

Abstract
For over two decades, research groups have been developing methods to engineer three-dimensional skeletal muscle tissues. These tissues hold great promise for use in disease modeling and pre-clinical drug development, and have potential to serve as therapeutic grafts for functional muscle repair. Recent advances in the field have resulted in the engineering of regenerative muscle constructs capable of survival, vascularization, and functional maturation in vivo as well as the first-time creation of functional human engineered muscles for screening of therapeutics in vitro. In this review, we will discuss the methodologies that have progressed work in the muscle tissue engineering field to its current state. The emphasis will be placed on the existing procedures to generate myogenic cell sources and form highly functional muscle tissues in vitro, techniques to monitor and evaluate muscle maturation and performance in vitro and in vivo, and surgical strategies to both create diseased environments and ensure implant survival and rapid integration into the host. Finally, we will suggest the most promising methodologies that will enable continued progress in the field.
Keywords: Tissue engineering, skeletal muscle, satellite cells, animal implantation
1. Introduction
Skeletal muscle is a highly complex organ with robust contractile and regenerative capacity contributed to a variety of cell types and intra- and extracellular mechanisms. Muscle contractions, the source of voluntary movement of our body, are produced by myofibers; aligned, multi-nucleated cells with registered sarcomeric structures powering force generation. The myofibers are embedded in a specific extracellular matrix (ECM), metabolically supported by a dense capillary network, and temporally coordinated by specialized neuronal inputs. A unique feature of the muscle tissue is its ability to regenerate in response to acute injuries over the course of a lifetime [1, 2]. Responsible for this feature are the resident muscle stem cells, or satellite cells (SCs). Upon injury, SCs will activate, proliferate, and differentiate to form new, or repair damaged, muscle. After completion of the repair process, SCs will reestablish their numbers and quiescent state by homing back to highly specialized niches, thus allowing future rounds of regeneration [1].
Although a robust trait in healthy conditions, the self-repair capacity of skeletal muscle can be seriously impaired in different types of congenital myopathies or large muscle loss following trauma [3, 4]. Furthermore, myofiber atrophy and reduced functionality and regenerative capacity are inevitable consequences of aging [5]. Over the years, extensive research on skeletal muscle biology and regeneration has been carried out to develop cellular and genetic therapies aimed to recover or rejuvenate damaged, diseased, or aged muscle. One solution that combines engineering and biological concepts is the construction of whole muscle tissues in vitro or in vivo. The ability to recreate the cellular complexity, function, and regenerative properties of muscle offer potential for on-site tissue reconstruction as well as the development of physiologically accurate in vitro models of human muscle disorders.
Over the last 25 years, researchers in the field have developed a variety of methods for engineering of functional three-dimensional (3D) skeletal muscle tissues. In particular, recent reports have demonstrated construction of rodent muscle tissues that recreate the functional [6-8] and regenerative [6] properties of native muscle in vitro and survive and contribute to contractile muscle function in vivo [9-12]. Most recently, Madden et al., reported engineering of the first functional human muscle tissues able to contract in response to electrical and chemical stimuli [13]. Here we will review procedures used by multiple groups in the field of skeletal muscle tissue engineering that involve generation of a myogenic cell pool, fabrication of 3D tissue constructs, structural and functional characterization in vitro, and implantation of tissue grafts in vivo.
2. Preparation of myogenic cell source
In vitro generation of tissues for use as therapeutic implants or screening models requires a large numbers of cells. Considering the myonuclei within myofibers are post-mitotic, the only suitable source of expandable primary myogenic cells are the satellite cells (SCs), a putative muscle stem cell source capable of myogenesis [1]. Myogenesis, the process of forming new muscle, begins with activation of the quiescent SCs, marked by transcription factor Pax7, and the ensuing expression of myogenic regulatory factor MyoD. Activated SCs will proliferate and either commit to a myogenic fate yielding a population of precursor cells termed myoblasts or self-renew and return to the quiescent state. Myoblasts will continue to proliferate and later differentiate, losing Pax7 and gaining expression of myogenin, a mature muscle marker found in myonuclei. At this point, the cells will either undergo primary fusion, in which they will generate new multi-nucleated myofibers, or secondary fusion, in which they will fuse to existing, damaged ones [1]. Unfortunately, the percentage of SCs in adult muscle is relatively low, amounting to roughly 4% of the total muscle nuclei [14]. Therefore, the common paradigm in muscle tissue engineering is to isolate the resident muscle stem cells, stimulate their continuous proliferation while maintaining their purity and differentiation potential, and utilize them in engineered tissue formation. In this section we will review various methods to generate large quantities of myogenic cells for use in skeletal muscle tissue engineering (reviewed in Figure 1).
Fig 1. Methods to isolate and expand myogenic cells.
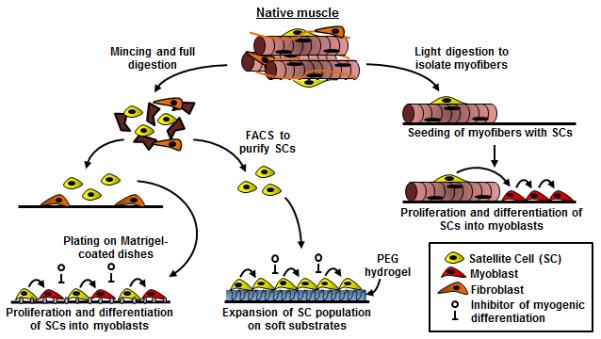
Starting from whole native muscle, the tissue can be lightly enzymatically digested (right) to isolate and plate intact myofibers from which satellite cells (SCs) will outgrow, proliferate, and differentiate into myoblasts. Alternatively (left), native muscle can be fully digested to yield a suspension of mononuclear cells. These cells can be FACS sorted for extracellular markers or preplated to purify the SC population. The purified SCs can be either plated on Matrigel-coated dishes or on a soft, laminin-coated hydrogel substrate. Supplementation of culture media with differentiation inhibitors prevents cell fusion and allows SC expansion with preserved regenerative capacity.
2.1. Satellite cell isolation
Isolation of SCs from skeletal muscle has been achieved via two main routes, cellular outgrowth and whole tissue digestion. The outgrowth method involves gentle separation of myofibers from the extracellular matrix, physical trituration to separate individual muscle fibers, and subsequent seeding of the fibers onto protein-coated dishes. This method relies on maintaining SCs within their niche on the myofiber periphery and their activation following plating. SCs will initiate the process of myogenesis and ‘outgrow’ from the myofiber onto the dish. Depending on the micro-environment, the cells will continue to proliferate and commit down the myogenic pathway, yielding a population of myoblasts capable of generating myofibers [15]. Alternatively, whole muscle tissue can be extensively minced and fully digested to release the SCs from their home on the myofiber [16]. The digested solution can be strained to filter out the mixture of fragmented myofibers, resulting in a single cell suspension that can be plated and expanded.
A number of methods may be used to purify the myogenic, and specifically SC, population following extraction. Pre-plating is a simple strategy used to remove the population of fast-adhering cells, primarily fibroblasts, from the culture. Myogenic cells are re-suspended in a growth media following digestion and plated on non-coated tissue culture plastic; a surface in which fibroblasts, but not SCs, will attach [17]. After roughly 2 hours, the supernatant, consisting of a purified cell mixture, can be transferred to a different flask to encourage myogenic cell attachment [6, 7]. Myogenic cells with a higher SC fraction can be obtained using fluorescence-activated cell sorting (FACS) with antibodies against the extracellular domains of SC-specific membrane proteins [18], including SM/C-2.6 [19, 20], α7-integrin [21, 22], β1-integrin [21], CD34 [22, 23], CXC chemokine receptor 4 (CXCR4) [21], neural cell adhesion molecule (NCAM/CD56) [24], Syndecan-3 and -4 [25], and vascular cell adhesion protein 1 (VCAM-1) [26]. For further purification, negative markers have also been identified to remove non-myogenic cells, including CD31 [19, 20, 22], CD45 [21-23], CD11b [22], Sca-1 [19-21, 23], and Mac-1 [21]. Multiple antibodies can be used to sort for a distinct combination of positive and negative markers to ensure a proliferative muscle stem cell pool. For example, the population of CD45−/CD31−/CD11b−/Sca1−/CD34+/integrin-α7+ cells from mouse muscle purified through two rounds of FACS was shown to contain 100% pure Pax7+/Myf5+ SCs that were capable of self-renewal and expansion following transplantation [22]. Similarly, in a recent report [27], Pax7+ SCs were enriched from human fetal and adult muscle isolates based on positive and negative expression of the aforementioned marker proteins.
2.2. Myoblast cell culture
As commonly acknowledged [22, 28], traditional passaging of isolated SCs results in their rapid differentiation towards a myoblast phenotype. Myoblasts, typically expressing transcription factor MyoD, are at a proliferating state and, under particular environmental stimuli, can be induced to fuse and form multi-nucleated myotubes [1]. For tissue engineering purpose, these myogenic cells are maintained at a proliferative state in vitro in order to expand cell numbers prior to differentiation, at which point they become post-mitotic. Critical for the sustained growth of myoblasts are both the growth surfaces and media constituents used during culture.
Collagen I coated plates were among the first 2D culture substrates identified to be supportive of both myogenic cell growth [29] and robust differentiation into cross-striated, contractile myotubes [30]. Recent efforts have identified Matrigel as a superior candidate for myogenic attachment and expansion [7, 31]. Matrigel is a commercially-available protein cocktail primarily consisting of laminin and collagen IV; the two main constituents of muscle basal lamina matrix [32]. Compared to collagen I, muscle-derived cells cultured on Matrigel-coated flasks experienced enhanced proliferation, expression of myogenic markers, and fusion capacity [31]. Laminin, alone, promotes myoblast growth as well, and stimulates cell locomotion [33]. To be discussed later, these results are likely attributable to outside-in integrin signaling due to binding between myoblasts and muscle-specific proteins.
In addition to protein-coated culture substrates, two media formulations are traditionally used for growth and differentiation of skeletal myoblasts. Generally, growth media contains high serum levels, typically 10-20% fetal bovine or calf serum, to stimulate cell proliferation by high doses of growth factors [34]. Identified as an inhibitor of myogenic differentiation, basic fibroblast growth factor (bFGF) is commonly used as a supplement to further inspire proliferation and prevent fusion events [29]. Likewise, epidermal growth factor (EGF) [35] and isoforms of platelet-derived growth factor [36] can also be supplemented to culture media to enhance myoblast growth. Sub-confluent cell culture (60-70% before cell passage) is another requisite condition to support cell proliferation and prevent spontaneous cell fusion events during expansion [37, 38]. Differentiation media is used to drive fusion of myoblasts to myotubes, which can be robustly achieved through use of low serum (e.g. 2% horse serum [29, 39]) or serum-free [40-42] media. In general, in high serum growth medium, myoblasts will express the inhibitor of DNA-binding/differentiation protein (Id), which prevents MyoD activation of downstream genes to trigger further myogenic differentiation [43]. Switching to low serum differentiation medium, significantly decreases Id expression and drives myoblast fusion into myotubes [44]. Additionally, specific growth factors, e.g. insulin-like growth factor 1 (IGF-1) or transforming growth factor-beta 1 (TGFβ-1) [17, 45], can be used to accelerate myotube formation.
2.3. Maintaining regenerative capacity in culture
Maintenance of the SC niche within native muscle involves both physical and biochemical interactions with the abutting myofiber membrane and the basal lamina that collectively support the capacity of SCs to self-renew and repair damaged muscle [28]. Due to the established contribution of satellite cells (SCs) to natural regeneration and muscle growth, a significant body of work over the last two decades has been devoted to developing methods for expansion of these cells for use in regenerative therapy. Supporting the rationale for this effort have been the studies that show the ability of SCs with maintained regenerative capacity (Pax7+) to robustly engraft and form new muscle following transplantation [22, 23]; a feat not attained by cultured myoblasts. However, when removed from their natural environment, in vitro cultured SCs differentiate into myoblasts and lose their capacity to self-renew as early as few days of culture [22, 23, 46] or a single passage [47]. Strategies to retain the stem cell nature (“stemness”) of a SC population outside the body have thus focused around the recreation of the natural niche in vitro.
In particular, recent work has identified culture conditions partially supportive of in vitro expansion of SCs with maintained regenerative potential. Specifically, the use of soft culture substrates (e.g. polyethylene glycol (PEG) hydrogels) with stiffness mimicking that of natural skeletal muscle (~12 kPa) has shown promise in the expansion of mouse SCs [46]. When cultured on such hydrogels functionalized with laminin to encourage cell adhesion, SCs showed enhanced proliferation, self-renewal, and maintenance of Pax7 expression compared to rigid surfaces and those mimicking the stiffness of non-muscle tissues [46]. After a week of in vitro culture and expansion, the cells were able to fill the SC niche following intramuscular transplantation and contribute to continual rounds of regeneration. Additionally, collagen VI has recently been identified as a coating protein that provides niche-like local mechanical properties leading to enhanced proliferation and Pax7 expression of SCs [48].
Supplementation of biochemical factors can also simulate SC niche-like environment in vitro. Recently, SCs from aged donors have been shown to upregulate p38α/β mitogen-activated protein kinase activity, typically induced by stress-related pathways. Supplementation of an inhibitor of p38α/β, SB202190, in conjunction with the expansion on a laminin-coated PEG hydrogels, resulted in enhanced SC proliferation and Pax7 expression in vitro and improved regeneration of injured muscle in vivo [49]. Additionally, high-throughput screening in zebrafish was recently used to identify forskolin, an adenylyl cyclase activator, and BIO, a GSK3β inhibitor, as small molecule enhancers of myogenesis. Forskolin was further found to promote expansion of mouse SCs in vitro, while the combination of the two chemicals augmented muscle differentiation of human induced pluripotent stem cells (iPSCs) [50]. Remarkably, Fu et al. [47] have recently identified four cytokines present in pro-myogenic, T cell-conditioned media (IL-1α, IL-13, TNF-α, IFN-γ) that when supplemented during expansion of mouse SCs supported their functional phenotype (>80% Pax7+ cells) for up to 22 passages. Following implantation, the expanded SCs displayed similar engraftment potential as freshly isolated cells and successfully integrated into the endogenous SC pool to aid muscle repair following secondary injury [47]. To our knowledge, this is the largest in vitro amplification of mouse SCs (1×1018 fold) with maintained “stemness”. It remains to be studied if a similar cytokine cocktail can be identified for use with humans SCs.
3. Engineering functional muscle constructs
Formation of in vitro skeletal muscle tissues requires the exposure of myogenic cells to a 3D environment conducive to their growth, differentiation, and function [51]. Concurrent with advances in the field of tissue engineering, scaffolds and techniques for generating muscle constructs have evolved over the years as groups have aimed to optimize cell maturation, tissue structure, contractile function, in vivo survival, and regenerative ability [51]. In this section we will discuss various methods of generating and culturing 3D constructs to optimize their structural organization and functional performance.
3.1. Generating 3D muscle
Engineering a biomimetic skeletal muscle tissue in vitro generally refers to the recreation of highly aligned and contractile myofibers within a 3D environment. Traditionally, there have been two approaches to develop such constructs. The first relies on the ‘self-assembly’ of differentiated myogenic cells into anchored cylindrical tissue bundles or free-floating planar tissue sheets as the cell monolayer is detached from a 2D surface. The second requires the initial or continuous presence of a supporting 3D matrix, where undifferentiated myogenic cells are either seeded into a polymer scaffold or encapsulated within a hydrogel and later induced to fuse and differentiate into myofibers. Both methodologies are still in use.
Self-assembly of in vitro muscle was first introduced by Strohman and colleagues in 1990 [52]. This method typically requires preparation of a protein-coated polydimethylsiloxane (PDMS) surface within a cell culture dish [8, 9, 53, 54]. The PDMS allows for the pinning of ‘anchors’ (e.g. silk sutures [8, 53, 54]), which act to secure the ends of the construct and provide passive tension to induce cellular alignment. To create more biomimetic systems, spinal cord explants [55], tendon structures [56], and engineered bone anchors [10] have also been used in attempt to establish neuromuscular or myotendinous junctions. Once the dish is prepared, a mixture of myogenic precursors (usually myoblast) and fibroblasts are seeded in a high serum growth medium to stimulate cell proliferation. The cells are grown into a confluent layer (4-7 days) and the media is switched to a low-serum differentiation medium to initiate myoblast fusion. After a few days, the dish is comprised of a population of contracting, multi-nucleated myotubes and fibroblasts that will compact the cell layer inward resulting in gradual detachment from the dish. Once completely separated from the surface, the cellular composite will assemble around the anchors in the dish to create a cylindrical 3D tissue structure. This procedure can be further shortened by coating a hydrogel layer (e.g. fibrin) atop the PDMS surface to facilitate cell detachment [8]. Alternatively, myogenic cell monolayers can be detached via the use of thermo-responsive polymer coatings [57], however, unlike the anchored cylindrical tissues, the resultant free-floating cell sheets are not amenable to physiological testing.
Encapsulating cells in 3D scaffolds is the most commonly utilized method for skeletal muscle tissue engineering. Here, the primary (freshly isolated [6, 7, 58-60] or expanded [13, 61, 62]) or immortalized myogenic cells [63-65] are embedded into 3D synthetic polymer scaffolds [65] or natural hydrogels (fibrin, collagen I) [58-63, 66]. For hydrogels, the cell/gel mixture is cast into a silicone tubing or PDMS mold coated in a pluronic solution to prevent cell adhesion. Cylindrical molds are typically used to generate a simple, cable-like tissue geometry, with porous felts at the ends of the mold that serve to anchor the hydrogel and provide passive tension for guiding unidirectional cell alignment [61]. For more complex planar tissue structures, mesoscopic hydrogel molding methods, in which soft lithography techniques are applied to precisely manufacture sub-millimeter PDMS posts [67], can be used to vary the local alignment of cells across a large tissue area [59, 66]. Once the cell/gel mixture is molded, the hydrogel is polymerized and the construct is placed in growth medium for a few days to increase cell number and density. During this period, fibroblasts present in the primary cell population will typically migrate to the periphery of the construct. Cell-mediated gel compaction will result in detachment of the construct from the mold and further constriction along the free tissue boundaries [7]. Switching to differentiation medium will induce rapid fusion of myoblasts into myofibers followed by progressive structural and functional maturation. In general, under optimized conditions, the 3D encapsulation of myogenic cells in hydrogels has yielded engineered muscle constructs with higher total and specific force generation [6-8, 51] and more pronounced sarcomeric structures [6, 7] compared to use of scaffold-less approaches based on the 2D cell self-assembly. This might be attributed to enhanced myogenic fusion and larger myofiber size resulting from higher density of cell-cell contacts in 3D compared to 2D culture environment.
Due to a number of advantages, naturally derived hydrogels, fibrin and collagen I are the most commonly used biomaterials for the engineering of functional muscle [58]. In particular, they: 1) support uniform 3D cell seeding, 2) contain plentiful cell attachment sites that support cell-mediated gel compaction and spreading, leading to the formation of highly dense and aligned tissues, and 3) have appropriate mechanical compliance to withstand (but not impede) forceful muscle contractions [66]. Collagen I gels are formed through thermally induced polymerization following pH neutralization (i.e. addition of NaOH), while fibrin gels are formed through the cross-linking of fibrinogen and thrombin, with both gel types typically being supplemented with Matrigel [32] for muscle engineering applications. Recently, in a direct comparison, engineered skeletal muscle tissues exhibited higher contractile forces when made using fibrin vs. collagen I gel [58]. This was attributed to stronger binding of integrins expressed by mature myotubes (α7, αV) to fibrin [68, 69] than collagen I (e.g. a collagen I-specific integrin α2 is not expressed in muscle [68]) which likely resulted in better transmission of contractile force between myofibers and the surrounding ECM. Furthermore, increased concentrations of Matrigel and its associated basal lamina proteins, additionally enhanced muscle contractile function. As previously described, this was likely due to the enhanced interactions between α7β1 integrin and laminin, also shown to regulate force transmission in native muscle [70]. More recently, the muscle ECM-specific proteins found in Matrigel (laminin and collagen IV) were shown to localize to the myofiber periphery, forming a native-like basal lamina in vitro [6].
3.2. Maturation of engineered muscle in vitro
Once the engineered muscle is formed, it is cultured in conditions that promote myofiber maturation and survival. Exogenous biophysical and biochemical stimulations are traditionally applied to mimic natural environmental cues and encourage further cellular growth and have been reviewed in detail elsewhere [71, 72]. Mechanical stimulation, in the form of progressive or cyclic stretch to mimic growth [73] and exercise [74, 75] respectively, has been proven to enhance alignment [73, 75], fusion [73, 75], myofiber hypertrophy [62], and force generation [74, 75] in engineered constructs. Bioreactors capable of such stimulation commonly consist of an elastic cell culture chamber or a “floating anchor” to which the construct is attached and a computer-controlled linear actuator [74, 75] or stepper motor [62] that controls applied strain. Typical stimulation regimes include slow continuous elongation (1% strain every hour [73]) and intermittent cyclic tension (10% strain, 3 times a minute for 5 minutes every hour [74, 75]). Similar to mechanical stimulation, electrical stimulation, used to mimic neuronal input to muscle, has also been shown to promote cell alignment, enhance fusion, improve maturation, and even guide fast-to-slow fiber type switching in engineered muscle [76-78]. Systems designed for exogenous electrical stimulation typically apply graphite, stainless steel, or platinum electrodes parallel to the engineered muscle within the culture dish to induce a field shock [76-79].
In addition to biophysical cues, defined growth factors, identified as drivers of myogenic differentiation in vivo (e.g. IGF-1, TGF-β, Wnt [1]) can be supplemented to culture media to further promote maturation and contractile function [40, 45]. The use of dynamic culture conditions for engineered muscle culture has recently been shown as an important contributor to engineered muscle survival and force generation [6, 7]. The total cross-sectional muscle area, myofiber size, and contractile force amplitude were all significantly increased when engineered muscle constructs were agitated during culture [7]. The observed effects were attributed to enhanced transport of oxygen and nutrients and the potential upregulation of IGF-1 signaling due to the application of shear [7] on the construct surface.
3.3 The satellite cell niche within engineered muscle
In addition to the abundance of mature and functional myofibers, maintenance of functional satellite cells (SCs) within engineered muscle would be required to ensure tissue survival and continued regeneration following implantation [22, 23, 80]. Recently, starting from a population of primary rat myogenic cells, our group has established a method to recreate the native cellular heterogeneity within engineered muscle that contained both differentiated myofibers and a pool of functioning SCs residing in well-defined niches [7]. Described in Figure 2, this has been achieved by utilizing activated SCs, co-expressing MyoD and Pax7, that have the ability to both differentiate into myoblasts and revert back to their quiescent state [1]. For best results, we used the cells isolated from rat hindlimb muscle left to adhere onto Matrigel-coated flasks for 36h. These ‘adherent fraction’ cells were gently detached by dispase to preserve their integrin expression and were embedded at high density within a fibrin/Matrigel matrix. This resulted in rapid formation of aligned myofibers that expressed M-cadherin, an essential cell-cell adhesion protein known to link SCs with myofibers in their native niche [2]. Rapid formation of M-cadherin+ myofibers facilitated the homing of the undifferentiated Pax7+ cells to myofiber surface reverting them to a quiescent state, marked by the loss of Ki67 expression. Over the following two weeks, the SCs remained non-proliferative and, as in the native niche, were attached to a layer of laminin and collagen IV at the basal surface and expressed M-cadherin at the apical surface. ‘Freshly-isolated’ myogenic cells directly encapsulated in hydrogels initially had more Pax7+ cells than the adherent fraction of cells, however, they failed to undergo rapid fusion into myofibers and eventually yielded engineered muscle with less quiescent SCs and inferior functionality [7].
Fig 2. The formation of SC niche within engineered muscle tissue.
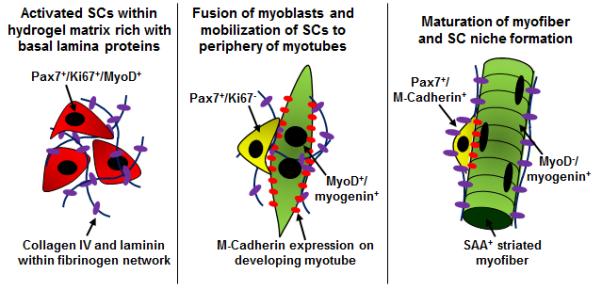
Left, activated SCs, when embedded in a myoconductive 3D environment, will rapidly fuse. Middle, a subset of the cells will home to the membrane of the developing myotubes, link to M-cadherin, and cease to proliferate. Right, with time, the SCs will further build their niche on the mature myofiber by homing beneath the basal lamina, expressing M-cadherin, and becoming quiescent.
4. In vitro assessment of engineered muscle
In vitro analysis of engineered muscle constructs involves both structural and functional evaluation to assess maturation level of the construct and predict its ability to repair tissue damage following implantation. With more emphasis on the development of screening assays using engineered tissues, these quality control in vitro assays can be adapted for higher throughput testing of drug effects on myogenic maturation, viability, and regeneration. In this section, we will review the most common in vitro methodologies to evaluate engineered muscle structure, function, and potential for regeneration.
4.1. Muscle structure and morphology
Assessment of muscle morphology, structure, and protein expression via qualitative and quantitative immunohistology and western blot analysis are routinely used to evaluate myogenic maturation of in vitro engineered muscle and its resemblance to native tissue. Gross structure of engineered muscle can be evaluated by assessment of myofiber alignment [60, 66], density [60], and presence of various muscle and non-muscle cell types [6, 7, 55, 56, 58, 64] in longitudinal and transverse histological cross-sections. The percent of cells that underwent fusion can be quantified by counting a fraction of construct nuclei expressing myogenin [7, 9]. Myofiber diameter is commonly measured to evaluate cellular hypertrophy through analysis of transverse cross-sectional stainings [6, 7, 9, 13, 81, 82]. The fraction of myofibers expressing sarcomeres, muscle contractile units typically labeled by sarcomeric α-actinin (SAA) and myosin heavy chain (MyHC) antibodies, has been quantified to gauge myogenic differentiation [6, 7, 13, 58]. Additionally, the relative expression of MyHC isoforms (embryonic, neonatal, and adult) and types (I, IIa, IIx) has been analyzed to assess myofiber maturation and metabolic phenotype [78, 81, 83]. Additionally, the density and morphology of acetylcholine receptors have been identified through staining with bungarotoxin [59, 83]. We recently reported a direct structural comparison of engineered muscle to native neonatal muscle fascicles revealing similar myofibril appearance (SAA+ sarcomeric units), basal lamina composition and distribution (collagen IV and laminin along periphery of myofibers), peripheral epimysial-like connective tissue layer (vimentin+ fibroblasts), and quiescent SC pool (Pax7+ cells) [6]. The dynamics of in vitro myogenesis in engineered muscle can be also tracked by immunostaining at different time points during culture to assess the shift from activated (Pax7+/Ki67+) to quiescent SCs (Pax7+/Ki67−) or from fusing (myogenin+/MyoD+) to mature myonuclei (myogenin+/MyoD−) [6, 7].
4.2. Contractile function
Recording contractile force generation is considered one of the most valuable methods to dissect functional maturity of engineered muscle and its therapeutic potential in vivo. As freshly fused myotubes transition to mature myofibers and hypertrophy, their force generating capacity is augmented via both the increase in number of contractile units in the cross-section and the transition of MyHC isoforms from embryonic to adult [84]. First reviewed as early as 1999 [85], contractile force measurement in engineered muscle typically involves use of a custom testing platform. Several groups have developed such systems utilizing similar equipment configurations [53, 54, 58, 82]. Typically, a tissue sample is placed in a temperature-controlled bath that houses a pair of electrodes connected to a stimulator. One end of the sample is secured to a fixed anchor while a floating anchor links the other end of the sample to a force transducer. Following a field shock delivered by electrodes (~3-5 V/mm for maximum force [6, 58]), the muscle will undergo isometric contraction, pulling on the floating anchor and generating a force that will be recorded by the transducer. For precise control of construct length, the transducer is usually mounted on a computer-controlled linear actuator.
The most common reported outputs from contractile force measurements are the amplitudes of twitch (induced by a single stimulus pulse) and tetanus (induced by a few hundred ms train of stimuli applied at 20-100 Hz). Furthermore, kinetic parameters of twitch and tetanus, e.g. time-to-peak force and relaxation time, can be calculated from force response traces [6, 13, 54, 58]. These contractile parameters may suggest the maturity and metabolic phenotype of engineered muscle as differences in myosin types and isoforms underlie differences in these properties [86]. Passive tension and elastic modulus of the construct can be also recorded from the difference between baseline forces at different elongations of the construct [6, 13, 58]. By varying construct elongation (strain), researchers can validate physiologic biphasic and monotonic increases in active and passive force, respectively, with increased construct length [6, 13, 82]. The force-frequency response, a sigmoidal relationship dependent on calcium sensitivity [87], can also be measured by varying stimulus frequency [7, 13, 54, 82]. Sensitivity of twitch force to extracellular calcium concentration can also be quantified by measuring the sensitivity curve, fitting the Hill’s equation, and calculating the Hill coefficient and the concentration of calcium required for half maximal activation [59]. Finally, contractile force measurements enable assessment of the engineered muscle excitability through application of stimuli of different duration and amplitude and derivation of the rheobase and chronaxie [53, 54, 82].
Assessment of contractile force in engineered muscle can be adapted for use in drug screening applications. A high throughput system, described in detail by Vandenburgh and colleagues [81], utilized a 96-microwell plate and a ‘myoforce analysis device,’ or MAD, to test the effects of various soluble factors on engineered muscle function [88]. The hydrogel-based muscle bundles were cast within the wells of a 96-well plate each containing two PDMS microposts (0.8 mm in diameter, 4 mm in height). In this platform, the MAD automatically places electrodes into the wells to induce tetanic contractions of the muscle. These contractions result in micropost deflections that are recorded by a camera and can later be analyzed to derive corresponding active force amplitudes (in μN) using linear beam theory and an a priori calibration. In our recent study, the contractile responses of human engineered muscle (“myobundle”) to statins, chloroquine, and clenbuterol were comparable to those in clinical reports [13], thus suggesting the utility of this system as a preclinical drug testing platform.
4.3. Calcium handling
Intracellular calcium handling is another critical parameter describing the maturity and functionality of the engineered muscle. Fluorescence recording of calcium-sensitive dyes (e.g., Fura-2, Rhod-2, Rhod-ff) and, more recently, genetically encoded calcium indicators (e.g. GCaMP) has been the classical method for measuring intracellular calcium transients in muscle cells. The GCaMP molecule, first developed by Nakai et al. [89], consists of fused permutated green fluorescent protein (GFP), calcium binding molecule, calmodulin, and M13, a peptide sequence from myosin light chain kinase. Binding of the free calcium to calmodulin causes a conformational shift of the molecule enabling rapid de-protonation of the GFP and bright fluorescence. The main advantage of these genetic indicators is the ability to perform cell type-specific, non-invasive, long-term calcium imaging in vitro and in vivo [13, 90, 91]. Usually, the system for intracellular calcium recordings requires the use of a sensitive photomultiplier tube or fluorescent camera, an adequate light source and filter set, and highly efficient light-collection optics. Amplitude of calcium transient determined from single-wavelength non-ratiometric measurement is usually expressed as the ratio between change in signal amplitude and baseline fluorescence, referred to as dF/F [6, 13]. Recent studies in human engineered muscle have shown good correlation between dF/F calcium signals measured non-invasively by lentivirally expressed GCaMP6 [91] and corresponding amplitudes of contractile force for both twitch and tetanus contractions under a variety of pharmacological conditions [13]. Kinetics of calcium transients (rise and decay time) can be further used to assess both the functional maturation of engineered muscle as well as potential dominance of slow vs. fast fiber types.
4.4. Regenerative potential
Functionality of satellite cells within engineered muscle constructs can be analyzed by testing the ability of muscle to regenerate after injury. We recently developed an in vitro injury assay (Fig 3) based on classic in vivo muscle regeneration studies using cardiotoxin (CTX, a toxin derived from Naja mossambica mossambica) [92]. Two-week constructs were exposed to 0.2 μM CTX for 6h, and assessed histologically and functionally at 6 hours, 5 days, and 10 days post-injury in attempt to record the regeneration process. Prior to injury, engineered muscle in its homeostatic state contained a pool of quiescent Pax7+/Ki67− satellite cells, a small fraction of MyoD+ nuclei, and a robust presence of cross-striated myogenin+ myofibers that generated forces comparable to those of native neonatal muscle. Following 6 hours of CTX exposure, severe myofiber disruption, loss of sarcomeric structures, and ~75% decrease in contractile function was observed. Five days after injury, the number of Pax7+, Pax7+/Ki67+, MyoD+, and myogenin+ cells was significantly increased, suggesting an expansion of the SC pool and the creation of new myonuclei. This was accompanied by an increase in myofibril density and a modest increase in force generation. By 10 days post-injury, the SC pool returned to near pre-injury levels with very little presence of MyoD+ cells. Myonuclei amounts were restored to healthy levels and sarcomeres were reconstructed, resulting in the significant recovery of contractile function. This ability to model and study skeletal muscle regeneration in vitro could aid the development of new pro-regeneration drugs for a variety of muscle disease.
Fig 3. In vitro assessment of engineered muscle regeneration.
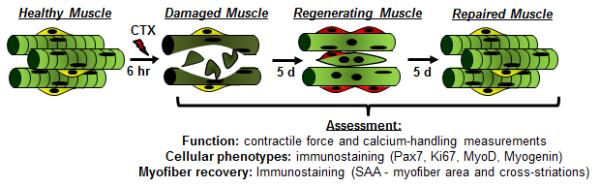
Cardiotoxin (CTX) is applied for 6h to induce myofiber fragmentation and response to injury was assessed by evaluating muscle function, cellular makeup, and myofiber recovery at 6 hours, 5 days, and 10 days post injury.
5. Implantation and evaluation of engineered muscle in vivo
Understanding prospects of tissue engineering therapies for human skeletal muscle injury and disease will require well-designed animal studies to examine the therapeutic efficacy of engineered muscle grafts in vivo. Ideally, engineered muscle implants would rapidly integrate into the host neurovascular system, restore function of the damaged muscle, and be able to self-regenerate to ensure long-term survival, maturation, and resistance to injury [51]. Surgical techniques and implantation methods have evolved over recent years to effectively recreate clinically relevant skeletal muscle defects and enable real-time monitoring of implant viability and function. Furthermore, established assays exist to evaluate engineered muscle performance in vivo, including vascularization, innervation, myofibril health and maturation, and function. In this section we will discuss various methods to assess the ability of engineered muscle to serve as an on-site therapeutic in vivo.
5.1. Implantation models and strategies
Surgical implantation methods (reviewed in Figure 4) are chosen based on critical endpoints of the particular study or to recreate a specific disease condition of relevance to human pathology. Advanced surgical techniques have also been applied to improve implant survival and integration. Described below are typical implantation models used to study performance of engineered muscle in vivo.
Fig 4. Methods for implantation of engineered muscle.
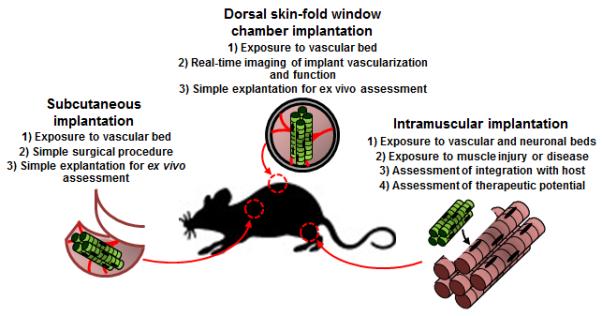
Three animal implantation methods (subcutaneous, intramuscular, and dorsal skin-fold window chamber) along with their main advantages.
5.1.1. Subcutaneous implantation
Subcutaneous implantation is a simple surgical method that exposes grafts to vascular beds beneath the animal skin and is employed to evaluate the ability of engineered muscle to vascularize and survive. In a recent study, subcutaneous implantation was used to determine optimal cell ratios of fibroblasts, endothelial cells, and myoblasts within a polymeric scaffold for enhanced construct survival and vascularization [65]. These studies revealed the importance of endothelial cells to pre-vascularize the construct and fibroblasts to promote vasculogenesis and stabilize the formed vessels [65]. Similarly, another study has identified the benefit of co-culturing endothelial cells and myoblasts on a urinary bladder matrix prior to subcutaneous implantation for tissue formation and vascular ingrowth in vivo [93].
5.1.2. Intramuscular implantation
Intramuscular implantations create more realistic models of corrective surgery and aim to assess engineered muscle’s ability to integrate with host musculature and contribute to its contractile function. In some of these procedures, tissue construct is placed either alongside the rodent hindlimb muscle [65, 94] or within a small void left by excising portion of the muscle [9, 80] or abdominal wall [64]. More drastic surgical procedures are needed to model volumetric muscle loss (VML) where the damage of more than 20% of muscle volume hinders the natural repair process and leads to scar accumulation [3]. One of the standardized VML procedures involves precise excision of a 10 mm×7 mm×3 mm (length×width×depth) volume from the middle third of the rat tibialis anterior (TA) muscle [95]. This results in a significant decrease in muscle weight, volume, and function and allows histological and functional studies of VML repair using tissue engineered constructs or acellular matrices [12, 95]. Large full-thickness abdominal wall defects (~1 cm2) have also been surgically created to evaluate the ability of engineered muscle to repair soft tissue defects [11]. In addition to muscle resection, another model of severe muscle damage in mice combined myotoxin injection to induce local muscle injury and, 6 days later, femoral artery ligation to induce ischemia [96].
Surgical strategies are also employed to encourage implant integration after insertion into the defect site. For example, for neurovascular integration, the ends of exposed portions of the sural nerve have been resected and, along with a neighboring vessel, placed in the middle of the muscle construct implanted alongside the femur [94]. In attempt to develop a mature vascular supply prior to grafting engineered muscle at the injury site, Shandalov et al. first wrapped constructs around femoral artery and vein and left them to vascularize in vivo. Vascularized constructs were then transferred to a muscle defect in the same animal, where the pre-formed vessels were able to anastomose to local vasculature and support implant survival [11]. In addition to re-routing local vessels and nerves, muscle implants can be made with engineered bone anchors at the ends and, to encourage integration with the host, implanted using tunnels drilled into the tibial bone [10].
5.1.3 Window chamber implantation
Engineered muscle tissues can be also implanted inside dorsal skin-fold window chambers to enable non-destructive, real-time monitoring of the implant survival and vascularization in live animals. Originally described for studies of tumor vascularization [97], the method involves removal of the front skin layer to expose the rearward-facing subcutaneous tissue. Implants are then laid against this tissue, covered by a glass top, and secured within a chamber sutured to the skin to lock its position and prevent contamination. As described recently, when combined with intravital microscopy, this method can be used to track real-time vascular ingrowth and calcium handling in implanted engineered muscle [6]. Moreover, the implant and underlying skin can be readily explanted for ex vivo functional and histological assessment.
5.2. Assessment of implant survival and regeneration
Crucial to the overall therapeutic effectiveness of an engineered muscle implant is its ability to add new functional muscle and improve the regenerative capacity at the site of muscle injury. Typical methods to evaluate the viability and growth of the muscle implant in vivo involve comparing its volume [6, 10, 12] or weight [80] to the values measured pre-implantation. Non-invasively, survival and size of muscle implant have been assessed by viral labeling of cells with a luciferase reporter gene followed by bioluminescence imaging in live animals [65]. Myogenic maturation and hypertrophy in vivo can be further assessed by quantifying the fraction of cross-striated myofibers and myofiber diameter. This has been accomplished in a number of studies using immunostaining [6, 10, 12], PCR [12], and western blots [75] for SAA or MyHC.
For the assessment of implant’s regenerative potential in vivo, positive staining for Pax7 can indicate the maintenance of the resident stem cell pool after implantation [6]. However, a more rigorous assay involves demonstration that the implanted engineered muscle can structurally and functionally regenerate after injury. In the study by Rossi et al. [80], bupivacaine was injected locally to induce damage to engrafted GFP-labeled engineered muscle 6 weeks post-implantation. Three days later, EdU, a thymidine analog that incorporates only in dividing cells, was injected and 48h later the animals were sacrificed. EdU+/GFP+ cells found in the implants represented the pool of proliferative myogenic cells contributing to muscle regeneration.
5.3. Assessment of implant vascularization and perfusion
Rapid and robust blood vessel perfusion within implanted engineered muscle is an important requirement for their long-term survival and integration in vivo. Immunostaining for endothelial cell markers, CD31 and von Willebrand factor (vWF), is a simple method for validating presence of vasculature within an implant region [6, 10, 11, 65, 93, 94, 98] and can be quantified in terms of total blood vessel length or lumen density to track ingrowth of host vessels over time [6, 11, 65, 93, 98]. For pre-vascularized implants, fluorescent (e.g. GFP) labeling of implanted cells can be used to distinguish donor and host vessels [64]. Detection of pericyte-associated vessels, through staining of CD146, allows for quantification of stable, mature vasculature [93]. To assess the perfusion of formed vessels, lipophilic dyes, typically dextran or lectin conjugated to a fluorophore, can be injected into the tail vein of the animal and allowed to shortly circulate before the animal is sacrificed. Positive staining of the vessels in the implant is then used to confirm its vascular integration and quantify perfused vessel density and size as shown for muscle implants made from a mixture of myoblasts, endothelial cells, and fibroblasts [64, 65].
Beside destructive methods, angiogenesis and blood flow can also be non-invasively monitored by use of laser Doppler imaging, a technique that detects dynamic light scattering as a result of blood movement. Use of this method allowed Borselli et al. to create blood flow maps and distinguish ischemic and vascularized tissue regions following engineered muscle implantation in a model of ischemic hindlimb injury [96]. These studies revealed the benefits of myogenic cell delivery coupled with scaffold-mediated VEGF and IGF-1 release for vascularization of an ischemic muscle [96]. Similarly, ultrasonographic evaluation of vessel patency and perfusion flow rate at the site of muscle implant has been performed by injecting microbubbles (as a nontargeted contrast agent) in the mouse tail vein [11]. In these studies, an ultrasound pulse was initially applied to destroy the injected microbubbles after which the microbubbles refilled the vessels at a rate that only depends on the capillary flow rate and was independent of the injection rate of the microbubbles. The perfusion volume (ratio of the mean intensity of the signal after the injection vs. after the initial disruption pulse) and flow rate (time until peak signal) was then quantified to non-invasively distinguish temporal changes in perfusion among the experimental groups [11]. Lastly, for the direct tracking of implant vascularization and perfusion in vivo, the window chamber model [6, 99] can be used to quantify total capillary area within the implanted muscle [6, 97]. Specifically, total hemoglobin concentration and oxygen saturation can be quantified through hyperspectral imaging using hemoglobin absorption as the primary source of contrast [97]. Furthermore, with use of a tunable filter to enhance hemoglobin contrast, the motion of red blood cells can be tracked in real time [6] to quantify blood flow direction and velocity [99]. Using this method, our group reported a progressive ingrowth of host vessels into the implanted engineered muscle and further demonstrated the perfusion of the newly-formed capillaries with red blood cells [6].
5.4. Assessment of implant innervation
Successful innervation of engineered muscle implant by host motor neurons is required for its synchronization with host muscle and continued maturation in vivo. Although less studied than engineered muscle vascularization, formation of host-donor neuromuscular junctions can be inferred from the co-localization of immunostained neurofilament markers (e.g. NF-200) that label host neurons [98] and bungarotoxin that identifies acetylcholine receptors in muscle implant [10, 94]. To date, different groups have reported the ingrowth of neurons [10, 94, 98], presence of acetylcholine receptors [10, 94], and formation of neuromuscular junctions [10] within engineered muscle implants. While promising, these studies have not unequivocally shown functional integration of implanted engineered muscle within host neurovascular system. One method to unambiguously demonstrate functional innervation of implanted muscle constructs would be to detect fluorescence signals from GCaMP-labeled implants in response to localized stimulation of an upstream nerve.
5.5. Assessment of implant function
The most therapeutically relevant assessment following engineered muscle implantation concerns the implant’s ability to improve force generation at the injury site. For the basic tests of contractile function, the whole muscle is explanted followed by ex vivo isometric force measurements in treated compared to untreated samples [9, 64, 94, 96, 98]. Except for the special case of window chamber implantation [6], these ex vivo measurements do not separate contributions of implanted vs. host muscle to measured force generation. In addition, the size of the muscle to be tested ex vivo is significantly limited by risk of ischemia, while the use of electrical stimulation does not test the true physiological state with neuromuscular involvement. Thus, more relevant and complex methods for measurement of in situ force generation have been recently developed and applied to assess engineered muscle repair of TA injury in rats [10]. Specifically, in these measurements the tested muscle was carefully isolated from the surrounding tissue without disrupting its neuronal and vascular supply. With the hindlimb fixed at the knee, the distal tendon of the TA was attached to a servomotor for force recordings while muscle contractions were induced in a physiological fashion by activating the sciatic nerve via a wire electrode. It was found that tissue-engineered skeletal muscle with bone-tendon interface was able to partially restore contractile function in this model of volumetric muscle loss [10].
As described for vascularization studies, the concept of non-invasive functional measurements in vivo offers additional advantages compared to ex vivo or in situ tests by enabling multiple recordings in the same animal. One such method is the hindlimb grip-strength test, recently utilized to evaluate functional outcomes following implantation of engineered muscle [9]. For this assessment, the animal is lowered until it forms a grip around a grasping bar connected to a force transducer. The animal is then pulled backwards with consistent force until it can no longer hold on, generating its peak grip-strength. Another more complex and quantitative method that isolates the effect of muscle contraction alone has utilized a custom setup to measure torque following stimulation of the peroneal nerve and activation of the anterior crural muscles [12]. Here, the animal was anesthetized and positioned with its knee fixed and foot placed on a custom-made plate attached to a servomotor. Upon stimulation, torque was generated by the extension of the foot, primarily contributed by the extensor digitorum longus (EDL) muscle and injured or treated TA muscles, and recorded. This specific study showed the benefits of myogenic cell seeding on a bladder matrix scaffold vs. the use of the scaffold alone in recovery of muscle function following injury [12]. Finally, in conjunction with the window chamber model, functionality of the engineered muscle implant can be assessed non-invasively in anesthetized animals by fast camera recordings of spontaneous calcium transients reported by the GCaMP reporter. Over time, the amplitude of the calcium transients in muscle implants increased, consistent with the increase in force generation found in ex vivo measurements [6].
6. Summary
A number of methodological advancements in the fabrication and functional and therapeutic evaluation of engineered skeletal muscle tissues have brought their practical applications closer to reality. Recent progress in creating the muscle constructs that can regenerate in vitro and survive, vascularize, and mature in vivo as well as the first-time engineering of functional human muscle are particularly exciting. While these advances, along with the progress in iPSC [100] and genome editing technologies [101], will enable the development of new in vitro platforms for predictive drug screening and disease modeling, a number of challenges remain to be addressed before engineered muscle tissues could be directly utilized in clinical applications. In particular, scaling-up human muscle constructs to therapeutically relevant sizes will require overcoming significant hurdles. Currently, large-scale derivation of satellite cells from human iPSCs [102] and integration of biological and bioengineering techniques to maintain and expand functional SCs in vitro [46, 49] are the most promising methodologies to provide sufficient numbers of regenerative myogenic cells. Furthermore, improved understanding of embryonic muscle development may yield approaches to encourage significant growth of engineered muscle implants in vivo. The continued advances in genetic and device-based systems for noninvasive monitoring of muscle differentiation and function will allow optimization and standardization of in vitro tissue engineering processes, including the use of real-time quality controls and biophysical and biochemical stimulation feedbacks. Finally, developing methods to enhance cellular complexity of muscle constructs (by including tendons, vascular networks, and neuronal inputs) will enable design of more physiological models of skeletal muscle tissue and facilitate implant integration for muscle repair applications.
HIGHLIGHTS.
Description of methods to isolate and expand muscle satellite cells in vitro.
Overview of methods to engineer highly functional tissues and maintain the satellite cell pool.
Discussion of methods for in vitro and in vivo assessment of muscle viability, structure, function, maturation, and regeneration.
Techniques to generate animal models and accelerate and study graft integration.
Acknowledgements
This study was supported by the National Science Foundation’s Graduate Research Fellowship to M. Juhas and grants AR055226 and AR065873 from National Institute of Arthritis and Musculoskeletal and Skin Diseases to N. Bursac. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- [1].Charge SB, Rudnicki MA. Physiological reviews. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- [2].Yin H, Price F, Rudnicki MA. Physiological reviews. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Turner NJ, Badylak SF. Cell and tissue research. 2012;347:759–774. doi: 10.1007/s00441-011-1185-7. [DOI] [PubMed] [Google Scholar]
- [4].Ruegg MA, Glass DJ. Annual review of pharmacology and toxicology. 2011;51:373–395. doi: 10.1146/annurev-pharmtox-010510-100537. [DOI] [PubMed] [Google Scholar]
- [5].Brack AS, Rando TA. Stem cell reviews. 2007;3:226–237. doi: 10.1007/s12015-007-9000-2. [DOI] [PubMed] [Google Scholar]
- [6].Juhas M, Engelmayr GC, Jr., Fontanella AN, Palmer GM, Bursac N. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1402723111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Juhas M, Bursac N. Biomaterials. 2014 doi: 10.1016/j.biomaterials.2014.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang YC, Dennis RG, Larkin L, Baar K. J Appl Physiol. 2005;98:706–713. doi: 10.1152/japplphysiol.00273.2004. Epub 2004 Oct 2008. [DOI] [PubMed] [Google Scholar]
- [9].Carosio S, Barberi L, Rizzuto E, Nicoletti C, Del Prete Z, Musaro A. Scientific reports. 2013;3:1420. doi: 10.1038/srep01420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].VanDusen KW, Syverud BC, Williams ML, Lee JD, Larkin LM. Tissue engineering. Part A. 2014 [Google Scholar]
- [11].Shandalov Y, Egozi D, Koffler J, Dado-Rosenfeld D, Ben-Shimol D, Freiman A, Shor E, Kabala A, Levenberg S. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6010–6015. doi: 10.1073/pnas.1402679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Corona BT, Ward CL, Baker HB, Walters TJ, Christ GJ. Tissue engineering. Part A. 2014;20:705–715. doi: 10.1089/ten.tea.2012.0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Madden L, Juhas M, Kraus WE, Truskey GA, Bursac N. eLife. 2015;4 doi: 10.7554/eLife.04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Snow MH. Cell and tissue research. 1977;185:399–408. doi: 10.1007/BF00220299. [DOI] [PubMed] [Google Scholar]
- [15].Rosenblatt JD, Lunt AI, Parry DJ, Partridge TA. In vitro cellular & developmental biology. Animal. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- [16].Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Developmental biology. 2006;294:50–66. doi: 10.1016/j.ydbio.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gaster M, Kristensen SR, Beck-Nielsen H, Schroder HD. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2001;109:735–744. doi: 10.1034/j.1600-0463.2001.d01-140.x. [DOI] [PubMed] [Google Scholar]
- [18].Fukada S, Ma Y, Ohtani T, Watanabe Y, Murakami S, Yamaguchi M. Frontiers in physiology. 2013;4:317. doi: 10.3389/fphys.2013.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fukada S, Higuchi S, Segawa M, Koda K, Yamamoto Y, Tsujikawa K, Kohama Y, Uezumi A, Imamura M, Miyagoe-Suzuki Y, Takeda S, Yamamoto H. Experimental cell research. 2004;296:245–255. doi: 10.1016/j.yexcr.2004.02.018. [DOI] [PubMed] [Google Scholar]
- [20].Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto H, Miyagoe-Suzuki Y, Takeda S. Stem cells. 2007;25:2448–2459. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- [21].Sherwood RI, Christensen JL, Conboy IM, Conboy MJ, Rando TA, Weissman IL, Wagers AJ. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- [22].Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- [24].Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, Li S, Belicchi M, Peretti G, Chamberlain JS, Wright WE, Torrente Y, Ferrari S, Bianco P, Cossu G. Nature cell biology. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- [25].Cornelison DD, Filla MS, Stanley HM, Rapraeger AC, Olwin BB. Developmental biology. 2001;239:79–94. doi: 10.1006/dbio.2001.0416. [DOI] [PubMed] [Google Scholar]
- [26].Rosen GD, Sanes JR, LaChance R, Cunningham JM, Roman J, Dean DC. Cell. 1992;69:1107–1119. doi: 10.1016/0092-8674(92)90633-n. [DOI] [PubMed] [Google Scholar]
- [27].Castiglioni A, Hettmer S, Lynes MD, Rao TN, Tchessalova D, Sinha I, Lee BT, Tseng YH, Wagers AJ. Stem cell reports. 2014;2:92–106. doi: 10.1016/j.stemcr.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cosgrove BD, Sacco A, Gilbert PM, Blau HM. Differentiation; research in biological diversity. 2009;78:185–194. doi: 10.1016/j.diff.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rando TA, Blau HM. The Journal of cell biology. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vandenburgh HH, Karlisch P, Farr L. In vitro cellular & developmental biology : journal of the Tissue Culture Association. 1988;24:166–174. doi: 10.1007/BF02623542. [DOI] [PubMed] [Google Scholar]
- [31].Grefte S, Vullinghs S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW. Biomedical materials. 2012;7:055004. doi: 10.1088/1748-6041/7/5/055004. [DOI] [PubMed] [Google Scholar]
- [32].Sanes JR. The Journal of biological chemistry. 2003;278:12601–12604. doi: 10.1074/jbc.R200027200. [DOI] [PubMed] [Google Scholar]
- [33].Goodman SL, Deutzmann R, Nurcombe V. Development. 1989;105:795–802. doi: 10.1242/dev.105.4.795. [DOI] [PubMed] [Google Scholar]
- [34].Gaster M, Beck-Nielsen H, Schroder HD. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2001;109:726–734. doi: 10.1034/j.1600-0463.2001.d01-139.x. [DOI] [PubMed] [Google Scholar]
- [35].Ham RG, St Clair JA, Webster C, Blau HM. In vitro cellular & developmental biology : journal of the Tissue Culture Association. 1988;24:833–844. doi: 10.1007/BF02623656. [DOI] [PubMed] [Google Scholar]
- [36].Yablonka-Reuveni Z, Balestreri TM, Bowen-Pope DF. The Journal of cell biology. 1990;111:1623–1629. doi: 10.1083/jcb.111.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Krauss RS, Cole F, Gaio U, Takaesu G, Zhang W, Kang JS. Journal of cell science. 2005;118:2355–2362. doi: 10.1242/jcs.02397. [DOI] [PubMed] [Google Scholar]
- [38].Chowdhury SR, Muneyuki Y, Takezawa Y, Kino-oka M, Saito A, Sawa Y, Taya M. Journal of bioscience and bioengineering. 2010;109:310–313. doi: 10.1016/j.jbiosc.2009.09.042. [DOI] [PubMed] [Google Scholar]
- [39].Blau HM, Webster C. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:5623–5627. doi: 10.1073/pnas.78.9.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gawlitta D, Boonen KJ, Oomens CW, Baaijens FP, Bouten CV. Tissue engineering. Part A. 2008;14:161–171. doi: 10.1089/ten.a.2007.0095. [DOI] [PubMed] [Google Scholar]
- [41].McAleer CW, Rumsey JW, Stancescu M, Hickman JJ. Biotechnology progress. 2015 doi: 10.1002/btpr.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fujita H, Endo A, Shimizu K, Nagamori E. Biotechnology and bioengineering. 2010;107:894–901. doi: 10.1002/bit.22865. [DOI] [PubMed] [Google Scholar]
- [43].Murre C, McCaw PS, Baltimore D. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- [44].Benezra R, Davis RL, Lassar A, Tapscott S, Thayer M, Lockshon D, Weintraub H. Annals of the New York Academy of Sciences. 1990;599:1–11. doi: 10.1111/j.1749-6632.1990.tb42359.x. [DOI] [PubMed] [Google Scholar]
- [45].Weist MR, Wellington MS, Bermudez JE, Kostrominova TY, Mendias CL, Arruda EM, Larkin LM. Journal of tissue engineering and regenerative medicine. 2013;7:562–571. doi: 10.1002/term.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fu X, Xiao J, Wei Y, Li S, Liu Y, Yin J, Sun K, Sun H, Wang H, Zhang Z, Zhang BT, Sheng C, Wang H, Hu P. Cell research. 2015;25:655–673. doi: 10.1038/cr.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Urciuolo A, Quarta M, Morbidoni V, Gattazzo F, Molon S, Grumati P, Montemurro F, Tedesco FS, Blaauw B, Cossu G, Vozzi G, Rando TA, Bonaldo P. Nature communications. 2013;4:1964. doi: 10.1038/ncomms2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Nature medicine. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Xu C, Tabebordbar M, Iovino S, Ciarlo C, Liu J, Castiglioni A, Price E, Liu M, Barton ER, Kahn CR, Wagers AJ, Zon LI. Cell. 2013;155:909–921. doi: 10.1016/j.cell.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Juhas M, Bursac N. Current opinion in biotechnology. 2013 doi: 10.1016/j.copbio.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Strohman RC, Bayne E, Spector D, Obinata T, Micou-Eastwood J, Maniotis A. In vitro cellular & developmental biology : journal of the Tissue Culture Association. 1990;26:201–208. doi: 10.1007/BF02624113. [DOI] [PubMed] [Google Scholar]
- [53].Dennis RG, Kosnik PE., 2nd In vitro cellular & developmental biology. Animal. 2000;36:327–335. doi: 10.1290/1071-2690(2000)036<0327:EAICPO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- [54].Dennis RG, Kosnik PE, 2nd, Gilbert ME, Faulkner JA. American journal of physiology. Cell physiology. 2001;280:C288–295. doi: 10.1152/ajpcell.2001.280.2.C288. [DOI] [PubMed] [Google Scholar]
- [55].Larkin LM, Van der Meulen JH, Dennis RG, Kennedy JB. In vitro cellular & developmental biology. Animal. 2006;42:75–82. doi: 10.1290/0509064.1. [DOI] [PubMed] [Google Scholar]
- [56].Larkin LM, Calve S, Kostrominova TY, Arruda EM. Tissue engineering. 2006;12:3149–3158. doi: 10.1089/ten.2006.12.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Takahashi H, Shimizu T, Nakayama M, Yamato M, Okano T. Biomaterials. 2013;34:7372–7380. doi: 10.1016/j.biomaterials.2013.06.033. [DOI] [PubMed] [Google Scholar]
- [58].Hinds S, Bian W, Dennis RG, Bursac N. Biomaterials. 2011;32:3575–3583. doi: 10.1016/j.biomaterials.2011.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bian W, Bursac N. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26:955–965. doi: 10.1096/fj.11-187575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bian W, Juhas M, Pfeiler TW, Bursac N. Tissue engineering. Part A. 2012;18:957–967. doi: 10.1089/ten.tea.2011.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shansky J, Del Tatto M, Chromiak J, Vandenburgh H. In vitro cellular & developmental biology. Animal. 1997;33:659–661. doi: 10.1007/s11626-997-0118-y. [DOI] [PubMed] [Google Scholar]
- [62].Powell CA, Smiley BL, Mills J, Vandenburgh HH. American journal of physiology. Cell physiology. 2002;283:C1557–1565. doi: 10.1152/ajpcell.00595.2001. [DOI] [PubMed] [Google Scholar]
- [63].Rhim C, Cheng CS, Kraus WE, Truskey GA. Tissue engineering. Part A. 2010;16:3589–3597. doi: 10.1089/ten.tea.2009.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Koffler J, Kaufman-Francis K, Shandalov Y, Egozi D, Pavlov DA, Landesberg A, Levenberg S. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14789–14794. doi: 10.1073/pnas.1017825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D'Amore PA, Langer R. Nature biotechnology. 2005;23:879–884. doi: 10.1038/nbt1109. [DOI] [PubMed] [Google Scholar]
- [66].Bian W, Bursac N. Biomaterials. 2009;30:1401–1412. doi: 10.1016/j.biomaterials.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bian W, Liau B, Badie N, Bursac N. Nature protocols. 2009;4:1522–1534. doi: 10.1038/nprot.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mayer U. The Journal of biological chemistry. 2003;278:14587–14590. doi: 10.1074/jbc.R200022200. [DOI] [PubMed] [Google Scholar]
- [69].Katagiri Y, Hiroyama T, Akamatsu N, Suzuki H, Yamazaki H, Tanoue K. The Journal of biological chemistry. 1995;270:1785–1790. doi: 10.1074/jbc.270.4.1785. [DOI] [PubMed] [Google Scholar]
- [70].Paul AC, Sheard PW, Kaufman SJ, Duxson MJ. Cell and tissue research. 2002;308:255–265. doi: 10.1007/s00441-002-0526-y. [DOI] [PubMed] [Google Scholar]
- [71].Cheng CS, Davis BN, Madden L, Bursac N, Truskey GA. Experimental biology and medicine. 2014;239:1203–1214. doi: 10.1177/1535370214538589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Rangarajan S, Madden L, Bursac N. Ann Biomed Eng. 2014;42:1391–1405. doi: 10.1007/s10439-013-0966-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Vandenburgh HH, Karlisch P. In vitro cellular & developmental biology : journal of the Tissue Culture Association. 1989;25:607–616. doi: 10.1007/BF02623630. [DOI] [PubMed] [Google Scholar]
- [74].Moon du G, Christ G, Stitzel JD, Atala A, Yoo JJ. Tissue engineering. Part A. 2008;14:473–482. doi: 10.1089/tea.2007.0104. [DOI] [PubMed] [Google Scholar]
- [75].Corona BT, Machingal MA, Criswell T, Vadhavkar M, Dannahower AC, Bergman C, Zhao WX, Christ GJ. Tissue Eng Pt A. 2012;18:1213–1228. doi: 10.1089/ten.tea.2011.0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Langelaan ML, Boonen KJ, Rosaria-Chak KY, van der Schaft DW, Post MJ, Baaijens FP. Journal of tissue engineering and regenerative medicine. 2011;5:529–539. doi: 10.1002/term.345. [DOI] [PubMed] [Google Scholar]
- [77].Serena E, Flaibani M, Carnio S, Boldrin L, Vitiello L, De Coppi P, Elvassore N. Neurological research. 2008;30:207–214. doi: 10.1179/174313208X281109. [DOI] [PubMed] [Google Scholar]
- [78].Khodabukus A, Baehr LM, Bodine SC, Baar K. Journal of cellular physiology. 2015 doi: 10.1002/jcp.24985. [DOI] [PubMed] [Google Scholar]
- [79].van der Schaft DW, van Spreeuwel AC, Boonen KJ, Langelaan ML, Bouten CV, Baaijens FP. Journal of visualized experiments : JoVE. 2013:e4267. doi: 10.3791/4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rossi CA, Flaibani M, Blaauw B, Pozzobon M, Figallo E, Reggiani C, Vitiello L, Elvassore N, De Coppi P. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:2296–2304. doi: 10.1096/fj.10-174755. [DOI] [PubMed] [Google Scholar]
- [81].Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, Valentini R, Crawford G. Muscle & nerve. 2008;37:438–447. doi: 10.1002/mus.20931. [DOI] [PubMed] [Google Scholar]
- [82].Lee PH, Vandenburgh HH. Tissue engineering. Part A. 2013;19:2147–2155. doi: 10.1089/ten.TEA.2012.0597. [DOI] [PubMed] [Google Scholar]
- [83].Wang L, Shansky J, Vandenburgh H. Molecular neurobiology. 2013;48:397–403. doi: 10.1007/s12035-013-8412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Davis TA, Fiorotto ML. Curr Opin Clin Nutr. 2009;12:78–85. doi: 10.1097/MCO.0b013e32831cef9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Faulkner JA, Brooks SV, Dennis RG. Methods in molecular medicine. 1999;18:155–172. doi: 10.1385/0-89603-516-6:155. [DOI] [PubMed] [Google Scholar]
- [86].Close R. J Physiol-London. 1964;173:74–95. doi: 10.1113/jphysiol.1964.sp007444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Roszek B, Baan GC, Huijing PA. Journal of applied physiology. 1994;77:2115–2124. doi: 10.1152/jappl.1994.77.5.2115. [DOI] [PubMed] [Google Scholar]
- [88].Vandenburgh H, Shansky J, Benesch-Lee F, Skelly K, Spinazzola JM, Saponjian Y, Tseng BS. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23:3325–3334. doi: 10.1096/fj.09-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Nakai J, Ohkura M, Imoto K. Nature biotechnology. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- [90].Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Nature methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Couteaux R, Mira JC, d'Albis A. Biology of the cell / under the auspices of the European Cell Biology Organization. 1988;62:171–182. [PubMed] [Google Scholar]
- [93].Criswell TL, Corona BT, Wang Z, Zhou Y, Niu G, Xu Y, Christ GJ, Soker S. Biomaterials. 2013;34:140–149. doi: 10.1016/j.biomaterials.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Williams ML, Kostrominova TY, Arruda EM, Larkin LM. Journal of tissue engineering and regenerative medicine. 2013;7:434–442. doi: 10.1002/term.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wu X, Corona BT, Chen X, Walters TJ. BioResearch open access. 2012;1:280–290. doi: 10.1089/biores.2012.0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Borselli C, Cezar CA, Shvartsman D, Vandenburgh HH, Mooney DJ. Biomaterials. 2011;32:8905–8914. doi: 10.1016/j.biomaterials.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Palmer GM, Fontanella AN, Shan S, Hanna G, Zhang G, Fraser CL, Dewhirst MW. Nature protocols. 2011;6:1355–1366. doi: 10.1038/nprot.2011.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Machingal MA, Corona BT, Walters TJ, Kesireddy V, Koval CN, Dannahower A, Zhao W, Yoo JJ, Christ GJ. Tissue engineering. Part A. 2011;17:2291–2303. doi: 10.1089/ten.tea.2010.0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Fontanella AN, Schroeder T, Hochman DW, Chen RE, Hanna G, Haglund MM, Rajaram N, Frees AE, Secomb TW, Palmer GM, Dewhirst MW. Microcirculation. 2013;20:724–735. doi: 10.1111/micc.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Darabi R, Perlingeiro RC. Journal of stem cell research & therapy. 2013;3 doi: 10.4172/2157-7633.1000e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Gaj T, Gersbach CA, Barbas CF., 3rd Trends in biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M, Perlingeiro RC. Cell stem cell. 2012;10:610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]