

Abstract
Objectives:
To describe response to treatment in a patient with autoantibodies against voltage-gated calcium channels (VGCCs) who presented with autoimmune cerebellar degeneration and subsequently developed Lambert-Eaton myasthenic syndrome (LEMS), and to study the effect of the patient's autoantibodies on Purkinje cells in rat cerebellar slice cultures.
Methods:
Case report and study of rat cerebellar slice cultures incubated with patient VGCC autoantibodies.
Results:
A 53-year-old man developed progressive incoordination with ataxic speech. Laboratory evaluation revealed VGCC autoantibodies without other antineuronal autoantibodies. Whole-body PET scans 6 and 12 months after presentation detected no malignancy. The patient improved significantly with IV immunoglobulin G (IgG), prednisone, and mycophenolate mofetil, but worsened after IV IgG was halted secondary to aseptic meningitis. He subsequently developed weakness with electrodiagnostic evidence of LEMS. The patient's IgG bound to Purkinje cells in rat cerebellar slice cultures, followed by neuronal death. Reactivity of the patient's autoantibodies with VGCCs was confirmed by blocking studies with defined VGCC antibodies.
Conclusions:
Autoimmune cerebellar degeneration associated with VGCC autoantibodies may precede onset of LEMS and may improve with immunosuppressive treatment. Binding of anti-VGCC antibodies to Purkinje cells in cerebellar slice cultures may be followed by cell death. Patients with anti-VGCC autoantibodies may be at risk of irreversible neurologic injury over time, and treatment should be initiated early.
Autoimmune cerebellar degeneration (ACD) is characterized by progressive ataxia, dysarthria, dysphagia, and diplopia and, in adults, often indicates the presence of underlying malignancy. Sera and CSF of affected patients frequently contain antibodies directed against cerebellar Purkinje cells.1,2 These include anti-Hu antibodies in patients with small cell lung cancers, anti-Yo antibodies in patients with gynecological or breast cancers, and anti-Tr antibodies in patients with Hodgkin disease.1,2
Although antibodies to P/Q-type voltage-gated calcium channels (VGCCs) are most commonly associated with Lambert-Eaton myasthenic syndrome (LEMS), occasionally patients with VGCC autoantibodies may present with ACD, with or without accompanying LEMS.3–11 While LEMS may improve with immunosuppressive therapy, treatment of patients with ACD and VGCC autoantibodies has usually been unsuccessful.3,7,8
Herein, we present a patient with ACD and VGCC autoantibodies who initially improved with IV immunoglobulin G (IgG) and immunosuppression but who subsequently developed LEMS despite immunosuppressive therapy. The patient's IgG bound to Purkinje cell calcium channels in rat cerebellar slice cultures and produced Purkinje cell death.
CASE REPORT
A 53-year-old man presented with 4 months of progressive gait instability, falls, and difficulty performing fine motor tasks. Examination revealed dystaxic speech and severe appendicular ataxia but was otherwise normal.
Blood counts and erythrocyte sedimentation rate were normal. Antinuclear antibody was not detected. CSF showed 7 white blood cells/mm3 and 62.1 mg/dL protein. Oligoclonal bands were absent. Extensive serum and CSF evaluations for infectious, toxic, and metabolic causes were negative. Serum contained antibodies against P/Q-type VGCCs (483.5 pmol/L; normal range 0.0–24.5 pmol/L) (ARUP Laboratories, Salt Lake City, UT). Other antineuronal autoantibodies were not detected. Contrasted brain MRI was unremarkable at presentation and 3 months later. Contrasted CT of the chest, abdomen, and pelvis were without evidence of malignancy as were PET scans at 6 and 12 months after onset of ataxia.
The patient was treated with IV IgG 2 g/kg over 3 days followed by methylprednisolone 1 g IV daily for 3 days, an oral prednisone taper beginning at 60 mg daily, and mycophenolate mofetil 500 mg twice daily. Speech, coordination, and ambulation initially improved significantly. A repeat trial of IV IgG, however, was discontinued after he developed aseptic meningitis. The patient's ataxia and dysarthria subsequently worsened despite plasma exchange and an increase in mycophenolate dosage to 750 mg twice daily. Initially his strength was normal, but 11 months later he developed progressive weakness. Electrophysiologic studies at that time demonstrated low amplitude and unexpectedly variable motor response compound motor action potentials in several nerve distributions. With 10 seconds of voluntary exercise, there was 50% to 300% facilitation in amplitude and area under the waveform curve. The findings were thought to represent evidence of a correctable presynaptic defect in neuromuscular junction transmission consistent with LEMS. Treatment with 3,4-diaminopyridine resulted in improvement of dysphagia but not coordination. The patient declined further therapy and succumbed 18 months after onset of his ataxia.
METHODS
The patient's pretreatment serum was confirmed by ARUP Laboratories to have VGCC autoantibodies, without other paraneoplastic or other antineuronal autoantibodies. Organotypic cultures of cerebellums from 24-day-old Sprague-Dawley rat pups were incubated with 1:400 dilutions of patient sera as previously described.12 At intervals from 24 to 144 hours, cultures were coincubated for 2 hours with SYTOX cell viability dyes, fixed with 2% paraformaldehyde, labeled postfixation with Cy5-conjugated goat anti-human IgG, and analyzed by confocal microscopy.12 Controls were sera from neurologically normal individuals without malignancy. To confirm that the reactivity of the patient's IgG for Purkinje cells represented specific binding to VGCCs, normal adult rat tissue was fixed with paraformaldehyde, placed in paraffin blocks, and sectioned onto slides. The normal tissue was treated with an antigen retrieval system before blocking for 1 hour with serial concentrations of commercially obtained rabbit anti-VGCC antibodies directed against the α1A subunit of the VGCC (Thermo PA5-19598, Rockford, IL), and then was subsequently exposed to patient sera (1:200) for 24 hours. Sections were fixed and labeled with Cy5-conjugated donkey anti-human IgG to detect the patient's antibodies and fluorescein isothiocyanate–conjugated anti-rabbit IgG to detect the commercial anti-VGCC antibody.
Standard protocol approvals, registrations, and patient consents.
The patient's pretreatment serum was obtained under appropriate institutional review board guidelines.
RESULTS
Cerebellar slices exposed to the patient's serum for 48 hours showed presence of antibody predominantly within the neuropil, with labeling of occasional Purkinje cells (figure 1). By 72 hours, Purkinje cells exhibited surface binding of antibody, and by 144 hours, Purkinje cells also showed cytoplasmic labeling, indicating IgG uptake (figure 1; videos 1 and 2 at Neurology.org/nn). Most antibody-labeled Purkinje cells excluded SYTOX dyes, indicating that these cells were viable (figure 1). At 144 hours, however, scattered Purkinje cells showed staining with SYTOX dyes, indicative of cell death (figures 1 and 2). Pretreatment of rat tissue with commercial anti-VGCC antibody inhibited binding of the patient's IgG to Purkinje cells in a concentration-dependent manner, confirming the specificity of the patient's antibody for VGCCs (figure e-1). Immunohistologic evaluation of the patient's serum using composite frozen sections did not detect any other characterized neuron-specific antibody patterns (data not shown).
Figure 1. VGCC antibody uptake in cerebellar slice culture.

Slice culture of rat cerebellum incubated with patient serum containing VGCC autoantibodies and examined at different points in time. (A) Slice culture incubated with normal serum for 144 hours, treated with SYTOX dyes to detect cell death, and labeled with Cy5-conjugated anti-human IgG, showing absence both of neuronal antibody binding and of SYTOX staining indicative of cell death. (B, C) Slice cultures incubated for 48 and 72 hours with serum containing VGCC autoantibodies. In these images, binding of the patient's IgG is labeled red; rare dead cells outside the Purkinje cell layer are labeled green. At 48 hours, IgG can be detected throughout the neuropil with labeling of occasional Purkinje cells (arrows) (B). By 72 hours, there is extensive binding of IgG to Purkinje cell membranes (arrows) (C). (D–F) A slice culture incubated with patient serum for 144 hours and labeled with Cy5-conjugated anti-human IgG (D), stained with SYTOX dyes to detect cell death (E), and as a merged image (F). In cultures at this time point, some Purkinje cells continued to show surface binding of antibody panels (G–I). In other areas, however, Purkinje cells showed uptake of IgG with cytoplasmic and nuclear labeling (D); some IgG-containing Purkinje cells demonstrated staining with SYTOX dyes, indicating cell membrane disruption and death (E, F), whereas others excluded SYTOX dyes, indicating antibody uptake by viable Purkinje cells. Purkinje cell antibody binding and cell death at 144 hours are shown at higher power in panels G through I. Arrows indicate the same cells in panels D–F and in G–I. Scale bars = 20 μm. IgG = immunoglobulin G; VGCC = voltage-gated calcium channel.
Figure 2. Binding of the patient's IgG to Purkinje cells is inhibited by pretreatment of rat tissue with anti-VGCC antibodies, confirming specificity of the patient's antibody response.

Adult rat tissue was fixed, sectioned, and treated with an antigen retrieval system before blocking for 1 hour with serial concentrations of commercially obtained rabbit anti-VGCC antibodies directed against the α1A subunit of the VGCC receptor before being exposed to a 1:200 dilution of patient serum for 24 hours. Tissue was then labeled with Cy5-conjugated donkey anti-human IgG to detect the patient's antibodies and fluorescein isothiocyanate–conjugated anti-rabbit IgG to detect the commercial anti-VGCC antibody. Pretreatment with commercial anti-VGCC antibody inhibited binding of the patient's IgG to Purkinje cells in a dose-dependent manner, confirming the specificity of the patient's antibody for the VGCC. Scale bars = 20 μm. IgG = immunoglobulin G; VGCC = voltage-gated calcium channel.
DISCUSSION
Although VGCC autoantibodies are usually associated with LEMS, they have also been detected in 21% to 41% of patients with paraneoplastic cerebellar degeneration associated with lung cancer6,7 and in 11% of patients with cerebellar degeneration in the absence of malignancy.10 The present case provides further evidence that adult patients presenting with subacute cerebellar ataxia should be tested for VGCC autoantibodies.
Although VGCC autoantibodies have been shown to be pathogenic in LEMS, their role in paraneoplastic cerebellar injury is less well defined. Other investigators have demonstrated that a single cisternal injection of mice with IgG from a VGCC-positive patient with LEMS and cerebellar degeneration produced temporary motor ataxia, whereas injection of IgG from a patient with LEMS alone produced no cerebellar impairment.13 These authors postulated that VGCC autoantibodies can cause cerebellar dysfunction as well as LEMS, but that anti-VGCC antibodies in patients with LEMS and ACD may react against a slightly different epitope than do those in patients with LEMS alone.13 These investigators did not detect histologic evidence of neuronal death in brains examined after 24 hours or 15 days after this single infusion. In contrast, our studies, using more prolonged exposure of cerebellar neurons to patient anti-VGCC serum, demonstrated surface labeling of Purkinje cells followed by antibody uptake and scattered Purkinje cell death. It is possible that Purkinje cell death might have been due to another unidentified antibody in the patient's serum, directed toward an unknown antigen of Purkinje cells, in addition to VGCCs. However, adsorption of the patient's serum against the α1A subunit of the VGCC antibodies effectively abolished all IgG labeling by our anti-human secondary antibody, making the presence of an additional autoantibody less likely. Although the possibility also exists that Purkinje cell death might have been due to some other nonantibody factor present in our patient's serum, our prior studies using other patient paraneoplastic antisera have demonstrated that neuronal death is specifically dependent on anti-neuronal IgG and not on other serum components.14,15 Our data, together with those of the investigators discussed above suggest that anti-VGCC antibodies may initially cause functional impairment of Purkinje cells, but that longer exposure to VGCC autoantibodies may result in irreversible Purkinje cell injury and death.
Although autoimmune neurologic disorders associated with cell-surface antibodies frequently respond to immunosuppressive treatment, the condition of patients with anti-VGCC antibodies and ACD has often failed to improve, even when there was improvement in accompanying LEMS (table).3,5,7,8 Our patient's ataxia improved during IgG therapy but subsequently worsened after IV IgG could no longer be administered. Another group described a similar case of ACD with VGCC antibodies, without an underlying malignancy; this patient also improved significantly following treatment with IV IgG.11 In both cases, IV IgG was administered relatively early in the course of disease, suggesting that prompt intervention may be essential and that early administration of IV IgG may be an effective means of treatment.
Table.
Previously reported cases of ACD in patients with VGCC autoantibodies, showing initial presentation, presence or absence of LEMS and/or malignancy, treatment used, and treatment response
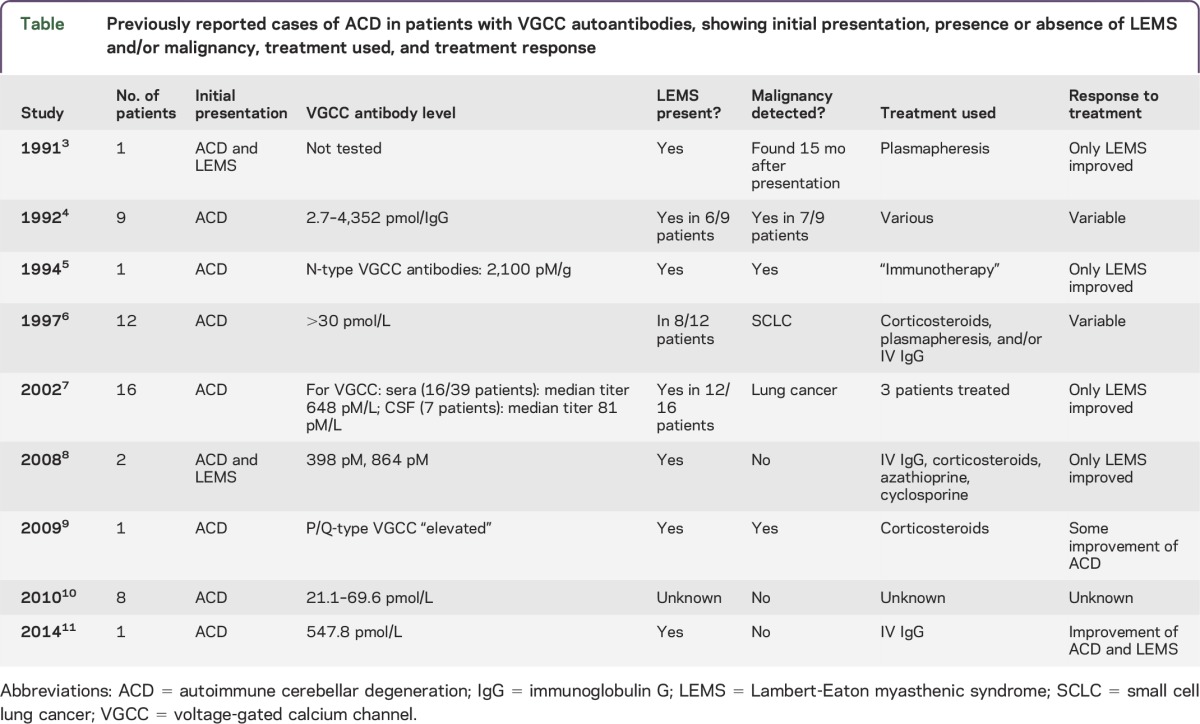
Paraneoplastic neurologic symptoms may antedate tumor diagnosis by as long as 5 years in as many as 70% of patients.1,2 For this reason, follow-up screening for malignancy is essential in these patients if initial studies are negative. As many as 6% to 20% of patients with VGCC autoantibodies and ACD may develop LEMS.6,7 Because manifestations of LEMS may be masked by severe cerebellar symptoms, electrophysiologic testing should be considered if there is any question of motor weakness.
VGCC autoantibodies may be associated with ACD as well as LEMS, and patients presenting with cerebellar signs and symptoms should be tested for VGCC autoantibodies as part of a comprehensive evaluation for neuronal antibodies, including the use of screening immunofluorescence to detect potentially unclassified antibodies. Although early treatment of ACD in patients with VGCC autoantibodies may result in clinical improvement, VGCC autoantibodies may cause Purkinje cell death over time. Patients with ACD and anti-VGCC antibodies should undergo electrophysiologic evaluation for LEMS, even if LEMS is not clinically evident, and should also undergo malignancy screening, with subsequent repeat evaluation if initial studies do not detect neoplasia.
Supplementary Material
GLOSSARY
- ACD
autoimmune cerebellar degeneration
- IgG
immunoglobulin G
- LEMS
Lambert-Eaton myasthenic syndrome
- VGCC
voltage-gated calcium channel
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Marilyn McKasson: data collection, analysis, writing or editing assistance. Stacey L. Clardy: data collection, analysis, writing or editing assistance, review of manuscript. Susan A. Clawson: data collection, analysis. Kenneth E. Hill: data collection, analysis. Blair Wood: data collection, analysis. Noel Carlson: writing or editing assistance, review of manuscript. Mark Bromberg: writing or editing assistance, review of manuscript. John E. Greenlee: data collection, analysis, writing or editing assistance, review of manuscript.
STUDY FUNDING
No targeted funding.
DISCLOSURE
M. McKasson is employed by Josephson Wallack Munshower Neurology, PC. S.L. Clardy received research support from Western Institute for Biomedical Research. S. Clawson, K.E. Hill, and B. Wood report no disclosures. N. Carlson received research support from Biogen, AstraZeneca, Department of Veterans Affairs Merit Review. M. Bromberg serves on the scientific advisory board for Accordant Health Care, Baxter Bioscience, received travel funding and speaker honoraria from Walgreens Infusion, Grifols Biotherapeutics, Baxter Bioscience, is an assistant editor for Muscle & Nerve, Clinical Neurophysiology, Journal of Clinical Neuromuscular Disease, receives royalties from Taylor & Francis, Cambridge, is on the speakers bureau for Walgreens Infusion, Grifols Biotherapeutics, Baxter Bioscience, Genzyme. J.E. Greenlee is a senior associate editor for MedLink, an associate editor for Clinical Neurology and Neurosurgery, receives publishing royalties from MedLink, receives compensation for editing and reviewing chapters for MedLink and the Merck Manual, received research support from United States Department of Veterans Affairs. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008;7:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenlee JE. Treatment of paraneoplastic neurologic disorders. Curr Treat Options Neurol 2010;12:212–230. [DOI] [PubMed] [Google Scholar]
- 3.Blumenfeld AM, Recht LD, Chad DA, DeGirolami U, Griffin T, Jaeckle KA. Coexistence of Lambert-Eaton myasthenic syndrome and subacute cerebellar degeneration: differential effects of treatment. Neurology 1991;41:1682–1685. [DOI] [PubMed] [Google Scholar]
- 4.Clouston PD, Saper CB, Arbizu T, et al. Paraneoplastic cerebellar degeneration: III: cerebellar degeneration, cancer, and the Lambert-Eaton myasthenic syndrome. Neurology 1992;42:1944–1950. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JM, Waxman SG, Vollmer TL, Lang B, Johnston I, Newsom-Davis J. Subacute cerebellar degeneration and Lambert-Eaton myasthenic syndrome associated with antibodies to voltage-gated calcium channels: differential effect of immunosuppressive therapy on central and peripheral defects. J Neurol Neurosurg Psychiatry 1994;57:1138–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mason WP, Graus F, Lang B, et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain 1997;120:1279–1300. [DOI] [PubMed] [Google Scholar]
- 7.Graus F, Lang B, Pozo-Rosich P, Saiz A, Casamitjana R, Vincent A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002;59:764–766. [DOI] [PubMed] [Google Scholar]
- 8.Lorenzoni PJ, Scola RH, Lang B, et al. Cerebellar ataxia in non-paraneoplastic Lambert-Eaton myasthenic syndrome. J Neurol Sci 2008;270:194–196. [DOI] [PubMed] [Google Scholar]
- 9.Iwanami M, Odaka M, Nakamura T, Hirata K. Paraneoplastic cerebellar degeneration and Lambert-Eaton myasthenic syndrome associated with anti P/Q-type voltage-gated calcium channel antibody in a patient with primary double lung cancer [in Japanese]. Brain Nerve 2009;61:1083–1087. [PubMed] [Google Scholar]
- 10.Burk K, Wick M, Roth G, Decker P, Voltz R. Antineuronal antibodies in sporadic late-onset cerebellar ataxia. J Neurol 2010;257:59–62. [DOI] [PubMed] [Google Scholar]
- 11.Rigamonti A, Lauria G, Stanzani L, Mantero V, Andreetta F, Salmaggi A. Non-paraneoplastic voltage-gated calcium channels antibody-mediated cerebellar ataxia responsive to IVIG treatment. J Neurol Sci 2014;336:169–170. [DOI] [PubMed] [Google Scholar]
- 12.Greenlee JE, Clawson SA, Hill KE, Wood BL, Tsunoda I, Carlson NG. Purkinje cell death after uptake of anti-Yo antibodies in cerebellar slice cultures. J Neuropathol Exp Neurol 2010;69:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Garcia E, Mannara F, Gutierrez-Cuesta J, et al. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J Neuroimmunol 2013;261:53–59. [DOI] [PubMed] [Google Scholar]
- 14.Greenlee JE, Clawson SA, Hill KE, et al. Anti-Yo antibody uptake and interaction with its intracellular target antigen causes Purkinje cell death in rat cerebellar slice cultures: a possible mechanism for paraneoplastic cerebellar degeneration in humans with gynecological or breast cancers. PLoS One 2015;10:e0123446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenlee JE, Clawson SA, Hill KE, et al. Neuronal uptake of anti-Hu antibody, but not anti-Ri antibody, leads to cell death in brain slice cultures. J Neuroinflammation 2014;11:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.