

Abstract
The nuclear lamina is primarily composed of type-V intermediate filaments, the A- and B-type lamins, which give mechanical support to the nuclear envelope, contribute to heterochromatin organization and regulate a myriad of nuclear processes. Over a dozen human diseases, collectively named laminopathies, are associated with mutations in the genes that encode the nuclear lamins. Although the etiology of the laminopathies is well understood, the molecular mechanisms by which they lead to disease remain unclear. Also poorly understood are the mechanisms by which the lamins contribute to a variety of nuclear and cellular functions. The identification of proteins associated with the lamins is likely to provide insight into these fundamental mechanisms. In recent years a unique method for identifying protein-protein and proximity-based interactions has emerged, called BioID (proximity-dependent biotin identification). BioID utilizes a mutant biotin ligase from bacteria that is fused to a protein of interest (bait). When expressed in living cells and stimulated with excess biotin, this BioID fusion protein promiscuously biotinylates directly interacting and vicinal endogenous proteins. Following biotin-affinity capture, the biotinylated proteins can be identified using mass spectrometry (MS). BioID thus enables screening for physiologically relevant protein associations that occur over time in living cells. The application of BioID is amenable to insoluble proteins such as lamins that are often refractory to study by other methods and is capable of identifying weak and/or transient interactions. In this review, we discuss the use of BioID as a powerful tool to help elucidate novel interacting partners of lamins.
Keywords: Lamins, BioID, Biotinylation, Protein-protein interactions, Proximity, Labeling
1. INTRODUCTION
1.1 Nuclear Lamina
One distinguishing feature of eukaryotes is the nuclear envelope (NE) that delineates the nucleus and partitions the DNA from other cellular structures. The major conserved components of the NE include the nuclear membranes, transmembrane proteins of the NE, and nuclear pore complexes (NPCs). As a specialized extension of the endoplasmic reticulum (ER), the NE is composed of a double lipid bilayer whose outer membrane (outer nuclear membrane, ONM) appears compositionally similar to the ER. The inner nuclear membrane (INM) contains a myriad of nuclear envelope transmembrane proteins (NETs), likely retained via associations with nuclear constituents (Wong et al., 2014). A special class of these NETs, called SUN-domain proteins, translumenally associate with ONM KASH-domain transmembrane proteins to physically link the nucleoskeleton with the cytoskeleton (LINC-complex) (Kim et al., 2015; Sosa et al., 2013). NPCs are massive protein complexes integrated with annulate lamellae of the NE. These nuclear pores are sophisticated portals that regulate the trafficking of macromolecules such as proteins, RNA, and transcription factors between the nucleus and cytoplasm (Ptak et al., 2014).
In metazoan species, structural stability of the NE is provided by a network of intermediate filaments on the inner face of the nuclear membrane called the nuclear lamina. The nuclear lamina is composed of A- and B-type lamins. The single LMNA gene encodes lamin-A and lamin-C (LaA and LaC), whereas LMNB1 and LMNB2 encode for lamin-B1 and lamin-B2 (LaB1 and B2). The basic lamin structure includes a central rod composed of coiled-coil α-helices, a nuclear localization sequence, and a C-terminal immunoglobulin (Ig)-like fold (Dhe-Paganon et al., 2002; Fisher et al., 1986; Krimm et al., 2002; McKeon et al., 1986). With the exception of LaC, lamins contain a C-terminal CaaX motif that is post-translationally prenylated and carboxylmethylated. LaA is further processed by the removal of the C-terminal 15 residues from pre-LaA to generate mature LaA (Kitten and Nigg, 1991; McKeon et al., 1986; Sinensky et al., 1994). Via their coiled-coil domains lamins form stable homodimers that can assemble via head-tail associations into filaments (Heitlinger et al., 1992). Additional lateral associations of these filaments may lead to filaments with larger diameters (10 nm filaments, (Min et al., 1996)). In addition to forming the nuclear lamina, a subset of the lamins also resides in the nucleoplasm. Early in mitosis, the nuclear lamins are disassembled via specific phosphorylation that occurs in conjunction with NE disassembly (Kochin et al., 2014; Ottaviano and Gerace, 1985). With this variable distribution it is easy to see how lamins are able to impact a myriad of cellular events including cell proliferation and senescence, DNA transcription, DNA repair, mRNA transcription, organization and anchoring of the peripheral heterochromatin, epigenetic chromatin modifications, nuclear envelope assembly and disassembly, and even nuclear dimensions (Broers et al., 2006; Dreesen et al., 2013; Holaska et al., 2002; Mahen et al., 2013; Verstraeten et al., 2007). And during mitosis, when the NE and nuclear lamina are disassembled, lamins have been implicated in the function of the mitotic spindle matrix (Tsai et al., 2006).
Mutations in LMNA have been found in 11 rare clinically distinct degenerative diseases, including muscular dystrophy, cardiomyopathy, lipodystrophy, and progeria (Chi et al., 2009). Duplication of LMNB1 is associated with adult-onset leukodystrophy and mutations in LMNB2 lead to partial acquired lipodystrophy (Gao et al., 2012; Hegele et al., 2006; Padiath et al., 2006). The diverse tissue-specific pathologies associated with laminopathies suggest lamins may function in a cell-type dependent manner. Mutations within genes that encode many other constituents of the NE, several of which are known to associate with the nuclear lamins, often lead to diseases collectively called envelopathies (Dauer and Worman, 2009; Worman et al., 2010). Many of these envelopathies phenotypically overlap with the laminopathies suggesting shared mechanisms. Given that lamins and their associated proteins are important in several diseases and function to regulate many normal cellular processes, it is of high interest to identify interacting partners of lamins to uncover the fundamental mechanisms by which lamins contribute to both normal cellular function and human disease.
1.2 Protein-protein Interactions
Studying protein-protein interactions (PPIs) has become a critical component of biological research because more often than not, proteins function collaboratively in cells to drive cellular signaling cascades, regulation, differentiation and proliferation. Proteins can also become “rogue” if they are mutated or modified, which commonly occurs in diseases. Rogue proteins have the potential to interact with other cellular proteins, organelles, and/or structures in which they wouldn’t interact normally, potentially leading to disease-states.
One hurdle to overcome in studying lamins and their associated proteins is the highly insoluble nature of the nuclear lamina itself. Use of ultra-sonification with high concentrations of ionic- or chaotropic detergents to solubilize the lamina can disrupt lamin-protein interactions (Kubben et al., 2010). Various strategies have been employed to identify lamin-associated proteins, including immuoprecipitation, crosslinking, fractionation of isolated nuclear membranes coupled with mass spectrometry (MS) analysis, and yeast two-hybrid (Y2H) (Dreger et al., 2001; Holaska and Wilson, 2007; Kubben et al., 2010; Wilkinson et al., 2003). These strategies have all been partially successful but also have specific drawbacks. Immunoprecipitation is performed under non-physiological conditions that can be insensitive, lead to loss of weak or transient interactions, and may identify non-specific interacting proteins due to mild solublization and/or washes. Crosslinking techniques prior to harsh lysis are attractive but potentially incur protein aggregation and complicate interpretation of the degree of protein associations, especially for a filamentous network such as the lamina. Fractionation techniques can be useful but large amounts of sample is required and obtaining pure membrane fractions can be difficult and time consuming and these approaches do not reveal the nature of protein associations. The Y2H approach can be very sensitive but can also produce many false-positives. Additionally, some of these techniques may not identify proteins if they contain post-translational modifications (PTMs) or require them for their protein interactions. In the last few years a novel technique has been developed to screen for potential PPIs in living cells, named BioID for proximity-dependent biotin identification, that provides an effective addition to the toolbox of methods for studying lamin-associated proteins (Roux et al., 2012).
1.3 BioID
BioID is a recently developed approach used for identifying PPIs by using a “bait” protein fused to a mutant biotin ligase which promiscuously biotinylates directly interacting or nearby primary amines of “prey” proteins over a period of time. BioID takes advantage of an enzyme found in prokaryotic cells called biotin ligase (BirA), a 35 kD protein from E. coli. Biotin is a water-soluble molecule found in living cells in small amounts as an enzyme co-factor. Under normal conditions, BirA activates biotin to biotinyl-AMP (bioAMP) by utilizing ATP, allowing bioAMP to react with lysine residues of very specific protein sequences (Chapman-Smith and Cronan, 1999; Lane et al., 1964). The BioID approach utilizes a mutant biotin ligase (hereafter called BioID) which prematurely releases bioAMP, allowing it to react with any free primary amines nearby (Kwon and Beckett, 2000; Roux et al., 2012). This promiscuous BioID protein (mutant biotin ligase + bait) can biotinylate directly interacting and/or vicinal proteins, leaving a modified protein trail or history of its interactions over time.
Figure 1 displays a schematic overview of the BioID method. BioID can be used to screen potential interacting partners of the bait in virtually any genetically tractable cell type. A gene of interest can be recombined in-frame with the BioID sequence in an expression vector plasmid. A stable cell line expressing the BioID protein is then established using common transfection methods or transduction using virus-mediated DNA transfer. Once cells are prepared for analysis, they can be incubated with excess biotin (typically 50 μM). Most conventional cell-culture media do not contain biotin so the low level of biotin needed by the cells is supplied by serum. Under these conditions there is minimal biotinylation by the BioID-fusion protein. The level of biotinylation can be regulated in a time-dependent manner by varying the biotin incubation time. Incubation with an excess of biotin for 15-18 hours is typically sufficient to saturate the biotinylation signal and it is estimated that the range of biotinylation around the BioID protein is ~10 nm (Kim et al., 2014; Roux et al., 2012). The cells are then subject to a stringent SDS/Triton X-100 lysis followed by sonication. A one-step purification process by affinity-capture is then achieved using magnetic beads coated with streptavidin, which has a well-known high affinity for biotin. These biotinylated proteins can then be analyzed using MS and/or immunoblot (IB) analysis. Additionally, fluorescently labeled streptavidin can be used to visualize the biotinylated proteins by immunofluorescence (IF). If no antibody is commercially available against the bait, a MYC or HA tag may be included for detection of the BioID fusion protein (Evan et al., 1985; Field et al., 1988). Furthermore, there is an anti-BirA antibody available which is effective for detection of the BioID fusion protein by IF and IB (see methods).
Figure 1. Application of BioID method to LaA.
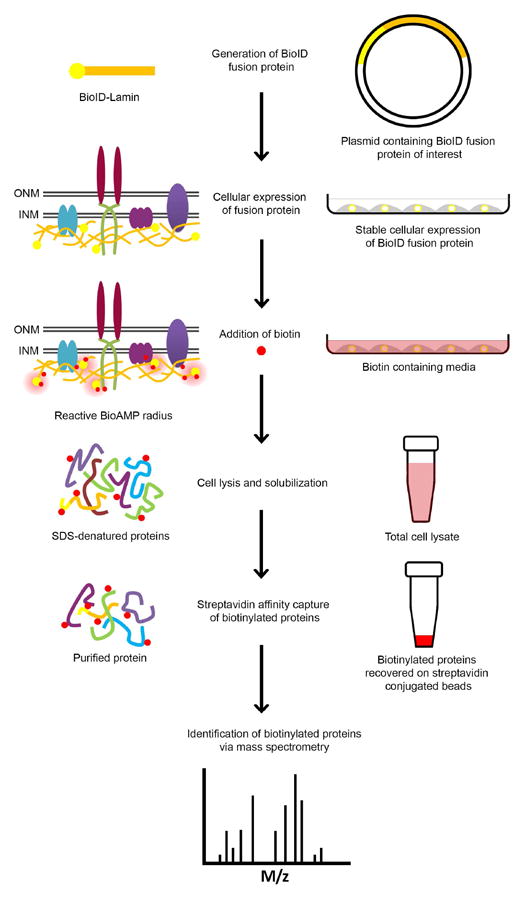
The N-terminus of a lamin can be fused to a promiscuous biotin-ligase, which prematurely releases BioAMP. Cells stably expressing the fusion protein in biotin containing media leads to the selective biotinylation of proteins within proximity of the BioID-lamin in living cells. After cell lysis and solubilization, biotinylated proteins are affinity purified using streptavidin-conjugated beads, allowing for stringent wash conditions. Candidate proteins are subsequently analyzed by IB analysis and identified by MS.
This novel technique has the ability to screen for protein-protein interactions in living cells. The candidate interacting partners are covalently modified in their normal cellular environment, thus, a stringent lysis can be employed which is beneficial to completely solubilize and denature the proteins. The ability to selectively enrich and identify the candidate interacting partners using affinity-capture and MS is also a major strength. BioID provides an alternative method for overcoming the challenges of studying lamins and their associated proteins. To date BioID has been used successfully to screen for constituents of the relatively insoluble mammalian nuclear lamina (Roux et al., 2012), nuclear pore complexes (Kim et al., 2014), chromatin-associated protein complexes (Lambert et al., 2014), the trypanosome bilobe (Morriswood et al., 2013), cell junction complexes (Fredriksson et al., 2015; Guo et al., 2014; Steed et al., 2014; Ueda et al., 2015; Van Itallie et al., 2013; Van Itallie et al., 2014), and centrosomes (Comartin et al., 2013; Firat-Karalar et al., 2014). The method also has been used to screen for proteins involved in the Hippo signaling pathway (Couzens et al., 2013). Additionally, BioID has been applied to a secreted protein of Chlamydia psittaci that targets the nuclear envelope (Mojica et al., 2015), used to identify novel components of the inner membrane complex of Toxoplasma gondii (Chen et al., 2015), analyzing HIV-1 Gag protein interactions (Ritchie et al., 2015), and utilized in detecting c-MYC interacting partners in cultured cells and mice xenograft tumors (Dingar et al., 2014).
2. BioID METHOD
2.1 Construction of a BioID fusion protein
The most important aspect of BioID is the fusion protein itself. In many scenarios, a functional copy of the endogenous protein of interest is desired. Thus, it is imperative to decide on the appropriate position within the protein to incorporate the biotin ligase. It may be beneficial to consider current research and determine if there has previously been a fusion protein generated for the bait with a similarly sized protein, like GFP, which retains localization and function. This may help guide the decision on where to incorporate the ligase. Care must be taken not to disrupt any PTMs of the N- or C-terminus, such as C-terminal prenylation or N-terminal signal peptides. If it is unknown how an N- or C-terminal fusion may affect the bait, it is recommended to try both. Proper targeting is often a good starting point to assess basic function of the fusion protein. If the protein is an enzyme, it may be necessary to assay enzyme activity. The subcellular compartment in which the BioID will be located must be taken into consideration, especially with integral membrane proteins. BioID does work in both the cytoplasmic/nuclear compartments and the lumen of the ER, but is markedly reduced in activity in the latter (personal experience).
2.2 Cell Choice
After generation of a BioID expression plasmid and preliminary validation of the fusion protein, it is important to select an appropriate cell-type in which to express the BioID fusion protein. Though the choice is largely dependent upon the circumstances, it is prudent to consider the uniformity and level of expression. For best results, it is important to have similar expression of fusion proteins in all cells within the population to be used for large-scale BioID pull-down and MS analysis. Thus, it is not advised to do a traditional transient transfection aside from preliminary validation of the functionality and targeting of the fusion protein. More useful is generation of a stable cell line expressing the fusion protein, as accomplished by standard random integration, various viral expression systems or genome editing. In most situations, a low level of expression is ideal to ensure physiological relevance while providing sufficient candidate proteins. On the contrary, overexpression may lead to aggregation of the fusion protein and false associations complicating the subsequent analysis. Biotinylation itself is inducible by the addition of excess biotin. However, if the bait is toxic, inducible expression of the bait must be considered.
2.3 Functional assessment of the fusion protein
Perhaps one of the most important aspects of the fusion protein is its functionality. Preferably, the fusion protein will behave the same as its endogenous counterpart, although, this may remain unclear unless the function of the protein is well understood. Phenotypic rescue of a knockout/knockdown or mutant system may be useful to these ends. In the least, it is typically possible to compare the localization of the fusion protein to the endogenous protein using IF microscopy. Again, particular attention to expression levels is imperative as overexpression systems can lead to erroneous localization. In general, expression levels at or below the level of the endogenous protein is often sufficient to identify potential interacting and/or vicinal proteins.
2.4 Establishing a stable BioID cell line
The following protocol describes the creation, characterization, and generation of a stable cell line expressing a BioID fusion protein in mammalian cells. PCR cloning and stable transfection techniques will not be described, as they vary dramatically depending on the situation. The methods needed to test the targeting of the fusion protein, expression, and activity in cells prior to a large-scale BioID pull-down are described.
Materials
Expression plasmid for BioID fusion protein (see Internet Resources)
cDNA for bait protein
Cells of choice for transfection and appropriate medium
1 mM (20X) biotin (see recipe)
Fixative, e.g., paraformaldehyde (PFA)
Triton X-100 (TX-100)
Streptavidin-Alexa Fluor (Invitrogen, currently Life Technologies)
DNA labeling reagent for IF (e.g., Hoechst, DAPI)
Phosphate-buffered saline (PBS)
SDS-PAGE sample buffer (see recipe)
BSA blocking buffer (see recipe)
Streptavidin-HRP (High Sensitivity Streptavidin-HRP, Abcam)
ABS blocking buffer (see recipe)
Enhanced chemiluminescence (ECL) reagent (commercially available, or see recipe)
Quenching solution (see recipe)
Antibodies specific to BioID fusion protein (e.g., anti-myc/HA) or chicken anti-BirA (Abcam, ab14002)
Secondary antibodies (Alexa Fluor form and HRP-conjugated form)
6-well plates
Glass coverslips
Sonicator
SDS-PAGE electrophoresis unit (Mini-PROTEAN II Electrophoresis Cell, Bio-Rad)
Semi-dry transfer cell (Trans-Blot, Bio-Rad)
Additional reagents and equipment for PCR cloning (Elion et al., 2007), transfection (Mortensen et al., 2003), SDS-PAGE separation, protein transfer, and IB detection
Generation and validation of BioID fusion protein
Prepare the expression plasmid for the BioID fusion protein. This typically requires the use of PCR cloning (Elion et al., 2007) to generate an expression vector in which the bait protein is fused in-frame with the BioID ligase. This requires plasmid DNA that is of sufficient quality and quantity for cellular transfection. To obtain mammalian expression plasmids for BioID, see Internet Resources.
For each experimental condition (e.g., BioID-only control, BioID fusion protein) plate two wells of cells in a standard 6-well plate, one with a single glass coverslip.
In the cells of choice, express the fusion protein by doing a transient transfection (Mortensen et al., 2003). Lipofectamine 2000 (Life Technologies) reagent works well with the manufacturer’s suggested protocol. Process mock or nontransfected cells in parallel. Apply biotin (50 μM final concentration) to cells at time of transfection. If transfection protocol requires replacement of medium shortly after transfection, add biotin at that time instead. The presence of excess biotin promotes biotinylation by the BioID fusion protein (usually over a period of 18 to 24 hr). This permits simultaneous analysis of expression, targeting, and biotinylation capability of the BioID fusion protein by IF microscopy and IB.
Process the cells on coverslips for analysis by IF microscopy ~24 hr after transient transfection. It is recommended to use PFA fixation (3% PFA in PBS) for 10 min, followed by TX-100 permeabilization (0.4% TX-100 in PBS) for 15 min. Use of methanol fixation will result in a strong mitochondrial signal observed with fluorescently labeled streptavidin (Streptavidin–Alexa Fluor). Use the labeled streptavidin at 1:1000 with the Alexa Fluor secondary antibody and DNA labeling reagent (e.g., Hoechst, DAPI). For more information on IF staining, see (Donaldson, 2001).
Process the cells for IB analysis approximately 24 hr following transient transfection and addition of biotin. To do this, briefly rinse cells in room-temperature PBS to remove serum proteins prior to lysis in SDS-PAGE sample buffer using a volume appropriate for the number of cells being lysed (e.g., 200 μl for 1 X 106 cells). Sonicate to shear DNA and heat to 98°C for 5 min to denature proteins.
Following whole-cell lysis, SDS-PAGE separation, and protein transfer, agitate the membrane in BSA blocking buffer for 20 to 30 min at room temperature. BSA is preferred for blocking to eliminate any free biotin that is potentially present in milk or serum. Free biotin will compete with biotinylated proteins for binding to streptavidin-HRP.
Agitate membrane in streptavidin-HRP at 1:40,000 in BSA blocking buffer for 40 min at room temperature. It may be necessary to optimize concentration depending on the source of streptavidin-HRP.
Quickly wash membrane in PBS 2 to 3 times to wash away unbound streptavidin-HRP.
Agitate membrane in ABS blocking buffer for 5 min. This step is critical to reduce background signal on membrane.
Quickly wash membrane in PBS 2 to 3 times.
Agitate membrane in PBS for 5 min.
Add enhanced chemiluminescence (ECL) reagent to observe biotinylated proteins.
Following successful analysis of biotinylated proteins, quench HRP signal on membrane by agitating for 20 min in quenching solution. Removal of streptavidin-HRP with standard membrane stripping methods is futile given the strength of the biotin-streptavidin interaction. The quenching permanently inactivates the HRP activity, allowing further probing with additional antibodies. Consider reapplication of ECL to the membrane and visualization to confirm success of quenching.
Wash membrane with PBS three times to remove quenching solution. This step removes residual hydrogen peroxide that may affect the following IB analysis.
Proceed to IB membrane with antibodies specific to the BioID fusion protein (e.g., anti-myc/HA/BirA and corresponding HRP-conjugated secondary antibody) to confirm its expression and migration by SDS-PAGE. Use standard protocol for this, beginning with blocking in ABS or equivalent (BSA blocking not required).
Generation of cells stably expressing BioID fusion protein
-
16.
Initiate generation of stable cell lines by stable transfection or viral transduction. This is a highly variable process depending on the strategy and cell type.
-
17.
If subcloning of cells is performed, screen subclones first by IF. Perform IB analysis on subclones that pass the IF screening. If viral infection is utilized, screen population of infected cells by IF and IB. It is important to add 50 μM biotin to the cells as in step 3 to monitor the biotinylation function of the BioID fusion protein.
-
18.
Freeze down multiple vials of stably expressing cells for future BioID experiments and store in liquid nitrogen.
2.5 Large scale BioID pull-down to identify interacting proteins
The following protocol describes the use of cells stably expressing a BioID fusion protein (along with nonexpressing parent or BioID-only control cells) to perform large-scale BioID pull-down experiments. The purpose of these experiments is to isolate sufficient amounts of proteins biotinylated by the BioID fusion protein to be identified by MS. The starting material for these experiments may vary depending on a number of factors. These include the efficiency of biotinylation by the BioID fusion protein and the number of desired candidate proteins. This protocol describes the analysis of four confluent 10-cm plates of cells/condition (4 X 107 cells). The protocol ends immediately prior to analysis by MS, since this is a service typically handled by a core facility.
Materials
Four 10-cm dishes of cells for each experimental condition (cells expressing BioID constructs or control cells)
Complete medium
1 mM (20X) biotin (see recipe)
Phosphate-buffered saline (PBS)
Lysis buffer (see recipe)
20% Triton X-100
50 mM Tris•Cl, pH 7.4
Dynabeads (MyOne Streptavidin C1, Life Technologies)
Wash buffer 1 (see recipe)
Wash buffer 2 (see recipe)
Wash buffer 3 (see recipe)
50 mM ammonium bicarbonate (NH4HCO3)
1X SDS -PAGE sample buffer (see recipe)
DNase/RNase-free tubes, 15 ml conical and 2 ml microcentrifuge
Tube cap opener
Sonicator (Branson Sonifier-250 or equivalent)
MagneSphere Technology Magnetic Separation Stand (Promega)
Rotator
Perform cell lysis
Unless otherwise specified, lysis and wash steps should be carried out at room temperature to avoid precipitation of SDS and deoxycholic acid. To reduce keratin contamination, use DNase/RNase-free tubes that have not previously been opened, wear gloves, and use a tube cap opener.
Begin with four 10-cm dishes for each experimental condition (cells expressing BioID constructs or control cells). The number of cells utilized will correlate with numbers of candidates identified (e.g., four 10-cm plates may yield 150 candidates while two 10-cm plates may yield 75 candidates) but the relative numbers of candidates identified varies depending on variables such as the biotin labeling time and expression level.
When cells reach approximately 80% confluency, change medium to fresh complete medium containing 50 μM biotin (1X).
Incubate cells with biotin. Incubation time and conditions may vary depending on the goals of the experiment (e.g., cell cycle stage-specific labeling). In general, 16 to 18 hr of biotin results in a maximal labeling whereas after 6 hr of biotin there is a considerably reduced level of biotinylated proteins (approximate 25%). Pilot studies with IF and IB analysis should be performed prior to large-scale experiments.
Remove medium completely by aspiration and rinse the cells twice at room temperature with 3 ml/dish of PBS. This step is important in that it removes residual free-biotin from the medium.
Add 540 μl of lysis buffer/dish and scrape cells gently to harvest the cells. Perform this step at room temperature. The purpose of this harsh lysis is to try to disrupt all protein interactions and completely denature/solubilize the proteins.
Transfer lysed cells to a 15-ml conical tube.
Add 240 μl of 20% Triton X-100 (final concentration 2%) and mix by trituration. Keep tube on ice during subsequent sonication. Adding a five-fold excess of Triton X-100 dilutes out the SDS and prevents its precipitation at 4°C.
Position the sonicator probe tip in the sample just above the tube bottom.
Apply sonication for two sessions with 30 pulses using a Branson Sonifier 250 (or equivalent) at 30% duty cycle and an output level of 4. Let the tube sit on ice for 2 min between each session to prevent overheating. If the sample is still viscous and cloudy after sonication, apply an additional period of sonication. Sonication also functions to shear DNA.
Add 2.4 ml of prechilled 50 mM Tris•Cl, pH 7.4, and mix well. This dilution provides more favorable conditions for affinity capture.
Apply one session of sonication (30 pulses at 30% duty cycle and an output level of 4, using a Branson Sonifier 250 or equivalent). This step helps solubilize any precipitated proteins that may be present due to reduced salt and detergent concentrations from the previous step and assists in mixing the sample.
Aliquot the sample evenly into three prechilled 2-ml tubes.
Spin down 10 min at 16,500 Xg, 4°C.
Perform affinity purification of biotinylated proteins
-
14.
During the centrifugation in step 13, place 15 ml conical tube in the magnetic separation stand and add 1 ml of room-temperature lysis buffer and 1 ml of room-temperature 50 mM Tris•Cl, pH 7.4, to each tube.
-
15.
Mix the stock of magnetic streptavidin beads well with gentle tapping to resuspend and add 300 μl of beads to each tube prepared in step 14. Wait for 3 min at room temperature. This step functions to equilibrate the beads in the binding buffer.
-
16.
Once the beads accumulate at one side of tube wall, remove the supernatant gently by pipetting. This step removes the buffer in which the beads equilibrated.
-
17.
After sample centrifugation (step 13), carefully transfer supernatant to the tubes prepared in step 16. Do not disturb the small insoluble pellet on the tube wall when removing the supernatant. Step 17 should be performed quickly to prevent the beads from drying out.
-
18.
Resuspend the samples and beads with gentle pipetting.
-
19.
Incubate the tube on a rotator at 4°C overnight.
-
20.
Place the conical tube on the magnetic separation stand and wait 3 min to collect beads at room temperature.
-
21.
Remove the supernatant gently by pipetting. Try not to disturb beads on the wall of the tube.
-
22.
Add 1 ml of wash buffer 1 to the tube and resuspend beads gently by pipetting, and transfer to a new 1.5 ml eppendorf.
-
23.
Place the tube on a rotator for 8 min at room temperature.
-
24.
Remove the supernatant as described in steps 20 and 21.
-
25.
Repeat steps 22-24 again using wash buffer 1.
-
28.
Wash once with 1 ml of wash buffer 2 as described in steps 22 through 24.
-
29.
Wash once with 1 ml of wash buffer 3 as described in steps 22 through 24.
-
30.
Add 1 ml of 50 mM Tris•Cl, pH 7.4, and resuspend gently with pipetting. This step is important to remove detergents in the sample that may interfere with MS.
-
31.
Save 100 μl (10% of total) of resupended beads for further analysis by IB.
-
32.
Spin beads (both 900- and 100-μl samples) for 5 min at 2,000 × g, room temperature, for 5 min to collect beads.
-
33.
Remove the supernatant completely. Do not disrupt beads at the tube bottom.
-
34.
Add 150 μl of 50 mM ammonium bicarbonate to the tube that contained 900 μl of sample (destined for mass-spec analysis) and resuspend gently with pipetting. Final wash buffer may vary depending on analysis method. If necessary, freeze the sample quickly in liquid nitrogen. Frozen samples can be stored at −80°C.
-
35.
For the 100-μl sample, remove supernatant; add 100 μl of 1× SDS-PAGE sample buffer and resuspend gently by pipetting. Heat samples at 98°C for 5 min. Samples can be stored at −20°C for further analysis by IB as described in Basic Protocol 1. Prior to sending samples for analysis by MS, it is recommended to perform IB analysis of ~5 μl from the protein in the SDS-PAGE sample buffer. This should follow the same protocol described above to check for evidence of biotinylated proteins from BioID fusion proteins. A considerable difference in the pattern of biotinylated proteins between the BioID-only and BioID fusion protein samples justifies proceeding to MS.
2.6 Reagents and solutions
Use Milli-Q purified water or equivalent in all recipes.
ABS blocking buffer
10% (v/v) adult bovine serum
0.2% (w/v) TritonX-100
Bring up to final volume with 1× PBS
Store up to 4 weeks at 4°C
Biotin, 1 mM (20X)
Dissolve 12.2 mg biotin (Sigma #B4501) in 50 ml of serum-free DMEM (for standard tissue culture medium). Pipetting may be required to dissolve biotin completely. Sterilize by passing through a 0.22-μm syringe-driven filter unit (Millex). Dispense into sterile 50-ml tube; cap tightly. Store up to 8 weeks at 4°C.
BSA blocking buffer
1% bovine serum albumin, fraction V
0.2% (w/v) Triton X-100
Bring up to final volume with 1× PBS
Store up to 4 weeks at 4°C
ECL reagent
Stock solutions
250 mM luminol; 90 mM coumaric acid; 1 M Tris·Cl, pH 8.5; 30%hydrogen peroxide
Store luminol and coumaric acid stock solutions in 500-μl aliquots at −20°C, others at 4°C
Solution 1
1 ml 1 M Tris·Cl, pH 8.5
45 μl coumaric acid
100 μl luminol
8.9 ml H2O
Solution 2
1 ml 1 M Tris·Cl, pH 8.5
6 μl 30% hydrogen peroxide
9 ml H2O
Store solutions 1 and 2 at 4°C for up to 1 month. Mix equal volumes of solutions 1 and 2 together and immediately add to membrane for 1 min prior to exposure to film or ChemiDoc detection
Lysis buffer
50 mM Tris·Cl, pH 7.4
500 mM NaCl
0.2% SDS (w/v)
Store up to 2 weeks at room temperature
1X protease inhibitor (Halt Protease Inhibitor Cocktail, EDTA-Free, Thermo Scientific; add just before use)
1 mM DTT (add just before use)
Quenching solution
30% (v/v) hydrogen peroxide
SDS-PAGE sample buffer
50 mM Tris·Cl, pH 6.8
12% sucrose
2% SDS
0.004% bromophenol blue
20 mM DTT (dithiothreitol)
Wash buffer 1
2% SDS (w/v)
Store up to 2 weeks at room temperature
Wash buffer 2
0.1% (w/v) deoxycholic acid
1% (w/v) Triton X-100
1 mM EDTA
500 mM NaCl
50 mM HEPES, pH 7.5
Store up to 2 weeks at room temperature
Deoxycholic acid stock solution must be protected from light.
Wash buffer 3
0.5% (w/v) deoxycholic acid
0.5% (w/v) NP-40
1 mM EDTA
250 mM LiCl
10 mM Tris·Cl, pH 7.4
Store up to 2 weeks at room temperature
50 mM ammonium bicarbonate
198 mgs ammonium bicarbonate
Fill up to 50 mL with distilled water
Invert several times to completely dissolve
Make fresh daily
3. Anticipated results
A typical BioID-lamin experiment starts with IF and IB analysis of stably expressing cells to look for fusion protein localization and the migration pattern of biotinylated proteins. Unlike BioID-only which is diffusely nucleoplasmic and cytoplasmic, BioID-LaA exhibits a subcellular localization predominantly at the NE with some nucleoplasmic distribution (Figure 2A). By IB analysis proteins biotinylated by BioID-LaA have different migration pattern and intensity as compared to the BioID-only or parental (non-transfected) control cells (Figure 2B). These results would support continuation on to a large-scale BioID pull-down for MS analysis. Typically anywhere from 50-250 unique proteins, those not substantially found in controls, are detected for a BioID-lamin experiment with 18 hrs of biotin labeling (Kim et al., 2014; Roux et al., 2012). These proteins represent candidate interactors or proteins that reside in close proximity (~10 nm) to the BioID-lamin protein.
Figure 2. Proximity dependent biotinylation by BioID-LaA.
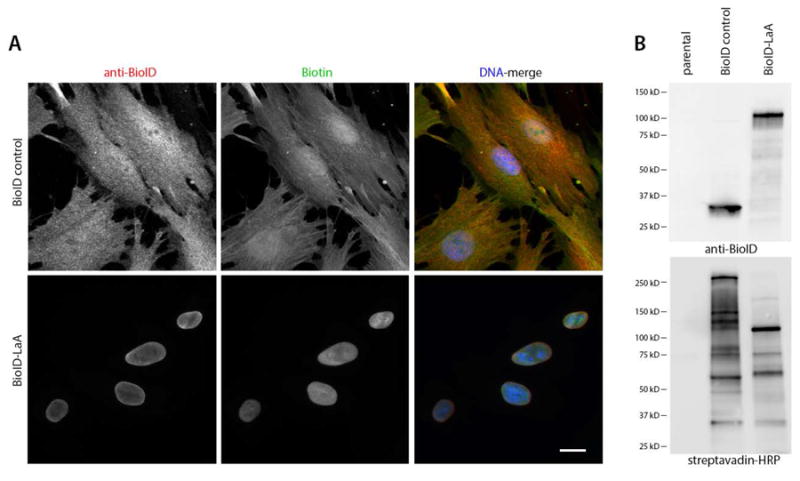
Control (non-transfected) human diploid foreskin fibroblast cells (BJ) or those cells stably expressing BioID-only or BioID-LaA (myc-BioID-LaA) by retroviral transduction were analyzed 18 hours after incubation in 50 μM biotin. (A) By IF analysis the BioID-only localizes to the nucleus and cytoplasm, whereas BioID-LaA (both red) localizes primarily to the nuclear envelope and minimally within the nucleoplasm. Biotinylated proteins, detected with labeled streptavidin (green), co-localize with the fusion proteins (red). DNA is labeled with Hoechst (blue). Bar, 10 μm. (B) IB analysis reveals migration of the fusion protein with anti-BioID and biotinylated endogenous proteins via streptavidin-conjugated HRP.
4. Conclusions
BioID is an effective alternative to help identify lamin-associated proteins. This unique method has several important advantages over conventional techniques. Traditionally, it has proven difficult to identify PPIs of insoluble or membrane-bound proteins due to their biochemical nature. However, BioID labels proteins in their natural environment (living cells) and has thus proved effective when applied to insoluble proteins of the nuclear lamina or other membrane proteins where stringent lysis is needed for complete cellular solubilization. Additionally, some protein interactions may be weak or transient in nature but due to the mechanism of the BioID approach, these interactions can be identified, aided by the temporal inducibility of biotin labeling. Lastly, the labeling and affinity-capture technique itself is advantageous for selective enrichment of modified proteins to be later identified by MS where successful protein identification is dependent on lowering ion suppression in complex lysates.
BioID is not without limitations. This technique is to be used to screen for nearby or potential protein-protein interactions, and may not to necessarily assess how abundant or strong these associations truly are. Whether an identified protein is incidentally interacting or directly interacting, or simply within its vicinity of the bait, would have to be further validated. Likewise, the very function of the bait may potentially be affected by altering its structure when fused to the biotin ligase. Also, biotinylation of proteins could influence the function by altering the charge of free amines or making lysine residues unavailable for alternate modifications. Lastly, if an interacting protein is only expressed or associated with the target protein in low concentrations, it could be overlooked by MS (a false negative) given the breadth of potential candidates.
Extensive characterization of the proteins of the inner nuclear membrane using lamin isoforms (A, B1, B2, and C) as well as some well-known lamin-associated proteins such as LBR, emerin, TMPO, MAN1, SUN1, SUN2, and LAP1 as “baits” in BioID experiments are currently ongoing (unpublished data, Roux lab). Also, development of a 2nd generation BioID with a smaller footprint that is less likely to affect function or targeting of the protein of interest has been developed (unpublished data, Roux lab). To date, most BioID experiments have been applied to a variety of proteins in cultured cells but future directions include development of an animal model. Towards this end, BioID has recently been applied to mice tumor xenografts to identify novel protein-protein interactions of the c-MYC oncoprotein and were able to identify >100 high confidence MYC interacting proteins including >30 known binding partners (Dingar et al., 2014). Genetically generated animal models incorporating BioID would help increase the confidence of PPIs previously identified at the nuclear envelope using the cell-based BioID. An in vivo BioID system may potentially lead to the identification of novel cell-and tissue-specific interacting partners. In conclusion, BioID is a complementary alternative method that can be used to elucidate novel protein-protein interactions of lamin-associated proteins and help better characterize their normal functions and how their dysfunction mechanistically leads to the laminopathies.
Acknowledgments
Generation of this publication was supported by the National Institutes of Health under award numbers RO1GM102203, RO1GM102486 and RO1EB014869. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Physiol Rev. United States: 2006. Nuclear lamins: laminopathies and their role in premature ageing; pp. 967–1008. [DOI] [PubMed] [Google Scholar]
- Chapman-Smith A, Cronan JE., Jr Molecular biology of biotin attachment to proteins. J Nutr. 1999;129:477S–484S. doi: 10.1093/jn/129.2.477S. [DOI] [PubMed] [Google Scholar]
- Chen AL, Kim EW, Toh JY, Vashisht AA, Rashoff AQ, Van C, Huang AS, Moon AS, Bell HN, Bentolila LA, Wohlschlegel JA, Bradley PJ. Novel components of the Toxoplasma inner membrane complex revealed by BioID. MBio. 2015;6:e02357–02314. doi: 10.1128/mBio.02357-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi YH, Chen ZJ, Jeang KT. The nuclear envelopathies and human diseases. Journal of Biomedical Science. 2009;16:96. doi: 10.1186/1423-0127-16-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comartin D, Gupta GD, Fussner E, Coyaud E, Hasegan M, Archinti M, Cheung SW, Pinchev D, Lawo S, Raught B, Bazett-Jones DP, Luders J, Pelletier L. CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Curr Biol. 2013;23:1360–1366. doi: 10.1016/j.cub.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, Wrana JL, Choi H, Gingras AC. Sci Signal. United States: 2013. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions; p. rs15. [DOI] [PubMed] [Google Scholar]
- Dauer WT, Worman HJ. The Nuclear Envelope as a Signaling Node in Development and Disease. Developmental Cell. 2009;17:626–638. doi: 10.1016/j.devcel.2009.10.016. [DOI] [PubMed] [Google Scholar]
- Dhe-Paganon S, Werner ED, Chi YI, Shoelson SE. Structure of the Globular Tail of Nuclear Lamin. Journal of Biological Chemistry. 2002;277:17381–17384. doi: 10.1074/jbc.C200038200. [DOI] [PubMed] [Google Scholar]
- Dingar D, Kalkat M, Chan PK, Srikumar T, Bailey SD, Tu WB, Coyaud E, Ponzielli R, Kolyar M, Jurisica I, Huang A, Lupien M, Penn LZ, Raught B. BioID identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. J Proteomics. 2014 doi: 10.1016/j.jprot.2014.09.029. [DOI] [PubMed] [Google Scholar]
- Donaldson JG. Immunofluorescence staining. Curr Protoc Cell Biol. 2001;Chapter 4(Unit 4 3) doi: 10.1002/0471143030.cb0403s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreesen O, Chojnowski A, Ong PF, Zhao TY, Common JE, Lunny D, Lane EB, Lee SJ, Vardy LA, Stewart CL, Colman A. J Cell Biol. United States: 2013. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence; pp. 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreger M, Bengtsson L, Schoneberg T, Otto H, Hucho F. Proc Natl Acad Sci U S A. United States: 2001. Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane; pp. 11943–11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Marina P, Yu L. Constructing recombinant DNA molecules by PCR. Curr Protoc Mol Biol. 2007;Chapter 3(Unit 3):17. doi: 10.1002/0471142727.mb0317s78. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firat-Karalar EN, Rauniyar N, Yates JR, 3rd, Stearns T. Proximity interactions among centrosome components identify regulators of centriole duplication. Curr Biol. 2014;24:664–670. doi: 10.1016/j.cub.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proceedings of the National Academy of Sciences. 1986;83:6450–6454. doi: 10.1073/pnas.83.17.6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson K, Van Itallie CM, Aponte A, Gucek M, Tietgens AJ, Anderson JM. PLoS One. United States: 2015. Proteomic analysis of proteins surrounding occludin and claudin-4 reveals their proximity to signaling and trafficking networks; p. e0117074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Li Y, Fu X, Luo X. A Chinese patient with acquired partial lipodystrophy caused by a novel mutation with LMNB2 gene. J Pediatr Endocrinol Metab. 2012:25, 375–377. doi: 10.1515/jpem-2012-0007. [DOI] [PubMed] [Google Scholar]
- Guo Z, Neilson LJ, Zhong H, Murray PS, Zanivan S, Zaidel-Bar R. American Association for the Advancement of Science. United States: 2014. E-cadherin interactome complexity and robustness resolved by quantitative proteomics, Sci Signal; p. rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the Reannotated LMNB2 Gene Reveals Novel Mutations in Patients with Acquired Partial Lipodystrophy. Am J Hum Genet. 2006:383–389. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitlinger E, Peter M, Lustig A, Villiger W, Nigg EA, Aebi U. J Struct Biol. United States: 1992. The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated B2 lamins; pp. 74–89. [DOI] [PubMed] [Google Scholar]
- Holaska JM, Wilson KL. An Emerin “Proteome”: Purification of Distinct Emerin-Containing Complexes from HeLa Cells Suggests Molecular Basis for Diverse Roles Including Gene Regulation, mRNA Splicing, Signaling, Mechanosensing, and Nuclear Architecture. Biochemistry. 2007;46:8897–8908. doi: 10.1021/bi602636m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska JM, Wilson KL, Mansharamani M. Curr Opin Cell Biol. United States: 2002. The nuclear envelope, lamins and nuclear assembly; pp. 357–364. [DOI] [PubMed] [Google Scholar]
- Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, Roux KJ. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci U S A. 2014;111:E2453–2461. doi: 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Kc B, Roux KJ. Making the LINC: SUN and KASH protein interactions. Biol Chem. 2015;396:295–310. doi: 10.1515/hsz-2014-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitten GT, Nigg EA. The CaaX motif is required for isoprenylation, carboxyl methylation, and nuclear membrane association of lamin B2. J Cell Biol. 1991;113:13–23. doi: 10.1083/jcb.113.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack CG, Melo-Cardenas J, Imanishi SY, Goldman RD, Eriksson JE. J Cell Sci. 2014. The Company of Biologists Ltd; England: 2014. Interphase phosphorylation of lamin A; pp. 2683–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimm I, Östlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, Bonne G, Courvalin JC, Worman HJ, Zinn-Justin S. The Ig-like Structure of the C-Terminal Domain of Lamin A/C, Mutated in Muscular Dystrophies, Cardiomyopathy, and Partial Lipodystrophy. Structure. 2002;10:811–823. doi: 10.1016/s0969-2126(02)00777-3. [DOI] [PubMed] [Google Scholar]
- Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T. Identification of differential protein interactors of lamin A and progerin. Nucleus. 2010;1:513–525. doi: 10.4161/nucl.1.6.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon K, Beckett D. Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 2000:9, 1530–1539. doi: 10.1110/ps.9.8.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics. 2014 doi: 10.1016/j.jprot.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MD, Rominger KL, Young DL, Lynen F. The Enzymatic Synthesis of Holotranscarboxylase from Apotranscarboxylase and (+)-Biotin: II. INVESTIGATION OF THE REACTION MECHANISM. Journal of Biological Chemistry. 1964;239:2865–2871. [PubMed] [Google Scholar]
- Mahen R, Hattori H, Lee M, Sharma P, Jeyasekharan AD, Venkitaraman AR. PLoS One. United States: 2013. Atype lamins maintain the positional stability of DNA damage repair foci in mammalian nuclei; p. e61893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature. 1986;319:463–468. doi: 10.1038/319463a0. [DOI] [PubMed] [Google Scholar]
- Min GW, Tong XJ, Chen B, Zhang B, Liu ZF, Ding MX, Zhai ZH. Assembly of lamins in vitro. Cell Research. 1996;6:11–22. [Google Scholar]
- Mojica SA, Hovis KM, Frieman MB, Tran B, Hsia RC, Ravel J, Jenkins-Houk C, Wilson KL, Bavoil PM. SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol Biol Cell. 2015 doi: 10.1091/mbc.E14-11-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, Havlicek K, Demmel L, Yavuz S, Sealey-Cardona M, Vidilaseris K, Anrather D, Kostan J, Djinovic-Carugo K, Roux KJ, Warren G. Eukaryot Cell. United States: 2013. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation; pp. 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen R, Chesnut JD, Hoeffler JP, Kingston RE. Selection of transfected mammalian cells. Curr Protoc Mol Biol. 2003;Chapter 9(Unit 9 5) doi: 10.1002/0471142727.mb0905s62. [DOI] [PubMed] [Google Scholar]
- Ottaviano Y, Gerace L. Phosphorylation of the nuclear lamins during interphase and mitosis. J Biol Chem. 1985;260:624–632. [PubMed] [Google Scholar]
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Nat Genet. United States: 2006. Lamin B1 duplications cause autosomal dominant leukodystrophy; pp. 1114–1123. [DOI] [PubMed] [Google Scholar]
- Ptak C, Aitchison JD, Wozniak RW. The multifunctional nuclear pore complex: a platform for controlling gene expression. Curr Opin Cell Biol. 2014;28:46–53. doi: 10.1016/j.ceb.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie C, Cylinder I, Platt EJ, Barklis E. J Virol. American Society for Microbiology. All Rights Reserved; United States: 2015. Analysis of HIV-1 Gag Protein Interactions via Biotin Ligase Tagging; pp. 3988–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. J Cell Biol. United States: 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells; pp. 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin. A J Cell Sci. 1994;107(Pt 1):61–67. doi: 10.1242/jcs.107.1.61. [DOI] [PubMed] [Google Scholar]
- Sosa BA, Kutay U, Schwartz TU. Structural insights into LINC complexes. Curr Opin Struct Biol. 2013;23:285–291. doi: 10.1016/j.sbi.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed E, Elbediwy A, Vacca B, Dupasquier S, Hemkemeyer SA, Suddason T, Costa AC, Beaudry JB, Zihni C, Gallagher E, Pierreux CE, Balda MS, Matter K. J Cell Biol. United States: 2014. MarvelD3 couples tight junctions to the MEKK1-JNK pathway to regulate cell behavior and survival; pp. 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MY, Wang S, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, Zheng Y. Science. United States: 2006. A mitotic lamin B matrix induced by RanGTP required for spindle assembly; pp. 1887–1893. [DOI] [PubMed] [Google Scholar]
- Ueda S, Blee AM, Macway KG, Renner DJ, Yamada S. Force Dependent Biotinylation of Myosin IIA by alpha-Catenin Tagged with a Promiscuous Biotin Ligase. PLoS One. 2015:e0122886. doi: 10.1371/journal.pone.0122886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Aponte A, Tietgens AJ, Gucek M, Fredriksson K, Anderson JM. J Biol Chem. United States: 2013. The N and C termini of ZO-1 are surrounded by distinct proteins and functional protein networks; pp. 13775–13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Tietgens AJ, Aponte A, Fredriksson K, Fanning AS, Gucek M, Anderson JM. J Cell Sci. England: 2014. Biotin ligase tagging identifies proteins proximal to E-cadherin, including lipoma preferred partner, a regulator of epithelial cell-cell and cell-substrate adhesion; pp. 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraeten VL, Broers JL, Ramaekers FC, van Steensel MA. The nuclear envelope, a key structure in cellular integrity and gene expression. Curr Med Chem. 2007;14:1231–1248. doi: 10.2174/092986707780598032. [DOI] [PubMed] [Google Scholar]
- Wilkinson FL, Holaska JM, Zhang Z, Sharma A, Manilal S, Holt I, Stamm S, Wilson KL, Morris GE. Eur J Biochem. Germany: 2003. Emerin interacts in vitro with the splicing-associated factor, YT521-B; pp. 2459–2466. [DOI] [PubMed] [Google Scholar]
- Wong X, Luperchio TR, Reddy KL. NET gains and losses: the role of changing nuclear envelope proteomes in genome regulation. Curr Opin Cell Biol. 2014;28:105–120. doi: 10.1016/j.ceb.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring. Harb Perspect Biol. 2010;2:a000760. doi: 10.1101/cshperspect.a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]