

Abstract
Axonal degeneration is a characteristic feature of neurodegenerative disease and nerve injury. Here, we characterize axonal degeneration in Caenorhabditis elegans neurons following laser-induced axotomy. We show that this process proceeds independently of the WLDS and Nmnat pathway, and requires the axonal clearance machinery that includes the conserved transmembrane receptor CED-1/Draper, the adaptor protein CED-6, the guanine nucleotide exchange factor complex Crk/Mbc/dCed-12 (CED-2/CED-5/CED-12) and the small GTPase Rac1 (CED-10). We demonstrate that CED-1 and CED-6 function non-cell-autonomously in the surrounding hypodermis, which we show acts as the engulfing tissue for the severed axon. Moreover, we establish a function in this process for CED-7, an ATP-binding cassette (ABC) transporter, and NRF-5, a lipid-binding protein, both associated with release of lipid-vesicles during apoptotic cell clearance. Thus, our results reveal the existence of a WLDS /Nmnat-independent axonal degeneration pathway, conservation of the axonal clearance machinery, and a function for CED-7 and NRF-5 in this process.
E-TOC Paragraph
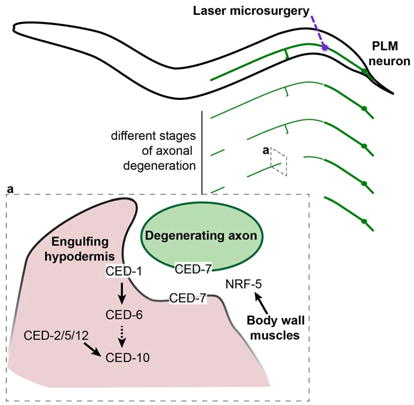
Axonal degeneration is a common hallmark of neurodegenerative disease and nerve injury. Using an axonal injury paradigm in C. elegans, Nichols et al. show that axonal degeneration in this organism is regulated by conserved components of the apoptotic engulfment machinery, and identify the epidermis as the engulfing tissue.
INTRODUCTION
The axon is a neuron’s longest neurite, requiring its structure to be actively maintained by support from the cell body through axonal transport, as well as from associated glial cells (Coleman, 2011). Following trauma or transection, the axon undergoes a stereotypical degenerative process termed Wallerian degeneration. This process consists of a latent phase followed by beading, thinning, fragmentation, and finally clearance of axonal debris by macrophages (Martini et al., 2008; Waller, 1850). Wallerian degeneration has been described across many vertebrate species as well as in Drosophila (MacDonald et al., 2006; Martin et al., 2010). The discovery of the Wallerian degeneration slow (WldS) mutation in mice, which drastically delays the onset of degeneration (Lunn et al., 1989), suggested that degeneration is the result of an active molecular pathway that can be manipulated. Expression of the murine WldS gene is able to robustly and potently delay axonal degeneration in every species tested, including rat, zebrafish, and Drosophila (Adalbert et al., 2005; Avery et al., 2009; Beirowski et al., 2008; Martin et al., 2010). WldS encodes a chimeric protein consisting of the first 70 N-terminal amino acids from the ubiquitin fusion degradation protein 2a (UFD2a), a unique 18 amino acid linker region, and the full in-frame sequence of nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1) (Conforti et al., 2000). Further studies have shown that it is the Nmnat portion that confers neuroprotection in WldS animals, and that the endogenous Nmnat2 protein in mice, and its Drosophila homolog dNmnat, are crucial axon survival factors (Fang et al., 2012; Gilley and Coleman, 2010). Upstream of dNmnat, the highly conserved E3 ubiquitin ligase Highwire (RPM-1 in C. elegans) promotes axonal degeneration while also acting through a parallel pathway involving the dual leucine kinase Wallenda (DLK-1 in C. elegans) (Xiong et al., 2012). In addition to its function in axonal degeneration in Drosophila, Wallenda/DLK-1 is also implicated in Wallerian degeneration in mice (Ghosh et al., 2011; Miller et al., 2009). Furthermore, a forward genetic screen in severed olfactory receptor neurons of Drosophila identified dSarm (TIR-1 in C. elegans) as an extremely potent promoter of axonal degeneration, an effect that is conserved in mice (Osterloh et al., 2012). Axonal degeneration ultimately ends with clearance of the axonal debris, and glial cells have been shown to play a key role in this process in both vertebrates and Drosophila (Doherty et al., 2009; Martini et al., 2008; Stoll et al., 1989). Importantly, despite axonal degeneration being distinct from apoptosis (Finn et al., 2000; Osterloh et al., 2012; Whitmore et al., 2003), the clearance of axonal debris shares molecular elements with the clearance of apoptotic cells. In particular, the apoptotic molecules Draper and dCed-6 (CED-1 and CED-6 in C. elegans) are required for the activation and recruitment of glial cells to degenerating Drosophila axons (MacDonald et al., 2006; Ziegenfuss et al., 2012). Furthermore, the guanine nucleotide exchange factor complex Crk/Mbc/dCed-12 and the small GTPase Rac1 (CED-2/CED-5/CED-12 and CED-10 in C. elegans) have been shown to function downstream of Draper and dCed-6 to promote the clearance of axonal debris (Ziegenfuss et al., 2012). However, many details of this clearance process, as well as possible alternative pathways and the molecules that regulate this event, remain unknown.
Although several studies have examined axonal degeneration in C. elegans in the context of neurodegenerative disease models and neuronal dysfunction (Calixto et al., 2012; Nagarajan et al., 2014; Neumann and Hilliard, 2014; Neumann et al., 2011), no Wallerian degeneration (i.e. severed axon) paradigm has been investigated. Here, we provide a detailed characterization of axonal degeneration in C. elegans neurons following laser-induced axotomy, and show that it proceeds independently from the WldS/Nmnat pathway. We identify a conserved function for the axonal clearance molecules CED-1 and CED-6, and the intracellular pathway that includes CED-2, CED-5, CED-12 and CED-10. Importantly, we also reveal a function in this process for the ATP-binding cassette (ABC) transporter CED-7 and the lipid-binding protein NRF-5, and identify the hypodermis as an engulfing tissue for the severed PLM axon fragments.
RESULTS
Characterization of axonal degeneration in C. elegans
The C. elegans mechanosensory neurons have been extensively studied as a model for both neurodegeneration and regeneration, and are amongst the best-characterized neurons in the nematode nervous system (Chalfie et al., 1985; Huang and Chalfie, 1994; Miyasaka et al., 2005; Neumann et al., 2015; Neumann et al., 2011; Zhang et al., 2008). We selected the posterior mechanosensory neurons (PLM left and right) in which to characterize axonal degeneration, and visualized these neurons with the zdIs5(Pmec-4∷GFP) transgene (Figure 1A). We then used laser-induced axotomy to selectively transect single PLM axons, allowing us to investigate the subsequent degeneration of the separated distal fragment. In wild-type animals, we found that the axon degenerated in a stereotypical manner, with similarities to Wallerian degeneration in other species, including thinning, beading, fragmentation, and clearance of the separated distal fragment (Figure 1A). However, unlike in other species, we did not observe a delay between the fragmentation and clearance phases of the degenerating axon, with these two processes apparently occurring simultaneously in different regions of the axon. To determine if the axonal degeneration phenotypes observed were indeed due to changes in axonal morphology, and not specific to the cytoplasmic GFP signal, we first analyzed animals expressing a membrane-bound fluorophore (Pmec-4∷MYR∷mCherry) together with cytoplasmic GFP. The degenerative phenotypes observed using the two different fluorophores were nearly identical (Figure S1A). Second, we obtained electron microscopy (EM) serial transverse sections of two unilaterally axotomized L4 (fourth larval stage) animals. We found that the thinning observed with epifluorescence corresponded to a decreased diameter of the axon compared to that on the contralateral uninjured side, with a highly reduced number of the mechanosensory neuron-specific large diameter microtubules (Figures 1B-1G and S1E-S1F).Furthermore, we found that the PLM axon on the injured side was discontinuous, matching the observation of axonal breaks made with epifluorescence (Figures 1B-1D and S2E-S2F). Based on these classical morphological indicators of degeneration, we developed a 5-point scale (Axonal Integrity Score) to score the degeneration of the distal fragment (Figure 1A), with 1 indicating complete clearance, and 5 indicating no signs of degeneration.
Figure 1. Characterization of axonal degeneration in C. elegans neurons following laser-induced axotomy.
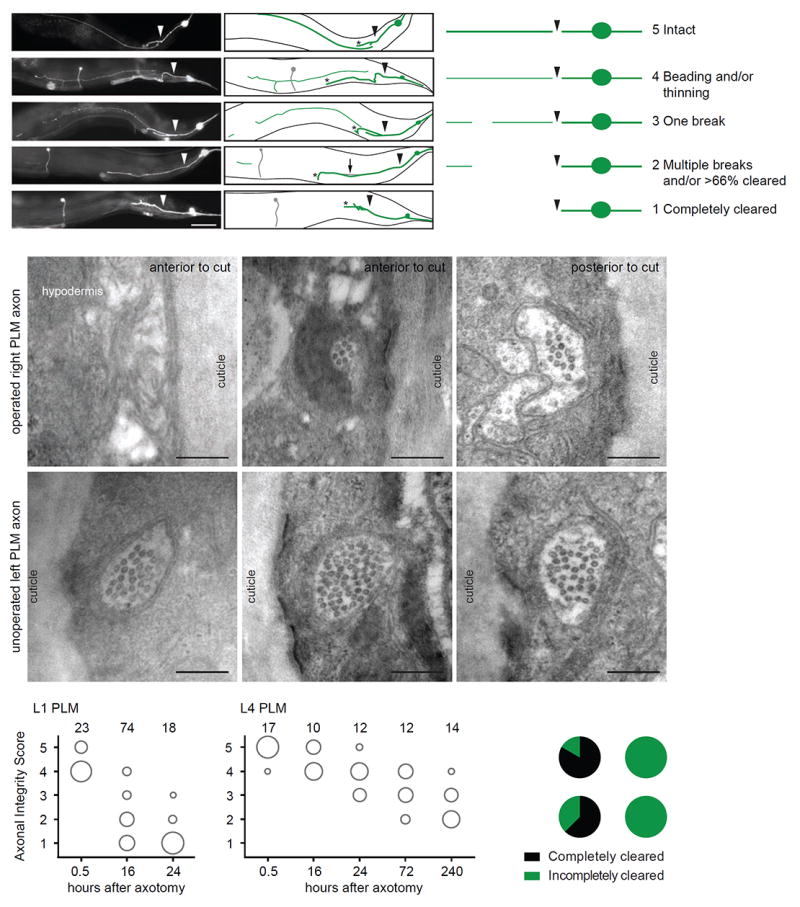
(A) Representative images and schematics of progressive stages of axonal degeneration after axotomy in PLM neurons in different L4 animals expressing the zdIs5(Pmec-4∷GFP) transgene. Arrowheads indicate location of axotomy, asterisks indicate the end of the regrowing axon, and arrow points to a distal fragment. The PVM neuron, which also expresses GFP, is represented in gray. Scale bar indicates 50 μm. Degeneration of the distal fragment classified as the Axonal Integrity Score with a value of 1 (completely cleared) to 5 (intact). The panel on the right is a schematic representation of these phenotypes. (B-G) Representative EM images of the degenerating PLM axon on the axotomized side (B-D) compared to the intact PLM axon on the non-operated side (E-G). The axon is intact upstream of the injury site (D), whereas in regions distal to the injury site it becomes severely reduced in diameter (C) or completely absent (B) (see also Figure S1E-S1F). On the contrary, on the unoperated contralateral side, PLM is intact along its entire length (E-G). (H,I) Axonal degeneration at multiple time points post-axotomy of the PLM neurons at L1 (H) or L4 (I) stages. The area of each circle represents the proportion of data points that fall into that category; n value indicated on top of each bubble graph. Each animal was imaged at only one time point. (J) Pie graph representing the complete clearance (score of 1) after 24 hours in PLM and DD neurons in L1 and L4 animals.
It has previously been suggested that axonal degeneration in C. elegans is developmentally dependent (Wu et al., 2007). To investigate this further, we compared the degeneration in L1 (first larval stage) and L4 animals at different time points after axotomy. In the majority of L1 animals, we found that the distal fragment was completely cleared by 24 hours post-axotomy (Figures 1H and 1J). This was in stark contrast to the L4 animals, in which the distal fragment was still present – although visibly degenerated – as late as 10 days post-axotomy (Figures 1I and 1J). We also performed axotomies at different time points after hatching and scored the degeneration 24 hours later, revealing that the rate of axonal degeneration was maximal within the first 10 hours after hatching, approximately corresponding to the duration of the L1 stage (Figure S1B).
To determine if the progression of degeneration and developmental stage-dependent differences were a general feature of the nervous system, we also performed axotomies on the GABAergic DD motor neurons (visualized with the ynIs37(Pflp-13∷GFP) transgene). Axonal degeneration proceeded rapidly in L1 animals, with complete clearance observed in the majority of animals 24 hours post-axotomy (Figures 1J and S1C). However, we again observed a far more gradual rate of degeneration in L4 animals, with complete clearance not observed until as late as 72 hours post-axotomy (Figures 1J and S1D). These results suggest that developmental stage-dependent differences in degeneration could be a general characteristic of C. elegans neurons, providing a platform to investigate axonal degeneration in this species.
WLDS and Nmnats do not delay laser-induced axonal degeneration in C. elegans
In both vertebrates and Drosophila, Wallerian degeneration has been shown to proceed through a highly conserved pathway that can be characterized by the protective effect of the WldS gene. To determine if this pathway is conserved in C. elegans, we generated transgenic strains that expressed the murine WldS gene in the six mechanosensory neurons. Surprisingly, and contrary to what has been observed in other organisms, we did not find any delay in axonal degeneration in WldS strains at either the L1 (Figures 2A and S2A) or L4 stage (Figure S2B). Moreover, we found that this lack of protection was not a function of neuronal class, as WLDS did not delay axonal degeneration in either the AVM mechanosensory neuron (Figure S2C) or the DD motor neurons of L4 animals (Figure S2D). However, WLDS did maintain function, as it protected against neurodegeneration caused by a dominant mutation in the degenerin channel MEC-4(d), similar to the overexpression of the C. elegans Nmnat2 (Calixto et al., 2012) (Figure S2E). Moreover, unlike in other species (MacDonald et al., 2006; Sasaki et al., 2009), overexpression of the C. elegans Nmnat genes (nmat-1 or nmat-2) in the six mechanosensory neurons could not delay axonal degeneration in L1 or L4 animals (Figures 2B and S2F-S2H). These results reveal that neither overexpression of WLDS nor the presence of endogenous Nmnat proteins is able to delay laser-induced axonal degeneration in C. elegans.
Figure 2. Overexpression of WldS or the endogenous Nmnat genes (nmat-1 and 2) has no protective effect against axonal degeneration after laser-induced axotomy in PLM neurons.

(A) Axonal degeneration of representative transgenic strains expressing Pmec-4∷WldS at low (5 ng/μL), medium (10 ng/μL), or high (20 ng/μL) concentrations, 16 hours post-axotomy at the L1 stage. (B) Axonal degeneration of the PLM neuron in representative Pmec-4∷nmat-1 and Pmec-4∷nmat-2 lines at low (5 ng/μL) and high (20 ng/μL) concentrations 16 hours post-axotomy at the L1 stage. The high concentration of Pmec-4∷nmat-2 induced an increase in degeneration. (C) Quantification of PLM axonal degeneration at the L1 stage in rpm-1(ju41), dlk-1(ju476), tir-1(tm3036) or tir-1(ok1052) animals compared to wild-type (WT) animals 16 hours post-axotomy. The area of each circle represents the proportion of data points that fall into that category; n value indicated on top of each bubble graph. * p<0.05 as determined by either a Mann-Whitney test or a Kruskal-Wallis and Dunn’s test.
In the past decade, three genes have been identified as promoters of the Wallerian degeneration pathway in Drosophila and/or mice: Sarm1 (Osterloh et al., 2012), Wallenda/Dlk1 (Ghosh et al., 2011; Miller et al., 2009; Xiong et al., 2010), and the E3 ubiquitin ligase Highwire (Xiong et al., 2012). We found that mutations in the C. elegans homologs of these genes, tir-1, dlk-1, and rpm-1, had no protective effect on axonal degeneration of PLM neurons (Figures 2C and S3A). Taken together, these results point to a possible divergence in C. elegans, such that the functions of key molecules identified in other species are not conserved in the context of axonal degeneration following axotomy, suggesting the existence of redundant pathways and/or other still unidentified molecules.
A role for CED-7 and NRF-5 in regulating axonal degeneration/clearance
As expected from studies in different species, we found no effect on axonal degeneration in mutants of genes of the canonical apoptosis pathway, ced-3 and ced-4 (Figures 3B and S3A), indicating that the axonal degeneration process in C. elegans, as in vertebrates, is not regulated by caspase function (Finn et al., 2000). However, in several species, clearance of severed axonal fragments by glial cells has been shown to use some of the same molecular components necessary for the engulfment of apoptotic cells. For example, the apoptotic engulfment genes Draper and dCed-6 are needed for axonal clearance in Drosophila and mice (Awasaki et al., 2006; MacDonald et al., 2006). Similarly, Crk/Mbc/dCed-12 and Rac1 have also been shown to function in this process (Ziegenfuss et al., 2012). To determine if this molecular machinery is also employed in C. elegans, we examined animals carrying mutations in the orthologous genes. We found that animals mutant for ced-6, ced-1, ced-12, or ced-10 all displayed a significantly decreased axonal degeneration phenotype following axotomy of PLM at the L1 stage (Figure 3A). As CED-12 acts in a molecular complex that includes CED-5 (mammalian Dock180) and CED-2 (mammalian CrkII) (Gumienny et al., 2001), we also tested animals carrying mutations in each of these genes, and found that each mutant displayed a defective degenerative phenotype compared to wild-type animals (Figure 3A). In other species, all of these apoptotic molecules function in glial cells, which are responsible for the clearance of the damaged axon, and thus their role is non-cell-autonomous with respect to the damaged neuron. However, the cells that can engulf the damaged axonal fragments in C. elegans have not previously been identified. At the L1 stage, PLM is tightly associated with the hypodermis, and later becomes embedded in this tissue (Chalfie and Sulston, 1981). We therefore hypothesized that the hypodermis might be responsible for engulfment of the distal fragment after axotomy. To test this, we generated transgenic ced-6 mutant animals in which wild-type CED-6 was tagged with mRFP and selectively expressed in the hypodermis using the dpy-7 promoter (Pdpy-7∷mRFP∷CED-6) (Gilleard et al., 1997). Strikingly, we found a strong rescue of the PLM defect (Figures 4A and 4B), indicating that the hypodermis is a key tissue in this process. In agreement with this conclusion, cell-autonomous expression of CED-6 (Pmec-4∷mRFP∷CED-6) failed to rescue the defect (Figure 4A). Similarly, expression of wild-type CED-1 tagged with mRFP in the hypodermis (Pdpy-7∷mRFP∷CED-1) was able to strongly rescue the defects of ced-1 mutant animals (Figures 4C and 4D), confirming that CED-1 and CED-6 both act within the engulfing cells. These results are consistent with our EM imaging data of L4 animals (Figures 1B-1D and S1E-S1F), where we found evidence of membrane whorls in the hypodermis in areas where the PLM axon was absent; these morphological changes in the hypodermis are strongly suggestive of a role of this tissue in the engulfment and clearance of the axon. In summary, we find that the molecular elements responsible for the clearance of severed axon fragments are conserved across species, and reveal that the hypodermal cells play a role in this process in C. elegans.
Figure 3. Conserved components of the apoptotic cell clearance machinery regulate the axonal degeneration phenotype of the PLM neurons.

(A) Mutations in the apoptotic cell clearance genes ced-6(n1813), ced-1(e1735), ced-12(bz187), ced-10(n3246), ced-5(n1812), ced-2(e1752), ced-7(n2094), ced-7(n2690), and nrf-5(sa513) delay axonal degeneration in L1 animals. (B) Mutations in the other apoptotic-related genes ced-3(n2452), ced-4(n1162), ttr-52(sm211), psr-1(ok714), ina-1(gn144), tat-1(tm3117), and scrm-1(tm805) have no effect on axonal degeneration following axotomy in L1 animals. The area of each circle represents the proportion of data points that fall into that category; n value indicated on top of each bubble graph. * p<0.05, ** p<0.005, *** p<0.0005 as determined by a Kruskal-Wallis and Dunn’s or a Mann-Whitney test.
Figure 4. CED-1 and CED-6 function in the hypodermis.
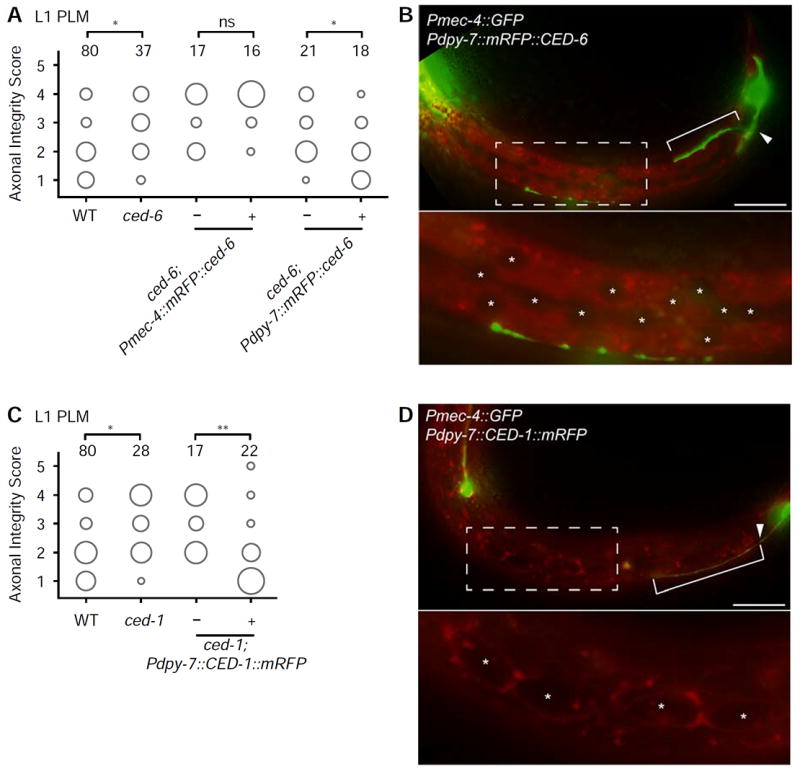
(A) Cell-specific expression of Pmec-4∷mRFP∷CED-6 indicates that the molecule does not act cell-autonomously in PLM neuron, whereas expression of Pdpy-7∷mRFP∷CED-6 in the hypodermis is sufficient to rescue the diminished degeneration and clearance in ced-6(n1813) mutants. This strain is representative for three independent rescue strains (data not shown). (B) Localization of Pdpy-7∷mRFP∷CED-6 in the hypodermis in an animal at the L1 stage, 16 hours after axotomy. (C) Cell-specific expression of Pdpy-7∷CED-1∷mRFP is sufficient to rescue the diminished degeneration and clearance in ced-1(e1735) mutants. This strain is representative of three independent rescue strains (data not shown). (D) Localization of Pdpy-7∷CED-1∷mRFP in the hypodermis and seam cells in an animal at the L1 stage, 16 hours after axotomy. The area of each circle in panels A and C represents the proportion of data points that fall into that category; n value indicated on top of each bubble graph. * p<0.05, ** p<0.005, as determined by a Kruskal-Wallis and Dunn’s or a Mann-Whitney test. Scale bars indicate 20 μm. Arrowheads indicate the location of axotomy, brackets indicate regrowth, and asterisks mark the hypodermal and seam nuclei.
During apoptosis, the CED-1 receptor present on an engulfing cell recognizes apoptotic cells through the exposure of phosphatidylserine (PS) on their membranes (Zhou et al., 2001). This process is facilitated by the ABC transporter CED-7, which promotes the generation of extracellular PS-containing vesicles (Mapes et al., 2012). To determine if CED-7 contributes to the axonal degeneration phenotype, we analyzed the PLM axon in ced-7 mutants after axotomy. Interestingly, these animals presented a striking defect at the L1 stage, resembling that observed for the other apoptotic engulfment genes analyzed (Figure 3A). Given the possible role of PS in this process, we then tested if similar phenotypes were also present in animals carrying mutations in nrf-5, which encodes a lipid-binding protein that functions with CED-7 to mediate PS transfer from apoptotic cells to phagocytes (Zhang et al., 2012). We found a significant defect in the PLM axonal degeneration phenotype following axotomy in nrf-5 mutant animals (Figure 3A). On the contrary, we observed no such effect in animals lacking TTR-52 (a secreted transthyretin-like protein that acts as a bridging molecule between CED-1 and PS (Wang et al., 2010)), PSR-1 or INA-1 (both PS membrane receptors (Hsu and Wu, 2010; Wang et al., 2003)) (Figure 3B), suggesting that these molecules are either not involved or have redundant roles in this process. Axonal degeneration/clearance also appeared normal in mutants of genes encoding for known PS-regulating molecules such as TAT-1 (an ATPase required for regulating PS asymmetry (Zullig et al., 2007)), and SCRM-1 (a phospholipid scramblase required for PS exposure (Wang et al., 2007)) (Figure 3B). Furthermore, using a mRFP-tagged version of Annexin V (sAnxV∷mRFP) that binds PS (Mapes et al., 2012), we could not detect PS on the axonal fragment in L1, and its overexpression did not affect the axonal degeneration phenotype (Figure S3C), as would be expected if PS exposure functioned in this process. Our data therefore suggest that PS might have, at most, only a minor role in axonal degeneration/clearance, or that CED-7 and NRF-5 could facilitate the transport of other lipids or molecules.
Axonal degeneration/clearance is modulated by CED-7 and mitochondria
The role of CED-7 appears to change with developmental stage, as compared with wild-type animals ced-7 mutants exhibited a delayed degenerative phenotype at the L1 stage (Figure 3A) but an increased rate at the L4 stage (Figure S3B). This developmental switch may be explained by a time-dependent role of CED-7 in regulating the secretion of lipids or other molecules. For example, in the context of apoptotic clearance, CED-7 not only promotes the generation of PS-containing extracellular vesicles, but is also required to remove externalized PS from unengulfed apoptotic cells over time (Mapes et al., 2012). This dual function of CED-7, with early production of lipid-containing vesicles promoting clearance and later removal of lipids from the degenerating axon preventing clearance, could explain its different effects at the L1 and L4 stages.
We also tested a number of other factors that potentially alter the axonal degeneration phenotype. Presynaptic loci are present on the PLM axon at the L4 stage but not at the L1 stage, as revealed by the absence of the synaptic vesicle-associated small guanosine triphosphatase RAB-3 on the PLM synaptic branch in L1 animals (Figure S4A and S4B). To investigate whether the presence of synapses stabilized the axon, we severed both the PLM synaptic branch and the main PLM axon in L4 animals; however, we found no difference in the degenerative phenotype between axons deprived of their synaptic branch and those with an intact branch (Figure S4C). The PLM axon is positioned adjacent to both the hypodermis and the muscle during the L1 stage, becoming separated from the muscle by the embedding hypodermis in L4 animals (Emtage et al., 2004). To test for a possible positional effect of the axon, we carried out axotomies in L4 mec-1 mutant animals in which the hypodermis fails to establish this separation (Emtage et al., 2004). Our results showed no significant difference in the degenerative phenotype between wild-type and mec-1 mutant animals (Figure S4D), indicating that the location of the PLM axon with respect to the surrounding tissues is not responsible for the differences in the degeneration between L1 and L4 stage animals.
A recent study has shown that the presence of mitochondria in C. elegans axons protects from degeneration following axotomy (Rawson et al., 2014). To ascertain whether mitochondria might similarly modulate the rate of degeneration in PLM, we performed axotomies in animals carrying a mutation in the ric-7 gene that disrupts mitochondrial transport, resulting in the accumulation of mitochondria in neuronal cell bodies and a near complete absence in neurites (Rawson et al., 2014) (Figures 5A and S5). We found that L4 ric-7 mutant animals displayed a very rapid axonal degenerative phenotype, with more than a third of axons completely cleared 24 hours post-axotomy (Figure 5B), a rate of degeneration comparable to that of L1 wild-type animals. To determine if this effect was specifically due to the lack of mitochondria in the PLM axon, we rescued the normal localization of mitochondria by tethering them to a kinesin protein (Pmec-4∷UNC-116∷GFP∷TOMM-7), thereby bypassing the requirement of RIC-7 for the transport of these organelles (Rawson et al., 2014). We found a strong rescue of the axonal degeneration defect in ric-7 animals expressing this construct (Figure 5B), supporting a specific, cell-autonomous role for mitochondria in regulating the rate of axonal degeneration in PLM.
Figure 5. Mitochondria are largely absent from the PLM axon in ric-7 mutant animals and are important for delaying axonal degeneration.
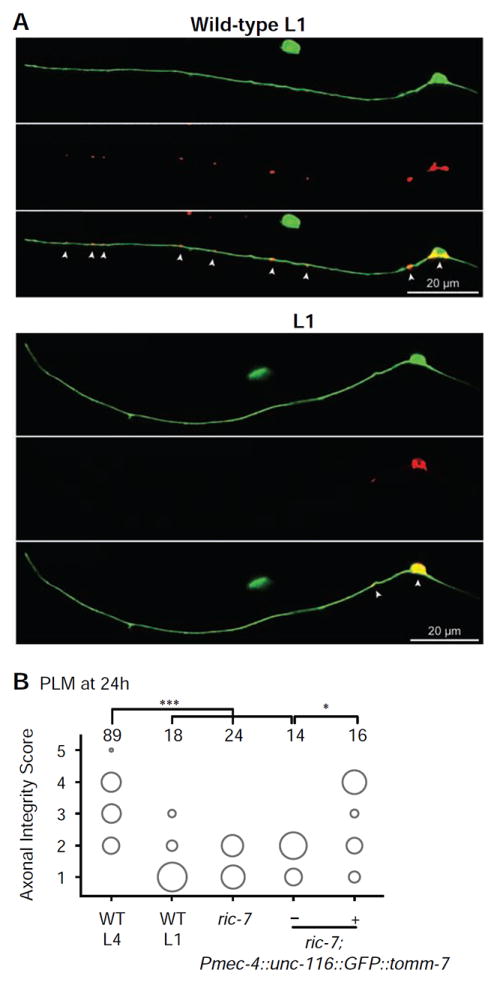
(A) Mitochondria visualized through expression of TOMM-20∷mRFP at the L1-stage in wild-type (top panels) and ric-7(nu447) (bottom panels) animals, highlighting the lack of mitochondria within the axons of PLM in ric-7 mutants. (B) PLM axons of L4 ric-7(nu447) animals show a significant increase in degeneration and clearance compared to wild-type L4 animals after 24 hours, closely resembling the degree of degeneration and clearance in wild-type L1 animals. This defect can be rescued with cell-autonomous expression of UNC-116∷GFP∷TOMM-7 to restore mitochondrial localization in the axon. The area of each circle represents the proportion of data points that fall into that category; n value indicated on top of each bubble graph. * p<0.05, *** p<0.0005 as determined by a Kruskal-Wallis and Dunn’s or a Mann-Whitney test.
DISCUSSION
Model organisms have been instrumental in our understanding of the molecular mechanisms behind axonal degeneration and the active Wallerian degeneration pathway (Coleman and Freeman, 2010). We have characterized axonal degeneration in C. elegans, investigated the molecules involved in this process, and examined whether the Wallerian degeneration pathway is functionally conserved in this species. We show that following laser-induced axotomy, C. elegans PLM mechanosensory and DD motor axons degenerate with morphological similarity to Wallerian degeneration, although there is no delay between fragmentation and clearance of the transected axon as in other organisms. Surprisingly, we also demonstrate that WldS expression does not delay axonal degeneration in different C. elegans neurons. We show that this lack of effect is not due to a lack of function, as the same transgene can protect against necrotic neurodegeneration caused by mec-4(d). In mammalian systems, WLDS has been shown to increase mitochondrial flux and calcium buffering capacities (Avery et al., 2012), whereas the mec-4(d) mutation causes calcium channels to be constitutively open, leading to necrosis (Bianchi et al., 2004; Driscoll and Chalfie, 1991). Therefore, WLDS could protect against mec-4(d) neurodegeneration by enhancing the capacity of mitochondria to buffer the influx of calcium. Also unexpectedly, but consistent with the inability of WLDS to protect against axonal degeneration, we found that the orthologs of dNmnat/Nmnat1, dsarm/Sarm1, Wallenda/Dlk1 and Highwire/Phr, which are involved in the conserved Wallerian degeneration program in Drosophila and vertebrates (Fang et al., 2012; Gilley and Coleman, 2010; Itoh et al., 2009; Osterloh et al., 2012; Xiong and Collins, 2012; Xiong et al., 2012), had no significant role in axonal degeneration following laser axotomy in C. elegans mechanosensory neurons.
In contrast, we found a conserved function in C. elegans for CED-1, CED-6, CED-2, CED-5, CED-12 and CED-10, all molecules that are known to be involved in axonal clearance in different species. Their effects appear to be restricted to the L1 stage during which the rate of axonal degeneration/clearance is extremely fast, with no effect detected at the L4 stage where the PLM axonal degenerative phenotype proceeds very slowly. Notably, tissue-specific rescue experiments with CED-1 and CED-6, and analysis of EM serial sections, demonstrated that the hypodermis is responsible for the clearance of the severed PLM axon. During apoptosis, the normally internally localized PS is externalized to the outer leaflet and serves as an “eat me” signal for engulfing phagocytes. The ABC transporter, CED-7, has previously been shown to promote the generation of PS-containing extracellular vesicles (Mapes et al., 2012). Interestingly, our data reveal a function for CED-7 in promoting axonal degeneration/clearance in L1 animals, whereas it appears to switch to a suppressive mode in L4 animals (Figure S3B). This resembles the function of CED-7 in apoptotic clearance. In apoptosis, CED-7 has two functions, the first involving early production of lipid-containing vesicles that promote clearance, and the second involving later removal of lipids from the apoptotic cell. In axonal degeneration, CED-7 may be acting early in L1 to promote the secretion of lipids from the PLM axon, which function as “eat me” signals leading to clearance. Conversely, in the L4 stage, during which degeneration proceeds over a longer timeline, the main function of CED-7 may switch to removal of lipids, thereby preventing clearance. Therefore, loss of CED-7 at the L4 stage leads to an accumulation of lipids on the axon, causing increased clearance. How exactly CED-7 can switch between these two modes and how this is regulated are currently unknown. A possible involvement of lipids is supported by our results with NRF-5, a lipid-binding protein that functions with CED-7 to mediate PS transfer from apoptotic cells to phagocytes, which also affects the axonal degenerative phenotype. However, our results from mutants in the tat-1, scrm-1, ttr-52, psr-1 and ina-1 genes, in which the axonal degenerative phenotype is not altered, could indicate either that CED-7 also has a function that is independent of PS, or that PS is produced and recognized by other molecules in the context of degeneration. Overall, our results suggest conservation of the molecular machinery involved in axonal clearance, and raise the possibility that CED-7 and NRF-5 have an important role in this process in species other than nematodes.
In C. elegans, a severed axon can present a new growth cone at the end of the proximal fragment within six hours of surgery (Wu et al., 2007), and this regrowing axon can reconnect to its separated distal fragment to restore the original axon tract, in a process known as axonal fusion (Ghosh-Roy et al., 2010; Neumann et al., 2015; Neumann et al., 2011; Wu et al., 2007). Remarkably, we have shown that some of the same molecules involved in axonal degeneration, such as CED-7, NRF-5, and CED-6, also regulate axonal fusion in L4 animals (Neumann et al., 2015). This suggests that, in a number of C. elegans neurons, axotomy triggers a tug-of-war between the axonal repair event occurring by way of axonal fusion, and degeneration/clearance of the distal axon fragment mediated by the surrounding hypodermis (Hilliard, 2009). For example, the nuclear receptor FAX-1 has been shown to simultaneously increase axonal regeneration and decrease degeneration in C. elegans AVK interneurons (Yaniv et al., 2012). In contrast, the presence of the distal fragment is inhibitory for regeneration and neuroplasticity in vertebrates (Martin et al., 2010). Delaying axonal degeneration through WldS expression has been shown to substantially delay the locomotory recovery of mice following partial spinal cord lesions from six to sixteen days (Zhang et al., 1998). This may explain why vertebrates have evolved to rapidly clear the inhibitory axonal fragments through an active degeneration pathway. The evolutionary pressure dictating why vertebrates have reduced regeneration in the presence of distal fragments, as well as in the central nervous system, is still a point of speculation. However, whereas it is presumably advantageous for mammals to actively promote axonal degeneration in order to undergo regeneration, there seems to be no such benefit for C. elegans. Even so, the need and mechanisms to clear axons once they have degenerated appear widely conserved.
MATERIALS AND METHODS
Strains
Animals were maintained at room temperature (~22°C) on nematode growth medium (NGM) plates seeded with Escherichia coli OP50 according to standard methods. The following strains were obtained from the Caenorhabditis Genetic Center (CGC): RB1085 [tir-1(ok1052)], CB3203 [ced-1(e1735)], MT8347 [ced-3(n2452)], MT2547 [ced-4(n1162)], MT4433 [ced-6(n1813)], MT9258 [ced-7(n2690)], VC537 [psr-1(ok714)], NG144 [ina-1(gm144)], CB3257 [ced-2(e1752)], MT4434 [ced-5(n1812)], ZB547 [ced-12(bz187)], MT9958 [ced-10(n3246)], CB1066 [mec-1(e1066)], JT513 [nrf-5(sa513)], CZ1251 [rpm-1(ju41)], CZ5730 [dlk-1(ju476)], KP2048 [ric-7(nu447)], NY2037 [ynIs37(Pflp-13∷GFP)] and the National Bioresource Project (NBRP): FX0805 [scrm-1(tm805)], FX3036 [tir-1(tm3036)] and FX3117 [tat-1(tm3117)].
The following transgenes were generated by microinjection (concentrations used for the microinjection mix are indicated in brackets; all injection mixes had a total concentration of 100ng/μL and empty pSM plasmid was used as a filler): vdEx289/vdEx290/vdEx291 [Pmec-4∷WldS (5ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx295/vdEx296/vdEx297 [Pmec-4∷WldS (10ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx298/vdEx299 [Pmec-4∷WldS (20ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx429/vdEx430/vdEx431 [Pflp-13∷WldS (5ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx717/vdEx718/vdEx720 [Pflp-13∷WldS (10ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx722/vdEx723/vdEx724 [Pflp-13∷WldS (20ng/μL); Podr-1∷DsRed (10ng/μL)], vdEx294 [Pmec-4∷WldS ∷mCherry (5ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx422 [Pmec-4∷nmat-1 (5ng/μL); Podr-1∷DsRed (30ng/μL)]; vdEx424 [Pmec-4∷nmat-1 (20ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx426/vdEx427/vdEx428 [Pmec-4∷nmat-2 (5ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx437/vdEx438/vdEx439 [Pmec-4∷nmat-2 (10ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx502 [Pmec-4∷nmat-2 (20ng/μL); Podr-1∷DsRed (30ng/μL)], vdEx1058 [Pdpy-7∷ced-1∷mRFP (1 ng/μL); Podr-1∷DsRed (30 ng/μL)], vdEx1025/vdEx1027 [Pdpy-7∷ced-1∷mRFP (5 ng/μL); Podr-1∷DsRed (30 ng/μL)], vdEx895 [Pmec-4∷mRFP∷ced-6 (5 ng/μL) ; Podr-1∷DsRed (30 ng/μL)], vdEx1059 [Pdpy-7∷mRFP∷ced-6 (1 ng/μL) ; Podr-1∷DsRed (30 ng/μL)], vdEx239[Pmec-4∷mCherry∷rab-3 (0.5 ng/μL); Podr-1∷DsRed (30 ng/μL)], vdIs13[Pmec-4∷tomm-20∷mRFP (0.5 ng/μL); Punc-22∷GFP (25 ng/μL)], vdEx919[Pmec-4∷myr∷mCherry (5ng/μL); Podr-1∷DsRed (60ng/μL)]; vdEx1124[Pmec-4∷unc-116∷GFP∷tomm-7 (10ng/μL); Podr-1∷DsRed (60ng/μL)]. smIs65[Phsp∷sAnxV∷mRFP] was used for the Annexin experiments.
Molecular biology
Molecular biology was performed using standard techniques (Sambrook et al., 1989). All primers were sourced from GeneWorks Pty Ltd (Hindmarsh, Adelaide). WldS was amplified from the WldS plasmid (a gift from Michael Coleman; Babraham Institute, University of Cambridge, UK) and cloned into pSM plasmids containing either Pmec-4 (Pmec-4∷WldS), P mec-4∷mCherry (Pmec-4∷mCherry∷WldS) or Pflp-13 (Pflp-13∷WldS) using 5’-TCAGTGGGATCCATGGAGGAGCTGAGCGCTG-3’ paired with either 5’-TACGGAATTCTCACAGAGTGGAATGGTTGTGC-3’ (for cloning into the Pmec-4 plasmid using BamHI and EcoRI sites) or 5’ – TCAGTGGGATCCGCCAGAGTGGAATGGTTGTGCT-3’ (for cloning into the Pmec-4∷mCherry plasmid using BamHI). Genomic sequences of nmat-1 and nmat-2 were each cloned into pSM plasmids containing the mec-4 promoter using the 5’-TCAGTGGCTAGCCTATGGGGACCGAAAAAGTTG-3’ and 5’-TCAGTGACCGGTGCTCATTCGTAGAGTCTATGAT-3’ or the 5’-TCAGTGGCTAGCACGATGAAACGAGTCGCTCT-3’ and 5’-GGGGGACCGGTATTAAATTTTCTGATACAGATTATTCT-3’ primers respectively, which include NheI and AgeI sites, and genomic DNA from mixed-stage animals as a template (whole animal lysates performed as previously described (Wicks et al., 2001)).
The dpy-7 promoter (Gilleard et al., 1997) was amplified from genomic C. elegans DNA using 5’-AAAAAAGGATCCTTATCTGGAACAAAATGTAAG-3’ and 5’-CGACGCCTGCCTTGAAACACTAC-3’ which include BamHI and HindIII sites for insertion into the pSM plasmid. pSM plasmids carrying Pmec-4∷ced-1∷mRFP and Pmec-4∷mRFP∷ced-6 were used to replace the mec-4 promoter with the dpy-7 promoter.
The Pmec-4∷UNC-116∷GFP∷TOMM-7 plasmid was built as follows: UNC-116∷GFP∷TOMM-7 was amplified from the pRR125 plasmid (a gift from Erik Jorgensen, University of Utah, Salt Lake City, USA) using primers 5’-CGCGGATCCCAAAAAAGCAGGCTGCATGG-3’ and 5’-ATTGGGCCCGCCAAGCGAGGACAATTCTC-3’, and cloned into Pmec-4 pSM using BamHI and ApaI restriction sites.
To build Pmec-4∷MYR∷mCherry, the insert MYR∷mCherry (from the plasmid pNV myr∷mCherry) was cloned into pSM Pmec-4∷GFP using MscI and EcoRI restriction sites (replacing GFP with MYR∷mCherry).
Microscopy
Fluorescence microscopy was performed using an upright Zeiss AxioImager Z1 equipped with epifluorescence, DIC, and a Cool SNAP HQ2 camera (Photometrics) or an AxioCam MRm camera (Carl Zeiss). Images were acquired and processed using either Metamorph or AxioVision Release 4.8 software. Additional processing was completed using ImageJ 64. Worms were anesthetized with 0.01–0.05 % tetramisole on 4 % agar pads.
Mitochondria were visualized using the vdIs13(Pmec-4∷tomm-20∷mRFP; Punc-122∷GFP) transgene and a LSM 710 META confocal microscope, equipped with GaAsP detectors and Zen 2012 software. Green cytoplasmic fluorescence was visualized with a 488 nm laser (0.15% power, with a gain of 650 and 8x averaging) and red fluorescence was visualized with a 561 nm laser (1.5% power, with a gain of 600 and 8x). Animals were analyzed at the L1 and L4 stages as indicated.
Laser axotomy
A MicroPoint 337 UV laser mounted onto an upright Zeiss AxioImager A1 equipped for fluorescence and DIC microscopy was used to perform individual axon transections. Animals were operated at the L1 (six hours after hatching) or L4 stage, while anesthetized with 0.01–0.05 % tetramisole on 4 % agar pads unless otherwise specified. The surgery was performed on the axon of the PLM neuron approximately 50 μm anterior to the cell body in the case of L4 animals and approximately 10 μm anterior to the cell body in L1 animals. In the case of the DD motor neurons, axotomies were performed approximately halfway across the commissure for DD3 and DD5, and cell bodies were ablated post-axotomy to prevent regeneration that would complicate visualization of the distal fragments. Animals were recovered on NGM plates seeded with E. coli, and observed at the single time point indicated. For the Annexin experiments, L1 animals receiving the heat shock (wild-type and Phsp∷sAnxV) were placed at 30°C for 30 minutes, 4 hours prior to axotomy.
Electron microscopy
72 hours post-axotomy of PLM in L4 animals, fluorescence imaging was used to determine the axonal degeneration phenotype of the PLM distal axonal fragment. Fixation of single animals was achieved by transection of the animal in the middle of the anterior half of the body in buffered aldehydes, followed by incubation in osmium tetroxide, uranyl acetate, and positioning in groups in agar cubes prior to embedding in Epon resin (Hall, 1995). Serial thin sections were collected onto plastic-coated slot grids (Pioloform) from multiple animals at once, and post-stained with uranyl acetate. Digital Transmission EM images were collected using iTEM software on an Olympus Morada camera mounted on a Philips CM10 electron microscope.
Data analysis
Degeneration data were gathered by qualitative measurements. Degeneration of each axotomized PLM was classified on a scale of 1 (complete clearance of the distal fragment) to 5 (no degeneration) (Figure 1A). Data were analyzed using the Mann-Whitney test for single comparisons and a Kruskal-Wallis and Dunn’s post-hoc test for multiple comparisons. In the case of the mec-4(d) phenotypic data, proportions were compared by calculating z-ratios for the significances of the differences between the independent proportions.
Supplementary Material
HIGHLIGHTS.
WLDS/Nmnat cannot delay axonal degeneration following axotomy in C. elegans.
Axonal degeneration is regulated by the apoptotic engulfment machinery.
The epidermis functions as the axonal engulfing tissue, through CED-1 and CED-6.
Lipid-vesicle associated molecules CED-7 and NRF-5 regulate axonal degeneration.
Acknowledgments
We thank Robert Parton, Rick Webb, and Robyn Webb for their invaluable help with electron microscopy. We thank Rowan Tweedale for comments on the manuscript, members of the Hilliard lab for discussions and comments, Michael Coleman and Erik Jorgensen for sharing reagents, and Erik Jorgensen and Randi Rawson for sharing unpublished results. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), and the International C. elegans Gene Knockout Consortium. This work was supported by NHMRC Project Grants 1067461 and 1068871, NIH R01 NS060129, and ARC Future Fellowship FT110100097 to M.A.H.; an NIH OD 010943 to David H. Hall; a University of Queensland Postdoctoral Fellowship to E.M.; The University of Queensland Research Scholarship to C.L.; a HFSP Long-Term Fellowship LT000762/2012 to R.G.S.; a University of Queensland, International Postgraduate Award to A.D.; and an ARC LIEF grant LE130100078 for microscopy.
Footnotes
AUTHOR CONTRIBUTIONS
A.N. and E.M. designed and carried out most experiments, and wrote the paper. C.L. and R.G.S. designed and contributed experiments, and edited the paper. A.D. and C.N. contributed some experiments. R.S. and D.H.H. contributed experiments and interpreted results. D.X. provided reagents and edited the paper. B.N. designed and interpreted experiments, and edited the paper. M.A.H. designed and interpreted experiments, and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adalbert R, Gillingwater TH, Haley JE, Bridge K, Beirowski B, Berek L, Wagner D, Grumme D, Thomson D, Celik A, et al. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci. 2005;21:271–277. doi: 10.1111/j.1460-9568.2004.03833.x. [DOI] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR. WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR. WldS requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol. 2009;184:501–513. doi: 10.1083/jcb.200808042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasaki T, Tatsumi R, Takahashi K, Arai K, Nakanishi Y, Ueda R, Ito K. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Coleman MP, Martin KR. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci. 2008;28:1166–1179. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Gerstbrein B, Frokjaer-Jensen C, Royal DC, Mukherjee G, Royal MA, Xue J, Schafer WR, Driscoll M. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci. 2004;7:1337–1344. doi: 10.1038/nn1347. [DOI] [PubMed] [Google Scholar]
- Calixto A, Jara JS, Court FA. Diapause formation and downregulation of insulin-like signaling via DAF-16/FOXO delays axonal degeneration and neuronal loss. PLoS Genet. 2012;8:e1003141. doi: 10.1371/journal.pgen.1003141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Molecular signaling how do axons die? Adv Genet. 2011;73:185–217. doi: 10.1016/B978-0-12-380860-8.00005-7. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, WldS, and Nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci U S A. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Logan MA, Tasdemir OE, Freeman MR. Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci. 2009;29:4768–4781. doi: 10.1523/JNEUROSCI.5951-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- Emtage L, Gu G, Hartwieg E, Chalfie M. Extracellular proteins organize the mechanosensory channel complex in C. elegans touch receptor neurons. Neuron. 2004;44:795–807. doi: 10.1016/j.neuron.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Fang Y, Soares L, Teng X, Geary M, Bonini NM. A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr Biol. 2012;22:590–595. doi: 10.1016/j.cub.2012.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleard JS, Barry JD, Johnstone IL. cis regulatory requirements for hypodermal cell-specific expression of the Caenorhabditis elegans cuticle collagen gene dpy-7. Mol Cell Biol. 1997;17:2301–2311. doi: 10.1128/mcb.17.4.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- Hall DH. Electron microscopy and three-dimensional image reconstruction. Meth Cell Biol. 1995;48:395–436. doi: 10.1016/s0091-679x(08)61397-7. [DOI] [PubMed] [Google Scholar]
- Hilliard MA. Axonal degeneration and regeneration: a mechanistic tug-of-war. J Neurochem. 2009;108:23–32. doi: 10.1111/j.1471-4159.2008.05754.x. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Wu YC. Engulfment of apoptotic cells in C. elegans is mediated by integrin alpha/SRC signaling. Curr Biol. 2010;20:477–486. doi: 10.1016/j.cub.2010.01.062. [DOI] [PubMed] [Google Scholar]
- Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994;367:467–470. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Comm. 2009;383:258–262. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mapes J, Chen Y-Z, Kim A, Mitani S, Kang B-H, Xue D. CED-1, CED-7, and TTR-52 regulate surface phosphatidylserine expression on apoptotic and phagocytic cells. Curr Biol. 2012;22:1267–1275. doi: 10.1016/j.cub.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SM, O’Brien GS, Portera-Cailliau C, Sagasti A. Wallerian degeneration of zebrafish trigeminal axons in the skin is required for regeneration and developmental pruning. Development. 2010;137:3985–3994. doi: 10.1242/dev.053611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Fischer S, Lopez-Vales R, David S. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia. 2008;56:1566–1577. doi: 10.1002/glia.20766. [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka T, Ding Z, Gengyo-Ando K, Oue M, Yamaguchi H, Mitani S, Ihara Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol Dis. 2005;20:372–383. doi: 10.1016/j.nbd.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Nagarajan A, Ning Y, Reisner K, Buraei Z, Larsen JP, Hobert O, Doitsidou M. Progressive degeneration of dopaminergic neurons through TRP channel-induced cell death. J Neurosci. 2014;34:5738–5746. doi: 10.1523/JNEUROSCI.4540-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Coakley S, Giordano-Santini R, Linton C, Lee ES, Nakagawa A, Xue D, Hilliard MA. EFF-1-mediated regenerative axonal fusion requires components of the apoptotic pathway. Nature. 2015;517:219–222. doi: 10.1038/nature14102. [DOI] [PubMed] [Google Scholar]
- Neumann B, Hilliard MA. Loss of MEC-17 leads to microtubule instability and axonal degeneration. Cell Rep. 2014;6:93–103. doi: 10.1016/j.celrep.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Nguyen KCQ, Hall DH, Ben-Yakar A, Hilliard MA. Axonal regeneration proceeds through specific axonal fusion in transected C. elegans neurons. Dev Dyn. 2011;240:1365–1372. doi: 10.1002/dvdy.22606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RL, Yam L, Weimer RM, Bend EG, Hartwieg E, Horvitz HR, Clark SG, Jorgensen EM. Axons degenerate in the absence of mitochondria in C. elegans. Curr Biol. 2014;24:760–765. doi: 10.1016/j.cub.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch E, Maniatis T. Molecular cloning: A laboratory manual. 2. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Trapp BD, Griffin JW. Macrophage function during Wallerian degeneration of rat optic nerve: clearance of degenerating myelin and Ia expression. J Neurosci. 1989;9:2327–2335. doi: 10.1523/JNEUROSCI.09-07-02327.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller AD. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations pruduced thereby in the sturcture of their primitive fibres. Phil Trans Royal Soc London Series B. 1850;140:7. [Google Scholar]
- Wang X, Li W, Zhao D, Liu B, Shi Y, Chen B, Yang H, Guo P, Geng X, Shang Z, et al. Caenorhabditis elegans transthyretin-like protein TTR-52 mediates recognition of apoptotic cells by the CED-1 phagocyte receptor. Nat Cell Biol. 2010;12:655–664. doi: 10.1038/ncb2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang J, Gengyo-Ando K, Gu L, Sun CL, Yang C, Shi Y, Kobayashi T, Shi Y, Mitani S et al. C. elegans mitochondrial factor WAH-1 promotes phosphatidylserine externalization in apoptotic cells through phospholipid scramblase SCRM-1. Nat Cell Biol. 2007;9:541–549. doi: 10.1038/ncb1574. [DOI] [PubMed] [Google Scholar]
- Wang X, Wu YC, Fadok VA, Lee MC, Gengyo-Ando K, Cheng LC, Ledwich D, Hsu PK, Chen JY, Chou BK, et al. Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science. 2003;302:1563–1566. doi: 10.1126/science.1087641. [DOI] [PubMed] [Google Scholar]
- Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet. 2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104:15132–15137. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Collins CA. A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J Neurosci. 2012;32:610–615. doi: 10.1523/JNEUROSCI.3586-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA. The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012;10:e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaniv SP, Issman-Zecharya N, Oren-Suissa M, Podbilewicz B, Schuldiner O. Axon regrowth during development and regeneration following injury share molecular mechanisms. Curr Biol. 2012;22:1774–1782. doi: 10.1016/j.cub.2012.07.044. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bianchi L, Lee WH, Wang Y, Israel S, Driscoll M. Intersubunit interactions between mutant DEG/ENaCs induce synthetic neurotoxicity. Cell Death Differ. 2008;15:1794–1803. doi: 10.1038/cdd.2008.114. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Kage-Nakadai E, Mitani S, Wang X. C. elegans secreted lipid-binding protein NRF-5 mediates PS appearance on phagocytes for cell corpse engulfment. Curr Biol. 2012;22:1276–1284. doi: 10.1016/j.cub.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Guth L, Steward O. Mechanisms of motor recovery after subtotal spinal cord injury: insights from the study of mice carrying a mutation (WldS) that delays cellular responses to injury. Exp Neurol. 1998;149:221–229. doi: 10.1006/exnr.1997.6717. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Ziegenfuss JS, Doherty J, Freeman MR. Distinct molecular pathways mediate glial activation and engulfment of axonal debris after axotomy. Nat Neurosci. 2012;15:979–987. doi: 10.1038/nn.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullig S, Neukomm LJ, Jovanovic M, Charette SJ, Lyssenko NN, Halleck MS, Reutelingsperger CP, Schlegel RA, Hengartner MO. Aminophospholipid translocase TAT-1 promotes phosphatidylserine exposure during C. elegans apoptosis. Curr Biol. 2007;17:994–999. doi: 10.1016/j.cub.2007.05.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.