

Abstract
The major cholesterol metabolite in brain, 24S-hydroxycholesterol (24S-HC), serves as a vehicle for cholesterol removal. Its effects on neuronal function, however, have only recently begun to be investigated. Here we review that nascent work. Our own studies have demonstrated that 24S-HC has potent positive modulatory effects on NMDA receptor (NMDAR) function. This could have implications for brain plasticity but also for pathological NMDAR overuse. Other work has demonstrated effects of 24S-HC on neuronal survival and as a possible biomarker of neurodegenerative disease. Depending on circumstances, both upregulation/mimicry of 24S-HC signaling and downregulation/antagonism may have therapeutic potential. We are interested in the possibility that synthetic analogues of 24S-HC with positive effects at NMDARs may hold neurotherapeutic promise, given the role of NMDA receptor hypofunction in certain neuropsychiatric disorders.
Keywords: glutamate, cholesterol. 24(S)-hydroxycholesterol, NMDA, apoptosis, Alzheimer’s disease
Introduction
Oxysterols are products of cholesterol oxidation that can be produced enzymatically and non-enzymatically. A host of roles for oxysterols in signaling, development, metabolism, membrane homeostasis, inflammation, and immune function have been demonstrated (Calkin and Tontonoz 2012; Cyster and others 2014; Leoni and Caccia 2013a; Meaney 2014; Olsen and others 2012; Poli and others 2013; Spann and Glass 2013). Here we focus on the most abundant oxysterol in brain, 24(S)-hydroxycholesterol (24S-HC; Figure 1A, B), and the effects of 24S-HC within the brain.
Figure 1.
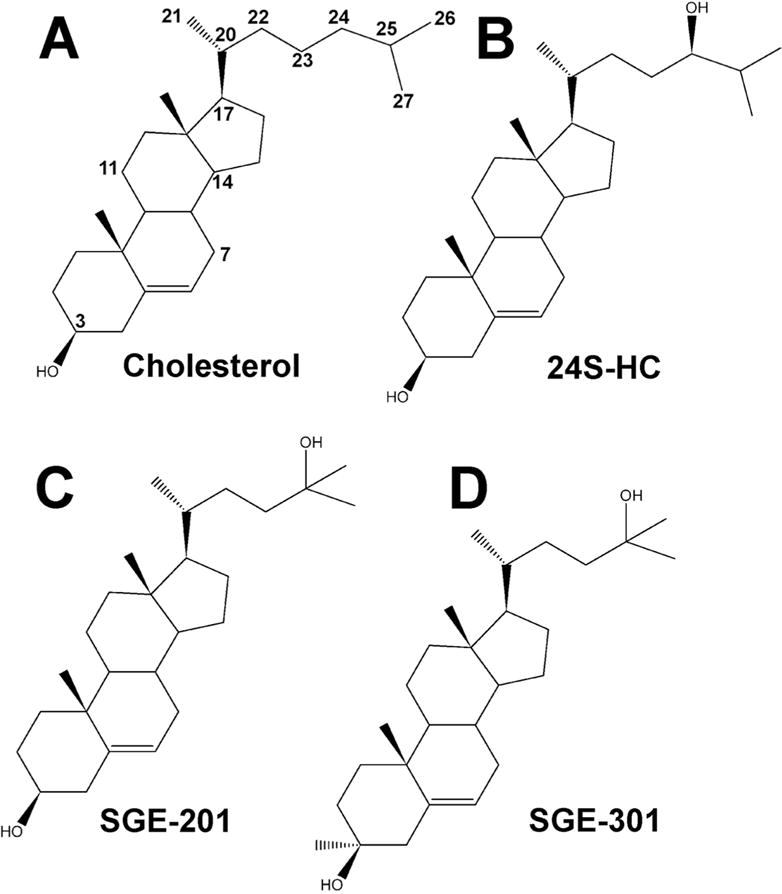
Structures of cholesterol (A) and 24S-HC (B). Numbers of select carbons on the steroid rings and side-chain have been labeled. C and D show structures of two synthetic 24S-HC analogues with positive modulatory effects on NMDARs.
The brain is unusual among organ systems outside the liver in that it synthesizes cholesterol, the immediate precursor of 24S-HC (Dietschy and Turley 2004). Peripheral circulating cholesterol, mainly bound by lipoproteins, does not cross the blood-brain barrier. There is still controversy over the primary cell types in brain that produce cholesterol, with neurons, astrocytes, and oligodendrocytes all likely participants. The relative contributions of the cell types responsible for cholesterol synthesis may shift with development, and astrocytes appear to be a major supplier of neuronal cholesterol in maturity (Pfrieger and Ungerer 2011), although excitatory neurons express the machinery for cholesterol synthesis and homeostasis (Valdez and others 2010). Astrocyte-derived cholesterol is delivered to neurons mainly by apolipoprotein E (ApoE). In neurons, membrane cholesterol has important roles in ion channel function (Allen and others 2007; Bennett and Simmonds 1996; Rankin and others 1997), vesicle exocytosis/endocytosis (Lang and others 2001; Wasser and others 2007), and other functions.
24S-HC production
The majority of brain cholesterol resides in a metabolically stable pool, mostly in myelin. About 30% of neuronal and glial cholesterol is labile, turning over at a low rate. Although much of this labile pool is recycled locally within the brain, some efflux of cholesterol metabolites occurs across the blood-brain barrier to maintain homeostasis. Metabolites include cholesterol oxidation products that are produced both enzymatically and non-enzymatically to generate a potentially bewildering array. However, one metabolite dwarfs all others in mature brain: the side-chain oxidation product 24S-HC (Meljon and others 2012; Russell and others 2009). 24S-HC is so prevalent in brain that it is referred to as “cerebrosterol.”
24S-HC is produced by the cytochrome P450 enzyme CYP46A1 (cholesterol 24-hydroxylase). Unlike cholesterol itself, enzymatic 24S-HC synthesis is strictly neuronal (Figure 2A, B). Genetic deletion of CYP46A1 reduces brain 24S-HC levels by ~95% or more, indicating that there is normally little non enzymatic 24S-HC production (Lund and others 2003). Plasma levels are reduced less, suggesting some peripheral production through alternative pathways. Interestingly, CYP46A1 expression is restricted to certain neuronal cell types. In the hippocampus principal neurons and some interneuron classes express CYP46A1 (Ramirez and others 2008). The signaling and metabolic implications of cell-selective 24S-HC synthesis are unknown. Within neurons that express the enzyme, CYP46A1 protein is found in the endoplasmic reticulum of postsynaptic compartments (soma and dendrites, schematized in Figure 2C). This could have implications for signaling effects of 24S-HC, some of which, as described below, may impact postsynaptic signaling. Several antifungal agents, including voriconazole, are high-affinity inhibitors of CYP46A1 (Mast and others 2010; Shafaati and others 2010). These compounds can pass the blood-brain barrier and may prove useful experimentally or clinically to probe 24S-HC function in the brain.
Figure 2.
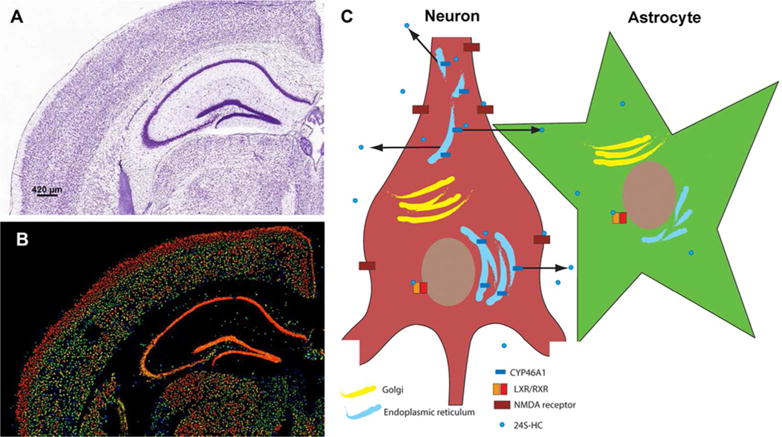
Nissl stain (A) and expression heat map for CYP46A1 mRNA expression (B) in coronal brain sections of mouse, showing strong neuronal expression. Images are taken from the Allen Brain Atlas, ©2014 Allen Institute for Brain Science. Allen Mouse Brain Atlas, available from http://mouse.brainmap.org/. C. Schematic of neuronal production of 24S-HC from endoplasmic reticulum and subsequent release. For simplicity, interaction of 24S-HC with only two targets is shown: LXR/RXRs of neurons and astrocytes and NMDARs on neuronal plasma membrane.
24S-HC passes through membranes more readily than cholesterol itself because of the side-chain oxidation (Bjorkhem and Meaney 2004). It is unknown whether 24S-HC release is regulated in other ways. Passive flux across membranes is believed to be the mechanism by which 24S-HC is released from cells and passes through the blood-brain barrier, although 24S-HC may be actively transported across the blood-brain barrier by organic anion transporting polypeptide 2 (Ohtsuki and others 2007). Once in the periphery, 24S-HC binds lipoproteins and is eventually converted to bile acids.
Brain 24S-HC levels are developmentally regulated. In rodents, brain levels are quite low during early stages, and several other oxysterols reach nearly comparable levels (Meljon and others 2012). With maturation into young adulthood, 24S-HC levels greatly exceed other metabolites. Free concentrations in mature human brain homogenates reach approximately 30 μM, and aging reduces 24S-HC levels relative to cholesterol (Lutjohann and others 1996). As discussed below, various neurodegenerative diseases of aging, such as Alzheimer’s disease and Parkinson’s disease, exhibit altered levels of 24S-HC compared to age-matched controls (Bjorkhem and others 2013; Leoni and others 2013c). These observations suggest that 24S-HC may be a clinically useful biomarker of neurodegenerative disease. It is interesting to speculate that these altered levels could have a role in pathology (or possibly as palliative compensatory mechanisms) through signaling effects. Supporting evidence for this speculation is offered below. Thus, in addition to biomarker status, 24S-HC could become a nexus for therapeutic development.
Factors that control CYP46A1 activity and 24S-HC synthesis are poorly understood. Although feedback regulation of cholesterol synthesis is important as described above, it is unclear whether postranscriptional regulators modify CYP46A1 activity. In addition to these potential direct influences, other cholesterol metabolic pathways may indirectly influence 24S-HC synthesis by shunting cholesterol away from 24S-HC. For instance, the mitochondrial cholesterol side-chain cleavage enzyme presumably competes with CYP46A1 and sends cholesterol down a different metabolic pathway of steroid synthesis, with different implications for neuronal signaling (e.g. Chisari and others 2010). Because CYP46A1 and cholesterol side-chain cleavage enzyme reside in different intracellular organelles, cholesterol trafficking to specific organelles could control which pathway is favored under certain conditions. Study of the modulation of neuronal 24S-HC synthesis by direct and indirect mechanisms remains in its infancy and could be important, given the signaling roles described below.
Oxysterols, transcription, and metabolism
The side-chain oxidized oxysterols 25-hydroxycholesterol (25-HC), 27-hydroxycholesterol, and 24S-HC are enzymatically generated. Forty years ago, prior to discovery of the synthetic enzymes, it was demonstrated that oxysterols inhibit sterol synthesis (Brown and Goldstein 1974; Kandutsch and Chen 1974), suggesting a negative feedback role. This remains the classical signaling role for oxysterols. Other roles in lipid metabolism were later discovered. These include transcriptional control of genes required for lipid metabolism, substrates for bile acid synthesis, mediators of sterol transport (Russell 2000) and regulators of cholesterol accessibility in membranes (Bielska and others 2014). We review some of these targets here, with an emphasis on brain signaling.
As regulators of transcription, oxysterols have several targets. 24S-HC and other oxysterols bind the nuclear hormone liver X receptors (LXRs), LXRα and LXRβ. LXRs are transcription factors that control the expression of gene products involved in cholesterol homeostasis. Various aspects of cholesterol uptake, transport, efflux and excretion are controlled by LXRs in a tissue dependent manner (Hong and Tontonoz 2014). 24S-HC has a particularly strong role in modulating cholesterol transporter and lipoprotein expression in astrocytes (Abildayeva and others 2006), a major cellular source of neuronal cholesterol. Thus, a complicated communication system appears to exist between neurons and astrocytes, with 24S-HC serving as an intercellular signal that controls the delivery of astrocytically derived cholesterol to neurons.
LXR subunits heterodimerize with a retinoid X receptor (RXR) to form a functional transcriptional regulator. The unliganded receptor appears to repress transcription in most cases. Another transcription level control on lipid homeostasis is through the sterol regulatory element-binding proteins (SREBPs). SREBP activity is key to important effects of oxysterols on lipogenesis and glucose metabolism. It is beyond the scope of this review to catalogue all of the transcriptional changes related to cholesterol and lipid homeostasis, but several excellent reviews have been published (Hong and others 2014; Wang and others 2002).
In the brain, LXRβ is ubiquitously expressed while LXRα is more sporadically expressed in selected cell types. As in the periphery, brain LXRs regulate cholesterol homeostasis and regulate anti-inflammatory pathways. Knockout of the LXRs produces severe phenotypes in ventricle development, neuronal loss in brain and spinal cord, astrocyte proliferation, and lipid accumulation (Wang and others 2002).
Side-chain oxysterols such as 24S-HC also alter the accessibility of membrane cholesterol, thereby altering membrane structure and indirectly influencing neuronal excitability (Bielska and others 2014; Bielska and others 2012). 25-HC, and presumably also 24S-HC, expands model lipid bilayers as it partitions. This effect is not shared by ring-modified oxysterols and is not enantioselective. This indicates that there is unlikely to be a protein binding site for the oxysterol to explain its membrane effects, and these effects can be explained entirely by incorporation into the achiral membrane environment. The membrane effect of the side-chain oxysterols renders resident cholesterol more accessible to external cholesterol sensors, and alters membrane cholesterol content (Bielska and others 2014; Bielska and others 2012). This latter effect could have implications for the function of ion channels that are dependent on membrane cholesterol content and membrane structure.
Lessons from CYP46A1−/− knockout mice
CYP46A1−/− mice show strongly depressed levels of 24S-HC, indicating that the vast majority of it is enzymatically produced. Interestingly, brain cholesterol levels are unaltered in the mice, suggesting a robust feedback mechanism to homeostatically reduce cholesterol synthesis in the absence of its turnover (Lund and others 2003). It was recently shown that other oxysterol products are also not strongly altered by genetic deletion of CYP46A1 (Meljon and others 2014). The lack of strong phenotype of neurodegeneration or developmental abnormalities found in CYP46A1−/− mice suggest that 24S-HC may not be a sufficiently important brain LXR ligand to explain the LXR phenotypes described above.
Although neurodegeneration was not observed, neurological phenotypes in the CYP46A1−/− animals included strong spatial learning and memory deficits in the Morris water maze, a behavioral test of hippocampal-dependent function (Kotti and others 2006). Homozygous null mice were nearly completely unable to learn the location of a hidden platform over repeated trials. Heterozygotes showed an intermediate effect, suggesting a gene dosage effect.
These behavioral defects were paralleled in studies of long-term potentiation (LTP) in hippocampal slices (Kotti and others 2008; Kotti and others 2006). LTP is a long-term increase in synaptic efficacy following certain patterns of presynaptic stimulation and has been linked to associative memory formation. NMDAR activation is a key mediator of LTP induction. NMDARs are Ca2+ permeable and can trigger second messenger cascades that result in changes in synaptic efficacy. However, because NMDARs are normally blocked in a voltage dependent manner by physiological concentrations of extracellular Mg2+, their activation requires coincident release of glutamate and postsynaptic depolarization. This coincidence is generally thought to meet the requirements for a cellular substrate for associative memory formation (Collingridge and Bliss 1995).
LXRα/β double deletion failed to alter the effect of CYP46A1deletion, suggesting that LXRs were not to blame for the LTP and learning deficits. Other targets of 24S-HC besides LXR were not considered. Instead, geranylgeraniol, a byproduct of cholesterol synthesis via the mevalonate pathway, was found to rescue LTP deficits in CYP46−/− animals. This result was interpreted to suggest that loss of intermediates in the cholesterol synthesis pathway, as a result of the decrease in cholesterol synthesis, was responsible for the deficits (Kotti and others 2006).
Effects of 24S-HC and analogues on NMDAR function
The LTP and learning and memory deficits in CYP46−/− mice could also be consistent with loss of 24S-HC signaling through pathways other than those mediated by LXR. In fact our recent work suggests another possibility. We and our collaborators found that 24S-HC potentiates NMDAR function with an EC50 of ~1 μM (Figure 3A, B). Thus, although the possibility still requires more rigorous testing, loss of endogenous 24S-HC effects on NMDAR function could participate along with cholesterol intermediates in the phenotypes of CYP46A1−/− mice. The potentiating effect on NMDAR function occurs even at saturating NMDA and glycine concentration. This suggests that the oxysterol and analogues do not alter agonist or co-agonist affinity. Rather, the positive modulation appears to increase channel open probability of the fully liganded receptor. In rats, application of 24S-HC analogues reversed a form of metaplastic LTP inhibition induced by pretreatment with subanesthetic concentrations of the NMDAR channel blocker ketamine (Izumi and Zorumski 2014; Paul and others 2013) (Figure 3C, D). Oxysterol analogues, systemically administered, also reversed behavioral deficits in spontaneous alternation, novelty object recognition and social interactions induced by NMDAR channel blockers (Paul and others 2013) (Figure 3E, F).
Figure 3.
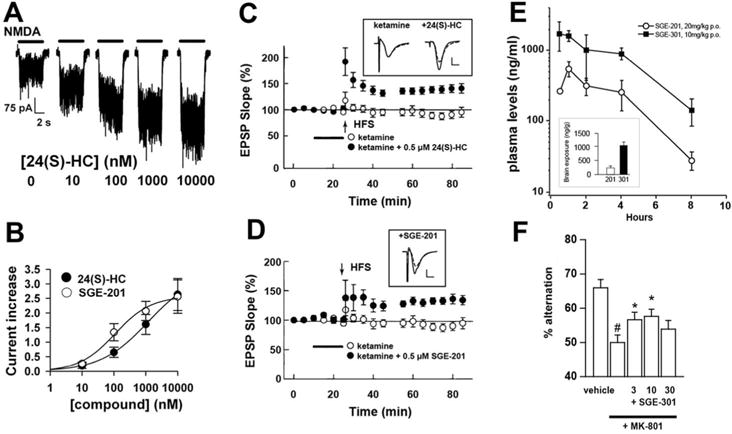
Modulation of NMDAR function, synaptic plasticity, and behavior. A. Examples of the effect of 24S-HC at varied concentrations on responses of a cultured hippocampal neuron to NMDA application. B. The concentration response relationship derived from experiments like that in A. The synthetic analogue SGE-201 shows higher potency for positive modulation. C, D. Synaptic efficacy, monitored by field EPSP slope was unchanged after high frequency stimulation 2 hr following ketamine exposure. HFS is typically sufficient to induce long-term potentiation (LTP) of the EPSP slope, but ketamine treatment prevents this. 0.5 μM 24S-HC or 0.5 μM SGE-201 overcomes the inhibition of LTP. E. Plasma levels of SGE-201 and SGE-301 following the indicated i.p. doses. The inset shows brain levels 60 min following administration. Figures panels are adapted from (Paul and others 2013). F. Spontaneous alternations are reduced by MK 801, a psychotomimetic NMDAR antagonist, and the MK 801 effect is partially abrogated by systemic SGE-301 at the indicated doses.
There are some peculiar aspects to 24S-HC modulation of NMDAR function that may yield insights into mechanisms (Linsenbardt and others 2014; Paul and others 2013). Although some positive and negative allosteric modulators exhibit selective effects on receptors containing certain GluN2 subunits (Hedegaard and others 2012), we failed to find any dependence of oxysterol potentiation on subunit composition. Potentiation is slow to develop and long lasting following 24S-HC removal, even when application and removal are rapid. Although this might suggest that 24S-HC initiates second messenger cascades to promote increased NMDAR channel function, potentiation is observed in excised outside out membrane patches, where second messenger cascades are unlikely (although membrane delimited second messenger pathways are possible). Potentiation by analogues of 24S-HC also exhibit slow onset, but effects are readily reversed by a molecular steroid scavenger, γ-cyclodextrin. Thus, it seems most likely that there is a direct effect of 24S-HC on the receptor. 24S-HC can access its target through membrane partitioning, since 24S-HC application to the bath in a cell attached recording is effective. On the other hand, analogues are not effective when applied to the intracellular face of an inside out membrane patch. This suggests that 24S-HC is only an effective potentiator of NMDARs if released from cells. Taken together, it seems likely that slow functional onset and offset are explained by slow membrane partitioning of extracellular 24S-HC into the plasma membrane, lipid diffusion to the receptor, and slow membrane departitioning.
Although it appears that 24S-HC has direct effects on NMDARs, the mechanism of potentiation appears distinct from that of other known lipophilic positive allosteric modulators. For instance, pregenolone sulfate and arachidonic acid potentiation is additive with that of 24S-HC (Paul and others 2013). By contrast 24S-HC potentiation occludes that of the synthetic analogues SGE-201 and SGE-301 (structures given in Figure 1C, D), suggesting a shared mechanism among these structural analogues. Another interesting observation is that 25-HC, an oxysterol synthesized by macrophages (and presumably brain microglia), very weakly potentiates NMDAR function but antagonizes the effects of 24S-HC and its analogues (Figure 4) through an apparently non-competitive mechanism (Linsenbardt and others 2014). Thus, there may be two separate oxysterol sites on NMDARs. In addition, another cholesterol metabolite was recently found to exhibit negative regulation of NMDAR function, increasing the complexity of possible interactions (Hu and others 2014).
Figure 4.
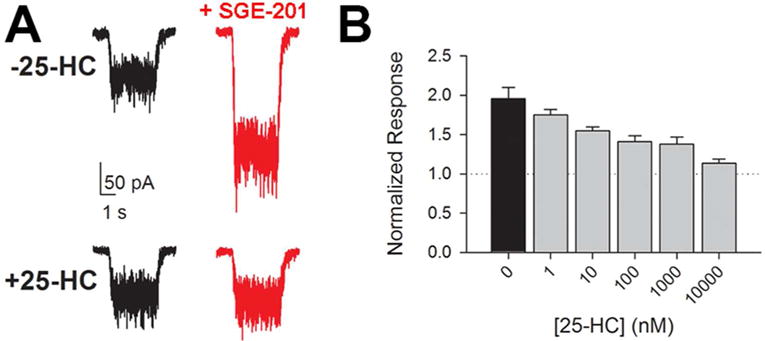
Antagonism of oxysterol positive modulation by another side-chain oxysterol, 25-HC. A. Top panel: example of effect of SGE-201 (0.2 μM) on neuronal response to NMDA application (horizontal bar). Bottom panel: block of SGE-201’s effect by 10 μM 25-HC. B. Effect of varied 25-HC concentrations on the potentiated NMDA response. NMDA current in the absence of modulator is indicated by the dotted line. Figure is adapted from (Linsenbardt and others 2014) and used with permission from Elsevier.
An alternative explanation is that potentiation results from local membrane perturbations such as those described above that alter cholesterol accessibility. This explanation is difficult to reconcile with the opposing effects of 24S-HC and 25-HC; both side chain oxysterols are expected to yield similar effects on membranes. The explanation is also hard to reconcile with enantioselective effects of 25-HC (Linsenbardt and others 2014). Regardless of whether modulation involves a specific binding site on the NMDAR, it is possible that physiologically relevant concentrations of 24S-HC could modulate the NMDAR. It is less likely that 25-HC has an important endogenous role given its lower levels in brain. On the other hand, local concentrations of 25-HC could reach physiological levels with microglial activation or other atypical situations.
24S-HC and Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the accumulation of brain amyloid-β (Aβ) plaques and fibrillary tangles of hyperphosphorylated tau protein. Evidence suggests a correlation between imbalanced cholesterol homeostasis and the symptoms and/or pathology of AD. For many years the field has known that genetic risk for AD is conferred by certain alleles of the ApoE gene, which encodes the major cholesterol carrying protein in the brain (reviewed in Gamba and others 2012). As the major cholesterol metabolite in brain, 24S-HC involvement in the etiology of AD has also been investigated (reviewed in Gamba and others 2012; Hughes and others 2013). Increased 24S-HC levels in both plasma and cerebrospinal fluid (CSF) have been observed at early stages of AD (Lutjohann and others 2000; Papassotiropoulos and others 2002). This phenomenon may result from elevated extracellular cholesterol levels caused by myelin breakdown (Bartzokis 2011; Hughes and others 2013). As the disease progresses, 24S-HC levels in plasma and CSF decline, possibly reflecting extensive neuronal loss and resultant loss of CYP46A1 enzyme (reviewed in Bjorkhem and others 2004). Taken together, these studies suggest a potential use of 24S-HC as a biomarker for AD diagnosis (Hughes and others 2013).
In normal brains CYP46A1 is selectively expressed in neurons, but in brains of AD patients, increased CYP46A1 expression in astrocytes has been observed (Bogdanovic and others 2001; Brown and others 2004). Moreover, CYP46A1 is preferentially expressed in degenerating neurites surrounding senile plaque in brains of AD patients (Brown and others 2004). Furthermore, association of CYP46A1 polymorphisms with risk of AD and elevated Aβ load has been reported (Garcia and others 2009; Papassotiropoulos and others 2003). The abnormal induction and distribution of CYP46A1 in brains of AD patients suggest that 24S-HC, in addition to being an indicator for AD diagnosis, may be more directly involved in AD pathogenesis. Based on findings from rat primary neuron cultures, 24S-HC inhibits Aβ and APP secretion. This inhibitory effect is accompanied by LXR activation and protein kinase C (PKC) inhibition (Brown and others 2004). Another study also reported down regulation of APP trafficking and a suppression of Aβ production in SH-SY5Y human neuroblastoma cells following 24S-HC treatment (Urano and others 2013). In the same cell line, 24S-HC inhibited Aβ production by increasing α-secretase activity, a mechanism favoring anti amyloidogenic effects of 24S-HC (Famer and others 2007).
Despite its beneficial effects on AD pathology, several contradictory findings have been reported, indicating that 24S-HC may exacerbate Aβ aggregation and enhance AD related neurotoxicity. 24S-HC potentiates Aβ-mediated neurotoxicity in the human differentiated neuroblastoma cell line MSN, in a manner involving generation of reactive oxygen species (ROS) (Ferrera and others 2008). Similarly, another study revealed that three oxysterols— 24S-HC, 27-HC and 7β-HC—each effectively potentiated Aβ peptide binding to cell membranes of human differentiated neuronal cell lines (SK-N-BE and NT-2). Most interestingly, only 24S-HC significantly exacerbated Aβ-mediated necrogenic and apoptotic effects on these cells (Gamba and others 2011; Gamba and others 2012). This selective neurotoxic effect of 24S-HC is determined by its pro-oxidant action that promotes ROS generation locally in neuronal cells, a behavior not mimicked by 27-OH and 7β-OH (Gamba and others 2011).
In addition to studies done in cell culture systems, the role of 24S-HC in AD pathogenesis has also been tested in vivo using CYP46A1 deficient/mutant APP mice. Surprisingly, reduction of endogenous 24S-HC level by CYP46A1 deletion does not affect amyloid formation or rates of amyloid deposition (Halford and Russell 2009). However, this study indicated an increased survival rate of APP-overexpressing mice when endogenous 24S-HC level is greatly reduced, suggesting that reduced 24S-HC levels may offer a protective effect in an AD-related context (Halford and others 2009). Assuming that the variety of contexts in which 24S-HC is pro survival vs. pro-pathological can be sorted out, controlling 24S-HC levels might one day be harnessed for therapeutic benefit in AD.
Parkinson’s disease
Parkinson’s disease (PD) is the second most prevalent progressive neurodegenerative disorder in humans. Clinical symptoms include tremor, bradykinesia, rigidity, postural instability, and cognitive changes. PD pathology is characterized by accumulation of α-synuclein, which is likely modulated by cholesterol through its cholesterol binding domains (Fantini and others 2011). The potential role of 24S-HC as a biomarker for PD diagnosis has also been investigated. Although one study showed markedly reduced 24S-HC levels in plasma of PD patients (Lee and others 2009), another study reported unchanged plasma levels, but significantly increased CSF levels of 24S-HC in PD patients (Bjorkhem and others 2013). The increased CSF levels of 24S-HC correlated with longer disease duration (Bjorkhem and others 2013), suggesting CSF levels of 24S-HC may reflect varying degrees of neurodegeneration.
Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by mutation of huntingtin (HTT) gene. Clinical symptoms of HD include motor impairment, neuropsychiatric symptoms, and cognitive decline. Similar to AD, disturbances in cholesterol metabolism have been found in HD patients. The overall rate of cholesterol synthesis was found reduced in inducible mutant HTT cell line, in the stratium and cortex of HTT mouse models, and in cortical tissue from HD patients (Leoni and Caccia 2014; Valenza and others 2007; Valenza and others 2005). As a result, a progressive reduction of plasma 24S-HC was observed in relation to the patient’s HD progression quantified by MRI measurements of striatal atrophy and volumes, and use of psychological and neurological tests (Leoni and others 2014; Leoni and others 2013b; Leoni and others 2011). These data suggest 24S-HC may serve as a biomarker for tracking HD progression, though its plasma levels may become less reliable under certain conditions (Leoni and others 2014).
Niemann-Pick C1 disease
Niemann-Pick type C1 (NPC1) disease is a rare neurodegenerative developmental disorder that causes progressive, massive neuron loss. A profound characteristic of this disease is the accumulation of free cholesterol in endolysosomes because of impaired intracellular trafficking of cholesterol (Ory 2000; Sturley and others 2009). In cultured NPC-1 deficient fibroblasts and murine NPC1−/− macrophages, this abnormal cholesterol accumulation is accompanied by elevated ROS, which promote nonenzymatic oxysterol production by directly attacking cholesterol (Zampieri and others 2009; Zhang and others 2008). Elevated levels of nonenzymatic oxysterols are observed in plasma and tissues of NPC1−/− mice. Unlike the nonenzymatic oxysterols, 24S-HC is produced exclusively through enzymatic cholesterol oxidation, and reduced level of 24S-HC is observed in whole brain tissues of NPC1−/− mice (Porter and others 2010). Similarly, 24S-HC plasma concentrations were significantly reduced in human NPC1 subjects (Porter and others 2010). The reduction of 24S-HC in plasma of NPC1 patients may reflect loss of CYP46A1 expressing neurons due to progressive neurodegeneration, supporting use of 24S-HC as a general marker for diagnosis of neurodegenerative diseases. However, the fact that reduced level of 24S-HC is observed in not only NPC1 but also other lysosomal storage diseases makes it alone less specific as a biomarker for NPC1 (Porter and others 2010). In contrast, a profile including both nonenzymatic and enzymatic oxysterols together could serve as more robust plasma biomarkers for diagnosis and treatment of NPC1 disease (Porter and others 2010).
Direct studies of 24S-HC and cell survival
Model systems have demonstrated that 24S-HC can be neurotoxic under some circumstances. At concentrations above 10 μM, it produces death of SH-SY5Y human neuroblastoma cells (Yamanaka and others 2011). Viability is reduced by half at 50 μM for 24 hr. Similar effects were observed with cortical neurons in primary dissociated culture (Yamanaka and others 2011). The features of cell death were not consistent with either pure necrotic or pure apoptotic cell death. Rather, death was ameliorated by pharmacological or genetic knockdown of receptor interacting serine/threonine kinase, required for a non-apoptotic form of cell death called necroptosis. In human T-lymphoma Jurkat cells, by contrast, purer apoptosis is induced by 24S-HC (Yamanaka and others 2014). Apoptosis in these cells is linked to activation of caspase 8, which is not found in SH-SY5Y cells. Thus, 24S-HC can induce cell death with prolonged exposure to high concentrations. Downstream pathways can vary by context. The direct targets of 24S-HC that trigger the ultimate demise of neurons are still unclear, although survival may depend on esterification of 24S-HC and subsequent lipid drop formation (Yamanaka and others 2014).
Neuroprotective potential of 24S-HC has also been revealed in SH-SY5Y cells in a preconditioning model (Okabe and others 2013). Ironically, 24S-HC protects against toxicity produced by another cholesterol oxidation product, 7-ketocholesterol. 7-ketocholesterol is among the oxidation products generated during cellular insults when oxygen radicals attack cholesterol. Pre-incubation with a sublethal 24S-HC concentration protects SH-SY5Y cells against subsequent 7-ketocholesterol induced damage. Unlike the situation with neurotoxicity, the trigger for 24S-HC protective effect seems clearer. Genetic knockdown of LXR, a known binding target of 24S-HC, reduces the adaptive preconditioning effect.
Our recent observation that 24S-HC and synthetic analogues potentiate NMDAR function raises the possibility that the oxysterol could exacerbate excitotoxicity. Excitotoxicity involves injury and death of neurons through overactivation of ionotropic glutamate receptors, particularly NMDARs (Hardingham and Bading 2010; Rothman and Olney 1987). Overactivation of NMDARs or selective activation of certain toxic populations of NMDARs, leads to Ca2+ influx that triggers cell death pathways. Excitotoxicity is believed to have a prominent role in neuronal injury following strokes and seizures. It may also participate in cell death in neurodegenerative conditions such as Alzheimer’s and Huntington’s diseases.
We examined the effect of oxysterols in the context of energy deprivation in primary cultures of hippocampal neurons. Synaptically mature, cultured hippocampal or cortical neurons respond to oxygen deprivation or oxygen/glucose deprivation with delayed cell death. This death can be largely prevented by incubation in an NMDAR antagonist during the insult (Hogins and others 2011; Rothman 1984). We recently showed that low micromolar concentrations of the 24S-HC analogue SGE-201 do not damage neurons when administered alone at baseline for a few hours. However, the same treatment exacerbates co-applied mild hypoxic damage (Emnett and others 2014). We have observed similar effects with 24S-HC (unpublished observations).
Positive allosteric modulation also alters the neuroprotective effect of use-dependent NMDAR channel blockers such as ketamine and memantine during excitotoxic insults or during physiological activity (Figure 5). These blockers require channel opening for access to and escape from their binding site, so allosteric modulators that increase channel open probability might be expected to alter the blockers’ effects (Emnett and others 2014). Although positive allosteric modulation does not alter the IC50 concentration for memantine and ketamine, it significantly speeds the drugs’ kinetics of action at the cellular level. In so doing, the positive modulator unmasks voltage dependent differences in the effects of memantine and ketamine that are not otherwise evident (Emnett and others 2014; Emnett and others 2013). 24S-HC analogues significantly reduce the physiological and neuroprotective effects of memantine, a drug that more rapidly re-equilibrates upon depolarization, relative to ketamine. Thus, both positive and negative endogenous modulators of NMDARs including 24S-HC, arachidonic acid, neurosteroids, pH, Zn2+ and others (Traynelis and others 2010) may alter the local actions of these clinically important drugs.
Figure 5.
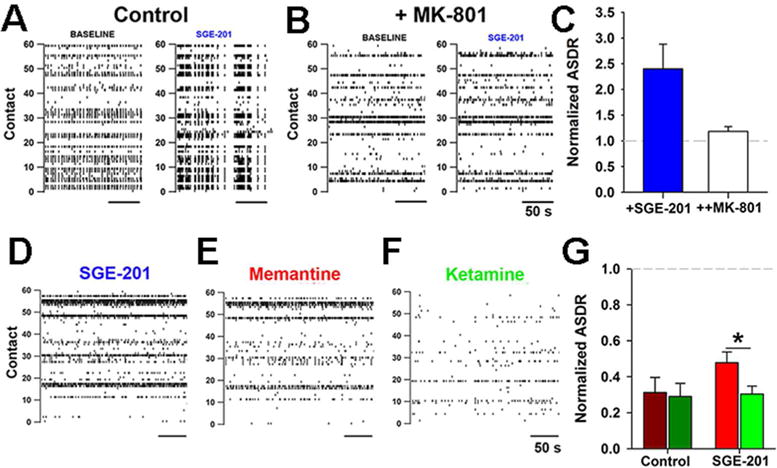
Interaction between positive modulators of NMDAR activity and NMDAR channel blockers. A–C. Effects of the 24S-HC analogue SGE-201 on neuronal activity can be accounted for by effects on NMDAR activity. A, B Raster plots of action potential activity in a dissociated hippocampal culture on a multi-electrode array. The y axis represents the electrodes. C. Under baseline conditions SGE-201 increases spiking activity (ASDR) by >2 fold (blue bar), but no significant increase occurs in the presence of 20 μM MK-801 (open bar), an NMDAR antagonist. D–G. In the presence of positive modulator, the open-channel blockers memantine and ketamine reduce activity. G. Summary data showing that in the absence of SGE-201 (Control), the reduction in activity by the two channel blockers is indistinguishable. In the presence of SGE-201, the effect of the more voltage-sensitive compound memantine is relieved more than that of ketamine. Figure is adapted from (Emnett and others 2014) and used with permission.
Endogenous roles and therapeutic potential of 24S-HC and analogues
Several possibilities arise as a result of newly recognized effects of 24S-HC on neuronal signaling and survival. First, when and to what degree does endogenous 24S-HC perform the signaling roles discovered by applying exogenous 24S-HC to model systems? Although circumstantial evidence suggests that there is sufficient endogenous 24S-HC at the right place (postsynaptic compartments) to modulate NMDAR function, direct tests of endogenous 24S-HC allosteric tone on NMDARs are still pending. It is possible that receptors are somehow protected from 24S-HC effects. Likewise, it remains to be proven whether local concentrations of 24S-HC are sufficiently high to promote preconditioning or toxic effects observed with exogenous application in SH-SY5Y cells. To address these questions, closer examination of mice genetically deficient in 24S-HC production should be helpful. Development of antagonists, such as 25-HC, acting against 24S-HC potentiation at NMDARs, may also be fruitful for testing hypotheses about the specific role of potential 24S-HC targets.
Regardless of whether basal nervous system functioning tone is affected by endogenous 24S-HC, it is possible that under some pathological conditions, excess 24S-HC may contribute to pathology. Under these conditions, intervening to dampen 24S-HC production or effects may be beneficial. Voriconazole or other CYP46A1-inihibiting compounds could work at the enzymatic source, while increased knowledge of 24S-HC targets might allow more specific disruption of these targets. For instance, to reduce pathological 24S-HC mediated potentiation of NMDAR function, analogues of 25-HC might be beneficial (Figure 4). The latter approach could promote neuroprotection in the absence of major side effects associated with direct NMDAR antagonists.
Increasing the actions of endogenous 24S-HC or administering exogenous 24S-HC agonists may also have therapeutic potential under certain conditions. In wild type rodents, clear behavioral effects of 24S-HC analogues were recently observed (Paul and others 2013). This indicates that relevant targets are not saturated basally, suggesting room for therapeutic intervention. Of particular interest to neuropsychiatry is positive allosteric modulation of NMDARs. The rationale for use in psychiatric illnesses is that NMDAR antagonists such as ketamine and PCP cause psychotic symptoms in humans. There is also evidence that NMDAR hypofunction, especially in certain classes of inhibitory interneurons, participates in disease progression in schizophrenia (Moghaddam and Javitt 2012; Mohn and others 1999). The interest in NMDARs as targets is driven partly by the idea that NMDARs may be particularly relevant for relieving cognitive symptoms associated with schizophrenia (Collingridge and others 2013), which are among the most debilitating effects of the disorder. Several classes of indirect and direct modulators of NMDAR function, notably glycine transport inhibitors and glycine-site agonists, are of current interest as therapies (Cioffi 2013). Selective ligands for the 24S-HC oxysterol site on NMDARs could have a place among novel therapeutic strategies. One obstacle is in developing selective ligands with actions solely at NMDARs, without effect on additional signaling pathways described above. To date, effects on neuronal activity of several-hour exposure of neurons in culture to the synthetic 24S-HC analogue SGE-201 can be entirely attributed to effects on NMDARs (Figure 5A–C) (Emnett and others 2014). Although promising, much more work is needed to test the selectivity of these ligands. In other contexts such as neurodegenerative illness, exogenous ligands that activate the adaptive, pro survival pathways triggered by 24S-HC might also prove productive.
Summary
In summary, in addition to classical feedback regulation of cholesterol homeostasis, the oxysterol 24S-HC has a number of potential signaling roles in the healthy and dysfunctional central nervous system. Some of these roles it may share with other side-chain oxysterols. Other effects, such as positive NMDAR modulation, appear to be specific for 24S-HC. Studies of oxysterol neuromodulatory functions are still in their infancy, and we anticipate future work to uncover additional roles.
Acknowledgments
The authors acknowledge funding from Sage Therapeutics, the Bantly Foundation, and NIH grants MH078823, MH101874, MH104506, MH077791, and AA017413. The authors thank Steve Paul and Sage Therapeutics for bringing the neuromodulatory potential of 24S-HC to our attention.
References
- Abildayeva K, Jansen PJ, Hirsch-Reinshagen V, Bloks VW, Bakker AH, Ramaekers FC, et al. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem. 2006;281(18):12799–808. doi: 10.1074/jbc.M601019200. [DOI] [PubMed] [Google Scholar]
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8(2):128–40. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32(8):1341–71. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PJ, Simmonds MA. The influence of membrane cholesterol on the GABAA receptor. Br J Pharmacol. 1996;117(1):87–92. doi: 10.1111/j.1476-5381.1996.tb15158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielska AA, Olsen BN, Gale SE, Mydock-McGrane L, Krishnan K, Baker NA, et al. Side-chain oxysterols modulate cholesterol accessibility through membrane remodeling. Biochemistry. 2014;53(18):3042–51. doi: 10.1021/bi5000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielska AA, Schlesinger P, Covey DF, Ory DS. Oxysterols as non-genomic regulators of cholesterol homeostasis. Trends Endocrinol Metab. 2012;23(3):99–106. doi: 10.1016/j.tem.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkhem I, Lovgren Sandblom A, Leoni V, Meaney S, Brodin L, Salveson L, et al. Oxysterols and Parkinson’s disease: evidence that levels of 24S-hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neuroscience Letters. 2013;555:102–5. doi: 10.1016/j.neulet.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806–15. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Bogdanovic N, Bretillon L, Lund EG, Diczfalusy U, Lannfelt L, Winblad B, et al. On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci Lett. 2001;314(1–2):45–8. doi: 10.1016/s0304-3940(01)02277-7. [DOI] [PubMed] [Google Scholar]
- Brown J, 3rd, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, Lee JM, et al. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem. 2004;279(33):34674–81. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J Biol Chem. 1974;249(22):7306–14. [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. 2012;13(4):213–24. doi: 10.1038/nrm3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari M, Eisenman LN, Covey DF, Mennerick S, Zorumski CF. The sticky issue of neurosteroids and GABAA receptors. Trends Neurosci. 2010;33(7):299–306. doi: 10.1016/j.tins.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi CL. Modulation of NMDA receptor function as a treatment for schizophrenia. Bioorg Med Chem Lett. 2013;23(18):5034–44. doi: 10.1016/j.bmcl.2013.07.019. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Bliss TVP. Memories of NMDA receptors and LTP. Trends in Neurosciences. 1995;18:54–56. [PubMed] [Google Scholar]
- Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, et al. The NMDA receptor as a target for cognitive enhancement. Neuropharmacology. 2013;64:13–26. doi: 10.1016/j.neuropharm.2012.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Dang EV, Reboldi A, Yi T. 25-hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol. 2014;14(11):731–43. doi: 10.1038/nri3755. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375–97. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Emnett CM, Eisenman LN, Mohan J, Taylor AA, Doherty JJ, Paul SM, et al. Interaction between positive allosteric modulators and trapping blockers of the NMDA receptor channel. Br J Pharmacol. 2014 doi: 10.1111/bph.13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emnett CM, Eisenman LN, Taylor AM, Izumi Y, Zorumski CF, Mennerick S. Indistinguishable synaptic pharmacodynamics of the N-methyl-D-aspartate receptor channel blockers memantine and ketamine. Mol Pharmacol. 2013;84(6):935–47. doi: 10.1124/mol.113.089334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famer D, Meaney S, Mousavi M, Nordberg A, Bjorkhem I, Crisby M. Regulation of alpha-and beta-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha-secretase pathway. Biochem Biophys Res Commun. 2007;359(1):46–50. doi: 10.1016/j.bbrc.2007.05.033. [DOI] [PubMed] [Google Scholar]
- Fantini J, Carlus D, Yahi N. The fusogenic tilted peptide (67–78) of alpha-synuclein is a cholesterol binding domain. Biochim Biophys Acta. 2011;1808(10):2343–51. doi: 10.1016/j.bbamem.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Ferrera P, Mercado-Gomez O, Silva-Aguilar M, Valverde M, Arias C. Cholesterol potentiates beta-amyloid-induced toxicity in human neuroblastoma cells: involvement of oxidative stress. Neurochem Res. 2008;33(8):1509–17. doi: 10.1007/s11064-008-9623-y. [DOI] [PubMed] [Google Scholar]
- Gamba P, Leonarduzzi G, Tamagno E, Guglielmotto M, Testa G, Sottero B, et al. Interaction between 24-hydroxycholesterol, oxidative stress, and amyloid-beta in amplifying neuronal damage in Alzheimer’s disease: three partners in crime. Aging Cell. 2011;10(3):403–17. doi: 10.1111/j.1474-9726.2011.00681.x. [DOI] [PubMed] [Google Scholar]
- Gamba P, Testa G, Sottero B, Gargiulo S, Poli G, Leonarduzzi G. The link between altered cholesterol metabolism and Alzheimer’s disease. Ann N Y Acad Sci. 2012;1259:54–64. doi: 10.1111/j.1749-6632.2012.06513.x. [DOI] [PubMed] [Google Scholar]
- Garcia AN, Muniz MT, Souza e Silva HR, da Silva HA, Athayde-Junior L. Cyp46 polymorphisms in Alzheimer’s disease: a review. J Mol Neurosci. 2009;39(3):342–5. doi: 10.1007/s12031-009-9227-2. [DOI] [PubMed] [Google Scholar]
- Halford RW, Russell DW. Reduction of cholesterol synthesis in the mouse brain does not affect amyloid formation in Alzheimer’s disease, but does extend lifespan. Proc Natl Acad Sci U S A. 2009;106(9):3502–6. doi: 10.1073/pnas.0813349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–96. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard M, Hansen KB, Andersen KT, Brauner-Osborne H, Traynelis SF. Molecular pharmacology of human NMDA receptors. Neurochem Int. 2012;61(4):601–9. doi: 10.1016/j.neuint.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogins J, Crawford DC, Jiang X, Mennerick S. Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol Dis. 2011;43(2):516–25. doi: 10.1016/j.nbd.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13(6):433–44. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- Hu H, Zhou Y, Leng T, Liu A, Wang Y, You X, et al. The major cholesterol metabolite cholestane-3beta,5alpha,6beta-triol functions as an endogenous neuroprotectant. Journal of Neuroscience. 2014;34(34):11426–38. doi: 10.1523/JNEUROSCI.0344-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TM, Rosano C, Evans RW, Kuller LH. Brain cholesterol metabolism, oxysterols, and dementia. J Alzheimers Dis. 2013;33(4):891–911. doi: 10.3233/JAD-2012-121585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Metaplastic effects of subanesthetic ketamine on CA1 hippocampal function. Neuropharmacology. 2014;86:273–81. doi: 10.1016/j.neuropharm.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandutsch AA, Chen HW. Inhibition of sterol synthesis in cultured mouse cells by cholesterol derivatives oxygenated in the side chain. J Biol Chem. 1974;249(19):6057–61. [PubMed] [Google Scholar]
- Kotti T, Head DD, McKenna CE, Russell DW. Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(32):11394–9. doi: 10.1073/pnas.0805556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(10):3869–74. doi: 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, et al. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20(9):2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Seet RC, Huang SH, Long LH, Halliwell B. Different patterns of oxidized lipid products in plasma and urine of dengue fever, stroke, and Parkinson’s disease patients: cautions in the use of biomarkers of oxidative stress. Antioxid Redox Signal. 2009;11(3):407–20. doi: 10.1089/ars.2008.2179. [DOI] [PubMed] [Google Scholar]
- Leoni V, Caccia C. Potential diagnostic applications of side chain oxysterols analysis in plasma and cerebrospinal fluid. Biochem Pharmacol. 2013a;86(1):26–36. doi: 10.1016/j.bcp.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Leoni V, Caccia C. Study of cholesterol metabolism in Huntington’s disease. Biochem Biophys Res Commun. 2014;446(3):697–701. doi: 10.1016/j.bbrc.2014.01.188. [DOI] [PubMed] [Google Scholar]
- Leoni V, Long JD, Mills JA, Di Donato S, Paulsen JS, group P-Hs Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol Dis. 2013b;55:37–43. doi: 10.1016/j.nbd.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni V, Mariotti C, Nanetti L, Salvatore E, Squitieri F, Bentivoglio AR, et al. Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci Lett. 2011;494(3):245–9. doi: 10.1016/j.neulet.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Leoni V, Solomon A, Lovgren-Sandblom A, Minthon L, Blennow K, Hansson O, et al. Diagnostic power of 24S-hydroxycholesterol in cerebrospinal fluid: candidate marker of brain health. J Alzheimers Dis. 2013c;36(4):739–47. doi: 10.3233/JAD-130035. [DOI] [PubMed] [Google Scholar]
- Linsenbardt AJ, Taylor A, Emnett CM, Doherty JJ, Krishnan K, Covey DF, et al. Different oxysterols have opposing actions at N-methyl-d-aspartate receptors. Neuropharmacology. 2014;85C:232–242. doi: 10.1016/j.neuropharm.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278(25):22980–8. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- Lutjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9799–804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutjohann D, Papassotiropoulos A, Bjorkhem I, Locatelli S, Bagli M, Oehring RD, et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41(2):195–8. [PubMed] [Google Scholar]
- Mast N, Charvet C, Pikuleva IA, Stout CD. Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem. 2010;285(41):31783–95. doi: 10.1074/jbc.M110.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney S. Epigenetic regulation of cholesterol homeostasis. Front Genet. 2014;5:311. doi: 10.3389/fgene.2014.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meljon A, Theofilopoulos S, Shackleton CH, Watson GL, Javitt NB, Knolker HJ, et al. Analysis of bioactive oxysterols in newborn mouse brain by LC/MS. J Lipid Res. 2012;53(11):2469–83. doi: 10.1194/jlr.D028233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meljon A, Wang Y, Griffiths WJ. Oxysterols in the brain of the cholesterol 24-hydroxylase knockout mouse. Biochem Biophys Res Commun. 2014;446(3):768–74. doi: 10.1016/j.bbrc.2014.01.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37(1):4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98(4):427–36. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Ito S, Matsuda A, Hori S, Abe T, Terasaki T. Brain-to-blood elimination of 24S-hydroxycholesterol from rat brain is mediated by organic anion transporting polypeptide 2 (oatp2) at the blood-brain barrier. Journal of Neurochemistry. 2007;103(4):1430–8. doi: 10.1111/j.1471-4159.2007.04901.x. [DOI] [PubMed] [Google Scholar]
- Okabe A, Urano Y, Itoh S, Suda N, Kotani R, Nishimura Y, et al. Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol. 2013;2:28–35. doi: 10.1016/j.redox.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen BN, Schlesinger PH, Ory DS, Baker NA. Side-chain oxysterols: from cells to membranes to molecules. Biochim Biophys Acta. 2012;1818(2):330–6. doi: 10.1016/j.bbamem.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory DS. Niemann-Pick type C: a disorder of cellular cholesterol trafficking. Biochim Biophys Acta. 2000;1529(1–3):331–9. doi: 10.1016/s1388-1981(00)00158-x. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Lutjohann D, Bagli M, Locatelli S, Jessen F, Buschfort R, et al. 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res. 2002;36(1):27–32. doi: 10.1016/s0022-3956(01)00050-4. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, Nicosia F, et al. Increased brain beta-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol. 2003;60(1):29–35. doi: 10.1001/archneur.60.1.29. [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, et al. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. Journal of Neuroscience. 2013;33(44):17290–300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res. 2011;50(4):357–71. doi: 10.1016/j.plipres.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Poli G, Biasi F, Leonarduzzi G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013;1(1):125–30. doi: 10.1016/j.redox.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med. 2010;2(56):56ra81. doi: 10.1126/scitranslmed.3001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol. 2008;507(5):1676–93. doi: 10.1002/cne.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin SE, Addona GH, Kloczewiak MA, Bugge B, Miller KW. The cholesterol dependence of activation and fast desensitization of the nicotinic acetylcholine receptor. Biophys J. 1997;73(5):2446–55. doi: 10.1016/S0006-3495(97)78273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. Journal of Neuroscience. 1984;4(7):1884–91. doi: 10.1523/JNEUROSCI.04-07-01884.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- Russell DW. Oxysterol biosynthetic enzymes. Biochim Biophys Acta. 2000;1529(1–3):126–35. doi: 10.1016/s1388-1981(00)00142-6. [DOI] [PubMed] [Google Scholar]
- Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem. 2009;78:1017–40. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaati M, Mast N, Beck O, Nayef R, Heo GY, Bjorkhem-Bergman L, et al. The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase in vitro and in vivo. J Lipid Res. 2010;51(2):318–23. doi: 10.1194/jlr.M900174-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. 2013;14(9):893–900. doi: 10.1038/ni.2681. [DOI] [PubMed] [Google Scholar]
- Sturley SL, Patterson MC, Pentchev P. Unraveling the sterol-trafficking defect in Niemann Pick C disease. Proc Natl Acad Sci U S A. 2009;106(7):2093–4. doi: 10.1073/pnas.0812934106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62(3):405–96. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano Y, Ochiai S, Noguchi N. Suppression of amyloid-beta production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013;27(10):4305–15. doi: 10.1096/fj.13-231456. [DOI] [PubMed] [Google Scholar]
- Valdez CM, Smith MA, Perry G, Phelix CF, Santamaria F. Cholesterol homeostasis markers are localized to mouse hippocampal pyramidal and granule layers. Hippocampus. 2010;20(8):902–5. doi: 10.1002/hipo.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza M, Leoni V, Tarditi A, Mariotti C, Bjorkhem I, Di Donato S, et al. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. 2007;28(1):133–42. doi: 10.1016/j.nbd.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, et al. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J Neurosci. 2005;25(43):9932–9. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA. Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13878–83. doi: 10.1073/pnas.172510899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser CR, Ertunc M, Liu X, Kavalali ET. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J Physiol. 2007;579(Pt 2):413–29. doi: 10.1113/jphysiol.2006.123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Saito Y, Yamamori T, Urano Y, Noguchi N. 24(S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J Biol Chem. 2011;286(28):24666–73. doi: 10.1074/jbc.M111.236273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Urano Y, Takabe W, Saito Y, Noguchi N. Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:cholesterol acyltransferase 1. Cell Death Dis. 2014;5:e990. doi: 10.1038/cddis.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri S, Mellon SH, Butters TD, Nevyjel M, Covey DF, Bembi B, et al. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J Cell Mol Med. 2009;13(9B):3786–96. doi: 10.1111/j.1582-4934.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JR, Coleman T, Langmade SJ, Scherrer DE, Lane L, Lanier MH, et al. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J Clin Invest. 2008;118(6):2281–90. doi: 10.1172/JCI32561. [DOI] [PMC free article] [PubMed] [Google Scholar]