

Abstract
Pulmonary arterial hypertension (PAH) is an often fatal disorder resulting from several causes including heterogeneous genetic defects. While mutations in the bone morphogenetic protein receptor type II (BMPR2) gene are the single most common causal factor for hereditary cases, pathogenic mutations have been observed in approximately 25% of idiopathic PAH patients without a prior family history of disease. Additional defects of the transforming growth factor beta (TGF-β) pathway have been implicated in disease pathogenesis. Specifically, studies have confirmed activin A receptor type II-like 1 (ACVRL1), endoglin (ENG) and members of the SMAD family as contributing to PAH both with and without associated clinical phenotypes. Most recently, next-generation sequencing has identified novel, rare genetic variation implicated in the PAH disease spectrum. Of importance, several identified genetic factors converge on related pathways and provide significant insight into the development, maintenance and pathogenetic transformation of the pulmonary vascular bed. Together, these analyses represent the largest comprehensive compilation of BMPR2 and associated genetic risk factors for PAH, comprising known and novel variation. Additionally, with the inclusion of an allelic series of locus-specific variation in BMPR2, these data provide a key resource in data interpretation and development of contemporary therapeutic and diagnostic tools.
Keywords: BMPR2, ACVRL1, ENG, SMAD1, SMAD4, SMAD9, CAV1, KCNA5, KCNK3, EIF2AK4, haploinsufficiency, locus heterogeneity
Introduction
Heritable pulmonary arterial hypertension (HPAH) (PPH1; MIM# 178600) is a severe, progressive autosomal dominant vascular disorder, predominantly affecting the arterial circulation and, in particular, the pulmonary arterioles [Tuder et al., 2013]. Histopathological investigation reveals abnormal muscularization of these structures, which leads to a chronic elevation of pulmonary arterial pressure, often resulting in right heart failure 2-3 years post-diagnosis in the absence of the contemporary treatment protocols [Vonk-Noordegraaf et al., 2013]. Identification of mutations in the BMPR2 gene in probands with a family history of disease provided the first insight into the molecular pathogenesis of HPAH [Deng et al., 2000; Lane et al., 2000]. Subsequently, BMPR2 mutations were identified in a cohort of idiopathic patients (IPAH) [Thomson et al., 2000]. Since, causal variation has been described in nine additional genes, in cases that include PAH associated with other conditions (APAH). Here, we describe molecular genetic analyses of the 10 functionally characterized genes that cause PAH (Figure 1) and provide a compilation of all mutations identified to date. The continuing identification of genetic factors, as explored in this report, provides unique insight to the genetic mechanisms driving disorders of pulmonary vascular function. Furthermore, these studies offer the foundation for the discovery and delivery of novel therapeutic options.
Figure 1.
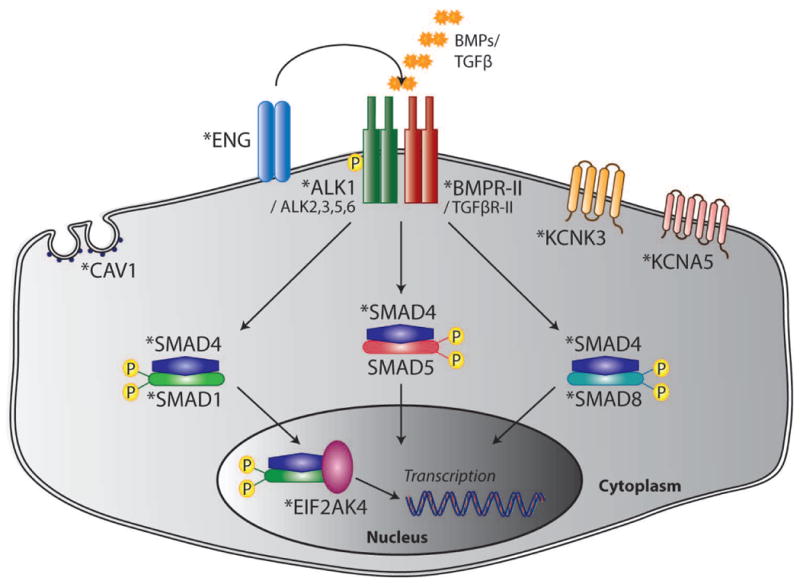
Schematic of canonical BMP signaling and additional pathways implicated in PAH pathogenesis by conventional and next-generation sequence analysis. Causal genes are indicated by the asterisks.
Key Components of the BMP Signaling Pathway
The BMPR2 gene (MIM# 600799) encodes a type II receptor of the TGF-β family of signaling molecules. The mature polypeptide is composed of a signal peptide (encoded by exon 1), an extracellular domain (exons 2-3), a single transmembrane domain (exons 4-5), a highly conserved eukaryotic protein kinase region (exons 6-11) and an unusually large cytoplasmic tail (exons 12-13) amongst TGF-β receptors species [Liu et al., 1995]. In the canonical pathway, BMPR-II binds ligand in a heteromeric complex with a type I receptor, which may be activin receptor-like kinase 1 (ALK1), -2 (ALK2), -3 (ALK3/BMPR1A) or -6 (ALK6/BMPR1B), to initiate activation of intracellular partners within a cell-specific context [David et al., 2009; Rigueur et al., 2015]. Phosphorylation of the receptor SMAD proteins (R-SMADs) 1, 5 and 8 leads to their association with the nuclear chaperone SMAD4. This signaling complex translocates to the nucleus, where it acts in combination with transcriptional co-activators and -repressors to effect control of target gene expression (Figure 1). BMPR-II signaling has been established as essential to a multitude of fundamental cellular processes including proliferation, apoptosis, differentiation and migration [Shi and Massague, 2003].
Mutations of BMPR2 Predispose to the Majority of Hereditary and Idiopathic Forms of PAH
Herein, we describe an additional 370 independent variants of BMPR2 in patients either previously excluded from or ascertained since the last comprehensive mutation update in 2009 [Machado et al., 2009]. Of these, 108 were identified as part of this study and were generated by specialist PAH centers based in Germany, France, North America and the UK (Table 1, Table 2). The research was prospectively reviewed and approved by a duly constituted ethics committee for each center. Probands were assessed for point mutations and large gene abnormalities using multiple screening technologies including Southern blotting, denaturing high-performance liquid chromatography, multiplex ligation-dependent probe amplification (MLPA), dye-terminator and next-generation sequencing (NGS). All variants considered to be pathogenic were absent from a control population of at least 200 chromosomes and public variation databases including dbSNP v142 (http://www.ncbi.nlm.nih.gov/SNP) and the 1000 genomes project (http://www.1000genomes.org) and/or have been previously demonstrated to have a functional impact. For a current estimation of population frequencies, we have additionally checked all point mutations reported here against the Broad Institute Exome Aggregation Consortium (ExAC) database v0.3 (http://exac.broadinstitute.org), comprising over 60,000 exomes derived from independent sequencing projects. Mutation nomenclature employs parameters set by the Human Genome Variation Society (http://www.hgvs.org/mutnomen). Taken together with previous reports [Machado et al., 2006; 2009], these data provide evidence of a total of 668 germline variants underlying PAH, thereby consolidating BMPR2 as the major causal gene for familial cases and subjects previously classified as IPAH. The spectrum and range of BMPR2 defects in this study comprise the major mutation categories which, in general, correlate with existing data [Machado et al., 2006; 2009]. Namely, we record missense variants leading to amino acid substitution (n=86, 23%), nonsense mutations (n=107, 29%), frameshift defects resulting from small insertions/deletions (n=79, 21%) and splice-site variation (n=33, 9%). However, and by contrast to earlier studies, we identified a significantly higher prevalence of major gene rearrangements (n=61, 16%) and single nucleotide mutations in the 5-prime untranslated region (5′ UTR) (Table 1, Table 2, Supp. Table S1). This, most likely, is a consequence of screening centers expanding the analysis of gene re-arrangements to include all exons of BMPR2, combined with a growing recognition that mutation short-fall within cohorts is potentially explained by defects harbored within non-coding regions of BMPR2 [Machado et al., 2006]. For example, we report a total of four recurrent 5′ UTR mutations resulting from a guanine to adenine change (c.−669G>A) likely to abolish specificity for an SP3 transcription factor binding site [Wang et al., 2009]. In combination, these genetic findings reinforce haploinsufficiency as the molecular mechanism for this disease [Machado et al., 2001; 2006; 2009]. Moreover, this report provides a comprehensive compilation of distinct variants (n=384) across the BMPR2 locus since the first identification of the gene (Supp. Table S2). A combination of genetic and functional studies have firmly established a large proportion of these to be likely pathogenic while others, although compelling, remain to be fully elucidated as disease-causing. These data have now been made available in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar).
Table 1. Novel pathogenic BMPR2 mutations identified in this analysis.
| Location | Mutation category | Nucleotide change | Amino acid change | Frequency in this study | Clinical classification |
|---|---|---|---|---|---|
| 5′UTR | Transition | c.-669G>A | p.? | 3 | I, NK, P |
| 5′UTR to exon 1 | Deletion | c. ?_-540_76+?del | p.? | 1 | NK |
| 5′UTR to exon 13 | Deletion | c? -540 3117+?del | p.? | 1 | NK |
| Exon 1 | Deletion | c.1-? 76+?del | p.? | [3] | I, NK (n=2) |
| Exon 1 | Frameshift | c.9dupC | p.S4Lfs*34 | 1 | NK |
| Exon 1 | Nonsense | c.16C>T | p.Q6* | 2 | I, NK |
| Exon 1 | Nonsense | c.38G>A | p.W13* | 1 | H |
| Exon 1 | Nonsense | c.48G>A | p.W16* | 1 | I |
| Intron 1 | Splice-site | c.76+1G>T | p.? | 1 | H |
| Intron 1 | Splice-site | c.76+2T>C | p.? | 1 | NK |
| Intron 1 | Splice-site | c.77-1G>A | p.A26Efs*9 | 1 | H |
| Exons 2-5 | Deletion | c.77-?_621+?del | p.? | 1 | H |
| Exons 2-9 | Deletion | c.77-?_1276+?del | p.? | 1 | NK |
| Exon 2 | Nonsense | c.82C>T | p.Q28* | 1 | NK |
| Exon 2 | Missense | c.178t>c | p.C60R | 1 | NK |
| Exon 2 | Missense | c.196t>g | p.C66G | 1 | NK |
| Exon 2 | Frameshift | c.236_238delinsAAAAGGGGACA | p.L79Qfs*5 | 1 | NK |
| Exon 2 | Frameshift | c.246dupA | p.G83Rfs*15 | 1 | H |
| Intron 2 | Splice-site | c.247+1G>A | p.? | 1 | NK |
| Exon 3 | Deletion | c.248-?_418+?del | p.? | [4] | I (n=3), NK |
| Exon 3 | Duplication | c.248-?_418+?dup | p.? | 1 | H |
| Exon 3 | Missense | c.280T>G | p.C94G | 1 | NK |
| Exon 3 | Frameshift | c.339_340insAA | p.R114Nfs*39 | 1 | H |
| Exon 3 | Frameshift | c.345_346delCT | p.F115Lfs*4 | 1 | NK |
| Exon 3 | Missense | c.350G>A | p.C117Y | 1 | NK |
| Exon 3 | Frameshift | c.353delG | p.C118Lfs*34 | 1 | NK |
| Exon 3 | Missense | c.354T>G | p.C118W | 1 | I |
| Exon 3 | Missense | c.377A>G | p.N126S | 1 | NK |
| Exon 4 | Deletion | c.419-?_529+?del | p.? | 1 | NK |
| Exons 4-7 | Deletion | c.419-?_967+?del | p.? | [2] | I, NK |
| Exons 4-10 | Deletion | c.419-?_1413+?del | p.? | 1 | NK |
| Exon 4 | Frameshift | c.435delT | p.F145Lfs*7 | 1 | NK |
| Exon 4 | Nonsense | c.439C>T | p.R147* | 1 | H |
| Exon 4 | Nonsense | c.482T>A | p.L161* | 1 | I |
| Exon 5 | Deletion | c.530-?_621+?del | p.? | 1 | NK |
| Exon 5 | Nonsense | c.541C>T | p.Q181* | 1 | H |
| Exon 6 | Nonsense | c.637C>T | p.R213* | 1 | H |
| Exon 6 | Nonsense | c.642T>G | p.Y214* | 1 | NK |
| Exon 6 | Frameshift | c.673 679delCGTCCAG | p.R225Lfs*3 | 1 | H |
| Exon 6 | Nonsense | c.727G>T | p.E243* | 1 | I |
| Exon 6 | Frameshift | c.795_796delinsTT | p.E265 L1038delinsD | 1 | NK |
| Intron 6 | Splice-site | c.853-2A>G | p.? | 1 | NK |
| Intron 6 | Splice-site | c.853-1G>A | p.? | 1 | NK |
| Exon 7 | Nonsense | c.860T>A | p.L287* | 1 | H |
| Exon 7 | Nonsense | c.872T>G | p.L291* | 1 | NK |
| Exon 7 | Nonsense | c.893G>A | p.W298* | 1 | NK |
| Exon 7 | Frameshift | c.894_895dupGG | p.V299Gfs*2 | 1 | H |
| Exon 7 | Frameshift | c.961delC | p.R321Efs*14 | 1 | NK |
| Intron 7 | Splice-site | c.967+2T>C | p.? | 1 | H |
| Intron 7 | Splice-site | c. 968-3 C>G | p.? | 1 | NK |
| Intron 7 | Splice-site | c.968-1G>T | p.? | 1 | NK |
| Exon 8 | Nonsense | c.994C>T | p.R332* | 1 | NK |
| Exon 8 | Frameshift | c.1011_1015delAAATG | p.R337Sfs*6 | 1 | I |
| Exon 8 | Frameshift | c.1060delC | p.L354Cfs*3 | 1 | NK |
| Exon 8 | Nonsense | c.1126G>T | p.E376* | 2 | I, NK |
| Exon 9 | Frameshift | c. 1129-1_1129dupGG | p.V377Gfs*13 | 1 | I |
| Exon 9 | Frameshift | c.1141dupA | p.R381Kfs*18 | 1 | H |
| Exon 9 | Missense | c.1156G>C | p.E386Q | 1 | NK |
| Exon 9 | Missense | c.1220a>c | p.Y407S | 1 | NK |
| Exon 9 | Nonsense | c.1221t>g | p.Y407* | 1 | H |
| Exon 9 | Frameshift | c.1268dupT | p.F424Lfs*24 | 1 | NK |
| Exon 9 | Missense | c.1276g>c | p.G426R | 1 | H |
| Intron 9 | Splice-site | c. 1277-9 A>C | p.? | 1 | H |
| Intron 9 | Splice-site | c.1277-8a>g | p.? | 1 | NK |
| Exon 10 | Frameshift | c.1279delG | p.E427Nfs*47 | 1 | NK |
| Exon 10 | Frameshift | c.1285_1286insGGATT | p.V429Gfs*47 | 2 | I, NK |
| Exon 10 | Frameshift | c.1293_1300delGTACCAGA | p.E431Dfs*14 | 1 | NK |
| Exon 10 | Frameshift | c.1371delA | p.K457Nfs*17 | 1 | H |
| Exon 10 | Nonsense | c.1398g>a | p.W466* | 1 | NK |
| Exon 10 | Nonsense | c.1402g>t | p.E468* | 1 | H |
| Exon 11 | Frameshift | c.1426_1450del | p.L476Gfs*22 | 1 | NK |
| Exon 11 | Nonsense | c.1441g>t | p.E481* | 1 | NK |
| Exon 11 | Nonsense | c.1451g>a | p.W484* | 1 | H |
| Exon 11 | Missense | c.1453g>a | p.D485N | 1 | P |
| Exon 11 | Nonsense | c.1456c>t | p.Q486* | 1 | H |
| Exon 11 | Frameshift | c.1477dupA | p.T493Nfs*6 | 1 | NK |
| Exon 11 | Missense | c.1486t>c | p.C496R | 1 | NK |
| Intron 11 | Splice-site | c. 1587-7_1587-4delCTTT | p.? | 1 | I |
| Exon 12 | Nonsense | c.1629t>g | p.Y543* | 1 | I |
| Exon 12 | Nonsense | c.1750c>t | p.R584* | 1 | NK |
| Exon 12 | Nonsense | c.1789C>T | p.R597* | 1 | NK |
| Exon 12 | Nonsense | c.1969C>T | p.Q657* | 2 | I, NK |
| Exon 12 | Nonsense | c.1981g>t | p.E661* | 1 | NK |
| Exon 12 | Frameshift | c.2291dupA | p.N764Kfs*49 | 1 | H |
| Exon 12 | Frameshift | c.2303 2309delAGCCCCG | p.E768Gfs*2 | 1 | NK |
| Exon 12 | Frameshift | c.2308delC | p.R770Gfs*2 | 2 | NK (n=2) |
| Exon 12 | Frameshift | c.2484delG | p.T829Qfs*10 | 1 | H |
| Exon 12 | Nonsense | c.2533G>T | p.E845* | 1 | I |
| Exon 12 | Nonsense | c.2695C>T | p.R899* | 3 | H (n=2), NK |
| Exon 12 | Nonsense | c.2730T>A | p.C910* | 1 | H |
| Exon 12 | Nonsense | c.2737C>T | p.Q913* | 1 | I |
GenBank reference sequence and version number for BMPR2: NM_001204.6; numbering is from +1 as A of the ATG initiation codon
Total number of independent cases. Frequencies in square brackets denote chromosomal rearrangements for which the breakpoints are unknown and may therefore represent distinct mutations
Key to abbreviations: H: heritable pulmonary arterial hypertension; I: idiopathic pulmonary arterial hypertension; NK: not known; P: pediatric pulmonary arterial hypertension
Table 2. BMPR2 variants of uncertain pathological significance.
| Location | Mutation category | Nucleotide change | Amino acid change | Clinical classification | Domain | Reference | Population frequency | a) PolyPhen-2 | b) PROVEAN | c) SIFT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prediction | Score | Prediction | Score | Prediction | Score | |||||||||||
| Exon 3 | Missense | c.266G>C | p.G89A | HPAH | ECD | Liu et al., 2012 | - | Possibly | 0.638 | Neutral | -1.785 | Tolerated | 0.137 | |||
| Exon 3 | Missense | c.276A>C | p.Q92H | HPAH | ECD | Kabata et al., 2013 | 0.0001071 | Benign | 0.001 | Neutral | -1.388 | Tolerated | 0.311 | |||
| Exon 3 | Missense | c.292G>A | p.E98K | IPAH | ECD | Wang et al., 2010 | - | Possibly damaging | 0.515 | Neutral | -1.792 | Tolerated | 0.092 | |||
| Exon 4 | Missense | c.461C>G | p.A154G | IPAH | TM | Pfarr et al., 2011 | - | Benign | 0.025 | Neutral | -1.994 | Damaging | 0.032 | |||
| Exon 6 | Missense | c.818T>G | p.M273R | IPAH | KD | Pfarr et al., 2011 | - | Benign | 0.004 | Deleterious | -2.716 | Tolerated | 0.213 | |||
| Exon 7 | Missense | c.901T>C | p.S301P | NK (n=2) | KD | Sztrymf et al., 2008; Girerd et al., 2010b | - | Benign | 0.046 | Neutral | -2.19 | Tolerated | 0.086 | |||
| Exon 7 | Missense | c.954A>C | p.E318D | HPAH | KD | This analysis | - | Benign | 0.089 | Neutral | -0.347 | Tolerated | 0.273 | |||
| Exon 8 | Missense | c.1042g>a | p.V348I | HPAH; IPAH (n=2) | KD | Wang et al., 2010; Liu et al., 2012 | 0.0004550 | Possibly damaging | 0.715 | Neutral | -0.45 | Damaging | 0.023 | |||
| Exon 8 | Missense | c.1066a>t | p.M356L | IPAH | KD | Wang et al., 2010 | - | Benign | 0.023 | Neutral | -0.497 | Tolerated | 0.417 | |||
| Exon 8 | Missense | c.1117g>c | p.A373P | IPAH | KD | Liu et al., 2012 | - | Probably damaging | 0.99 | Neutral | -2.493 | Tolerated | 0.087 | |||
| Exon 11 | Missense | c.1516a>g | p.M506V | IPAH | CD | This analysis | 0.0000412 | Benign | 0.008 | Neutral | -0.896 | Tolerated | 0.244 | |||
| Exon 12 | Missense | c.1598a>g | p.H533R | IPAH | CD | Pfarr et al., 2011 | - | Benign | 0.267 | Neutral | -2.049 | Damaging | 0.021 | |||
| Exon 12 | Missense | c.1766a>g | p.Y589C | CHD-APAH (exercise-induced) (n=2) | CD | Möller et al., 2010 | 0.0001320 | Probably damaging | 0.999 | Deleterious | -3.436 | Damaging | 0.001 | |||
| Exon 12 | Missense | c.2296A>G | p.T766A | IPAH; CTEPH | CD | Liu et al., 2012; Feng et al., 2014 | 0.0000083 | Benign | 0 | Neutral | -0.183 | Tolerated | 0.424 | |||
| Exon 12 | Missense | c.2618G>A | p.R873Q | NK (n=2) | CD | Sztrymf et al., 2008; Girerd et al., 2010b | 0.0001153 | Probably damaging | 0.966 | Neutral | -0.861 | Tolerated | 0.128 | |||
Population frequency data were obtained from the ExAC database (http://exac.broadinstitute.org/gene/ENSG00000204217). The likely pathogenicity of each missense variant was calculated by three in silico prediction methods, using the default parameters in each case. Ranges and cut-offs for output scores were as follows: a) PolyPhen-2 v2.2.2 (http://genetics.bwh.harvard.edu/pph2). Range: 0 – 1 [benign: 0 – 0.452; possibly damaging: 0.453 – 0.956; probably damaging: 0.957 – 1]; b) PROVEAN Human Protein v1.1 (http://provean.jcvi.org/protein_batch_submit.php?species=human). Cut-off: -2.5 [d
Distribution and Biological Significance of BMPR2 Variation
Although variation has been described across the entire coding structure of the gene, the mutation load differs significantly across exons, indicating both the likely existence of mutation hot-spots and potential regions of key functional importance. Taken together with previously reported findings, our analyses indicate that the majority of amino acid substitutions cluster in exons encoding the ligand-binding domain and key catalytic regions of the kinase domain, namely exons 2-3, 6-9 and 11 respectively. By contrast, exons 1, 4, 10 and 13, encoding receptor regions of uncertain importance to function, have a low frequency of missense mutation (Figure 2A). However, assessment of individual nucleotide defects within the BMPR2 open reading frame, relative to exon length, illustrates variant load by exon may be resultant on an abnormally high frequency of recurrence (e.g. exon 12) which corresponds to a low percentage of affected bases (Figure 2B). Conversely, exon 9 whilst harboring a relatively modest 6.7% of all reported coding variants, contains the highest proportion of independent nucleotide defects (n=30) relative to exon size (148 bp). In combination with previous reports, the majority of mutations predict incorporation of a premature termination codon in the mRNA (n=483, 72%) and, as previous functional studies have demonstrated, result in degradation of the message through the nonsense-mediated decay pathway [Aldred et al., 2007; Nasim et al., 2008].
Figure 2.
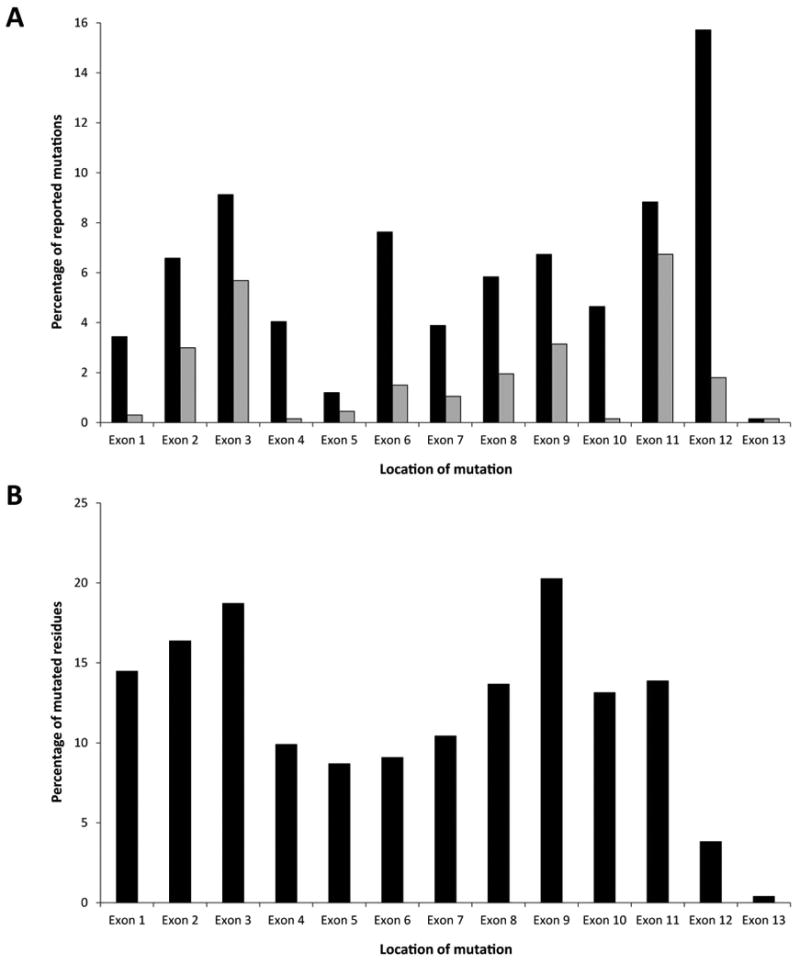
A) Distribution of reported exonic mutations across the BMPR2 gene. Black bars represent all mutation categories; grey bars indicate missense mutations only. This graph excludes data from non-coding regions and gene rearrangements for which start and/or end points have not been conclusively determined. B) Proportion of distinct point mutations relative to exon size. Multiple and/or recurrent variants at the same nucleotide were counted as a single event. The total number of mutated residues confined to the open-reading frame was calculated as a percentage of exon length in nucleotides.
Cysteine substitutions comprise the majority of missense mutations in the extracellular ligand-binding domain and are concentrated on 9 of 10 conserved residues, which are essential for the formation of five disulfide bridges necessary to maintain the integrity of this highly ordered three-dimensional structure (Figure 3) [Greenwald et al., 1999]. Moreover, this analysis has indicated the existence of additional critical residues. Notably, mutation of an asparagine (p.N126S), adjacent to the frequently mutated cysteine residue (p.C123R, p.C123S), is observed on seven independent occasions [Machado et al., 2009] (Table 1, Supp. Table S1), highlighting its putative significance to extracellular domain function. The majority of tested mutations in the kinase domain of BMPR-II abolish catalytic function as determined by in vitro BMP/SMAD luciferase reporter gene assays. By contrast, the significance of mutations within the cytoplasmic tail remains enigmatic, as these receptors retain significant capacity for downstream signaling through the SMAD family. Yet, these defects appear to perturb non-canonical pathways which include signaling through the cytoskeleton-associated factors LIMK-1 and Tctex-1 [Foletta et al., 2003; Machado et al., 2003]. In addition, studies have suggested that missense variants present across all the functional domains of BMPR-II trigger constitutive up-regulation of p38MAPK indicating a perturbation of one or more SMAD-independent pathways yet to be fully investigated [Nishihara et al., 2002; Rudarakanchana et al., 2002].
Figure 3.

Three-dimensional structure of the BMPR-II extracellular domain highlighting the location of substitutions impacting upon 9 of the 10 key cysteine residues responsible for disulfide bridge formation, indicated in dark blue. Defects in the Cys116 residue have not been identified in PAH thus far. Figure was reproduced from the crystal structure (PDB ID: 2HLQ) and processed using Cn3D v4.3 software.
Clinical Significance
Extracellular Domain Mutation Spectrum
Transient over-expression and subcellular localization of constructs harboring cysteine substitutions, specifically p.C60Y, p.C117Y, p.C118Y, p.C123R and p.C123S, have previously shown intracellular retention of these receptor species combined with a dramatic diminution of SMAD activation [Rudarakanchana et al., 2002]. Here, we report further cysteine substitutions likely to underlie structural variation in BMPR-II (p.C34R, p.C60G/R, p.C66G/R/Y, p.C84F/G/R, p.C94G/R, p.C99F/R/Y, p.C117R/S, p.C118W) (Figure 3). Utilization of these genetic observations with combinatorial functional studies facilitated an exploration of receptor rescue and restoration of signaling. By targeting the p.C118W mutant receptor with chemical chaperones, namely thapsigargin, glycerol or sodium 4-phenylbutyrate, a demonstrable and significant increase in plasma membrane localization of receptor species was observed concomitant with enhanced phosphorylation of SMADs 1 and 5. Rescued trafficking also led to an increase in the density of wild-type BMPR-II at the cell surface [Sobolewski et al., 2008]. This study provides an arresting example of how exploitation of genetic insights may lead to the potential development of future targeted therapeutic options in PAH.
Variants of Unknown Significance
Analyses of amino acid substitutions by PolyPhen, PROVEAN and SIFT bioinformatic tools [Kumar et al., 2009; Adzhubei et al., 2013; Choi and Chan, 2015] lead to ambiguous conclusions of pathogenicity in a proportion of observed missense variants reported herein. In the present dataset, a total of 13 variants are predicted by at least two algorithms to be non-damaging and/or benign (Table 2). In order to achieve greater confidence in assigning pathogenic status, it is important to assess such in silico predictions in the context of genomic data derived from substantive populations, for example the ExAC database. Comparison of missense variants against this combined cohort identified five variants with a population allele frequency greater than 0.000015, based on the most conservative measure of PAH prevalence in the literature (15 cases per million) [Archer et al., 2010]. Of interest, the two most commonly observed variants (p.V348I and p.Y589C) had been previously determined as damaging by at least two prediction methods and, therefore, had not been classified as variants of unknown significance (VUS). In the remaining three cases, the population data supported the in silico predictions which, taken together, provide further evidence for unclear pathogenicity (Table 2). One additional variant (p.T766A), previously designated VUS status by prediction algorithms, was present in the ExAC database but with a population allele frequency lower than our assigned threshold. Through these combined analytical techniques, we have identified a total of 15 variants that might be considered of uncertain significance from a genetic perspective (Table 2). While employing such analytical tools is of emerging value in mutation data interpretation, these analyses demonstrate that they must be treated with caution as: 1) the two approaches utilized may produce conflicting outputs, leading to ambiguity in interpretation; 2) large cohort datasets may contain study participants for whom the phenotype cannot be definitively assigned. Further, variants with a low population allele frequency in an apparently normal cohort may also be explained by the reduced penetrance of this condition. Together, this highlights a continuing role for functional studies, as a gold standard, to determine the true impact of observed variation in BMPR2, which is of value to both diagnostic and basic science understanding of the physiological role of this receptor. In addition, these observations indicate the likely existence of as yet unexplored pathways underlying pathogenesis.
Uncommon TGF-β Family Variation in PAH
Mutations of Receptor Species and Functional Outcomes
PAH infrequently clinically co-presents with the autosomal dominant vascular disorder hereditary hemorrhagic telangiectasia (HHT), characterized by the presence of mucocutaneous telangiectasia and visceral arteriovenous malformations. PAH-associated HHT is caused by molecular defects in ACVRL1, encoding a type I receptor of the TGF-β family, and to a lesser extent by mutations of the ENG gene which encodes a type III, or accessory receptor (Figure 1). In rare instances, ACVRL1 mutations have been identified in PAH patients without HHT but typically in early-onset disease thereby not precluding the development of the latter condition in later life [Harrison et al., 2003; Fujiwara et al., 2008]. Here we have compiled complete data on 66 mutations for both genes (ACVRL1, n=57; ENG, n=9), including 61 previously reported variants and 5 novel mutations underlying the development of PAH with and without HHT (Table 3). In ACVRL1, the majority of this variation occurs within the vital kinase domain of the protein (exons 6-10) resulting in pathogenic amino acid substitutions (n=42, 74%) in marked contrast to the BMPR2 pattern of predominantly truncating mutations and, indeed, the mutation spectrum in HHT alone. A recent study conducted in a cohort of 43 IPAH patients identified two missense variants in BMPR1B (p.S160N and p.F392L). However, contrary to previous reports, these variants induced SMAD9 signaling with concomitant induction of transcriptional activity [Chida et al., 2012a]. These studies suggest that further functional investigations are required for clarification of the pathogenic impact of these variants.
Table 3. PAH mutations identified in BMP pathway members.
| Gene name | Location | Mutation category | Nucleotide change | Amino acid change | Frequency in this study | Clinical classification | Reference(s) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ACVRL1 | Exon 2 | Frameshift | c.37delC | p.L13Cfs*2 | 1 | PAH-HHT | Trembath et al., 2001; Girerd et al., 2010a | |||
| ACVRL1 | Exon 3 | Missense | c.199C>T | p.R67W | 1 | PAH-HHT | Chen et al., 2013 | |||
| ACVRL1 | Exon 3 | Missense | c.293A>G | p.N98S | 1 | NK | This analysis | |||
| ACVRL1 | Exon 4 | Nonsense | c.430C>T | p.R144* | 1 | I | Machado et al., 2009 | |||
| ACVRL1 | Exon 5 | Missense | c.536A>C | p.D179A | 1 | I | Harrison et al., 2003ˆ | |||
| ACVRL1 | Exon 5 | Missense | c.593T>A | p.V198E | 1 | PAH-HHT | Chen et al., 2013 | |||
| ACVRL1 | Exon 5 | Missense | c.602A>G | p.Q201R | 1 | PAH-HHT | Girerd et al., 2010a | |||
| ACVRL1 | Exon 6 | Missense | c.632G>A | p.G211D | 1 | PAH-HHT | Harrison et al., 2003 | |||
| ACVRL1 | Exon 6 | Missense | c.653_654inv | p.R218P | 1 | PAH-HHT | Chen et al., 2013 | |||
| ACVRL1 | Exon 6 | Deletion | c.760_762delGAC | p.D254del | 1 | PAH-HHT | Trembath et al., 2001 | |||
| ACVRL1 | Exon 7 | Missense | c.788A>G | p.D263G | 1 | PAH-HHT | This analysis | |||
| ACVRL1 | Exon 7 | Missense | c.818T>C | p.L273P | 1 | PAH-HHT | Smoot et al., 2009 | |||
| ACVRL1 | Exon 7 | Missense | c.853C>T | p.L285F | 1 | NK | This analysis | |||
| ACVRL1 | Exon 7 | Missense | c.854T>C | p.L285P | 1 | P | Chida et al., 2012b | |||
| ACVRL1 | Exon 7 | Missense | c.936C>G | p.H312Q | 1 | P | Fujiwara et al., 2008ˆ | |||
| ACVRL1 | Exon 7 | Missense | c.950T>C | p.I317T | 1 | P | Pfarr et al., 2013 | |||
| ACVRL1 | Exon 7 | Missense | c.955G>C | p.G319R | 1 | NK | This analysis | |||
| ACVRL1 | Exon 7 | Missense | c.1031g>a | p.C344Y | 2 | PAH-HHT (n=2) | Harrison et al., 2003 | |||
| ACVRL1 | Exon 8 | Missense | c.1055c>a | p.A352D | 2 | PAH-HHT (n=2) | Smoot et al., 2009 | |||
| ACVRL1 | Exon 8 | Missense | c.1120C>T | p.R374W | 3 | PAH-HHT (n=2), NK | Harrison et al., 2003; Abdalla et al., 2004; This analysis | |||
| ACVRL1 | Exon 8 | Missense | c.1121G>A | p.R374Q | 2 | PAH-HHT (n=2) | Harrison et al., 2003; Chen et al., 2013 | |||
| ACVRL1 | Exon 8 | Missense | c.1124A>G | p.Y375C | 1 | PAH-HHT | Chen et al., 2013 | |||
| ACVRL1 | Exon 8 | Missense | c.1142T>C | p.L381P | 1 | P | Fujiwara et al., 2008ˆ | |||
| ACVRL1 | Exon 8 | Missense | c.1195T>C | p.W399R | 1 | PAH-HHT | Chen et al., 2013 | |||
| ACVRL1 | Exon 8 | Missense | c.1196G>C | p.W399S | 1 | PAH-HHT | Harrison et al., 2003 | |||
| ACVRL1 | Exon 8 | Missense | c.1196G>T | p.W399L | 1 | PAH-HHT | Ishiwata et al., 2014 | |||
| ACVRL1 | Exon 8 | Missense | c.1231c>t | p.R411W | 1 | PAH-HHT | Trembath et al., 2001 | |||
| ACVRL1 | Exon 8 | Missense | c.1232g>a | p.R411Q | 1 | PAH-HHT | Harrison et al., 2003 | |||
| ACVRL1 | Exon 9 | Missense | c.1270c>a | p.P424T | 1 | P | Fujiwara et al., 2008ˆ | |||
| ACVRL1 | Exon 9 | Missense | c.1280a>t | p.D427V | 1 | PAH-HHT | Girerd et al., 2010a | |||
| ACVRL1 | Exon 9 | Missense | c.1324g>a | p.V442M | 1 | PAH-HHT | Girerd et al., 2010a | |||
| ACVRL1 | Exon 10 | Nonsense | c.1385C>G | p.S462* | 1 | PAH-HHT | Abdalla et al., 2004 | |||
| ACVRL1 | Exon 10 | Frameshift | c.1388delG | p.G463Afs*2 | 1 | PAH-HHT | Girerd et al., 2010a | |||
| ACVRL1 | Exon 10 | Frameshift; Nonsense | c.1388delG; c.1390delC | p.G463Afs*2; p.L464* | 1 | PAH-HHT (mosaic) | Eyries et al., 2012 | |||
| ACVRL1 | Exon 10 | Missense | c.1433C>A | p.A478D | 1 | P | Chida et al., 2012b | |||
| ACVRL1 | Exon 10 | Nonsense | c.1435c>t | p.R479* | 2 | PAH-HHT (n=2) | Abdalla et al., 2004; Chen et al., 2013 | |||
| ACVRL1 | Exon 10 | Missense | c.1436g>a | p.R479Q | 1 | P | Fujiwara et al., 2008ˆ | |||
| ACVRL1 | Exon 10 | Missense | c.1436g>c | p.R479P | 1 | I | Machado et al., 2009 | |||
| ACVRL1 | Exon 10 | Missense | c.1450C>G | p.R484G | 1 | H | Jones et al., 2014 | |||
| ACVRL1 | Exon 10 | Missense | c.1450c>t | p.R484W | 2 | PAH-HHT (n=2) | Trembath et al., 2001; Girerd et al., 2010a | |||
| ACVRL1 | Exon 10 | Frameshift | c.1450delinsTG | p.R484Wfs*10 | 1 | PAH-HHT | Abdalla et al., 2004 | |||
| ACVRL1 | Exon 10 | Missense | c.1451g>a | p.R484Q | 7 | P (n=2); PAH-HHT (n=3; 1 mosaic); NK (n=2) | Harrison et al., 2005ˆ; Fujiwara et al., 2008ˆ; Best et al., 2011; Chen et al., 2013; Pfarr et al., 2013; This analysis | |||
| ACVRL1 | Exon 10 | Missense | c.1460a>c | p.K487T | 1 | PAH-HHT | Harrison et al., 2003 | |||
| ACVRL1 | Exon 10 | Nonsense | c.1468c>t | p.Q490* | 1 | PAH-HHT | Trembath et al., 2001; Girerd et al., 2010a | |||
| ENG | Exon 5 | Missense | c.640G>A | p.G214S | 1 | P | Pfarr et al., 2013 | |||
| ENG | Exon 5 | Frameshift | c.682_686delTCGGC | p.S228Rfs*104 | 1 | PAH-HHT | Harrison et al., 2003ˆ | |||
| ENG | Exon 6 | Missense | c.788T>A | p.I263N | 1 | PAH-HHT | Chen et al., 2013 | |||
| ENG | Exon 11 | Frameshift | c.1334delT | p.M445Rfs*46 | 1 | PAH-HHT | Harrison et al., 2003ˆ | |||
| ENG | Exon 11 | Frameshift | c.1410delG | p.Q471Sfs*20 | 1 | PAH-HHT + dexfenfluramine | Chaouat et al., 2004ˆ | |||
| ENG | Exon 12 | Missense | c.1633g>a | p.G545S | 1 | CHD-PAH | Pfarr et al., 2013 | |||
| ENG | Intron 13 | Branch-site | c.1742-22T>C | p.C582 R618del | 1 | PAH-HHT | Harrison et al., 2005ˆ; Mache et al., 2008ˆ | |||
| ENG | Exon 14 | Frameshift | c.1804delA | p.I602Sfs*38 | 1 | PAH-HHT | Chen et al., 2013 | |||
| ENG | Exon 15 | Missense | c.1853G>T | p.R618L | 1 | I | This analysis | |||
| SMAD1 | Exon 2 | Missense | c.8T>C | p.V3A | 1 | I | Nasim et al., 2011 | |||
| SMAD4 | Exon 2 | Missense | c.38A>G | p.N13S | 1 | I | Nasim et al., 2011 | |||
| SMAD4 | Intron 11 | Splice-site | c.1448-6t>c | p.? | 1 | I | Nasim et al., 2011 | |||
| SMAD9 | Exon 2 | Missense | c.127a>g | p.K43E | 1 | I | Nasim et al., 2011 | |||
| SMAD9 | Exon 3 | Nonsense | c.606C>A | p.C202* | 1 | I | Shintani et al., 2009 | |||
| SMAD9 | Exon 5 | Nonsense | c.880C>T | p.R294* | 1 | H | Drake et al., 2011 | |||
GenBank reference sequence and version number for ACVRL1: NM_000020.2; ENG: NM_001114753.2; SMAD1: NM_005900.2; SMAD4: NM_005359.5; SMAD9: NM_001127217.2
Total number of independent cases
Previously reported in Machado et al., 2009
Key to abbreviations: H: heritable pulmonary arterial hypertension; I: idiopathic pulmonary arterial hypertension; NK: not known; P: pediatric pulmonary arterial hypertension; PAH-HHT: pulmonary arterial hypertension with hereditary hemorrhagic telangiectasia
Mutations within Intracellular Partners of the BMP Signaling Pathway
Conventional functional candidate gene strategies conducted in Asian and European patient panels have subsequently identified independent mutations in the BMP-specific SMAD pathway, namely SMAD1 (n=1), SMAD4 (n=2) and SMAD9 (n=3) [Shintani et al., 2009; Drake et al., 2011; Nasim et al., 2011] (Table 3). Of these, the SMAD1 and -4 defects have been described as VUS due to in vitro luciferase SMAD responsive elements reporter assays demonstrating an unclear impact on the canonical pathways [Nasim et al., 2011]. However, these analyses did not investigate SMAD-independent pathways, implicated in disease pathogenesis, leaving open the possibility that the identified variants may deleteriously affect other BMP related systems. By contrast, SMAD9 mutations (PPH2; MIM# 615342) lead to a marked reduction of SMAD transcriptional activity and a down-regulation of the BMP target gene Id1 [Shintani et al., 2009; Nasim et al., 2011]. Of interest, heterozygous SMAD9 mutations have been observed to perturb non-canonical downstream pathways, in particular, micro-RNA (miRNA) processing. Examination of a human patient with a SMAD9 nonsense mutation (p.R294*) indicated a modest reduction of Id1 expression in contrast to a complete abrogation of miR-21. In vitro restoration of miR-21 by over-expression led to a reversal of the hyperproliferative mutation-positive phenotype. These data suggest a specific role for SMAD8 in PAH pathogenesis and SMAD4-independent signaling [Drake et al., 2011].
Genotype-Phenotype Correlation in Risk Alleles of the BMP Pathway
In 53-86% of patients with familial aggregation and 14-35% of IPAH patients mutations in the BMPR2 gene have been identified [Sztrymf et al., 2008; Girerd et al., 2010a; Pfarr et al., 2011; Liu et al., 2012; Kabata et al., 2013]. Patients who carry BMPR2 mutations differ in several important aspects from IPAH patients who are BMPR2-negative [Soubrier et al., 2013]. Investigators have reported that HPAH patients with pathogenic variants in BMPR2 develop this disorder at a younger age (38.53 ± 12.38 vs. 45.78 ± 11.32 years, p <0.001) [Girerd et al., 2010b; Pfarr et al., 2011], and have a more severe clinical and hemodynamic phenotype at diagnosis [Koehler et al., 2004; Sztrymf et al., 2008; Austin et al., 2009a; Pfarr et al., 2011]. BMPR2 mutation carriers undergo diagnostic catheterization almost 10 years earlier than patients with no identified BMPR2 defect. Furthermore, compared with BMPR2-negative IPAH patients, BMPR2-positive patients have a higher pulmonary vascular resistance measured at diagnostic catheterization, are less likely to demonstrate acute vasoreactivity [Elliott et al., 2006; Rosenzweig et al., 2008], and are more likely to progress to death or lung transplantation [Sztrymf et al., 2008]. These observations all suggest that BMPR2 mutations lead to a more severe PAH phenotype. Of note, patients with missense variants present with a higher degree of morbidity and mortality than those with truncating defects, suggestive of a more severe impact on the signaling pathway [Austin et al., 2009b]. However, BMPR2 mutations are not associated with a worse exercise capacity and prognosis. The younger age of BMPR2 mutation carriers may explain the similar survival and exercise capacity despite worse hemodynamics as compared with BMPR2-negative patients. Most recently, Girerd et al. indicated that BMPR2 mutation position may influence clinical phenotype. Specifically, PAH patients with a point mutation within the cytoplasmic tail of BMPR2 displayed a later age of onset, lower pulmonary vascular resistance and, of note, a higher proportion of acute vasodilator response by contrast to patients harboring mutations outside of this domain. In addition, in vitro assays suggested that cytoplasmic domain mutations tolerated activation of the Smad pathway, which is indicative of a lower degree of penetrance [Girerd et al., 2015].
These findings appear not to be influenced by gender [Girerd et al., 2010b]. However, there is a trend for more severe prognosis of the disease in males, particularly in male BMPR2 mutation carriers. This observation is consistent with the observation that PAH mortality is most closely associated with male gender [Humbert et al., 2010]. Even though no significant impact of gender was observed on age at diagnosis and outcomes, it should be emphasized that PAH mostly occurs in females, irrespective of BMPR2 status (sex ratio females:males = 2.4:1 in both BMPR2 mutation carriers and non-carriers). To explain over-representation of female patients it has been suggested that estrogens and estrogen metabolism might be involved in the pathogenesis of PAH [West et al., 2008a; Austin et al., 2009a; Mair et al., 2015]. These studies support the hypothesis that altered estrogen metabolism could contribute to the penetrance of PAH in women and suggest Cytochrome P450 1B1 (CYP1B1) as a sex-specific modifier gene.
Similar findings are observed with ACVRL1 mutations with a significant number of pediatric cases and a dismal prognosis [Girerd et al., 2010a]. In this study, ACVRL1 mutation carriers were shown to be characterized by a younger age at PAH diagnosis (21.8 ± 16.7 years) than BMPR2 mutation carriers and non-carriers (35.7 ± 14.9 and 47.6 ± 16.3 years, respectively; p <0.0001). However, ACVRL1-positive patients had better hemodynamic status at diagnosis, but none responded to acute vasodilator challenge. Thus, despite less severe initial hemodynamics and similar management, these patients had a worse prognosis than other patients with PAH, suggesting more rapid disease progression.
Most recently, a ‘two-hit’ model has been proposed, wherein digenic mutations may account for earlier occurrence, increased severity and more rapid deterioration of PAH patients [Wang et al., 2014].
Expansion of the Genetic Architecture of PAH by Next-Generation Sequence Analysis
Caveolin 1 (CAV1)
Whole-exome sequencing was used to study one large family with six PAH cases across three generations with autosomal dominant transmission and without known mutation. Specifically, the exomes of 4 of the 6 PAH patients were evaluated and following bioinformatic analyses 11 rare candidate variants were determined to be shared by all four patients in the family. Genetic analysis of an additional patient in the family supported the conclusion that a rare mutation in the CAV1 gene (PPH3; MIM# 615343) was of pertinence to disease. The observed mutation in exon 3 (c.474delA; p.L159Sfs*22), impacts a highly conserved region and predicts deleterious functional consequences.
An additional 62 independent PAH families and 198 unrelated idiopathic PAH patients, all without detectable TGF-β gene mutations, were screened for CAV1 mutations by Sanger sequencing. Of 260 patients one early-onset idiopathic patient harbored a de novo CAV1 mutation in exon 3 of the gene (c.473delC; p.P158Hfs*23) (Table 4). Of note, identified variants were not present in over 1000 ethnically-matched Caucasian controls [Austin et al., 2012].
Table 4. PAH mutations identified in non-canonical BMP pathways.
| Gene name | Location | Mutation category | Nucleotide change | Amino acid change | Frequency in this study | Clinical classification | Reference(s) |
|---|---|---|---|---|---|---|---|
| CAV1 | Exon 3 | Frameshift | c.473delC | p.P158Hfs*23 | 1 | I | Austin et al., 2012 |
| CAV1 | Exon 3 | Frameshift | c.474delA | p.L159Sfs*22 | 1 | H | Austin et al., 2012 |
|
| |||||||
| KCNA5 | Exon 1 | Missense | c.544G>A | p.G182R | 2 | I | Remillard et al., 2007 |
| KCNA5 | Exon 1 | Missense | c.633G>C | p.E211D | 2 | I | Remillard et al., 2007 |
| KCNA5 | Exon 1 | Frameshift | c.1448delA | p.Y483Sfs*4 | 1 | H | Wang et al, 2014ˆ |
|
| |||||||
| KCNK3 | Exon 1 | Missense | c.23C>A | p.T8K | 1 | I | Ma et al., 2013 |
| KCNK3 | Exon 2 | Missense | c.289G>A | p.G97R | 1 | H | Ma et al., 2013 |
| KCNK3 | Exon 2 | Missense | c.544G>A | p.E182K | 1 | I | Ma et al., 2013 |
| KCNK3 | Exon 2 | Missense | c.575A>G | p.Y192C | 1 | I | Ma et al., 2013 |
| KCNK3 | Exon 2 | Missense | c.608G>A | p.G203D | 1 | H | Ma et al., 2013 |
| KCNK3 | Exon 2 | Missense | c.661G>C | p.V221L | 1 | H | Ma et al., 2013 |
GenBank reference sequence and version number for CAV1: NM_001753.4; KCNA5: NM_002234.3; KCNK3: NM_002246.2
Total number of independent cases
Reported as a compound heterozygote with a BMPR2 c.1471C>T (p.R491W) mutation
Key to abbreviations: H: heritable pulmonary arterial hypertension; I: idiopathic pulmonary arterial hypertension
Potassium Channel, Subfamily K, Member 3 (KCNK3)
Most recently, whole-exome sequencing has led to the identification of KCNK3 as a risk factor for familial and idiopathic disease (PPH4; MIM# 615344). By screening three affected subjects from an autosomal dominant family negative for mutation in BMPR2, ACVRL1, ENG, SMAD9 and CAV1, Ma et al. detected a novel coding variant of KCNK3 that was shared among all three subjects and predicted to be pathogenic by in silico bioinformatic tools. Subsequent Sanger sequencing across the extended family confirmed co-segregation of the disease with the c.608G>A (p.G203D) variant, which was absent from 100 ethnically matched control individuals [Ma et al., 2013]. Analysis of exome sequence from 10 further HPAH probands identified two additional novel heterozygous variants, which also segregated with disease, providing strong evidence for a role of KCNK3 in PAH pathogenesis. To assess the frequency of KCNK3 variation in familial and idiopathic disease, an extended cohort of 82 HPAH and 230 IPAH cases were screened for mutation. Three novel heterozygous missense variants were detected in the idiopathic cohort, suggesting that KCNK3 mutation accounts for 1.3% of IPAH cases and 3.2% of HPAH families [Ma et al., 2013] (Table 4).
Eukaryotic Translation Initiation Factor 2 Alpha Kinase 4 (EIF2AK4)
Similarly, exome sequencing has revealed several disease causing mutations in the EIF2AK4 gene in pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH), together classified as group 1′ of PAH in the most recent diagnostic classification [Simonneau et al., 2013]. Both conditions are inherited in an autosomal recessive manner (PVOD2; MIM# 234810) and are mainly characterized by proliferation of capillaries in the lung leading to an occlusion of pulmonary vasculature. Eyries et al. assessed five families with PVOD and focused on rare variants (minor allele frequency <0.1% in control populations), which were homozygous or compound heterozygous in affected children and heterozygous in unaffected parents. Using this analysis strategy, mutations in EIF2AK4 were identified in all 13 families studied. The examination of 20 sporadic cases revealed EIF2AK4 mutations in 5 additional patients [Eyries et al., 2014]. Mutations were distributed throughout the gene and belonged to the major mutation categories (Table 5). The range of mutations situated upon this locus underlines its functional importance in the development of PVOD.
Table 5. PVOD/PCH mutations identified in EIF2AK4.
| Location | Mutation category | Nucleotide change | Amino acid change | Frequency in this study | Clinical classification | Reference(s) |
|---|---|---|---|---|---|---|
| Exon 3; Intron 9 | Frameshift; Splice-site | c.354 355delTG; c.1554-4C>A | p.C118Wfs*7; p.C519Dfs*17 | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 5 | Frameshift | c.560_564delAAGAA; c.560 564delAAGAA | p.K187Rfs*9; p.K187Rfs*9 | 1 | PVOD-S | Eyries et al., 2014 |
| Exon 5 | Frameshift | c.567dupG; c.567dupG | p.K190Efs*8; p.K190Efs*8 | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 7; Exon 12 | Nonsense; Frameshift | c.745C>T; c.2136 2139dupCACT | p.R249*; p.S714Hfs*21 | 1 | PVOD-F | Eyries et al., 2014 |
| Intron 7; Exon 25 | Splice-site; Nonsense | c.860-1G>A; c.3448C>T | p.?; p.R1150* | 1 | PCH | Best et al., 2014 |
| Exon 9; Exon 28 | Frameshift; Nonsense | c.1153dupG; c.3766C>T | p.V385Gfs*30; p.R1256* | 1 | PCH | Best et al., 2014 |
| Exon 9 | Nonsense | c.1387c>t; c.1387c>t | p.R463*; p.R463* | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 9; Exon 23 | Nonsense; Nonsense | c.1387C>T; c.3244C>T | p.R463*; p.Q1082* | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 9 | Frameshift | c.1392delT; c.1392delT | p.R465Vfs*38; p.R465Vfs*38 | 1 | PCH | Best et al., 2014 |
| Exon 9; Exon 28 | Frameshift; Nonsense | c.1392delT; c.3802C>T | p.R465Vfs*38; p.Q1268* | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 11 | Missense | c.1754g>a; c.1754g>a | p.R585Q; p.R585Q | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 12 | Missense | c.1928t>g; c.1928t>g | p.L643R; p.L643R | 1 | PVOD-F | Eyries et al., 2014 |
| Intron 13 | Splice-site | c.2319+1G>A; c.2319+1G>A | p.?; p.? | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 15 | Nonsense | c.2458C>T; c.2458C>T | p.R820*; p.R820* | 1 | PVOD-S | Eyries et al., 2014 |
| Exon 19; Intron 25 | Nonsense; Splice-site | c.2857C>T; c.3576+1G>T | p.Q953*; p.? | 1 | PVOD-S | Eyries et al., 2014 |
| Exon 21 | Splice-site | c.3159G>A; c.3159G>A | p.K975 K1053del; p.K975 K1053del | 2 | PVOD-F, -S | Eyries et al., 2014 |
| Exon 23 | Missense | c.3344C>T; c.3344C>T | p.P1115L; p.P1115L | 5 | H | Tenorio et al., 2014 |
| Exon 24 | Nonsense | c.3406C>T; c.3406C>T | p.R1136*; p.R1136* | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 25; Intron 36 | Nonsense; Splice-site | c.3448C>T; c.4728+1 4728+13delinsTTCT | p.R1150*; p.? | 1 | PVOD-F | Eyries et al., 2014 |
| Intron 29 | Splice-site | c.4065+1G>C; c.4065+1G>C | p.?; p.? | 1 | PVOD-F | Eyries et al., 2014 |
| Exon 31 | Frameshift | c.4205dupT; c.4205dupT | p.S1403Kfs*45; p.S1403Kfs*45 | 1 | PVOD-S | Eyries et al., 2014 |
GenBank reference sequence and version number for EIF2AK4: NM_001013703.3
Total number of independent cases
Key to abbreviations: H: heritable pulmonary arterial hypertension; PCH: pulmonary capillary hemangiomatosis; PVOD: pulmonary veno-occlusive disease (-F: familial; -S: sporadic)
In parallel, mutations in EIF2AK4 were independently identified by Best et al. in patients with heritable PCH. The exomes of two affected brothers were sequenced and filtered for variants with a minor allele frequency of less than 1% in public repositories of polymorphic data resulting in the identification of two pathogenic variants in the gene. The unaffected parents and sister were confirmed to be heterozygous carriers of one of the two mutations [Best et al., 2014]. Further sequencing of EIF2AK4 in 10 patients with pathologically verified sporadic PCH and one familial case detected three additional mutations in two sporadic patients (Table 5). The genetic correlation between PVOD and PCH further supports the likelihood that these diseases define different clinical spectra of the same underlying disorder.
Most recently, EIF2AK4 has also been investigated in an itinerant Iberian population with PAH [Tenorio et al., 2015]. The authors identified a homozygous c.3344C>T (p.P1115L) missense mutation in five patients from five independent families with HPAH. Likely ancestral, this mutation co-segregated with a more severe phenotype than previously reported for other EIF2AK4 mutations. The majority of affected subjects presented with an early onset, aggressive form of the disease resulting in an abnormally low survival rate post-lung transplantation (1.1 years).
Biological Significance of Mutations in Non-Canonical BMP Pathways
While rare, the biologic plausibility for CAV1 mutations in PAH is strong. CAV1 encodes Caveolin-1, a membrane protein required to form the flask-shaped invaginations of the cell membrane known as caveolae, abundant in lung endothelial and mesenchymal cells [Minshall et al., 2003; Xu et al., 2008]. Caveolae are critical to a number of cellular processes and receptor rich regions of the cell membrane [Nohe et al., 2005; Mercier et al., 2009; Chidlow and Sessa, 2010]. Intriguingly, mice haploinsufficient for Cav1 display pulmonary vascular disease analogous to PAH [Drab et al., 2001; Zhao et al., 2002; Murata et al., 2007; Maniatis et al., 2008]. Moreover, Caveolin-1 protein staining is reduced in lung endothelial cells from human PAH patients [Zhao and Malik, 2009]. Nevertheless, although under careful scrutiny, the precise mechanism(s) by which CAV1 mutations promote PAH remain unclear.
The KCNK3 gene is located on chromosome 2p24 and encodes a pH-sensitive potassium channel with a role in regulation of resting membrane potential in a variety of cell types. Electrophysiological analyses demonstrated that all identified mutations lead to a loss of function as measured by current density. A subset of identified mutations (p.T8K, p.E182K and p.G203D) exhibited a significant increase in potassium-channel current when treated with the phospholipase inhibitor ONO-RS-082. The identification of this gene provides an additional avenue of treatment strategies for PAH.
The EIF2AK4 gene encodes a kinase, which phosphorylates an initiation factor in protein synthesis that is primarily responsible for the translation of stress response proteins [Donnelly et al., 2013]. Interactions between the kinase and various members of the BMPR-II pathway have also been detected, but the exact link to the clinical manifestations of pulmonary veno-occlusive PH remains to be clarified [Eyries et al., 2014]. The EIF2AK4 protein belongs to a family of kinases that regulate angiogenesis in response to cellular stress. Of interest, these properties are compatible with the angiogenic pathology of PCH, a disorder characterized by uncontrolled proliferation of pulmonary microvessels.
Modifying Predisposition to PAH
Cerebellin 2 Precursor (CBLN2)
PAH demonstrates several complex traits including incomplete penetrance, sex bias and variable age of disease onset both within and across families. This has led to the hypothesis that modifier genes contribute to disease manifestation and/or progression. To examine the role of common variation in PAH predisposition, Germain et al. performed a genome-wide association study (GWAS) to identify susceptibility loci in IPAH and HPAH cases without BMPR2 mutation. The discovery dataset comprised 340 PAH patients and 1,068 healthy controls, genotyped for ∼470,000 variants, from which the 384 most significant variants were assessed in an independent replication cohort of 285 cases and 457 controls. This approach, which represents the best powered study to date, identified two variants 52 kb downstream of the CBLN2 gene that were associated with a two-fold increased risk of disease [Germain et al., 2013].
CBLN2 encodes a secreted neuronal glyocoprotein primarily expressed in the brain. However, real-time PCR studies demonstrated CBLN2 mRNA expression in the whole lung, significantly higher in explants from PAH patients comparative to histologically normal lung tissue. Similar results were obtained for pulmonary arterial endothelial cells. Furthermore, an inhibition of pulmonary artery smooth muscle cell (PASMC) proliferation was observed when treated with increasing concentrations of CBLN2 peptide [Germain et al., 2013].
Potassium Channel, Voltage Gated Shaker Related Subfamily A, Member 5 (KCNA5)
Membrane potential is essential for contraction and vasodilation of PASMCs. Expression of potassium channels is regulated by BMP signaling in vitro and in vivo and knockouts in Drosophila melanogaster result in defects that are strikingly similar to phenotypes that result from disrupted TGF-β/BMP signaling [Young et al., 2006; Dahal et al., 2012]. Variation of the potassium channel KCNA5 has been identified in IPAH patients [Remillard et al., 2007; Wang et al., 2014] suggesting a potential role in PAH development and penetrance. The potassium channel response in PASMCs of IPAH patients has been proven to be down regulated [Yuan et al., 1998] likely increasing pulmonary vasoconstriction and PASMC proliferation. Moreover, the channel, which is responsive to nitric oxide, is reduced in patients carrying a specific coding missense mutation [Remillard et al., 2007]. Mutations within KCNA5 have been identified as a so-called ‘second hit’ in an index patient additional to a BMPR2 missense mutation leading to an early onset and severe phenotype [Wang et al., 2014]. While these findings may represent a rare case of genetic modification in PAH, replication of this digenic genotype in an independent cohort has not been observed. Further, in the absence of comprehensive functional analysis, interpretation of the significance of this study requires caution.
Animal Models of PAH
Model systems of PAH serve a two-fold purpose in providing 1) a source of relevant, genetically modified cell types for in vitro studies, and 2) an in vivo platform for the analysis and refinement of direct therapeutic intervention. Several natural and engineered models of disease exist, for example, the fawn-hooded rat and the Bmpr2 transgenic mouse over-expressing the p.R899* mutant allele, the latter being generated by introduction of a smooth muscle-specific doxycycline-inducible mutant transgene [West et al., 2008b; Ryan et al., 2011]. Further, knock-out models lacking exons 4 and 5 of Bmpr2 generated by homologous recombination have been developed. These models develop mild to moderate disease phenotypes, often upon the application of environmental insults including exposure to hypoxia and 5-lipoxygenase as previously described [Machado et al., 2006]. Conditional targeting of mutant alleles to hallmark sites of damage in PAH, in particular the pulmonary artery endothelial or smooth muscle cell layers, has produced a more convincing in vivo reflection of the human disease state [West et al., 2004; Hong et al., 2008]. Most recently a heterozygous knock-in mouse model of the p.R899* mutation has been shown to develop age-related disease with close phenotypic relatedness to the human condition [Long et al., 2015]. Additionally a rat model of PAH (BMPR2Δ140Ex1/+), the first of its kind, has provided support to the hypothesis that endothelial-to-mesenchymal transition represents a pathophysiological process in PAH [Ranchoux et al., 2015]. Of note, mice representative of recently observed defects in previously uncharacterized genes, namely Cav1 and Smad9, support the emerging concept of greater than anticipated genetic heterogeneity in PAH. Homozygous knock-out models of both genes develop spontaneous indications of PAH providing a powerful correlation to the human studies described herein [Zhao et al., 2002; Huang et al., 2009].
Genetic Counseling
Current guidelines recommend offering molecular genetic analysis and genetic counseling for HPAH patients [Badesch et al., 2007; McLaughlin et al., 2009]. Genetic counseling typically offers a combinatorial approach of dealing with putative outcomes and consequences of the analysis, based upon structured protocols determined by specialist centers evaluates the family history, educates patients about the causes of PAH, discusses the risks and benefits of genetic testing and supports patients and families through the process of genetic testing and disclosure of results. In addition to HPAH patients, IPAH patients and their relatives may also benefit from these analyses since up to 25% will have a BMPR2 mutation; therefore, genetic testing in these populations should be considered [Badesch et al., 2007]. Current findings indicate that in patient populations with PVOD and PCH, autosomal recessive inheritance of the EIF2AK4 gene is the most likely mode of transmission. Hence, within the PAH spectrum of disease, based on current classification, genetic counseling should take account of both autosomal dominant and recessive inheritance models, as well as the higher penetrance of bi-allelic mutations in patients carrying this genetic defect.
Following identification of an established molecular defect in the proband, at-risk asymptomatic family members should be offered the option of genetic counseling and targeted mutation analysis within the parameters of full informed consent. Importantly, the data provided here offer an indication of the likely pathogenicity of identified variation. A significant factor to consider is the reduced penetrance of mutant alleles which must be addressed by informed pre-test counseling, comprising robust estimations of risk based on available population-specific epidemiological data and gender [Cogan et al., 2012; Larkin et al., 2012]. Of note, where a mutation is identified, implications for reproductive planning must be considered. In families harboring mutation, early mutational analysis of offspring is essential and should be combined with clinical assessment and initiation of treatment as deemed necessary by specialist centers. Additionally, a possible future avenue of medical care has been explored. Within a BMPR2-positive family, pre-implantation genetic analysis of blastomeres following in vitro fertilization led to the successful implantation and delivery of a mutation-negative offspring [Frydman et al., 2012]. Although in its earliest stages this strategy is of significant clinical potential in the context of considered, long-term genetic evaluation.
Diagnostic Strategies
Molecular diagnostics for PAH have traditionally focused around dideoxy Sanger sequencing methods to screen the BMPR2, ACVRL1 and ENG protein-coding regions for heterozygous mutation. However, based on these and previous studies, for a complete exploration of deletion and duplication across these three genes, the application of MLPA or targeted comparative genomic hybridization (CGH) array technology is required. The HHT/PPH1 MLPA panel was introduced by MRC-Holland in the mid-2000s, with the current version containing 51 probes across BMPR2, ACVRL1 and ENG, and has led to the successful detection of numerous gene rearrangements that would otherwise not be identified by sequence analysis [Aldred et al., 2006; Cogan et al., 2006].
Despite the success of this combined approach to mutation detection in PAH, the recent expansion of candidate disease genes has resulted in traditional sequencing methods becoming more labor-intensive and less cost-effective. Indeed, to comprehensively screen the 10 genes detailed here would involve sequencing approximately 105 coding exons, totaling over 19 kb of DNA sequence. Custom capture and NGS are now becoming chosen methodologies across screening centers globally for the analysis of established candidate genes.
Future Prospects
It is clear that the most significant advance in the identification of risk factors for PAH over recent years has been the advent of exome sequencing, which has led to the rapid identification of multiple novel genes using relatively small sample sets. Whilst exome sequencing is undoubtedly a powerful method for detecting rare, highly penetrant genes in families with multiple affected individuals, there remain important caveats in applying these technologies to the identification of novel genes across cohorts of unrelated subjects and genetically divergent patient groups. In PAH, complexities such as locus heterogeneity, incomplete penetrance, de novo mutation and late onset of disease introduce significant challenges to the interpretation of exome sequence data, highlighting the need for large homogeneous patient cohorts to detect pathogenic variation. In terms of technical limitations, exome sequencing also only focuses on the exonic regions. While these regions represent the most likely sites of functional mutations in PAH, there is increasing evidence supporting pathogenic variants in the promoter and 5′ UTR of BMPR2, suggesting this may also be true of other PAH genes. A recent report by Hinderhofer et al. has also identified an intronic mutation of BMPR2 that leads to aberrant splicing due to an insertion of an intronic Alu element 26 bp upstream from exon 6 [Hinderhofer et al., 2014]. This points to a potential role for other intronic variants or regulatory elements such as intra- or intergenic enhancer and repressor motifs in the pathogenesis of PAH.
To address some of these issues, it is likely that future avenues will include the use of more NGS technologies, the pinnacle of which is whole-genome sequencing. Harnessing the complete genetic information of individuals affected with PAH will not only offer opportunities to identify the causative mutation but will also provide an important tool to correlate phenotypic information to individual genotype data, allowing for tailored approaches to the clinical management of disease, or so-called personalized or precision medicine. While this technology remains financially prohibitive to some centers, custom capture of genes with defined causal links to disease offers an alternative, yet powerful, means of analyses. These studies would, ideally, include non-coding sections of the genes to determine the presence of regulatory mutations increasingly implicated in PAH etiology as described herein. The catalogue of mutations described in this report provides an important tool for the determination of deleterious mutation, both within the context of conventional and indeed NGS analyses.
For future gene identification and follow-up GWAS studies, it is of particular importance in PAH, which exhibits traits of complex disease, that phenotype is precisely assigned for this disorder. This degree of rigor is most likely to lead to further advances in both the understanding and treatment of PAH at a significantly accelerated pace.
Supplementary Material
Acknowledgments
The authors would like to sincerely thank all of the individuals with PAH for their participation in these studies. We also thank our many clinical and scientific collaborators who over a considerable period of time have ascertained patient samples complete with crucial phenotypic information. This work was supported by funding from the National Institutes of Health to EDA, JEL and JHN (PO1 HL108800), MAA (R01 HL098199) and WKC (R01 HL060056). Funding has also been provided within the 5th European Framework “Disposition to PPH” for the examination of patients from the center in Heidelberg.
Funding Sources: NIH: PO1 HL108800 (EDA, JEL, JHN); R01 HL098199 (MAA); R01 HL060056 (WKC). European Union: QLGI-CT-2002-01116 (EG)
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
References
- Abdalla SA, Gallione CJ, Barst RJ, Horn EM, Knowles JA, Marchuk DA, Letarte M, Morse JH. Primary pulmonary hypertension in families with hereditary haemorrhagic telangiectasia. Eur Respir J. 2004;23:373–377. doi: 10.1183/09031936.04.00085504. [DOI] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7. 2013;Unit7:20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, Galie N, Manes A, Corris P, Simonneau G, Humbert M, Morrell NW, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27:212–213. doi: 10.1002/humu.9398. [DOI] [PubMed] [Google Scholar]
- Aldred MA, Machado RD, James V, Morrell NW, Trembath RC. Characterization of the BMPR2 5′-untranslated region and a novel mutation in pulmonary hypertension. Am J Respir Crit Care Med. 2007;176:819–824. doi: 10.1164/rccm.200701-164OC. [DOI] [PubMed] [Google Scholar]
- Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045–2066. doi: 10.1161/CIRCULATIONAHA.108.847707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA., 3rd Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009a;34:1093–1099. doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED, Phillips JA, Cogan JD, Hamid R, Yu C, Stanton KC, Phillips CA, Wheeler LA, Robbins IM, Newman JH, Loyd JE. Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res. 2009b;10:87. doi: 10.1186/1465-9921-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, 3rd, Palomero T, Sumazin P, Kim HR, Talati MH, West J, Loyd JE, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131:1917–1928. doi: 10.1378/chest.06-2674. [DOI] [PubMed] [Google Scholar]
- Best DH, Vaughn C, McDonald J, Damjanovich K, Runo JR, Chibuk JM, Bayrak-Toydemir P. Mosaic ACVRL1 and ENG mutations in hereditary haemorrhagic telangiectasia patients. J Med Genet. 2011;48:358–360. doi: 10.1136/jmg.2010.088286. [DOI] [PubMed] [Google Scholar]
- Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, Rosenzweig EB, Bayrak-Toydemir P, Mao R, Cahill BC, Tazelaar HD, Leslie KO, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest. 2014;145:231–236. doi: 10.1378/chest.13-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaouat A, Coulet F, Favre C, Simonneau G, Weitzenblum E, Soubrier F, Humbert M. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59:446–448. doi: 10.1136/thx.2003.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Yang QH, Liu D, Liu QQ, Eyries M, Wen L, Wu WH, Jiang X, Yuan P, Zhang R, Soubrier F, Jing ZC. Clinical and genetic characteristics of Chinese patients with hereditary haemorrhagic telangiectasia-associated pulmonary hypertension. Eur J Clin Invest. 2013;43:1016–1024. doi: 10.1111/eci.12138. [DOI] [PubMed] [Google Scholar]
- Chida A, Shintani M, Nakayama T, Furutani Y, Hayama E, Inai K, Saji T, Nonoyama S, Nakanishi T. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ J. 2012a;76:1501–1508. doi: 10.1253/circj.cj-11-1281. [DOI] [PubMed] [Google Scholar]
- Chida A, Shintani M, Yagi H, Fujiwara M, Kojima Y, Sato H, Imamura S, Yokozawa M, Onodera N, Horigome H, Kobayashi T, Hatai Y, et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol. 2012b;110:586–593. doi: 10.1016/j.amjcard.2012.04.035. [DOI] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, Stanton KC, Wheeler LA, Phillips JA, 3rd, Loyd JE, Nichols WC. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:590–598. doi: 10.1164/rccm.200602-165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan J, Austin E, Hedges L, Womack B, West J, Loyd J, Hamid R. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation. 2012;126:1907–1916. doi: 10.1161/CIRCULATIONAHA.112.106245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal GR, Rawson J, Gassaway B, Kwok B, Tong Y, Ptacek LJ, Bates E. An inwardly rectifying K+ channel is required for patterning. Development. 2012;139:3653–3664. doi: 10.1242/dev.078592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L, Feige JJ, Bailly S. Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev. 2009;20:203–212. doi: 10.1016/j.cytogfr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci. 2013;70:3493–3511. doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Drake KM, Zygmunt D, Mavrakis L, Harbor P, Wang L, Comhair SA, Erzurum SC, Aldred MA. Altered MicroRNA processing in heritable pulmonary arterial hypertension: an important role for Smad-8. Am J Respir Crit Care Med. 2011;184:1400–1408. doi: 10.1164/rccm.201106-1130OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott CG, Glissmeyer EW, Havlena GT, Carlquist J, McKinney JT, Rich S, McGoon MD, Scholand MB, Kim M, Jensen RL, Schmidt JW, Ward K. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113:2509–2515. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- Eyries M, Coulet F, Girerd B, Montani D, Humbert M, Lacombe P, Chinet T, Gouya L, Roume J, Axford MM, Pearson CE, Soubrier F. ACVRL1 germinal mosaic with two mutant alleles in hereditary hemorrhagic telangiectasia associated with pulmonary arterial hypertension. Clin Genet. 2012;82:173–179. doi: 10.1111/j.1399-0004.2011.01727.x. [DOI] [PubMed] [Google Scholar]
- Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, Chelghoum N, Coulet F, Bonnet D, Dorfmuller P, Fadel E, Sitbon O, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet. 2014;46:65–69. doi: 10.1038/ng.2844. [DOI] [PubMed] [Google Scholar]
- Feng YX, Liu D, Sun ML, Jiang X, Sun N, Mao YM, Jing ZC. BMPR2 germline mutation in chronic thromboembolic pulmonary hypertension. Lung. 2014;192:625–627. doi: 10.1007/s00408-014-9580-y. [DOI] [PubMed] [Google Scholar]
- Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol. 2003;162:1089–1098. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman N, Steffann J, Girerd B, Frydman R, Munnich A, Simonneau G, Humbert M. Pre-implantation genetic diagnosis in pulmonary arterial hypertension due to BMPR2 mutation. Eur Respir J. 2012;39:1534–1535. doi: 10.1183/09031936.00185011. [DOI] [PubMed] [Google Scholar]
- Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, Imamura S, Uehara R, Nakayama T, Takao A, Nakazawa M, Saji T. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J. 2008;72:127–133. doi: 10.1253/circj.72.127. [DOI] [PubMed] [Google Scholar]
- Germain M, Eyries M, Montani D, Poirier O, Girerd B, Dorfmuller P, Coulet F, Nadaud S, Maugenre S, Guignabert C, Carpentier W, Vonk-Noordegraaf A, et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet. 2013;45:518–521. doi: 10.1038/ng.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jais X, Tregouet D, Reis A, Drouin-Garraud V, Fraisse A, Sitbon O, O'Callaghan DS, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010a;181:851–861. doi: 10.1164/rccm.200908-1284OC. [DOI] [PubMed] [Google Scholar]
- Girerd B, Montani D, Eyries M, Yaici A, Sztrymf B, Coulet F, Sitbon O, Simonneau G, Soubrier F, Humbert M. Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res. 2010b;11:73. doi: 10.1186/1465-9921-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girerd B, Coulet F, Jais X, Eyries M, Van Der Bruggen C, De Man F, Houweling A, Dorfmuller P, Savale L, Sitbon O, Vonk-Noordegraaf A, Soubrier F, et al. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest. 2015;147:1385–1394. doi: 10.1378/chest.14-0880. [DOI] [PubMed] [Google Scholar]
- Greenwald J, Fischer WH, Vale WW, Choe S. Three-finger toxin fold for the extracellular ligand-binding domain of the type II activin receptor serine kinase. Nat Struct Biol. 1999;6:18–22. doi: 10.1038/4887. [DOI] [PubMed] [Google Scholar]
- Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, Elliott CG, Robbins IM, Olschewski H, McLaughlin V, Gruenig E, Kermeen F, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40:865–871. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RE, Berger R, Haworth SG, Tulloh R, Mache CJ, Morrell NW, Aldred MA, Trembath RC. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation. 2005;111:435–441. doi: 10.1161/01.CIR.0000153798.78540.87. [DOI] [PubMed] [Google Scholar]
- Hinderhofer K, Fischer C, Pfarr N, Szamalek-Hoegel J, Lichtblau M, Nagel C, Egenlauf B, Ehlken N, Grünig E. Identification of a new intronic BMPR2-mutation and early diagnosis of heritable pulmonary arterial hypertension in a large family with mean clinical follow-up of 12 years. PLoS One. 2014;9:e91374. doi: 10.1371/journal.pone.0091374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–730. doi: 10.1161/CIRCULATIONAHA.107.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wang D, Ihida-Stansbury K, Jones PL, Martin JF. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum Mol Genet. 2009;18:2791–2801. doi: 10.1093/hmg/ddp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- Ishiwata T, Terada J, Tanabe N, Abe M, Sugiura T, Tsushima K, Tada Y, Sakao S, Kasahara Y, Nakanishi N, Morisaki H, Tatsumi K. Pulmonary arterial hypertension as the first manifestation in a patient with hereditary hemorrhagic telangiectasia. Intern Med. 2014;53:2359–2363. doi: 10.2169/internalmedicine.53.2850. [DOI] [PubMed] [Google Scholar]
- Jones G, Robertson L, Harrison R, Ridout C, Vasudevan P. Somatic mosaicism in ACVRL1 with transmission to several offspring affected with severe pulmonary arterial hypertension. Am J Med Genet A. 2014;164A:2121–2123. doi: 10.1002/ajmg.a.36568. [DOI] [PubMed] [Google Scholar]
- Kabata H, Satoh T, Kataoka M, Tamura Y, Ono T, Yamamoto M, Huqun, Hagiwara K, Fukuda K, Betsuyaku T, Asano K. Bone morphogenetic protein receptor type 2 mutations, clinical phenotypes and outcomes of Japanese patients with sporadic or familial pulmonary hypertension. Respirology. 2013;18:1076–1082. doi: 10.1111/resp.12117. [DOI] [PubMed] [Google Scholar]
- Koehler R, Grünig E, Pauciulo MW, Hoeper MM, Olschewski H, Wilkens H, Halank M, Winkler J, Ewert R, Bremer H, Kreuscher S, Janssen B, et al. Low frequency of BMPR2 mutations in a German cohort of patients with sporadic idiopathic pulmonary arterial hypertension. J Med Genet. 2004;41:e127. doi: 10.1136/jmg.2004.023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, Robbins IM, West JD, Phillips JA, 3rd, Hamid R, Loyd JE. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:892–896. doi: 10.1164/rccm.201205-0886OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Liu QQ, Eyries M, Wu WH, Yuan P, Zhang R, Soubrier F, Jing ZC. Molecular genetics and clinical features of Chinese idiopathic and heritable pulmonary arterial hypertension patients. Eur Respir J. 2012;39:597–603. doi: 10.1183/09031936.00072911. [DOI] [PubMed] [Google Scholar]
- Liu F, Ventura F, Doody J, Massague J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol Cell Biol. 1995;15:3479–3486. doi: 10.1128/mcb.15.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. 2015;21:777–785. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Tregouet DA, Borczuk A, Rosenzweig EB, Girerd B, Montani D, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–361. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RD, Rudarakanchana N, Atkinson C, Flanagan JA, Harrison R, Morrell NW, Trembath RC. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2003;12:3277–3286. doi: 10.1093/hmg/ddg365. [DOI] [PubMed] [Google Scholar]
- Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, Gruenig E, Janssen B, Koehler R, Seeger W, Eickelberg O, Olschewski H, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27:121–132. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- Machado RD, Eickelberg O, Elliott CG, Geraci MW, Hanaoka M, Loyd JE, Newman JH, Phillips JA, 3rd, Soubrier F, Trembath RC, Chung WK. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S32–42. doi: 10.1016/j.jacc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mache CJ, Gamillscheg A, Popper HH, Haworth SG. Early-life pulmonary arterial hypertension with subsequent development of diffuse pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia type 1. Thorax. 2008;63:85–86. doi: 10.1136/thx.2007.076109. [DOI] [PubMed] [Google Scholar]
- Mair KM, Yang XD, Long L, White K, Wallace E, Ewart MA, Docherty CK, Morrell NW, MacLean MR. Sex affects bone morphogenetic protein type II receptor signaling in pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. 2015;191:693–703. doi: 10.1164/rccm.201410-1802OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis NA, Shinin V, Schraufnagel DE, Okada S, Vogel SM, Malik AB, Minshall RD. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1-/- mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–873. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–1619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Mercier I, Jasmin JF, Pavlides S, Minetti C, Flomenberg N, Pestell RG, Frank PG, Sotgia F, Lisanti MP. Clinical and translational implications of the caveolin gene family: lessons from mouse models and human genetic disorders. Lab Invest. 2009;89:614–623. doi: 10.1038/labinvest.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1179–1183. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- Möller T, Leren TP, Eiklid KL, Holmstrøm H, Fredriksen PM, Thaulow E. A novel BMPR2 gene mutation associated with exercise-induced pulmonary hypertension in septal defects. Scand Cardiovasc J. 2010;44:331–336. doi: 10.3109/14017431.2010.525747. [DOI] [PubMed] [Google Scholar]
- Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasim MT, Ghouri A, Patel B, James V, Rudarakanchana N, Morrell NW, Trembath RC. Stoichiometric imbalance in the receptor complex contributes to dysfunctional BMPR-II mediated signalling in pulmonary arterial hypertension. Hum Mol Genet. 2008;17:1683–1694. doi: 10.1093/hmg/ddn059. [DOI] [PubMed] [Google Scholar]
- Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, Lord GM, Gibbs JS, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32:1385–1389. doi: 10.1002/humu.21605. [DOI] [PubMed] [Google Scholar]
- Nishihara A, Watabe T, Imamura T, Miyazono K. Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Biol Cell. 2002;13:3055–3063. doi: 10.1091/mbc.E02-02-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohe A, Keating E, Underhill TM, Knaus P, Petersen NO. Dynamics and interaction of caveolin-1 isoforms with BMP-receptors. J Cell Sci. 2005;118:643–650. doi: 10.1242/jcs.01402. [DOI] [PubMed] [Google Scholar]
- Pfarr N, Szamalek-Hoegel J, Fischer C, Hinderhofer K, Nagel C, Ehlken N, Tiede H, Olschewski H, Reichenberger F, Ghofrani AH, Seeger W, Grunig E. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir Res. 2011;12:99. doi: 10.1186/1465-9921-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfarr N, Fischer C, Ehlken N, Becker-Grunig T, Lopez-Gonzalez V, Gorenflo M, Hager A, Hinderhofer K, Miera O, Nagel C, Schranz D, Grunig E. Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Respir Res. 2013;14:3. doi: 10.1186/1465-9921-14-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O'Connor DT, Yuan JX. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C1837–1853. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- Rigueur D, Brugger S, Anbarchian T, Kim JK, Lee Y, Lyons KM. The Type I BMP Receptor ACVR1/ALK2 is Required for Chondrogenesis During Development. J Bone Miner Res. 2015;30:733–741. doi: 10.1002/jbmr.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig EB, Morse JH, Knowles JA, Chada KK, Khan AM, Roberts KE, McElroy JJ, Juskiw NK, Mallory NC, Rich S, Diamond B, Barst RJ. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant. 2008;27:668–674. doi: 10.1016/j.healun.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- Ryan J, Bloch K, Archer SL. Int J Clin Pract Suppl. 2011. Rodent models of pulmonary hypertension: harmonisation with the world health organisation's categorisation of human PH; pp. 15–34. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46:331–337. doi: 10.1136/jmg.2008.062703. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- Smoot LB, Obler D, McElhinney DB, Boardman K, Wu BL, Lip V, Mullen MP. Clinical features of pulmonary arterial hypertension in young people with an ALK1 mutation and hereditary haemorrhagic telangiectasia. Arch Dis Child. 2009;94:506–511. doi: 10.1136/adc.2007.133082. [DOI] [PubMed] [Google Scholar]
- Sobolewski A, Rudarakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, Morrell NW. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet. 2008;17:3180–3190. doi: 10.1093/hmg/ddn214. [DOI] [PubMed] [Google Scholar]
- Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D13–21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, Sitbon O, Montani D, Souza R, Simonneau G, Soubrier F, Humbert M. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med. 2008;177:1377–1383. doi: 10.1164/rccm.200712-1807OC. [DOI] [PubMed] [Google Scholar]
- Tenorio J, Navas P, Barrios E, Fernandez L, Nevado J, Quezada CA, Lopez-Meseguer M, Arias P, Mena R, Lobo JL, Alvarez C, Heath K, et al. A founder EIF2AK4 mutation causes an aggressive form of pulmonary arterial hypertension in Iberian Gypsies. Clin Genet. 2015 doi: 10.1111/cge.12549. [Epub: 7 Jan 2015] [DOI] [PubMed] [Google Scholar]
- Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, Nichols WC, Morrell NW, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4–12. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, et al. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013;62:D22–33. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- Wang G, Knight L, Ji R, Lawrence P, Kanaan U, Li L, Das A, Cui B, Zou W, Penny DJ, Fan Y. Early onset severe pulmonary arterial hypertension with ‘two-hit’ digenic mutations in both BMPR2 and KCNA5 genes. Int J Cardiol. 2014;177:e167–169. doi: 10.1016/j.ijcard.2014.08.124. [DOI] [PubMed] [Google Scholar]
- Wang H, Li W, Zhang W, Sun K, Song X, Gao S, Zhang C, Hui R, Hu H. Novel promoter and exon mutations of the BMPR2 gene in Chinese patients with pulmonary arterial hypertension. Eur J Hum Genet. 2009;17:1063–1069. doi: 10.1038/ejhg.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cui QQ, Sun K, Song L, Zou YB, Wang XJ, Jia L, Liu X, Gao S, Zhang CN, Hui RT. Identities and frequencies of BMPR2 mutations in Chinese patients with idiopathic pulmonary arterial hypertension. Clin Genet. 2010;77:189–192. doi: 10.1111/j.1399-0004.2009.01335.x. [DOI] [PubMed] [Google Scholar]
- West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94:1109–1114. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- West J, Cogan J, Geraci M, Robinson L, Newman J, Phillips JA, Lane K, Meyrick B, Loyd J. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics. 2008a;1:45. doi: 10.1186/1755-8794-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol. 2008b;295:L744–755. doi: 10.1152/ajplung.90255.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Buikema H, van Gilst WH, Henning RH. Caveolae and endothelial dysfunction: filling the caves in cardiovascular disease. Eur J Pharmacol. 2008;585:256–260. doi: 10.1016/j.ejphar.2008.02.086. [DOI] [PubMed] [Google Scholar]
- Young KA, Ivester C, West J, Carr M, Rodman DM. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2006;290:L841–848. doi: 10.1152/ajplung.00158.2005. [DOI] [PubMed] [Google Scholar]
- Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YY, Malik AB. A novel insight into the mechanism of pulmonary hypertension involving caveolin-1 deficiency and endothelial nitric oxide synthase activation. Trends Cardiovasc Med. 2009;19:238–242. doi: 10.1016/j.tcm.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.