

Abstract
The chemical nature of prions and the mechanism by which they propagate are now reasonably well understood. In contrast, much less is known about the identity of the toxic prion protein (PrP) species that are responsible for neuronal death, and the cellular pathways that these forms activate. In addition, the normal, physiological function of cellular PrP (PrPC) has remained mysterious, hampering efforts to determine whether loss or alteration of this function contributes to the disease phenotype. Considerable evidence now suggests that aggregation, toxicity, and infectivity are distinct properties of PrP that do no necessarily coincide. In this review, we will discuss several mutant forms of PrP that produce spontaneous neurodegeneration in humans and/or transgenic mice without the formation of infectious PrPSc. These include an octapeptide insertional mutation, point mutations that favor synthesis of transmembrane forms of PrP, and deletions encompassing the central domain whose neurotoxicity is antagonized by the presence of wild-type PrP. By isolating the neurotoxic effects of PrP from the formation of infectious prions, these mutants have provided important insights into possible pathogenic mechanisms. These studies suggest that prion neurotoxicity may involve subversion of a cytoprotective activity of PrPC via altered signaling events at the plasma membrane.
Introduction
Prion diseases are fatal neurodegenerative illnesses of man and animals. This group of disorders includes Creutzfeldt-Jakob Disease (CJD), kuru, fatal familial insomnia (FFI), Gerstmann-Sträussler syndrome (GSS), and new variant CJD in humans, as well as bovine spongiform encephalopathy in cattle, chronic wasting disease in deer and elk, and scrapie in sheep and goats (Prusiner, 2004). Patients affected with these disorders suffer from dementia and ataxia, and often display spongiform degeneration and amyloid deposition in their brains.
A wealth of evidence suggests that the central molecular event in prion diseases is the conformational conversion of PrPC, a normal cell-surface glycoprotein, into PrPSc, an abnormal isoform that is infectious in the absence of nucleic acid (Aguzzi et al., 2008; Prusiner, 1998). The precise structural differences between the two PrP isoforms remain to be defined, although it is clear that PrPSc contains significantly more β-sheet and is more protease-resistant and aggregated than PrPC. The conversion of PrPC to PrPSc is thought to involve a templating mechanism in which the two forms physically interact.
Although we now have a detailed understanding of how prions propagate, the cellular mechanisms by which they kill neurons, and the toxic forms of PrP responsible, are poorly understood (Chiesa and Harris, 2001; Harris and True, 2006). Important insights into this issue have been obtained by analysis of PrP molecules carrying neurotoxic mutations. Several kinds of mutant PrP molecules induce spontaneous neurological illness in human beings or transgenic mice in the absence of infection from exogenous sources (Table 1 and Fig. 1). One category of such molecules are those carrying point or insertional mutations linked to human familial prion diseases. These mutants generally display PrPSc-like biochemical properties, and at least part of their pathogenicity is likely to depend on the toxic properties of the oligomeric protein aggregates that they form. A second category of mutations are those in the N-terminal signal sequence and hydrophobic domain that influence the membrane topology of PrP. A third category includes a series of deletion mutations encompassing the central region of PrP that endow the protein with a powerful neurotoxic activity suppressible by co-expression of wild-type PrP. Mutants in the last two categories are not aggregated or protease-resistant, and their effects are likely due to alterations in a physiological activity of PrPC.
TABLE 1.
Properties of PrPC, PrPSc, and neurotoxic mutants of PrP
| Molecule | Protease-resistance/aggregation | Cellular localization | Infectivity | Disease onset (days)a | Pathological targets | Effect of wild-type PrP (expression level) |
|---|---|---|---|---|---|---|
| PrPC | − | PM/Rafts | − | N/A | None | N/A |
| PrPSc | +++ | PM, endosomes | + | Depends on strain/host | Neurons, WM | Exacerbation |
| PG14Spon1–3 | + | ER | − | 240 | CGN, dendrites, axons/myelin | No effect (1X) |
| PG14RML1, 2 | +++ | Unknown | + | 240 (post-inoculation) | CGN | Not tested |
| CtmPrP (L9R/3AV)4–6 | − | ER (CHO cells); Golgi (neurons) | − | 85–172b | CGN, HPN | Exacerbation (0.5–1X) |
| PrPΔ32–1217 | − | Not tested | − | 42–56c | CGN | Rescue (0.5X) |
| PrPΔ32–1347 | − | PM | − | 21–35c | CGN, WM | Rescue (0.5X) |
| PrPΔ94–1348 | − | Rafts | − | 14c | WM | Rescue (2–3X) |
| PrPΔ105–1259 | − | PM/Rafts | − | 7c | CGN, WM | Rescue (5X) |
Abbreviations: CHO, Chinese hamster ovary; CGN, cerebellar granule neurons; ER, endoplasmic reticulum; HPN, hippocampal neurons; N/A, not applicable; PM, plasma membrane; WM, white matter.
Disease onset varies with transgene expression level. Data are for highest expressing lines (1–3X).
For mice on the Prn-p+/+ background.
For mice on the Prn-p0/0 background.
References:
FIGURE 1. Schematic of wild-type and mutant PrP molecules, Doppel, and Shadoo.
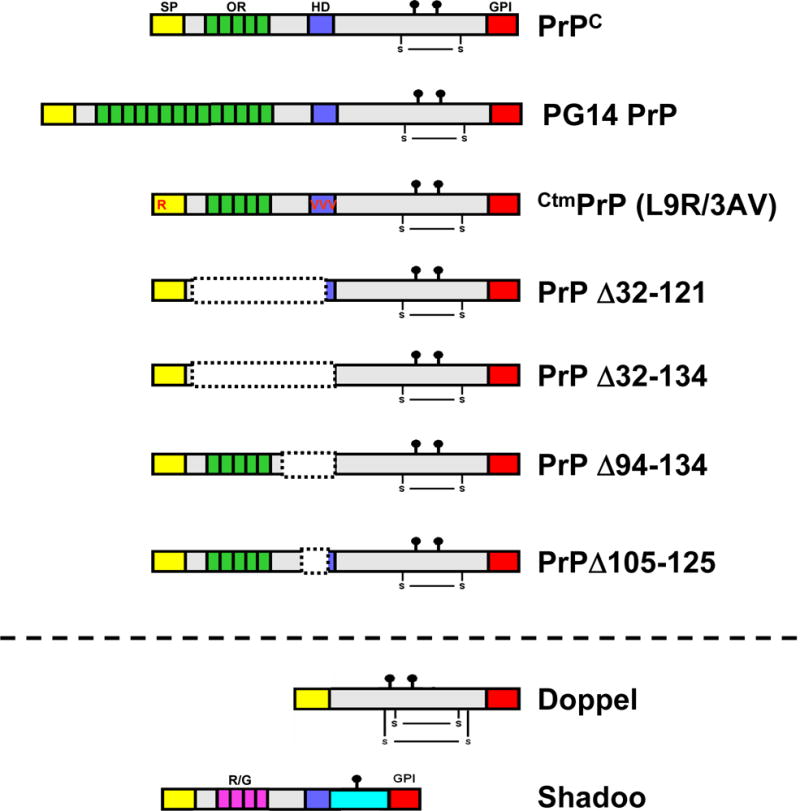
Structural domains are indicated by the colored blocks: SS (yellow), signal sequence; OR (green), octapeptide repeats; HD (blue), hydrophobic domain; GPI (red), glycosyl-phosphatidylinositol attachment signal; R/G (pink), arginine/glycine repeats of Sho. The lollipop symbols indicate sites of N-linked glycosylation, and the S—S symbols indicate disulfide linkages.
Importantly, none of these three categories of mutations is accompanied by the formation of infectious PrPSc. By isolating the neurotoxic effects of PrP from the propagation of infectious prions, these mutants have made it possible to focus on pathogenic mechanisms underlying the disease process. Some of these mechanisms turn out to be surprisingly similar to those associated with non-infectious neurodegenerative disorders such as Alzheimer’s disease.
In this article, we will discuss work form our laboratory utilizing each of these three categories of PrP mutants. As a prelude, we will first review what is currently known about the PrP forms and cellular pathways underlying prion neurotoxicity, as well as our current understanding of the physiological function of PrPC.
Prion neurotoxicity: what is the toxic molecule?
What form of PrP is responsible for killing neurons? It has commonly been assumed that PrPSc itself is the primary cause of neurodegeneration, based on the temporal and anatomical correlation between the accumulation of this form and the development of neuropathological changes. However, there are a number of situations where this correlation is weak or absent. In several kinds of transmission experiments, for example, significant pathology and/or clinical dysfunction develop with little accumulation of PrPSc (Flechsig et al., 2000; Lasmezas et al., 1997; Manson et al., 1999). In addition, some familial prion diseases are not transmissible, and are not accompanied by the accumulation of protease resistant PrP (Brown et al., 1994b; Piccardo et al., 2001; Tateishi and Kitamoto, 1995; Tateishi et al., 1990; Tateishi et al., 1996). On the other hand, there are sub-clinical infections in which there is abundant PrPSc but little symptomatology, for example after inoculation of hamster prions into mice (Hill et al., 2000; Race et al., 2001).
Taken together, these situations argue that PrPSc, the infectious form of PrP, may not be the proximate cause of neuronal dysfunction and degeneration in prion diseases. Several alternative forms of PrP, distinct from both PrPC and PrPSc, have therefore been hypothesized to be the primary neurotoxic species (designated PrPtoxic). In subsequent sections, we will discuss two possible candidates for PrPtoxic (PG14spon and CtmPrP).
Prion neurotoxicity: what are the cellular pathways?
How do PrPSc or other toxic forms of PrP induce neuronal death? There is now a growing body of evidence that PrPC, in addition to serving as a precursor of PrPSc, acts as a signal transducer or mediator of the neurotoxic effects of PrPSc (Harris and True, 2006). Support for this concept derives from several experimental situations in which expression of membrane-anchored PrPC in target neurons is essential for conferring sensitivity to PrPSc-induced neurodegeneration or toxicity. First, Prn-p0/0 neurons are resistant to the pathologic effects of PrPSc supplied from grafted brain tissue (Brandner et al., 1996) or from nearby astrocytes (Mallucci et al., 2003). Second, scrapie-inoculated mice expressing a glycolipid anchor-negative version of PrP develop minimal brain pathology and neurological dysfunction despite the accumulation of substantial amounts of PrPSc amyloid (Chesebro et al., 2005). Although interpretation of this experiment is complicated by the relatively low expression levels of anchorless PrP in these mouse lines, one possible implication is that PrP must be membrane-anchored to efficiently mediate a toxic signal (Aguzzi, 2005). Third, Prn-p0/0 neurons in culture have been found to be resistant to apoptosis induced by exposure to the synthetic peptide PrP106–126, which has been used as a model for PrPSc (Brown et al., 1994a). Finally, a recent study has shown that PrPC exerts a protective activity against cellular stress, and that PrPSc abrogates this activity by activating stress-related signaling cascades (Rambold et al., 2008).
Combined with evidence that PrPC normally serves as a neuroprotective molecule (see below), these results taken together suggest a “subversion-of-function” hypothesis to explain prion-induced pathology (Harris and True, 2006): specifically, interaction with PrPSc (or other pathogenic intermediates) alters a normal, physiological activity of PrPC in such a way that a neurotoxic stimulus is delivered. We will return to this mechanism later, when discussing the neurotoxicity of certain deleted forms of PrP.
Function of PrPC
In order to specify how PrPC function is altered or subverted in prion diseases, it is clearly essential to understand the normal, physiological role of the protein. Surprisingly, however, twenty-five years since PrPC was first identified as an endogenous cellular protein, its physiological activity remains obscure (reviewed in Westergard et al., 2007). A variety of functions have been proposed, including roles in metal ion trafficking (Pauly and Harris, 1998), cell adhesion (Mange et al., 2002), and signal transduction (Mouillet-Richard et al., 2000). However, the evidence in favor of each of these hypothesized functions is not definitive.
Attempts to deduce the function of PrPC from the phenotypes of PrP-null mice have been unrewarding. These mice display no major anatomical or developmental deficits, with the exception of lines in which the gene encoding Doppel (a PrP paralog) is artifactually up-regulated (Büeler et al., 1992; Manson et al., 1994). Subtle phenotypic abnormalities have been described in PrP knock-out mice at the cellular or behavioral level, but some of these have been controversial, and in any case they have not provided an unambiguous clue to the biological function of PrPC (Steele et al., 2007). A recent report suggests a role for PrPC in embryonic cell adhesion, based on the phenotype of zebrafish in which expression of a PrP homologue has been knocked down (Malaga-Trillo et al., 2009). Whether this purported function is conserved in mammalian PrP remains to be determined.
Several intriguing lines of evidence have emerged recently suggesting that PrPC may exert a cytoprotective activity, particularly against stresses (either internal or environmental) that initiate an apoptotic program (reviewed in Roucou et al., 2004; Roucou and LeBlanc, 2005). First, PrP over-expression rescues cultured neurons, some mammalian cell lines and yeast cells from pro-apoptotic stimuli, including Bax expression, serum withdrawal, and cytokine treatment (Bounhar et al., 2001; Diarra-Mehrpour et al., 2004; Kuwahara et al., 1999; Li and Harris, 2005; Roucou et al., 2005). Second, there is evidence that endogenous PrP protects cells against oxidative and pathologic stressors. For example, neurons cultured from Prn-p0/0 mice display several subtle abnormalities related to increased susceptibility to oxidative stress (Brown et al., 2002). Moreover, after ischemic or traumatic brain injury, lesion size is larger in Prn-p0/0 compared to wild-type mice (Hoshino et al., 2003; McLennan et al., 2004; Sakurai-Yamashita et al., 2005; Spudich et al., 2005). Finally, Prn-p0/0 mice are more susceptible to kainate-induced seizures (Rangel et al., 2007) and their retinal photoreceptors are more prone to light-induced degeneration (Frigg et al., 2006).
To explore the cytoprotective activity of PrPC, we recently attempted to reproduce several of the published assays for this activity (Christensen and Harris, 2008). In one set of experiments, we found that PrP over-expression had a minimal effect on the death of MCF-7 breast carcinoma cells treated with TNF-α, and on Prn-p0/0 immortalized hippocampal neurons (HpL3–4 cells) subjected to serum deprivation. In a second set of assays, we observed only a small difference in viability between cerebellar granule neurons cultured from PrP-null and control mice in response to activation of endogenous or exogenous Bax. Although our results do not rule out a cytoprotective activity of PrPC, they suggest that existing cell culture systems may be inadequate for modeling this activity.
Mutant PrP molecules associated with inherited prion diseases
Studies of mutant PrP molecules associated with inherited prion diseases have provided important clues to the molecular characteristics of PrPtoxic, and the mechanisms by which it is pathogenic. Approximately 10% of the cases of CJD and all cases of GSS and FFI are linked to dominantly inherited, germline mutations in the PrP gene on chromosome 20 (Mead, 2006). The mutations are presumed to favor spontaneous conversion of the protein to the PrPSc state without the necessity for contact with exogenous infectious agent. Point mutations occur in the C-terminal half of the PrP molecule, and are associated with either CJD, GSS or FFI. Insertional mutations, which are associated with a variable phenotype that can include features of CJD or GSS, consist of one to nine additional copies of a peptide repeat that is normally present in five copies in the N-terminal half of the protein.
A number of studies have used transfected cell lines to analyze the biochemical and cell biological properties of PrP molecules carrying disease-associated mutations (reviewed by Harris, 2003). These studies have revealed that some, but not all mutants, display PrPSc-like biochemical properties including aggregation propensity as well as resistance to proteases and a GPI-specific phospholipase (Lehmann and Harris, 1996; Priola and Chesebro, 1998). Correlating with these abnormal biochemical properties, some mutants display altered subcellular localization, including partial retention in the ER (Ivanova et al., 2001), or retrotranslocation into the cytoplasm with subsequent degradation by the proteasome (Lorenz et al., 2002). Interestingly, some disease-associated PrP mutants are identical to wild-type PrP in terms of their biochemical properties and cellular distribution (Harris, 2003), raising the possibility that these molecules are pathogenic, not because they misfold and aggregate, but because the mutation subtly alters a physiological activity normally associated with PrPC. Unfortunately, expression in transfected cells of PrP molecules carrying disease-associated mutations is generally not cytopathic, making it difficult to analyze the neurotoxic mechanisms underlying these mutants. For this reason, we and others have turned to transgenic models of familial prion diseases.
A transgenic model of an octapeptide insertion: Tg(PG14) mice
Tg(PG14) mice express the mouse homologue of a nine-octapeptide insertional PrP mutant associated with a familial prion disease of humans (Duchen et al., 1993; Krasemann et al., 1995; Owen et al., 1992) (Fig. 1). These animals display a spontaneous, progressive neurodegenerative disease with symptoms of ataxia, cerebellar granule cell loss, gliosis, and PrP deposition (Chiesa et al., 2000; Chiesa et al., 1998). Tg(PG14) mice spontaneously accumulate in their brains an insoluble and weakly protease-resistant form of the mutant protein (Chiesa et al., 2003). This form (designated PG14Spon) is highly neurotoxic, but is not infectious in animal bioassays. In contrast, when Tg(PG14) mice are inoculated with the RML strain of prions, they accumulate a different form of PG14 PrP (designated PG14RML) that is highly protease-resistant and infectious in animal transmission experiments (Chiesa et al., 2003). Consistent with other published work, these studies emphasize that infectivity and pathogenicity are two distinct properties of PrP.
To gain insight into the molecular determinants of infectivity and pathogenicity, we have undertaken a biochemical characterization the biochemical properties of PG14Spon and PG14RML. Our initial studies (Chiesa et al., 2003) indicated that these two forms, although conformationally related, differed in their quaternary structure, with PG14RML forming larger, more tightly packed aggregates. We found that the majority of PG14RML aggregates have a sedimentation coefficient of >50S, with ~30% of them having a sedimentation coefficient of >120S. If composed exclusively of PrP, the latter aggregates would contain >200 molecules of the protein. In contrast, 15–20% of PG14Spon PrP was monomeric (3.2S), with the rest sedimenting at 16–20S (corresponding to oligomers containing ~20–30 molecules of PrP). These results suggest that while highly aggregated polymers of PrP are a prerequisite for prion infectivity, small, β-rich oligomers are the species that are toxic to nerve cells (Chiesa et al., 2003). This conclusion is strikingly reminiscent of recent work in Alzheimer’s disease, which has demonstrated that small oligomers of the Aβ peptide (ranging in size from 2–100 subunits), rather than large amyloid fibers, are the primary toxic species (Haass and Selkoe, 2007; Walsh and Selkoe, 2007).
In a more recent study (Biasini et al., 2008), we have subjected PG14Spon and PG14RML to a panel of assays commonly used to distinguish infectious PrP (PrPSc) from cellular PrP (PrPC), including immobilized metal affinity chromatography, precipitation with sodium phosphotungstate, and immunoprecipitation with PrPC- and PrPSc-specific antibodies. Surprisingly, we found that aggregates of PG14Spon and PG14RML behave identically to each other, and to authentic PrPSc, in each of these biochemical assays. PG14Spon however, in contrast to PG14RML and PrPSc, was unable to seed the misfolding of PrPC in an in vitro protein misfolding cyclic amplification reaction. Collectively, these results suggest that infectious and non-infectious aggregates of PrP share common structural features accounting for their toxicity, and that self-propagation of PrP involves more subtle molecular differences that remain to be identified.
Gain vs. loss of function as a pathogenic mechanism
How do mutant PrP molecules induce neurodegeneration? For most dominantly inherited neurodegenerative disorders, a gain-of-function mechanism is invoked in which the mutant protein is presumed to acquire a toxic property unrelated to its normal physiological activity (Winklhofer et al., 2008). For example, aggregates of the mutant protein could disrupt important cellular processes and so impair neuronal viability.
Such a mechanism appears to play a role in the pathogenicity of the PG14 PrP, based on our studies of the localization of this mutant in neurons in the brains of transgenic mice. We generated transgenic mice expressing PG14-EGFP, a fluorescent fusion protein that can be directly visualized in vivo (Medrano et al., 2008). Tg(PG14-EGFP) mice develop an ataxic neurological illness characterized by astrogliosis, PrP aggregation, and accumulation of a partially protease-resistant form of the mutant PrP. Strikingly, PG14-EGFP forms numerous fluorescent aggregates in the neuropil and white matter of multiple brain regions. These aggregates are particularly prominent along axonal tracts in both brain and peripheral nerve, and similar intracellular deposits are visible along the processes of cultured neurons. Our results suggest that intra-axonal aggregates of mutant PrP could contribute to the pathogenesis of familial prion disease by disrupting axonal transport. A similar mechanism of axonal blockage has recently been proposed to explain the pathogenesis Alzheimer’s disease (Stokin et al., 2005).
To investigate whether a loss-of-function mechanism could also play a role in the neurotoxicity of PG14 PrP, we crossed Tg(PG14) mice with Tg(PrPΔ32–134) mice (Li et al., 2007c). Tg(PrPΔ32–134) mice, which express an N-terminally truncated form of PrP, spontaneously develop a neurodegenerative phenotype that is stoichiometrically reversed by co-expression of wild-type PrP. We found that, at equivalent expression levels, PG14 PrP is significantly less efficient than wild-type PrP in suppressing the development of clinical symptoms and neuropathology in Tg(PrPΔ32–134) mice. Thus, our results suggest that some features of the neurological illness associated with inherited PrP mutations may be attributable to a loss of PrP neuroprotective function.
Since PrP-null mice do not display features of a prion disease (Büeler et al., 1992; Manson et al., 1994), it is unlikely that these disorders are due exclusively to the simple loss of an essential physiological function of PrPC. A loss of function mechanism also appears to be incompatible with the dominant mode of inheritance of familial prion diseases. However, these facts do not rule out a contribution of PrP functional deficiency to prion-related neurodegeneration. For example, PrPSc or mutant PrP may sequester wild-type PrPC into aggregates that lack functional activity, thereby producing a dominant-negative effect. Moreover a biological activity of PrPC that is dispensable under normal conditions may become essential in the disease state due to cellular or organismal stress.
Taken together, our results also indicate that more than one pathogenic mechanism (both gain- and loss-of-function) may contribute to neurotoxicity in inherited prion diseases.
Topological variants of PrP
There is evidence that alterations in membrane topology can result in neurotoxic forms of PrP. While most PrP molecules are linked to the plasma membrane exclusively via a GPI anchor, three topological variants have been described: CytoPrP, NtmPrP, and CtmPrP (Chakrabarti et al., 2009). CytoPrP, in which the polypeptide chain lies entirely in the cytoplasm, is produced at low levels as a result of inefficient translocation into the ER (Drisaldi et al., 2003). The amount of CytoPrP can be regulated by cellular stress, a mechanism that has been termed “pre-emptive quality control” (Kang et al., 2006). Mice expressing a transgenically encoded form of CytoPrP show a severe neurodegenerative phenotype, indicating that this molecule possesses neurotoxic activity (Ma et al., 2002). It has been claimed that prion infection increases production of CytoPrP (Rane et al., 2008), thereby contributing to neurotoxicity, but this observation is not confirmed by out own data (Stewart and Harris, 2003). At present, the pathogenic role of CytoPrP in prion diseases remains unclear.
Two transmembrane variants of PrP have also been described. CtmPrP and NtmPrP span the lipid bilayer once via a stretch of hydrophobic amino acids in the central region of the protein (residues 111–135), with the N- or C-terminus, respectively, in the cytoplasm. These molecules were first observed in cell-free translation/translocation systems, where it was shown that their amounts could be altered by mutations in the central hydrophobic domain, as well as in an adjacent “stop transfer effector” segment that contains several positively charge amino acids (Hay et al., 1987; Hegde et al., 1998; Yost et al., 1990). Subsequent work from our laboratory (Stewart and Harris, 2003) and others (Kim et al., 2001) demonstrated that non-conservative substitutions in the core of the N-terminal signal sequence also increased the proportion of CtmPrP, and that combining these mutations with ones in the central domain resulted in almost exclusive synthesis of the CtmPrP. Cell biological analysis of one such compound mutant (L9R/3AV) (Fig. 1) led to the conclusion that CtmPrP contains an uncleaved signal peptide as well as a GPI anchor, and that it is retained intracellularly in either the ER or the Golgi (depending on the cell type) (Stewart et al., 2001; Stewart and Harris, 2005).
Transgenic mice expressing PrP molecules with CtmPrP-promoting mutations display a neurodegenerative phenotype, implying that CtmPrP has neurotoxic potential in vivo (Hegde et al., 1998; Stewart et al., 2005). PrPSc accumulation has been claimed to cause enhanced generation of CtmPrP, and on this basis it has been suggested that CtmPrP represents a key neurotoxic intermediate in prion disorders (Hegde et al., 1999). However, this hypothesis is called into question by our observations that scrapie infection does not significantly alter CtmPrP levels in cultured cells or brain (Stewart and Harris, 2003), and that pathogenic mutations outside of the central, hydrophobic domain do not alter the membrane topology of PrP (Stewart and Harris, 2001).
Whether or not CtmPrP itself plays a role in naturally occurring prion diseases, it is likely to be revealing PrP-related neurotoxic signaling mechanisms that could contribute to the pathological process. Our studies of Tg(L9R/3AV) mice exemplify this point. These animals develop a fatal neurological illness characterized by ataxia and neuronal loss in the cerebellum and hippocampus (Stewart et al., 2005). Importantly, this phenotype is strongly dependent on co-expression of endogenous, wild-type PrP, suggesting a synergistic interaction between transmembrane and exclusively GPI-anchored forms of PrP in transducing a toxic signal.
Neurotoxicity of PrP deletion mutants
One of the most surprising and intriguing observations to have emerged concerning the neurotoxic effects of PrP was first made over 10 years ago by Shmerling et al. (1998) as part of a study to examine which parts of the PrP molecule were necessary for sustaining prion infection. These authors discovered that mice expressing PrP harboring either of two large, N-terminal deletions (Δ32–121 and Δ32–134) (Fig. 1) developed a spontaneous neurodegenerative illness even without inoculation with scrapie prions. This illness was characterized by ataxia and massive degeneration of cerebellar granule neurons. Mice with shorter deletions (Δ32–80, Δ32–93, Δ32–106) were phenotypically normal, implying a critical role for amino acids distal to residue 106. What was even more surprising, the neurodegenerative phenotype of the Tg(Δ32–121) and Tg(32–134) mice was only observed on the Prn-p0/0 background: co-expression of endogenous, wild-type PrP from even a single Prn-p allele completely abrogated clinical symptoms and neuropathology. To explain these puzzling observations, the authors proposed a loss-of-function model based on the existence of two hypothetical molecules, one of which (Lprp) is a ligand that binds to PrP and the other (π) a receptor that binds the ligand when PrP is absent. It was suggested that binding of Lprp to wild-type PrP delivered an essential trophic signal, while binding to N-terminally deleted PrP failed to elicit this signal and also prevented interaction with π.
To more precisely map the region of PrP responsible for the spontaneous neurodegenerative phenotype, we created Tg(ΔCR) mice expressing PrP with a much smaller deletion, comprising residues 105–125 within the Central Region of the molecule (Li et al., 2007b) (Fig. 1). The deleted segment lies within a highly conserved, unstructured region of the protein, and encompasses a cluster of three positively charged amino acids (residues 105, 109, and 110) followed by a stretch of 15 hydrophobic residues (residues 111–135). There were three motivations for focusing on the 105–125 region as a possible toxicity-determining domain in PrP. First, the study by Shmerling et al. (1998) had pinpointed residues distal to position 106 as being critical determinants of neurotoxicity. Second, it was known that a synthetic peptide derived from this region (PrP106-126) was toxic when applied to cultured neurons from Prn-p+/+ but not from Prn-p0/0 mice (Brown et al., 1994a). Third, the 105–125 region is one of the most highly conserved portions of the sequence, being virtually identical in PrP molecules from zebrafish to humans (Rivera-Milla et al., 2006).
Tg(ΔCR) mice display a spontaneous neurodegenerative phenotype that is more severe than any we have ever encountered in other transgenic mice (Li et al., 2007b). This phenotype is observed at modest expression levels of the mutant protein (0.5–1X). On the Prn-p0/0 background, Tg(ΔCR) mice become ill and die within one week of birth. This phenotype is reversed in a dose-dependent fashion by co-expression of wild-type PrP: one Prn-p allele (0.5X expression level) delays death until 25 days, and two Prn-p alleles (1X expression level) delays death until 48 days. The presence of one Tga20 allele (5X expression level of wild-type PrP) strongly rescues the neurodegenerative phenotype, with mice remaining alive for well over one year.
Tg(ΔCR) mice show two major kinds of neuropathology (Li et al., 2007b). First, there is massive degeneration of cerebellar granule cells (with no effect on Purkinje cells), resulting in severe atrophy of the cerebellum. Degenerating granule neurons display extensive DNA fragmentation, but there is no activation of caspases 3 or 8 (Christensen et al., manuscript in preparation). By electron microscopy, granule cells display a unique, non-apoptotic ultrastructural morphology (Christensen et al., manuscript in preparation). In conjunction with evidence that neurodegeneration in Tg(ΔCR) mice is Bax-independent (Li et al., 2007a), our ultrastructural and biochemical data indicate that PrPΔCR is activating a novel form of neuronal death that is not related to apoptosis, necrosis, or autophagy.
The second major neuropathological feature of Tg(ΔCR) mice is vacuolar degeneration of white matter areas of the brain and spinal cord, particularly in older animals. This abnormality, which was also seen Tg(Δ32–134) mice (Radovanovic et al., 2005), may be due either to axonal or myelin damage.
The biochemical and cell biological properties of PrPΔCR suggest that the neurotoxicity of this molecule results from an alteration of a normal activity of PrPC, rather than from accumulation of misfolded protein aggregates or cellular mislocalization. PrPΔCR is not aggregated or protease-resistant like PrPSc (Li et al., 2007b). Moreover, the cellular localization of PrPΔCR is identical to that of wild-type PrP (Christensen and Harris, 2009). Like its wild-type counterpart, ΔCR PrP is present in lipid rafts on the plasma membrane, where it is attached by a GPI anchor, and it selectively localizes to the apical surface of polarized MDCK epithelial cells. Moreover, PrPΔCR is distributed in a punctate pattern on the surface of neuronal processes, similar to the wild-type protein.
Recently, Baumann et al. (Baumann et al., 2007) described lines of mice expressing PrP with a deletion of residues 94–134. These animals also display a neurodegenerative phenotype that is suppressible by co-expression of wild-type PrP. It is instructive to compare the features of mice expressing the four different pathogenic deletions thus far published (Δ32–121, Δ32–134, Δ94–134, and Δ105–125) (Fig. 1). Each of these deletions encompasses the 105–125 region, demonstrating the importance of these residues in determining the neurotoxic activity of PrP. Interestingly, the four kinds of mice differ in the severity of the phenotypes they display (as indicated by age at symptom onset), and there is a correlation between the severity of the phenotype and the amount of wild-type PrP required for rescue (Table 1). Tg(ΔCR) mice, which have the strongest phenotype (illness onset at <1 week on the Prn-p0/0 background), require >5X expression of wild-type PrP for complete rescue, while Tg(PrPΔ34–134) mice (illness onset at 3–5 weeks) are almost completely rescued by 0.5X co-expression of wild-type PrP. This quantitative relationship between neurotoxic potency and the ability to be suppressed by wild-type PrP provides important clues to the underlying molecular mechanisms (see below).
Doppel and shadoo
Studies of two proteins with structural similarities to PrP have provided additional insights into the neurotoxic activities of PrP. Doppel (Dpl) is a PrP paralog that resembles the C-terminal half of PrP, but lacks residues homologous to the flexible N-terminal domain (Mo et al., 2001) (Fig. 1). Dpl thus structurally resembles the deleted forms of PrP that produce neurodegeneration in transgenic mice. The Dpl gene, which is normally expressed primarily in testis, is expressed ectopically in the brain in certain lines of Prn-p0/0 mice as a result of intergenic splicing events between the adjacent PrP and Dpl genes (Moore et al., 1999). These lines, as well as transgenic lines expressing elevated levels of Dpl in the brain, display a neurodegenerative phenotype that is stoichiometrically rescued by wild-type PrP (Moore et al., 2001; Rossi et al., 2001). Dpl-induced neuronal death involves Purkinje cells as well as granule cells in the cerebellum, and is partially Bax-independent (Dong et al., 2007; Heitz et al., 2007). Taken together, the available data make it very likely that the neurotoxicity of Dpl involves the same molecular pathways as those activated by deleted forms of PrP.
While Dpl is structurally related to the C-terminal half of PrP, the recently described protein, Shadoo (Sho), resembles a GPI-anchored version of the flexible, N-terminal tail of PrP, including the highly conserved central region (reviewed in Watts and Westaway, 2007) (Fig. 1). Sho is expressed in many of the same regions of the brain as PrPC, although the cellular distribution of the two proteins is complementary (Watts et al., 2007). In the cerebellum, for example, Sho is most abundant in Purkinje cell bodies and dendrites, while PrPC is present at highest levels in granule neurons and their axons in the molecular layer. In a neuronal culture assay, Sho had no toxic activity on its own, but was able to suppress the toxicity of co-expressed Dpl and PrPΔ32–121 (Watts et al., 2007). Thus, Sho displays a neuroprotective activity like that of wild-type PrPC. The protective activity of PrPC presumably resides in the N-terminal half of the protein, based on the observation that fusion of this region to Dpl prevents neurodegeneration in transgenic mice (Yoshikawa et al., 2008). Interestingly, Sho protein levels are dramatically down-regulated in scrapie-infected mice, raising the possibility that loss of Sho neuroprotection may play a role in prion diseases (Watts et al., 2007). It has been suggested that Sho is a candidate for π, the hypothetical receptor that has been postulated to deliver an essential neurotrophic signal in the absence of PrP (Watts et al., 2007).
A model for PrP neurotoxicity: subversion of function
Studies of the biological activities of ΔCR and other deleted forms of PrP, along with Dpl and Sho, provide powerful mechanistic insights into the normal function of PrPC, and how it can be subverted to produce neurotoxic effects. The dose-dependent, opposing effects of truncated and wild-type PrP are most easily explained by supposing that both proteins compete for binding to a common molecular target, with binding of truncated PrP eliciting a neurotoxic signal, and binding of wild-type PrP silencing the signal (or restoring a non-toxic, physiological signal) (Fig. 2). The molecular target for PrP is presumably a receptor or other cell-surface complex capable of transducing a signal to the interior of the cell. We hypothesize that binding to the target involves at least two sites on PrP: the CR region (residues 105–125), as well as a more C-terminal region. In the absence of the CR region (as in PrPΔCR), binding of the C-terminal region alone would elicit a neurotoxic signal. Binding at both sites (wild-type PrP) would produce a physiological signal or no signal. We suggest that an identical mechanism is responsible for the neurotoxicity of all four deleted forms of PrP as well as Dpl, with variations in the toxic potency of these molecules being attributable to differences in their affinity for the putative binding target. The cytoprotective effect of Sho could be due to its ability to occupy the CR binding site on the putative transducer molecule.
FIGURE 2. Model for the neuroprotective activity of PrPC, and subversion of this activity by neurotoxic forms of PrP.

The structured, C-terminal half of PrP is shown in green and the flexible, N-terminal tail as a blue line. The CR segment of PrP (residues 105–125) is shown as a red rectangle. Tr, hypothetical signal transducing protein that normally generates a neuroprotective signal (solid pink), but which can assume an altered conformation (crosshatched pink) that generates a neurotoxic signal. Two binding sites between PrP and Tr are shown, one involving the C-terminal half of PrP and the other CR segment of PrP. When both binding sites are occupied, Tr elicits a non-essential neuroprotective signal (PrPC). When only the C-terminal site is occupied, as would be the case when the CR segment is absent (PrPΔCR), embedded in the lipid bilayer (CtmPrP), or conformationally altered (PG14 and PrPSc), the transducer delivers a neurotoxic signal. The toxicity of CtmPrP requires the cooperation of wild-type PrPC.
Although ΔCR and other deleted forms of PrP are artificial molecules, working out how they kill neurons will likely provide crucial insights into neurodegeneration in naturally occurring prion diseases. Like PrPΔCR, PrPSc (or other toxic forms of PrP that accumulate during prion infection) appears to act by subverting a normal function of PrPC (see above). It is therefore possible that PrPSc/PrPtoxic interacts with the same putative membrane target as PrPΔCR, resulting in generation of a similar neurotoxic signal (Fig. 2). Interestingly, there is evidence that PrPSc is conformationally altered in the CR region (Peretz et al, 1997), perhaps preventing binding interactions involving this site. Thus, PrPSc/PrPtoxic, like PrPΔCR, may be able to interact with its target only via C-terminal binding sites. The same may be true for PG14 PrP, which is aggregated and conformationally altered in the CR region (Biasini et al., 2008) (Fig. 2).
The model outlined here may also explain the toxicity of CtmPrP, as well as the synthetic peptide PrP106–126. PrP106–126 has been reported to be toxic to neurons cultured from Prn-p+/+ but not Prn-p0/0 mice (Brown et al., 1994a; Fioriti et al., 2005; Forloni et al., 1993). This result suggests that the peptide alters interaction between PrP and the hypothetical transducer by competitively blocking binding within the 105–125 region of PrP. This would then produce a toxic signal equivalent to the one elicited by PrPΔCR which lacks the 105–125 domain. In the absence of PrPC, no signal would be delivered and the peptide would have no effect. In CtmPrP, the CR region is partially embedded in the lipid bilayer, and so would be unavailable for binding to the transducer (Fig. 2). In contrast to the situation for PrPΔCR, however, activation of the transducer by CtmPrP is presumably facilitated by the presence of wild-type PrP, which potentiates the toxicity of CtmPrP.
Conclusions and perspective
Table 1 summarizes the properties of the non-infectious, neurotoxic mutants of PrP discussed in this review, and compares them to the properties of PrPC and infectious PrPSc. Studies of neurotoxic PrP mutants have provided a number of important insights into the pathogenic species and cellular pathways that may underlie prion-induced neurodegeneration. An important principle exemplified by all of these mutants is the distinction between prion infectivity and neurotoxicity. For some of the mutants associated with familial prion disorders, such as octapeptide insertions, small, β-rich aggregates are likely to represent a key neurotoxic intermediate. There is evidence that these aggregates act via both gain- and loss-of-function mechanisms. In contrast, transmembrane mutants as well as mutants harboring deletions within the central region display biochemical and cell biological properties indistinguishable from those of wild-type PrP. The neurotoxicity of these mutants is likely to depend on alteration or subversion of a neuroprotective function of PrPC.
Further understanding prion neurotoxicity will require progress on several fronts. First, it will be necessary to identify the molecular components of the cellular signaling pathways activated by neurotoxic forms of PrP. This will involve searching for PrP-interacting proteins via biochemical methods, as well as via genetic means using model organisms. The availability of highly potent neurotoxic forms like PrPΔCR should facilitate this search, since these molecules should have an especially high affinity for putative cell surface transducers. Second, it will be important to develop cell culture systems that allow the neurotoxic effects of PrP to be studied in vitro. Unfortunately, the mutant PrP molecules discussed here, all of which are neurotoxic in transgenic mice, have only modest effects on cell viability when expressed in cultured cells (our unpublished data) (Ashok and Hegde, 2008; Drisaldi et al., 2004; Rambold et al., 2008; Watts et al., 2007). Presumably, some factor present in the brain milieu required for maximal toxicity of these molecules must be missing in cell culture systems. Finally, and perhaps most importantly, we will need to demonstrate a connection between the neurotoxic mechanisms activated by artificial mutants like PrPΔCR and those operative in “natural” prion diseases of humans and animals. The most direct way to achieve this goal will be to knock down expression of the relevant signaling components in vivo, and see if this mitigates the disease phenotype.
Most current approaches to treatment of prion diseases are based on inhibiting accumulation of PrPSc (Trevitt and Collinge, 2006). Identification of the cellular pathways activated by neurotoxic forms of PrP would allow development of an entirely new class of anti-prion therapeutics based on blocking these pathways. Assuming appropriate cell culture models can be developed, some of the mutant forms of PrP described in this review may provide particularly good substrates for a drug screening approach to identify inhibitors of neurotoxic pathways. Such compounds may have wide applicability, given some of the underlying similarities between prion diseases and other neurodegenerative disorders in terms of pathogenic mechanisms. Indeed, recent evidence that PrPC is a cell-surface receptor for the Alzheimer’s Aβ peptide provides a direct link between neurotoxic mechanisms involved in these two diseases (Laurén et al., 2009).
Acknowledgments
Work in the Harris laboratory is supported by grants from the National Institutes of Health (NS052526 and NS040975) and the Dana Foundation.
References
- Aguzzi A. Cell biology. Prion toxicity: all sail and no anchor. Science. 2005;308:1420–1421. doi: 10.1126/science.1114168. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- Ashok A, Hegde RS. Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol Biol Cell. 2008;19:3463–3476. doi: 10.1091/mbc.E08-01-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini E, Medrano AZ, Thellung S, Chiesa R, Harris DA. Multiple biochemical similarities between infectious and non-infectious aggregates of a prion protein carrying an octapeptide insertion. J Neurochem. 2008;104:1293–1308. doi: 10.1111/j.1471-4159.2007.05082.x. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Brown DR, Herms J, Kretzschmar HA. Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. Neuroreport. 1994a;5:2057–2060. doi: 10.1097/00001756-199410270-00017. [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res. 2002;67:211–224. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- Brown P, Gibbs CJ, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994b;35:513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Chakrabarti O, Ashok A, Hegde RS. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem Sci. 2009 doi: 10.1016/j.tibs.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, Harris DA. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci USA. 2000;97:5574–5579. doi: 10.1073/pnas.97.10.5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, Harris DA. Prion diseases: what is the neurotoxic molecule? Neurobiol Dis. 2001;8:743–763. doi: 10.1006/nbdi.2001.0433. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol. 2003;77:7611–7622. doi: 10.1128/JVI.77.13.7611-7622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen HM, Harris DA. Prion protein lacks robust cytoprotective activity in cultured cells. Mol Neurodegener. 2008;3:11. doi: 10.1186/1750-1326-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen HM, Harris DA. A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J Neurochem. 2009;108:44–56. doi: 10.1111/j.1471-4159.2008.05719.x. [DOI] [PubMed] [Google Scholar]
- Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, et al. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- Dong J, Li A, Yamaguchi N, Sakaguchi S, Harris DA. Doppel induces degeneration of cerebellar Purkinje cells independently of Bax. Am J Pathol. 2007;171:599–607. doi: 10.2353/ajpath.2007.070262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, Chishti MA, Marvi M, Windl O, Ahrens R, et al. Genetic mapping of activity determinants within cellular prion proteins: N-terminal modules in PrPC offset pro-apoptotic activity of the Doppel helix B/B’ region. J Biol Chem. 2004;279:55443–55454. doi: 10.1074/jbc.M404794200. [DOI] [PubMed] [Google Scholar]
- Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–21743. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Poulter M, Harding AE. Dementia associated with a 216 base pair insertion in the prion protein gene: clinical and neuropathological features. Brain. 1993;116:555–567. doi: 10.1093/brain/116.3.555. [DOI] [PubMed] [Google Scholar]
- Fioriti L, Quaglio E, Massignan T, Colombo L, Stewart RS, Salmona M, Harris DA, Forloni G, Chiesa R. The neurotoxicity of prion protein (PrP) peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol Cell Neurosci. 2005;28:165–176. doi: 10.1016/j.mcn.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron. 2000;27:399–408. doi: 10.1016/s0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- Frigg R, Wenzel A, Samardzija M, Oesch B, Wariwoda H, Navarini AA, Seeliger MW, Tanimoto N, Reme C, Grimm C. The prion protein is neuroprotective against retinal degeneration in vivo. Exp Eye Res. 2006;83:1350–1358. doi: 10.1016/j.exer.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull. 2003;66:71–85. doi: 10.1093/bmb/66.1.71. [DOI] [PubMed] [Google Scholar]
- Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Hay B, Barry RA, Lieberburg I, Prusiner SB, Lingappa VR. Biogenesis and transmembrane orientation of the cellular isoform of the scrapie prion protein. Mol Cell Biol. 1987;7:914–920. doi: 10.1128/mcb.7.2.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, Defea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402:822–826. doi: 10.1038/45574. [DOI] [PubMed] [Google Scholar]
- Heitz S, Lutz Y, Rodeau JL, Zanjani H, Gautheron V, Bombarde G, Richard F, Fuchs JP, Vogel MW, Mariani J, et al. BAX contributes to Doppel-induced apoptosis of prion-protein-deficient Purkinje cells. Dev Neurobiol. 2007;67:670–686. doi: 10.1002/dneu.20366. [DOI] [PubMed] [Google Scholar]
- Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci USA. 2000;97:10248–10253. doi: 10.1073/pnas.97.18.10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Inoue K, Yokoyama T, Kobayashi S, Asakura T, Teramoto A, Itohara S. Prions prevent brain damage after experimental brain injury: a preliminary report. Acta Neurochir Suppl. 2003;86:297–299. doi: 10.1007/978-3-7091-0651-8_64. [DOI] [PubMed] [Google Scholar]
- Ivanova L, Barmada S, Kummer T, Harris DA. Mutant prion proteins are partially retained in the endoplasmic reticulum. J Biol Chem. 2001;276:42409–42421. doi: 10.1074/jbc.M106928200. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, McGovern G, Barmada S, Medrano AZ, Harris DA. Prion protein with an insertional mutation accumulates on axonal and dendritic plasmalemma, and is associated with distinctive ultrastructural changes. Am J Pathol. 2009 doi: 10.2353/ajpath.2009.090125. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a preemptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Rahbar R, Hegde RS. Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J Biol Chem. 2001;276:26132–26140. doi: 10.1074/jbc.M101638200. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Zerr I, Weber T, Poser S, Kretzschmar H, Hunsmann G, Bodemer W. Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Mol Brain Res. 1995;34:173–176. doi: 10.1016/0169-328x(95)00175-r. [DOI] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science. 1997;275:402–405. doi: 10.1126/science.275.5298.402. [DOI] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann S, Harris DA. Mutant and infectious prion proteins display common biochemical properties in cultured cells. J Biol Chem. 1996;271:1633–1637. doi: 10.1074/jbc.271.3.1633. [DOI] [PubMed] [Google Scholar]
- Li A, Barmada SJ, Roth KA, Harris DA. N-terminally deleted forms of the prion protein activate both Bax-dependent and Bax-independent neurotoxic pathways. J Neurosci. 2007a;27:852–859. doi: 10.1523/JNEUROSCI.4244-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007b;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Harris DA. Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem. 2005;280:17430–17434. doi: 10.1074/jbc.C500058200. [DOI] [PubMed] [Google Scholar]
- Li A, Piccardo P, Barmada SJ, Ghetti B, Harris DA. Prion protein with an octapeptide insertion has impaired neuroprotective activity in transgenic mice. EMBO J. 2007c;26:2777–2785. doi: 10.1038/sj.emboj.7601726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz H, Windl O, Kretzschmar HA. Cellular phenotyping of secretory and nuclear prion proteins associated with inherited prion diseases. J Biol Chem. 2002;277:8508–8516. doi: 10.1074/jbc.M110197200. [DOI] [PubMed] [Google Scholar]
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- Malaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7:e55. doi: 10.1371/journal.pbio.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mange A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002;514:159–162. doi: 10.1016/s0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 1999;18:6855–6864. doi: 10.1093/emboj/18.23.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–281. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- Medrano AZ, Barmada SJ, Biasini E, Harris DA. GFP-tagged mutant prion protein forms intra-axonal aggregates in transgenic mice. Neurobiol Dis. 2008;31:20–32. doi: 10.1016/j.nbd.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo H, Moore RC, Cohen FE, Westaway D, Prusiner SB, Wright PE, Dyson HJ. Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci USA. 2001;98:2352–2357. doi: 10.1073/pnas.051627998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA. 2001;98:15288–15293. doi: 10.1073/pnas.251550798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Collinge J, Leach M, Lofthouse R, Crow TJ, Harding AE. A dementing illness associated with a novel insertion in the prion protein gene. Mol Brain Res. 1992;13:155–157. doi: 10.1016/0169-328x(92)90056-h. [DOI] [PubMed] [Google Scholar]
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, et al. Prion proteins with different conformations accumulate in Gerstmann-Sträussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol. 2001;158:2201–2207. doi: 10.1016/S0002-9440(10)64692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola SA, Chesebro B. Abnormal properties of prion protein with insertional mutations in different cell types. J Biol Chem. 1998;273:11980–11985. doi: 10.1074/jbc.273.19.11980. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, editor. Prion Biology and Diseases. Second. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- Race R, Raines A, Raymond GJ, Caughey B, Chesebro B. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. J Virol. 2001;75:10106–10112. doi: 10.1128/JVI.75.21.10106-10112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci. 2005;25:4879–4888. doi: 10.1523/JNEUROSCI.0328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Muller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27:1974–1984. doi: 10.1038/emboj.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell. 2008;15:359–370. doi: 10.1016/j.devcel.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel A, Burgaya F, Gavin R, Soriano E, Aguzzi A, Del Rio JA. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J Neurosci Res. 2007;85:2741–2755. doi: 10.1002/jnr.21215. [DOI] [PubMed] [Google Scholar]
- Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R, Zhou Y, Solis GP, Stuermer CA, Malaga-Trillo E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J. 2006;20:317–319. doi: 10.1096/fj.05-4279fje. [DOI] [PubMed] [Google Scholar]
- Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J Neurosci Res. 2004;75:153–161. doi: 10.1002/jnr.10864. [DOI] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–795. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- Roucou X, LeBlanc AC. Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med. 2005;83:3–11. doi: 10.1007/s00109-004-0605-5. [DOI] [PubMed] [Google Scholar]
- Sakurai-Yamashita Y, Sakaguchi S, Yoshikawa D, Okimura N, Masuda Y, Katamine S, Niwa M. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience. 2005;136:281–287. doi: 10.1016/j.neuroscience.2005.06.095. [DOI] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, et al. Expression of aminoterminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005;20:442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion. 2007;1:83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RS, Drisaldi B, Harris DA. A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic reticulum. Mol Biol Cell. 2001;12:881–889. doi: 10.1091/mbc.12.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RS, Harris DA. Most pathogenic mutations do not alter the membrane topology of the prion protein. J Biol Chem. 2001;276:2212–2220. doi: 10.1074/jbc.M006763200. [DOI] [PubMed] [Google Scholar]
- Stewart RS, Harris DA. Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection. J Biol Chem. 2003;278:45960–45968. doi: 10.1074/jbc.M307833200. [DOI] [PubMed] [Google Scholar]
- Stewart RS, Harris DA. A transmembrane form of the prion protein is localized in the Golgi apparatus of neurons. J Biol Chem. 2005;280:15855–15864. doi: 10.1074/jbc.M412298200. [DOI] [PubMed] [Google Scholar]
- Stewart RS, Piccardo P, Ghetti B, Harris DA. Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J Neurosci. 2005;25:3469–3477. doi: 10.1523/JNEUROSCI.0105-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T. Inherited prion diseases and transmission to rodents. Brain Pathol. 1995;5:53–59. doi: 10.1111/j.1750-3639.1995.tb00577.x. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T, Doh-ura K, Sakaki Y, Steinmetz G, Tranchant C, Warter JM, Heldt N. Immunochemical, molecular genetic, and transmission studies on a case of Gerstmann-Sträussler-Scheinker syndrome. Neurology. 1990;40:1578–1581. doi: 10.1212/wnl.40.10.1578. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology. 1996;46:532–537. doi: 10.1212/wnl.46.2.532. [DOI] [PubMed] [Google Scholar]
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain. 2006;129:2241–2265. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, Sy MS, Yoong L, Young R, Mastrangelo P, et al. The CNS glycoprotein Shadoo has PrPC-like protective properties and displays reduced levels in prion infections. EMBO J. 2007;26:4038–4050. doi: 10.1038/sj.emboj.7601830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Westaway D. The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta. 2007;1772:654–672. doi: 10.1016/j.bbadis.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrPC): its physiological function and role in disease. Biochim Biophys Acta. 2007;1772:629–644. doi: 10.1016/j.bbadis.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa D, Yamaguchi N, Ishibashi D, Yamanaka H, Okimura N, Yamaguchi Y, Mori T, Miyata H, Shigematsu K, Katamine S, et al. Dominant-negative effects of the N-terminal half of prion protein on neurotoxicity of prion protein-like protein/doppel in mice. J Biol Chem. 2008;283:24202–24211. doi: 10.1074/jbc.M804212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost CS, Lopez CD, Prusiner SB, Myers RM, Lingappa VR. Non-hydrophobic extracytoplasmic determinant of stop transfer in the prion protein. Nature. 1990;343:669–672. doi: 10.1038/343669a0. [DOI] [PubMed] [Google Scholar]