488-conjugated goat anti-rabbit secondary antibodies overnight at 4°C. Sections were finally washed three times in PBS + 0.1% Triton X-100 and one wash in PBS and then wet mounted on microscope slides and coverslipped. TH-positive neurons were viewed and photographed using an Olympus (Center Valley, PA, USA) BX51 fluorescence microscope. TH-positive neurons were counted manually as described above for microglial cell counting in the area of the parabrachial pigmented nucleus to encompass elements of the SNc and VTA. This area corresponds to the area dissected for immunoblot analysis of TH protein levels.
Data analysis
The effects of drug treatments on NAc DA content and cell counts (activated microglia and TH-positive neurons in the SNc/VTA) were tested for significance by ANOVA. Individual treatment groups were compared with appropriate controls for DA and cell counts with a one-way ANOVA followed by Tukey’s Multiple Comparison Test in GraphPad prism 5 (GraphPad Software Inc., San Diego, CA, USA). Differences were considered significant if p < 0.05.
Results
The effects of METH on NAc DA levels are presented in Fig. 1. METH alone caused only a minor and non-significant reduction (~15%) in NAc DA 2 days after treatment. This result contrasts sharply with the effects of the same METH treatment regimen in the CPu where DA reductions reach 65–75% over the same time-course (Thomas et al. 2008). The effects of METH on NAc DA showed a time-dependent response and these results are also presented in Fig. 1. It can be seen that METH itself caused a slight but significant reduction in DA levels (i.e. to 73% of control) at 7 days and this response recovered to control levels by 14 days. Drugs that increase the cytoplasmic (or METH-releasable) pool of DA significantly enhance METH-induced neurotoxicity and microglial activation in the CPu (Thomas et al. 2008). Therefore, the effects of L-DOPA, clorgyline, and reserpine on DA levels in the NAc were tested to determine if METH neurotoxicity is extended anatomically under these conditions as well. It can be seen in Fig. 1 that each treatment significantly potentiated the effects of METH 2 days after treatment. L-DOPA or clorgyline in combination with METH depleted NAc DA by ~50%, and reserpine combined with METH to deplete DA by more than 80%.
Fig. 1.
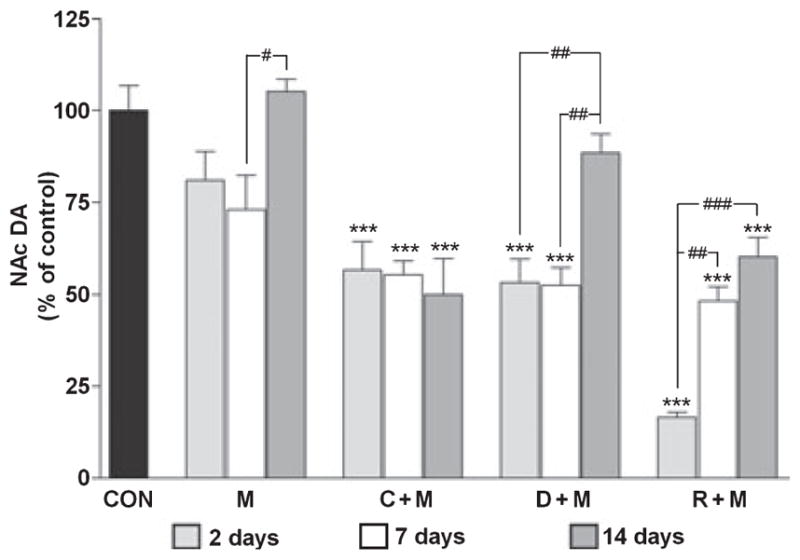
Effects of a neurotoxic METH regimen on NAc DA depletion when administered alone and in conjunction with clorgyline, L-DOPA, or reserpine. Mice (n = 5–8 per group) were treated with METH alone (M: 4 × 5 mg/kg; t = 0, 2, 4, and 6 h), or in conjunction with clorgyline (C + M: 10 mg/kg; t = −1 h), L-DOPA (D + M: 50 mg/kg, t = −1 and 3 h), or reserpine (R + M: 2.5 mg/kg; t = −24 h). NAc DA levels were determined at 2, 7, and 14 days post-METH. Results are presented as mean ± SEM relative to controls. Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test, and are indicated as follows: ***p < 0.001 relative to control (CON); #p < 0.05, ##p < 0.01, and ###p < 0.001 indicate significant differences between indicated treatment conditions.
The enhancement of METH toxicity caused by L-DOPA, clorgyline, and reserpine showed differential recoveries as well. At the 7 days time point, NAc DA levels in mice treated with clorgyline + METH or L-DOPA + METH remained at about the same levels (i.e. 50% of control) as seen at 2 days post-METH (Fig. 1) and each was significantly different from control (p < 0.001). By 14 days, the L-DOPA + METH group showed near-total recovery to control DA levels (i.e. 88% of control) whereas the clorgy-line + METH group did not show any recovery over the 2–14 days time span after treatment. DA levels in mice treated with reserpine + METH were intermediate to the L-DOPA and clorgyline pre-treatment groups. NAc DA content recovered to ~50% at 7 days and to ~60% at 14 days. DA levels in mice injected with clorgyline, L-DOPA or reserpine alone were not different from controls at 2, 7 or 14 days after treatments (data not shown).
In view of the novel response of the NAc DA system to METH by comparison to the CPu, it was important to confirm the effects of alterations in cytoplasmic DA on drug toxicity with the use of other markers for DA nerve ending status. Therefore, we measured TH and DAT protein levels in NAc but limited the analysis to groups treated with METH, reserpine + METH or clorgyline + METH because these latter drug combinations caused the most persistent enhancement of METH-induced depletion of DA (Fig. 1). In addition, mice treated with clorgyline + METH were studied only at the 2 days time point because recovery is not seen after this treatment. The results in Fig. 2a show that METH alone caused a slight reduction (~15%) in NAc TH content at 2 days. This response remained essentially the same at 7 days and returned to about 90% of control by 14 days. Although these effects trended toward reductions, they did not reach statistical significance at any time point. The combined treatment of mice with reserpine + METH resulted in a much greater reduction in NAc TH and these results are shown in Fig. 2a as well. It can be seen that reserpine + METH reduced TH levels by ~50% at 2 days. A partial recovery of TH expression was seen by 7–14 days when its levels reached about 70% of control. The effects of reserpine + METH were significantly different from control at 2 days (p < 0.001) and 14 days (p < 0.01). TH protein levels at 2 days were significantly lower than those at 7 and 14 days. The time-dependent recovery of TH in the reserpine + METH group was significant (p < 0.001). The effect of clorgyline + METH on NAc TH at 2 days post-treatment, included in Fig. 2a, was the same as reserpine in combination with METH and reduced TH by about 50% (p < 0.01). Reserpine or clorgyline alone did not change the expression of TH in NAc at any time point (data not shown).
Fig. 2.

Effects of reserpine or clorgyline on TH (a) and DAT (b) levels in NAc in mice treated with a neurotoxic METH regimen. Mice (n = 5–8 per group) were treated with METH alone, reserpine + METH, or clorgyline + METH as described. NAc TH and DAT protein levels were determined by western blot analysis at the indicated times post-METH and are presented as mean ± SEM relative to control. Mice treated with clorgyline + METH were tested only at the 2 days time point. Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test: **p < 0.01 and ***p < 0.001 relative to control; #p < 0.05 indicates significant differences between indicated treatment conditions.
The effects of reserpine or clorgyline in combination with METH on DAT levels in the NAc are shown in Fig. 2b. As seen with TH, METH alone did not significantly reduce DAT although there was a trend toward reduction at the 2 days time point only. The combined treatment of reserpine + METH caused a significant reduction in NAc DAT at 2 days (37% of control, p < 0.01), 7 days (29% of control, p < 0.01), and 14 days (35% of control, p < 0.01). Clorgy-line + METH reduced NAc DAT levels at the 2 days time point to the same extent as reserpine (35% of control, p < 0.01). NAc DAT levels did not show the tendency to recover as was seen for TH after the same treatments. Reserpine or clorgyline alone did not change the expression of DAT in NAc at any time point (data not shown).
Because DA and TH levels in the NAc show evidence of partial recovery after treatment with at least reserpine + METH, the effects of these treatments on CPu TH and DAT content was determined. Figure 3a shows that METH and reserpine + METH led to significant reductions in CPu TH content. METH alone depleted TH by 50–60% (p < 0.01), remaining relatively constant over the 2–14 days time course. The effect of reserpine + METH on CPu TH was slightly less pronounced (i.e. 40% depletion; p < 0.05) by comparison to METH alone, but the temporal pattern of response was the same. The effects of METH and reserpine + METH were significantly different from controls at all time points. The two treatment conditions did not differ with regard to the extent of TH depletion and the main effect of time was not significant, indicating that recovery of TH levels in the CPu did not occur. In contrast to the failure of reserpine to enhance METH-induced losses in CPu TH, clorgyline did potentiate the reduction in CPu TH caused by METH. TH was reduced to 25% of control (p < 0.001) 2 days after treatment with clorgyline + METH. The effects of reserpine or clorgyline + METH on CPu DAT levels are presented in Fig. 3b. METH alone caused a 75% reduction in CPu DAT (p < 0.001) and this effect was relatively constant over the 2–14 days time course. At no time did reserpine enhance the loss of CPu DAT caused by METH. Clorgyline had the same effect as reserpine when given in combination with METH, reducing CPu DAT to 35% of control at the 2 days time point (p < 0.01). Reserpine or clorgyline alone did not change the expression of TH or DAT in CPu at any time point (data not shown).
Fig. 3.

Effects of reserpine or clorgyline on TH (a) and DAT (b) levels in CPu in mice treated with a neurotoxic METH regimen. Mice (n = 5–8 per group) were treated with METH alone, reserpine + METH, or clorgyline + METH as described previously. TH and DAT levels in CPu were determined by western blot analysis at the indicated times post-METH and are presented as mean relative to control. Mice treated with clorgyline + METH were tested only at the 2 days time point. Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test: *p < 0.05, **p < 0.01, and ***p < 0.001 relative to control.
The effect of METH is generally thought to be restricted to the CPu, but some evidence of damage to the SNc has been reported at later times after higher-dose treatment (Sonsalla et al. 1996; Brown et al. 2006). Therefore, we examined the area of the SNc and VTA for evidence of microglial activation and TH content at 2, 7, and 14 days after METH treatment alone or in combination with reserpine. Neither treatment caused microglial activation in the SNc/VTA at any time point (data not shown). TH protein levels in the area of the SNc/VTA were assessed by immunoblotting and the results are presented in Fig. 4a. It is evident that METH or reserpine + METH did not cause reductions in TH levels at any time point. Figure 4b–d also shows TH-containing neurons in the area of the SNc and VTA 14 days after treatment of animals with control (Fig. 4b), METH (Fig. 4c) or reserpine + METH (Fig. 4d). None of the treatments changed the number of TH-containing neurons suggesting that the SNc and the VTA are relatively resistant to damage by conditions that significantly enhance METH effects in the NAc.
Fig. 4.

Effects of reserpine or clorgyline on TH (a) and DAT (b) levels in SNc/VTA in mice treated with a neurotoxic METH regimen. Mice (n = 5–8 per group) were treated with METH alone, or reserpine + METH as described previously. TH levels were determined by western analysis of SNc/VTA harvested at the indicated times post-METH and are presented as mean ± SEM relative to control (a). One-way ANOVA revealed no significant differences among the treatment conditions. (b–d) Representative fluorescent micrographs of TH-positive neurons in the area of the SNc and VTA 14 days after treatment with (b) control, (c) METH, or (d) reserpine + METH. Images captured in the area of the parabrachial pigmented nucleus, at the juncture of the SNc and VTA, reveal similar numbers (mean ± SEM) of TH-containing neurons (control = 102 ± 3; METH = 94 ± 2; and reserpine + METH = 97 ± 3). One-way ANOVA revealed no significant differences among the treatment conditions with regard to TH-positive cell number.
Microglial activation in the NAc after METH treatment showed a very different pattern by comparison to CPu where robust response is seen within 1–2 days (Thomas et al. 2004b). It can be seen in Fig. 5 that METH treatment alone (Fig. 5b) did not change NAc microglial status from control levels (Fig. 5a). However, when clorgyline (Fig. 5c), L-DOPA (Fig. 5d) or reserpine (Fig. 5e) was combined with METH, extensive microglial activation was evident in the NAc at 2 days. The increases in microglial activation caused by each pre-treatment drug in combination with METH were significantly different from controls (p < 0.001) and from METH alone (p < 0.001), but were not different from each other. Treatment with L-DOPA, clorgyline, or reserpine without METH did not cause changes in NAc microglial activation and a representative image from mice treated with reserpine alone is included in Fig. 5f. In agreement with previous results (Thomas et al. 2004b), microglial activation subsided to control levels by the 7 and 14 days time points (data not shown). It can also be seen in Fig. 5 that the enhancement of METH-induced microglial activation caused by L-DOPA, clorgyline, and reserpine extended in a ventral direction beyond the NAc and was evident in areas where DA fibers of passage in the median forebrain bundle and the olfactory tubercle traverse at the level of the NAc. These effects did not occur in control mice or in mice treated with METH alone (data not shown).
Fig. 5.

Effects of clorgyline, L-DOPA, and reserpine on microglial activation in NAc caused by a neurotoxic METH regimen. Mice (n = 3–5 per group) were treated as described and analyzed for microglial activation in the NAc 48 h after the last METH injection. Microglia counts were obtained as described in the Materials and Methods and are presented as mean ± SEM. Treatment conditions and microglia counts for each panel are (a) control (5 ± 1), (b) METH (15 ± 2), (c) clorgyline + METH (61 ± 5), (d) L-DOPA + METH (58 ± 6), (e) reserpine + METH (66 ± 6), and (f) reserpine only (8 ± 1). Significant differences were determined via one-way ANOVA followed by Tukey’s multiple comparison test: p < 0.001 for clorgyline + METH, L-DOPA + METH and reserpine + METH relative to control. No significant differences were determined for METH or reserpine only relative to control (p > 0.05).
The effects of the treatments described above on body temperature were identical to those reported previously (Thomas et al. 2008) using independent but identically treated groups of mice. Therefore, these data are not presented in this report.
Discussion
Methamphetamine causes long-term changes in CNS structure and function in humans (Volkow et al. 2001; Thompson et al. 2004; McCann et al. 2008) and in animal models of neurotoxicity (O’Callaghan and Miller 1994; Yamamoto and Bankson 2005; Cadet et al. 2007; Fleckenstein et al. 2007). The brain area most affected by METH is the CPu where persistent loss-of-function changes are seen in TH, DAT, and VMAT. Collectively, modification of these important proteins depletes DA from nerve endings of the CPu. Even though the CPu is a primary target for METH, its neurotoxicity is remarkably heterogeneous throughout striatum (Ricaurte et al. 1980; Fukui et al. 1986; Seiden et al. 1988). DA nerve endings in the NAc are much more resistant to the damaging effects of METH by comparison to the CPu. METH-induced reductions in NAc DA levels are 20–40% of what is seen in CPu (Morgan and Gibb 1980; O’Dell et al. 1991; Sabol et al. 2001; Wallace et al. 2001; Davidson et al. 2007) while DAT binding sites (Eisch et al. 1992, 1996) and TH activity (Haughey et al. 1999) are not changed in the NAc. The METH-induced formation of high molecular weight DAT complexes that occurs in CPu (Baucum et al. 2004) does not occur in the NAc (Hadlock et al. 2009), and a non-toxic METH dosing regimen that induces behavioral sensitization lowers DAT density in CPu but not in the NAc (Bjorklund et al. 2008). The evoked release of DA is also reduced in CPu but not in the NAc by prior exposure to a neurotoxic regimen of METH (Cass 1997). In contrast to these results showing greater sensitivity to METH toxicity in the CPu versus the NAc, Broening et al. (1997) found that METH reduces TH immunoreactivity within the core of the NAc and Pereira et al. (2006) reported that CPu TH levels were not reduced by METH. DA neurons of the SNc and VTA are generally thought to be resistant to the damaging effects of METH, although contrasting results have been published. For instance, METH causes losses of TH-containing neurons of the SNc in some studies (Sonsalla et al. 1996; Brown et al. 2006) while others show no losses of TH (Theodore et al. 2006; Boger et al. 2007) or DAT (Brunswick et al. 1992) and few, if any, signs of neuronal degeneration or gliosis (O’Callaghan and Miller 1994; Sriram et al. 2006). In vervet monkeys, nigrostriatal DA deficits caused by METH recover without SNc cell loss (Harvey et al. 2000b).
Dopamine is an essential participant in METH neurotoxicity. Reductions in cytoplasmic DA protect against METH neurotoxicity (Wagner et al. 1983; Schmidt et al. 1985; Albers and Sonsalla 1995). Increases in cytoplasmic DA caused by reserpine enhance METH toxicity in the CPu (Wagner et al. 1983; Albers and Sonsalla 1995; Thomas et al. 2008) as does L-DOPA, which increases DA levels, and clorgyline, which inhibits its enzymatic inactivation (Thomas et al. 2008). In the present studies, DA, TH, and DAT levels in NAc were not altered by a METH regimen that depletes them in the CPu by 65–75%. Thus, increases in the METH-releasable pool of DA compromises the resistance of the NAc to METH and reveals significant toxicity in this structure. Another element of METH action in the NAc is quite distinct from what is seen in the CPu. The enhanced depletion of DA and TH in the NAc after treatment of mice with reserpine + METH (and to a lesser extent after L-DOPA + METH) shows a partial recovery. By contrast, the greater depletions of DA, TH, and DAT from CPu seen after these same treatments show no evidence of recovery. Therefore, it appears that the NAc expresses a plasticity not seen in the CPu after METH treatment. The mechanisms regulating recovery from METH-induced neurotoxicity have not been investigated but it is known that sprouting of DA nerve terminals does occur in the CPu in response to MPTP (Bezard et al. 2000) or 6-hydroxydopamine (Georgievska et al. 2002; Stanic et al. 2003), or after penetrating injury (Batchelor et al. 1999). On the other hand, it may be possible that the differing responses of the NAc and CPu to reserpine + METH are dependent on how toxicity is assessed. Partial recoveries in DA and TH observed in NAc but not in CPu after treatment with reserpine + METH may reflect the ability of reserpine to up-regulate TH expression in the VTA but not in the SNc (Pasinetti et al. 1990). Extending this point of view, DAT levels may serve as a more sensitive marker of METH toxicity than TH and changes in DAT caused by reserpine (or clorgyline) + METH more closely reflect changes seen in DA than in TH. In those instances where reserpine might be altering expression of TH and complicating interpretation of data with regard to enhanced METH neurotoxicity, use of clorgyline in its place confirms that increases in the METH-releasable pool of DA enhances toxicity in NAc and CPu using either TH or DAT as the dependent variable.
We considered the possibility that increased cytoplasmic DA would extend the damaging effects of METH to the SNc and VTA. Immunoblot analysis of TH revealed that METH or reserpine + METH did not reduce TH levels. Similarly, immunocytochemical analysis indicated that TH-containing neurons of the SNc and VTA were not reduced in number. These results indicate that conditions leading to heightened METH-induced neurotoxicity in DA nerve ending areas of the brain are not extended to the SNc and the VTA. The differences between the present results and those showing neuronal losses in the SNc (Sonsalla et al. 1996; Brown et al. 2006) could be attributed to the use of higher drug doses in the latter studies. A brain-regional heterogeneity in microglial function could also explain these results in the event that activated microglia in the SNc are reparative of damage (Streit 2002) whereas activated microglia in the CPu and NAc are not.
Microglial activation is emerging as an important component of the METH neurotoxic cascade (LaVoie et al. 2004; Thomas et al. 2004a,b) and is now known to occur in brains of human METH abusers (Sekine et al. 2008). The time course and pharmacological profile of microglial activation seen after METH suggests that it is an active participant in the toxic process and is not merely responding to it (LaVoie et al. 2004; Thomas et al. 2004a; Thomas and Kuhn 2005). The anatomical sites at which microglial activation occurs after METH administration is limited to the CPu and its pattern parallels the heterogeneity of damage to DA nerve terminals (i.e. lateral greater than medial; Thomas et al. 2004b). However, increases in cytoplasmic DA lead to a significant increase in microglial activation in the NAc after METH, in parallel with increased neurotoxicity. Microglial activation also extends ventrally into the olfactory tubercle and the median forebrain bundle, both of which are rich in DA axons of passage. We did not test these areas for evidence of toxicity, but METH can damage DA terminals of the olfactory bulb (Deng et al. 2007). Clearly, alterations in cytoplasmic DA that lead to heightened neurotoxicity also cause parallel increases in microglial activation within the same anatomical elements of the DA neuronal system. While the cause-effect nature of METH-induced microglial activation with regard to its neurotoxicity has not been resolved fully (Bowyer et al. 1994, 2008), recent studies have addressed this issue. Sriram et al. (2006) showed that minocycline blocks METH-induced increases in expression of proinflammatory cytokines and chemokines (i.e. signs of microglial activation) without decreasing signs of METH-induced neurotoxicity and astrogliosis (Sriram et al. 2006). Therefore, the early signs of METH neurotoxicity in striatum can emerge even when microglial activation is partially suppressed by high-dose minocycline treatment.
The present results add substantiation to the stance that DA is an important mediator of METH neurotoxicity and point to interactions among METH, VMAT, DAT, and DA released into the synapse as critical for the expression of neurotoxicity. METH enters the pre-synaptic process and causes leakage of DA from vesicles into the cytoplasm via its weak-base properties (Sulzer et al. 2005). DA is then released in a calcium-independent manner by reverse transport through an outward-facing DAT (Sulzer et al. 1993; Erreger et al. 2008). Multiple injections of METH, as used presently, increase extracellular DA to extremely high levels (> 500% of basal levels) as measured by microdialysis (O’Dell et al. 1991). Manipulations that alter pre-synaptic DA homeostasis have effects on METH neurotoxicity that are entirely consistent with this model. For example, depletion of cytoplasmic DA prevents METH neurotoxicity (Wagner et al. 1983; Albers and Sonsalla 1995; Thomas et al. 2008). Reserpinized mice (Wagner et al. 1983; Albers and Sonsalla 1995; Thomas et al. 2008) or mice deficient in VMAT (Fumagalli et al. 1999; Larsen et al. 2002) show heightened neurotoxicity and astrogliosis (Guillot et al. 2008) in response to METH because these manipulations increase the drug-releasable pool of DA (Butcher et al. 1988; Fon et al. 1997). Pharmacological (Marek et al. 1990; Pu et al. 1994; Sandoval et al. 2003) or genetic inactivation of the DAT (Fumagalli et al. 1998) confers protection against METH toxicity by preventing drug-induced reverse transport of DA. L-DOPA potentiates METH-induced DA overflow and leads to toxicity after a single non-toxic dose of METH (Weihmuller et al. 1993) and enhances neurotoxicity in the CPu when multiple doses are given (Thomas et al. 2008).
When the factors just discussed are considered with regard to the NAc, possible reasons for its resistance to METH toxicity arise. The density and activity of the DAT is lower in NAc by comparison to the CPu (McElvain and Schenk 1992; Meiergerd and Schenk 1994; Povlock and Schenk 1997). The METH-induced inhibition of VMAT in CPu (Brown et al. 2000) is much less evident in the NAc (Volz et al. 2007) and amphetamine-induced release of DA is also smaller in the NAc than in the CPu (Hernandez et al. 1987; Cass 1997). Basal superoxide dismutase levels are higher in NAc than in CPu (Perumal et al. 1992) suggesting that regional variations in antioxidant capacity across the CPu and NAc may determine sensitivity to METH neurotoxicity. Taken together, METH-induced disruptions in pre-synaptic DA homeostasis are far less drastic in the NAc than in CPu, resulting in a lower net release of DA. Increases in the cytoplasmic pool of DA caused by reserpine, L-DOPA and clorgyline increase METH-induced release in the NAc to a much greater extent than METH alone, exceeding the threshold for neurotoxicity.
Acknowledgments
This work was supported by NIH grants DA020680 (DMT), DA010756 (DMK), DA017327 (DMK) and by the Department of Veteran’s Affairs (DMT and DMK). We thank Dr Mariana Angoa-Pérez for her expert assistance with SNc/VTA cell counts, and Samer Nuwayhid and Mrudang Shah for assistance with western blotting. We are also indebted to Dr Roxanne A. Vaughan for providing DAT antibodies.
Abbreviations used
- CPu
caudate–putamen
- DA
dopamine
- DAT
dopamine transporter
- HRP
horseradish peroxidase
- METH
metham-phetamine
- ILB4
isolectin B4
- NAc
nucleus accumbens
- PBS
phosphate-buffered saline
- SNc
substantia nigra
- TH
tyrosine hydroxylase
- VMAT
vesicular monoamine transporter
- VTA
ventral tegmentum
References
- Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ, II, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE. Methamphetamine increases dopamine transporter higher molecular weight complex formation via a dopamine- and hyperthermia-associated mechanism. J Neurosci. 2004;24:3436–3443. doi: 10.1523/JNEUROSCI.0387-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Dovero S, Imbert C, Boraud T, Gross CE. Spontaneous long-term compensatory dopaminergic sprouting in MPTP-treated mice. Synapse. 2000;38:363–368. doi: 10.1002/1098-2396(20001201)38:3<363::AID-SYN16>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Bjorklund NL, Sorg BA, Schenk JO. Neuronal dopamine transporter activity, density and methamphetamine inhibition are differentially altered in the nucleus accumbens and striatum with no changes in glycosylation in rats behaviorally sensitized to methamphetamine. Synapse. 2008;62:736–745. doi: 10.1002/syn.20528. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Patrick KS, Ramamoorthy S, Denehy ED, Zhu H, Pacchioni AM, Granholm AC, McGinty JF. Long-term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line-derived neurotrophic factor heterozygous mice. J Neurosci. 2007;27:8816–8825. doi: 10.1523/JNEUROSCI.1067-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- Bowyer JF, Robinson B, Ali S, Schmued LC. Neurotoxic-related changes in tyrosine hydroxylase, microglia, myelin, and the blood-brain barrier in the caudate–putamen from acute methamphetamine exposure. Synapse. 2008;62:193–204. doi: 10.1002/syn.20478. [DOI] [PubMed] [Google Scholar]
- Broening HW, Pu C, Vorhees CV. Methamphetamine selectively damages dopaminergic innervation to the nucleus accumbens core while sparing the shell. Synapse. 1997;27:153–160. doi: 10.1002/(SICI)1098-2396(199710)27:2<153::AID-SYN6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hanson GR, Fleckenstein AE. Methamphetamine rapidly decreases vesicular dopamine uptake. J Neurochem. 2000;74:2221–2223. doi: 10.1046/j.1471-4159.2000.0742221.x. [DOI] [PubMed] [Google Scholar]
- Brown JM, Gouty S, Iyer V, Rosenberger J, Cox BM. Differential protection against MPTP or methamphetamine toxicity in dopamine neurons by deletion of ppN/OFQ expression. J Neurochem. 2006;98:495–505. doi: 10.1111/j.1471-4159.2006.03902.x. [DOI] [PubMed] [Google Scholar]
- Brunswick DJ, Benmansour S, Tejani-Butt SM, Hauptmann M. Effects of high-dose methamphetamine on monoamine uptake sites in rat brain measured by quantitative autoradiography. Synapse. 1992;11:287–293. doi: 10.1002/syn.890110404. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW. Amphetamine-induced dopamine release in the rat striatum: an in vivo microdialysis study. J Neurochem. 1988;50:346–355. doi: 10.1111/j.1471-4159.1988.tb02919.x. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN, Jayanthi S, Lyles J. Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res. 2007;11:183–202. doi: 10.1007/BF03033567. [DOI] [PubMed] [Google Scholar]
- Cass WA. Decreases in evoked overflow of dopamine in rat striatum after neurotoxic doses of methamphetamine. J Pharmacol Exp Ther. 1997;280:105–113. [PubMed] [Google Scholar]
- Davidson C, Chen Q, Zhang X, Xiong X, Lazarus C, Lee TH, Ellinwood EH. Deprenyl treatment attenuates long-term pre- and post-synaptic changes evoked by chronic methamphetamine. Eur J Pharmacol. 2007;573:100–110. doi: 10.1016/j.ejphar.2007.06.046. [DOI] [PubMed] [Google Scholar]
- Deng X, Ladenheim B, Jayanthi S, Cadet JL. Methamphetamine administration causes death of dopaminergic neurons in the mouse olfactory bulb. Biol Psychiatry. 2007;61:1235–1243. doi: 10.1016/j.biopsych.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Gaffney M, Weihmuller FB, O’Dell SJ, Marshall JF. Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res. 1992;598:321–326. doi: 10.1016/0006-8993(92)90201-j. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, O’Dell SJ, Marshall JF. Striatal and cortical NMDA receptors are altered by a neurotoxic regimen of methamphetamine. Synapse. 1996;22:217–225. doi: 10.1002/(SICI)1098-2396(199603)22:3<217::AID-SYN3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J Neurosci. 2008;28:976–989. doi: 10.1523/JNEUROSCI.2796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Fukui K, Kariyama H, Kashiba A, Kato N, Kimura H. Further confirmation of heterogeneity of the rat striatum: different mosaic patterns of dopamine fibers after administration of methamphetamine or reserpine. Brain Res. 1986;382:81–86. doi: 10.1016/0006-8993(86)90113-7. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgievska B, Kirik D, Bjorklund A. Aberrant sprouting and downregulation of tyrosine hydroxylase in lesioned nigrostriatal dopamine neurons induced by long-lasting overexpression of glial cell line derived neurotrophic factor in the striatum by lentiviral gene transfer. Exp Neurol. 2002;177:461–474. doi: 10.1006/exnr.2002.8006. [DOI] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, Miller GW. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlock GC, Baucum AJ, King JL, Horner KA, Cook G, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE. Mechanisms underlying methamphetamine-induced dopamine transporter complex formation. J Pharmacol Exp Ther. 2009;329:169–174. doi: 10.1124/jpet.108.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey DC, Lacan G, Melegan WP. Regional heterogeneity of dopaminergic deficits in vervet monkey striatum and substantia nigra after methamphetamine exposure. Exp Brain Res. 2000a;133:349–358. doi: 10.1007/s002210000386. [DOI] [PubMed] [Google Scholar]
- Harvey DC, Lacan G, Tanious SP, Melega WP. Recovery from methamphetamine induced long-term nigrostriatal dopaminergic deficits without substantia nigra cell loss. Brain Res. 2000b;871:259–270. doi: 10.1016/s0006-8993(00)02439-2. [DOI] [PubMed] [Google Scholar]
- Haughey HM, Fleckenstein AE, Hanson GR. Differential regional effects of methamphetamine on the activities of tryptophan and tyrosine hydroxylase. J Neurochem. 1999;72:661–668. doi: 10.1046/j.1471-4159.1999.0720661.x. [DOI] [PubMed] [Google Scholar]
- Haughey HM, Brown JM, Wilkins DG, Hanson GR, Fleckenstein AE. Differential effects of methamphetamine on Na(+)/Cl(−)-dependent transporters. Brain Res. 2000;863:59–65. doi: 10.1016/s0006-8993(00)02094-1. [DOI] [PubMed] [Google Scholar]
- Hernandez L, Lee F, Hoebel BG. Simultaneous microdialysis and amphetamine infusion in the nucleus accumbens and striatum of freely moving rats: increase in extracellular dopamine and serotonin. Brain Res Bull. 1987;19:623–628. doi: 10.1016/0361-9230(87)90047-5. [DOI] [PubMed] [Google Scholar]
- Hirata H, Ladenheim B, Carlson E, Epstein C, Cadet JL. Autoradiographic evidence for methamphetamine-induced striatal dopaminergic loss in mouse brain: attenuation in CuZn-superoxide dismutase transgenic mice. Brain Res. 1996;714:95–103. doi: 10.1016/0006-8993(95)01502-7. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Billingsley ML. Tyrosine hydroxylase: purification from PC-12 cells, characterization, and production of antibodies. Neurochem Int. 1987;11:463–475. doi: 10.1016/0197-0186(87)90036-2. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Arthur RE, Jr, Thomas DM, Elferink LA. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: possible relevance to Parkinson’s disease. J Neurochem. 1999;73:1309–1317. doi: 10.1046/j.1471-4159.1999.0731309.x. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Ann NY Acad Sci. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson’s Disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res. 1990;513:274–279. doi: 10.1016/0006-8993(90)90467-p. [DOI] [PubMed] [Google Scholar]
- McCann UD, Kuwabara H, Kumar A, et al. Persistent cognitive and dopamine transporter deficits in abstinent methamphetamine users. Synapse. 2008;62:91–100. doi: 10.1002/syn.20471. [DOI] [PubMed] [Google Scholar]
- McElvain JS, Schenk JO. A multisubstrate mechanism of striatal dopamine uptake and its inhibition by cocaine. Biochem Pharmacol. 1992;43:2189–2199. doi: 10.1016/0006-2952(92)90178-l. [DOI] [PubMed] [Google Scholar]
- Meiergerd SM, Schenk JO. Striatal transporter for dopamine: catechol structure-activity studies and susceptibility to chemical modification. J Neurochem. 1994;62:998–1008. doi: 10.1046/j.1471-4159.1994.62030998.x. [DOI] [PubMed] [Google Scholar]
- Morgan ME, Gibb JW. Short-term and long-term effects of methamphetamine on biogenic amine metabolism in extrastriatal dopaminergic nuclei. Neuropharmacology. 1980;19:989–995. doi: 10.1016/0028-3908(80)90010-6. [DOI] [PubMed] [Google Scholar]
- Nappi AJ, Vass E. The effects of nitric oxide on the oxidations of L-dopa and dopamine mediated by tyrosinase and per-oxidase. J Biol Chem. 2001;276:11214–11222. doi: 10.1074/jbc.M009872200. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- O’Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- Park SU, Ferrer JV, Javitch JA, Kuhn DM. Peroxynitrite inactivates the human dopamine transporter by modification of cysteine 342: potential mechanism of neurotoxicity in dopamine neurons. J Neurosci. 2002;22:4399–4405. doi: 10.1523/JNEUROSCI.22-11-04399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinetti GM, Morgan DG, Johnson SA, Millar SL, Finch CE. Tyrosine hydroxylase mRNA concentration in midbrain dopaminergic neurons is differentially regulated by reserpine. J Neurochem. 1990;55:1793–1799. doi: 10.1111/j.1471-4159.1990.tb04970.x. [DOI] [PubMed] [Google Scholar]
- Pereira FC, Lourenco ES, Borges F, Morgadinho T, Ribeiro CF, Macedo TR, Ali SF. Single or multiple injections of methamphetamine increased dopamine turnover but did not decrease tyrosine hydroxylase levels or cleave caspase-3 in caudate–putamen. Synapse. 2006;60:185–193. doi: 10.1002/syn.20285. [DOI] [PubMed] [Google Scholar]
- Perumal AS, Gopal VB, Tordzro WK, Cooper TB, Cadet JL. Vitamin E attenuates the toxic effects of 6-hydroxydop-amine on free radical scavenging systems in rat brain. Brain Res Bull. 1992;29:699–701. doi: 10.1016/0361-9230(92)90142-k. [DOI] [PubMed] [Google Scholar]
- Povlock SL, Schenk JO. A multisubstrate kinetic mechanism of dopamine transport in the nucleus accumbens and its inhibition by cocaine. J Neurochem. 1997;69:1093–1105. doi: 10.1046/j.1471-4159.1997.69031093.x. [DOI] [PubMed] [Google Scholar]
- Pu C, Fisher JE, Cappon GD, Vorhees CV. The effects of amfonelic acid, a dopamine uptake inhibitor, on methamphetamine-induced dopaminergic terminal degeneration and astrocytic response in rat striatum. Brain Res. 1994;649:217–224. doi: 10.1016/0006-8993(94)91067-7. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Sabol KE, Roach JT, Broom SL, Ferreira C, Preau MM. Long-term effects of a high-dose methamphetamine regimen on subsequent methamphetamine-induced dopamine release in vivo. Brain Res. 2001;892:122–129. doi: 10.1016/s0006-8993(00)03244-3. [DOI] [PubMed] [Google Scholar]
- Sandoval V, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J Pharmacol Exp Ther. 2003;304:1181–1187. doi: 10.1124/jpet.102.045005. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Seiden LS, Commins DL, Vosmer G, Axt K, Marek G. Neurotoxicity in dopamine and 5-hydroxytryptamine terminal fields: a regional analysis in nigrostriatal and mesolimbic projections. Ann NY Acad Sci. 1988;537:161–172. doi: 10.1111/j.1749-6632.1988.tb42104.x. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Ouchi Y, Sugihara G, et al. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci. 2008;28:5756–5761. doi: 10.1523/JNEUROSCI.1179-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996;738:172–175. doi: 10.1016/0006-8993(96)00995-x. [DOI] [PubMed] [Google Scholar]
- Sriram K, Miller DB, O’Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006;96:706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- Stanic D, Finkelstein DI, Bourke DW, Drago J, Horne MK. Time course of striatal re-innervation following lesions of dopaminergic SNpc neurons of the rat. Eur J Neurosci. 2003;18:1175–1188. doi: 10.1046/j.1460-9568.2003.02800.x. [DOI] [PubMed] [Google Scholar]
- Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4) J Histochem Cytochem. 1990;38:1683–1686. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Theodore S, Cass WA, Maragos WF. Methamphetamine and human immunodeficiency virus protein Tat synergize to destroy dopaminergic terminals in the rat striatum. Neuroscience. 2006;137:925–935. doi: 10.1016/j.neuroscience.2005.10.056. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res. 2005;1050:190–198. doi: 10.1016/j.brainres.2005.05.049. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004a;367:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004b;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. Gene expression profile of activated microglia under conditions associated with dopamine neuronal damage. FASEB J. 2006;20:515–517. doi: 10.1096/fj.05-4873fje. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, Simon SL, et al. Structural abnormalities in the brains of human subjects who use methamphetamine. J Neurosci. 2004;24:6028–6036. doi: 10.1523/JNEUROSCI.0713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Volz TJ, Fleckenstein AE, Hanson GR. Methamphetamine-induced alterations in monoamine transport: implications for neurotoxicity, neuroprotection and treatment. Addiction. 2007;102(Suppl 1):44–48. doi: 10.1111/j.1360-0443.2007.01771.x. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Lucot JB, Schuster CR, Seiden LS. Alpha-methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- Wallace TL, Gudelsky GA, Vorhees CV. Methamphet-amine-induced neurotoxicity alters locomotor activity, stereotypic behavior, and stimulated dopamine release in the rat. J Neurosci. 1999;19:9141–9148. doi: 10.1523/JNEUROSCI.19-20-09141.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace TL, Gudelsky GA, Vorhees CV. Neurotoxic regimen of methamphetamine produces evidence of behavioral sensitization in the rat. Synapse. 2001;39:1–7. doi: 10.1002/1098-2396(20010101)39:1<1::AID-SYN1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Weihmuller FB, O’Dell SJ, Marshall JF. L-Dopa pre-treatment potentiates striatal dopamine overflow and produces dopamine terminal injury after a single methamphetamine injection. Brain Res. 1993;623:303–307. doi: 10.1016/0006-8993(93)91442-u. [DOI] [PubMed] [Google Scholar]
- Whitehead RE, Ferrer JV, Javitch JA, Justice JB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Bankson MG. Amphetamine neuro-toxicity: cause and consequence of oxidative stress. Crit Rev Neurobiol. 2005;17:87–117. doi: 10.1615/critrevneurobiol.v17.i2.30. [DOI] [PubMed] [Google Scholar]