

Abstract
Aims
During ischemia/reperfusion (I/R), ribosomal S6 kinase (RSK) activates Na+/H+ exchanger 1 (NHE1) by phosphorylating NHE1 at serine 703 (pS703-NHE1), which promotes cardiomyocyte death and injury. Pharmacologic inhibition of NHE1 effectively protects animal hearts from I/R. However, clinical trials using NHE1 inhibitors failed to show benefit in patients with acute myocardial infarction (MI). One possible explanation is those inhibitors block both agonist-stimulated activity (increasing I/R injury) and basal NHE1 activity (necessary for cell survival). We previously showed that dominant-negative RSK (DN-RSK) selectively blocked agonist-stimulated NHE1 activity. Therefore, we hypothesized that a novel RSK inhibitor (BIX02565) would blunt agonist-stimulated NHE1 and protect hearts from I/R.
Methods and Results
Serum/angiotensin II-stimulated pS703-NHE1 was significantly decreased by BIX02565 in cultured cells. Intracellular pH recovery assay showed that BIX02565 selectively inhibited serum-stimulated NHE1 activity. Ischemia/reperfusion decreased left ventricular–developed pressure (LVDP; inhibited) to 8.7% of the basal level in non-transgenic littermate control (NLC) mouse hearts, which was significantly improved (44.6%) by BIX02565. Similar protection was observed in vehicle-treated, cardiac-specific DN-RSK-Tg mice (43%). No additional protective effect was seen in BIX02565-treated DN-RSK-Tg hearts. BIX02565 also improved LVDP in cardiac-specific wild-type (WT)-RSK-Tg mouse hearts (7.4%-40.9%, P < .01). Finally, Western Blotting results confirmed DN-RSK and BIX02565 significantly decreased I/R-induced pS703-NHE1.
Conclusion
The RSK plays a crucial role in I/R-induced activation of NHE1 and cardiac injury. The RSK inhibition may provide an alternative target for patients with MI.
Keywords: I/R, NHE1, RSK inhibitor, cardioprotection
Introduction
Cardiovascular disease is the leading cause of death globally, of which about 50% is related to ischemia-induced heart failure. A critical factor in the development of heart failure is cardiomyocyte death. Intracellular pH (pHi) is essential to maintain cell viability. At low pHi, the cells are more susceptible to death. Under conditions of low oxygen supply (which occurs during ischemia/reperfusion [I/R] injury), transporters such as the sodium–hydrogen exchanger (NHE) are important regulators of pHi.1-3 The NHE1 is the major isoform expressed in heart3 pharmacologic or genetic inhibition of NHE1 activity and has been proven to protect mouse hearts from I/R injury.4-7 However, clinical trials using NHE1 in humans showed mixed results and side effects.8-10 This discrepancy may possibly be due to the inhibition of both basal and stimulated NHE1 function by the drugs chosen for the trials.
Our laboratory has reported that p90 ribosomal S6 kinase (RSK), a mitogen-activated serine threonine kinase, is highly activated in failing human hearts11 as well as in ischemic animal hearts.12 We have also shown that I/R-activated RSK phosphorylates NHE1 at serine 703 (S703). This phosphorylation site is responsible for the binding of protein 14-3-3, which maintains NHE1 in a highly activated state.13-15 The NHE1 S703A mutation abolishes serum-stimulated NHE1 activation while maintaining basal NHE1 activity.13 Previously we showed that H9C2 cells, a cardiac-derived cell line, transfected with Ad-NHE1S703A had lower apoptosis after anoxia/reoxygenation.15 Interestingly, overexpressing dominant-negative RSK (DN-RSK) blunts hydrogen peroxide (H2O2)-stimulated NHE1 activation while maintaining basal NHE1 activity.15 In addition, cardiac-specific DN-RSK transgenic (DN-RSK-Tg) mice exhibit cardioprotection in an infarction model.15 Hence, we hypothesized that RSK inhibitor will blunt I/R-induced activation of NHE1 (but maintain basal NHE1 activity) and protect hearts from I/R injury.
The RSK consists of 2 functional kinase domains, N-terminal kinase domain (NTKD) and C-terminal kinase domain (CTKD).16-18 A novel RSK inhibitor BIX02565 targeting the NTKD of RSK was recently reported by Boehringer-Ingelheim.19-21 BIX02565 represents more than 100-fold selectivity for RSK compared to over 220 kinases analyzed through Invitrogen Kinase Select Screen profiling, except for leucine-rich repeat kinase 2 (LRRK2) and protein kinase D1 (PRKD1).19 In addition, BIX02565 has a significantly higher in vitro affinity compared to the 2 most commonly used RSK inhibitors, fmk (15-fold) and BI-D1870 (30-fold).18 In this study, we investigated the ability of BIX02565 to inhibit agonist-induced NHE1 phosphorylation and activation in vitro and ex vivo. Our results showed that BIX02565 pretreatment significantly improved cardiac functional recovery and decreased infarct size in non-transgenic littermate control (NLC) and cardiac-specific wild-type (WT)-RSK-Tg mouse hearts after I/R. The protective effects were similar to what were observed in DN-RSK-Tg mouse hearts.
Materials and Methods
Cell Culture and Reagents
The myoblastic H9C2 cell line (originally derived from embryonic rat heart tissue) was purchased from the American Tissue Type Collection (ATTC; Manassas, Virginia; Catalog no. CRL-1446). PS127A cells (Chinese hamster lung fibroblasts that overexpress human NHE1) were a gift of Dr J. Pouysse'gur (University of Nice, France). Human embryonic kidney (HEK 293) cells were purchased from ATTC (Catalog no. CRL-1573). Primary culture of rat aortic smooth muscle (RASM) cells was prepared in our laboratory as described previously22 and used before passage 12. All cells were maintained in Dulbecco-modified Eagle medium (catalog no. 11995; Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin and 100 mg/mL streptomycin. BIX02565 was kindly provided by Boehringer-Ingelheim Pharmaceuticals Corporation (Ridgefield, Connecticut). The detailed specificity and potency of BIX02565 have been tested and reported in a separate article.19 Anti-phospho-NHE1 (S703) antibody detecting sequence “TMSRARIGSpD-PLAYE”14 was kindly provided by Boehringer-Ingelheim.
Preparation of Cell Lysates and Western Blotting
Prior to the start of experiments, cells were serum starved (0% FBS) overnight. BIX02565 and fmk were added 1.5 hours and calphostin C (catalog no. EI198; BIOMOL International LP, USA) was added 0.5 hour before the stimulation. Cells stimulated with 20% FBS for 5 to 20 minutes or 100 nmol/L angiotensin II (Ang II) for 5 minutes were harvested using lysis buffer (catalog no. #9803; Cell Signaling, USA). Mouse heart samples harvested at the end of 40 minutes ischemia and 10 minutes reperfusion were immediately frozen in liquid nitrogen (LN). Hearts were ground to powder in LN. Protein samples were obtained by adding lysis buffer to the heart powder. After sonication and centrifugation, supernatants were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted with anti-phospho-NHE1 (S703) antibody. Anti-NHE1 (Chemicon, USA) and anti-tubulin (Sigma, USA) were used as loading controls. Anti-phospho-ERK1/2 (Cell Signaling) was used as a positive control.
Intracellular pH Measurement
Activity of NHE1 was determined by pHi recovery following acid loading using the fluorescent pH-sensitive dye, acetoxymethyl ester of 2′,7′-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM; Molecular Probes, Inc, Eugene, Oregon) as described previously.23 All experiments were performed at 37°C. Briefly, H9C2 cells were grown in 96-well collagen-coated plates (BD 354649) and serum staved (0% FBS) for 24 hours prior to use. Cells were loaded with 5 μmol/L BCECF-AM for 30 minutes. After that, the cells were washed 2 times and prepulsed with acid loading buffer containing 10 mmol/L ammonium chloride (NH4Cl) for 30 minutes with or without different dosages of BIX02565. For the serum stimulation, 10% FBS was added during the final 10 minutes of acid loading. Initial baseline fluorescence was recorded for each well simultaneously using a fluorescence-imaging plate reader (FLIPRTETRA; Molecular Devices, USA) at 470 to 495 emission/515 to 575 excitation. Data were collected for 5 minutes for each treatment. Afterward, the nigericin/high K+ technique with 20 μmol/L nigericin in KCl solution (130 mmol/L KCl, 10 mmol/L Hydroxyethyl-Piperazine Ethanesulafonic Acid (HEPES), pH 7.4-6.5) was used to calibrate the relationship between excitation ratio and pHi. The rate of pHi recovery was converted to millimoles (mmol) of H+ per minute per liter cells (JH) by multiplying with the buffering power. Buffering power was calculated from the change in cell pHi observed upon NH4Cl addition. The data were then plotted by calibrating 10% FBS-stimulated NHE1 activity as 100%. The NHE1 activity under all the other treatments was calibrated as a ratio of the 10% FBS treatment.
Animals and Measurement of Left Ventricular Function by Langendorff Preparation
The animal experiments conform to the Guide for the Care and Use of Laboratory Animals that was published by the US National Institute of Health in 1996. The transgenic mouse strain that overexpresses WT-RSK or DN-RSK (with K94A/K447A) under the control of α-myosin heavy-chain promoter was generated as described previously.12,15 Mice were maintained by breeding to filial 1 of Friend Virus B-Type mouse (FVB F1) animals (Jackson Laboratory, Bar Harbor, Maine). All mice were used in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. All procedures were approved by the University of Rochester Animal Care Committee.
WT-RSK-Tg, DN-RSK-Tg, and NLC mice (10-12 weeks of age) were anesthetized with ketamine (130 mg/kg) and xylazine (8.8 mg/kg) intraperitoneally (IP) and heparinized (5000 U/kg) IP. Hearts were subjected to Langendorff preparation as described previously.12,24 Briefly, hearts were randomly excised in a blinded manner and immersed in a chamber maintained at 37°C in KH buffer (118 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 2.5 mmol/L CaCl2, 25 mmol/L NaHCO3, 0.5 mmol/L Na-EDTA, and 11 mmol/L glucose), which was saturated with 95% O2/5% CO2 (v/v, pH 7.4, 37°C) for 30 minutes. Isolated hearts were perfused through the aorta in a retrograde Langendorff apparatus with KH buffer in constant flow mode (12 mL/min/g tissue). A homemade water-filled balloon was inserted into the left ventricle through the left atrium and was adjusted to a left ventricular end-diastolic pressure of 2 mm Hg during initial equilibration. In the absence of a heart, we confirmed that the balloon was sufficiently large to allow it to be fully inflated to greater than the size of stretched ventricular lumen without itself exerting any pressure. Cardiac functionality was monitored by a water-filled left ventricular latex balloon linked to a pressure transducer with digital recording (Dataq, Akron, Ohio). Isolated hearts were subjected to 20 minutes perfusion with KH buffer for stabilization and then perfused for 10 minutes with vehicle (KH buffer) or BIX02565 (30, 100, 300 nmol/L) in KH buffer. BIX02565 was only perfused at this stage. Hearts from DN-RSK-Tg and NLC mice were then subjected to 40 minutes of no-flow normothermic global ischemia and hearts from WT-RSK-Tg mice were subjected to 20 minutes of ischemia (due to the susceptibility of WT-RSK-Tg mice to the I/R25), followed by 60 minutes of reperfusion.
Analysis of Infarct in the Hearts
Following I/R, hearts were sliced horizontally and stained with 2,3,5-triphenyltetrazolium chloride (TTC). Infarct area was analyzed with Scion Image (Scion Corporation, USA) as described previously.25
Statistical Analysis
The animal number for each group (n = 6) was determined by a power calculation. The formula for determining the sample sizes for 2 independent populations is ni = 2(zσ/E)2. Z is the value from the table of probabilities of the standard normal distribution for the desired confidence level (eg, Z = 1.96 for 95% confidence). σ is the standard deviation (SD) of the outcome of interest. E is the margin of error that the investigator specifies as important from a clinical or practical standpoint. Values are presented as mean ± standard error of the mean. Statistical differences between groups were determined by 1-way analysis of variance. Afterward, 2-tailed t test was run to compare each group. Values of P < .01 were considered statistically significant.
Results
Serum- and Ang II-Stimulated Phosphorylation of NHE1 at S703 is Inhibited by a Novel RSK Inhibitor, BIX02565
We have previously demonstrated that serum and Ang II activate RSK, which then phosphorylates NHE1 at S703, leading to a significant increase in its activity.13,14,22,26 To evaluate the efficacy of BIX02565 as an antagonist of RSK downstream signaling, we studied responses elicited by serum or Ang II and assayed NHE1 S703 phosphorylation (pS703-NHE1) using a phospho-specific antibody. As shown in Figure 1, treatment with 20% FBS for 5 minutes significantly induced NHE1 phosphorylation in H9C2 cells (5.0 ± 0.2-fold). BIX02565 inhibited FBS-stimulated NHE1 phosphorylation in a dose-dependent manner (Figure 1A and B). At 1 μmol/L BIX02565, pS703-NHE1 in response to 20% FBS was inhibited by 76%. In contrast, FBS-stimulated ERK1/2 phosphorylation was not affected by BIX02565, demonstrating both specificity and minimal effect on signaling upstream of RSK.
Figure 1.
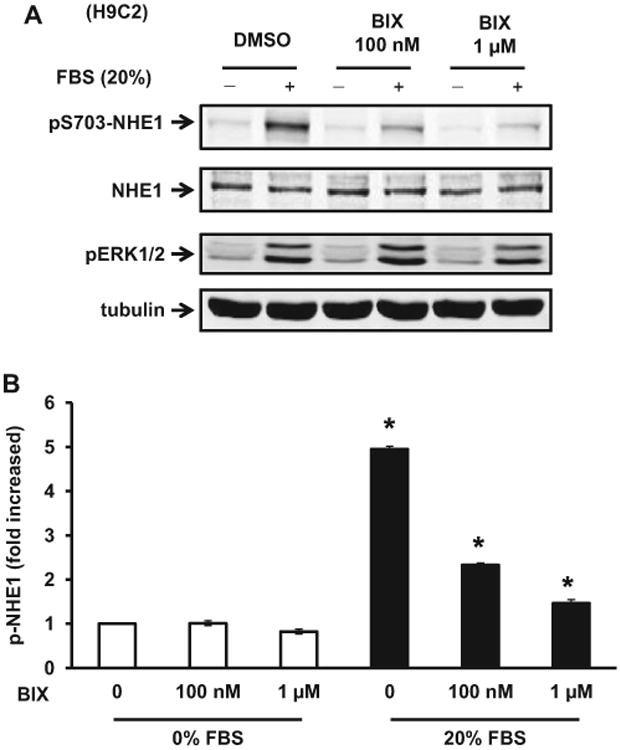
Serum-stimulated phosphorylation of Na+/H+ exchanger 1 (NHE1) at S703 is inhibited by ribosomal S6 Kinase (RSK) inhibitor BIX02565 in a dose-dependent manner. A, H9C2 cells were serum starved (0% fetal bovine serum [FBS]) overnight and stimulated with 20% FBS for 5 minutes. BIX02565 (100 nmol/L and 1 μmol/L) was added 1.5 hours before the serum stimulation. Protein lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with anti-phospho S703-NHE1 antibody. The same membrane was blotted with anti-phospho-ERK1/2 antibody to confirm the serum activation. Anti-NHE1 and antitubulin antibodies were used for the loading control. B, Quantified result of phospho S703-NHE1 normalized to total NHE1 protein levels (shown as mean ± SD, n = 3, *P < .01).
We next investigated whether BIX02565 was able to block RSK signaling in other cell types (Supplemental Figure S1A-C). As shown in Figure S1A, BIX02565 diminished serum-stimulated NHE1 phosphorylation (at 5 minutes) in PS127 cells in a dose-dependent manner. A similar effect was seen in HEK 293 cells (Figure S1B). Angiotensin II (100 nmol/L) significantly increased NHE1 phosphorylation in RASM cells, which was completely abolished by BIX02565 (Figure S1C). Fmk is another RSK inhibitor,27,28 which specifically inhibits RSK-CTKD, while BIX02565 inhibits RSK-NTKD. We found that fmk also decreased Ang II-stimulated NHE1 phosphorylation in RASM, similar to BIX02565 (Figure S1C). In contrast, the protein kinase C (PKC) inhibitor calphostin C had no effect on Ang II-stimulated NHE1 phosphorylation, which is consistent with our previous observation that NHE1 S703 is specifically phosphorylated by RSK but not by PKC upon Ang II stimulation.26
To confirm that the pS703-NHE1 antibody exhibited high specificity, we expressed WT full-length NHE1 (1-815aa), C-terminal NHE1 (516-815aa), and C-terminal NHE1 (516-815aa) S703A mutant in PS120 cells that lack endogenous NHE1 (Figure S2). After serum stimulation, pS703-NHE1 antibody detected both full length and C-terminal of WT-NHE (lanes 1 and 2) but not NHE1 S703A (lane 3). There were very few nonspecific bands. The band at 40 kD in the NHE1 (1-815) lane is likely a degraded NHE1 product that contains the C-terminal NHE1.29,30 These observations confirm that the pS703-NHE1 antibody is a specific reagent for detecting RSK activity.
BIX02565 Inhibits Serum-Enhanced But Not the Basal NHE1 Activity
Our previous study showed that NHE1 S703A mutant or DN-RSK prevented agonist stimulation of NHE1 without affecting its basal activity.13,15 To evaluate whether BIX02565 can selectively abolish agonist-stimulated NHE1 activity, we performed a pHi recovery assay (Figure 2). Ten percent FBS fully activated NHE1 (calibrated as 100%) compared to the 34% activation of NHE1 at the basal level. Pretreatment with BIX02565 inhibited serum-stimulated NHE1 activity in a dose-dependent manner. Serum-stimulated NHE1 activity was inhibited by BIX02565 with a half maximal inhibitory concentration 50 (IC50) of ∼1.2 μmol/L. These data are consistent with the inhibition of pS703-NHE1 by BIX02565 in Figure 1. Importantly, there was no inhibition of basal NHE1 activity up to 10 μmol/L BIX02565.
Figure 2.
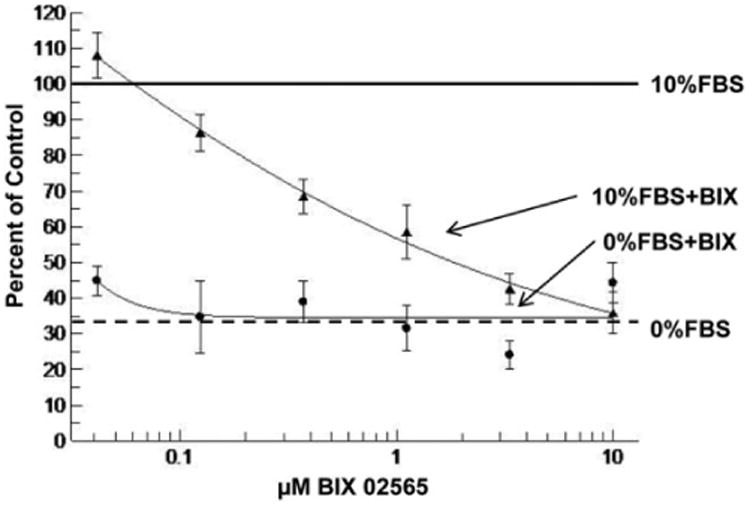
BIX02565 inhibits serum enhanced but not the basal Na+/H+ exchanger 1 (NHE1) activity. H9C2 cells were serum starved (0% fetal bovine serum [FBS]) for 24 hours and kept in the presence of 10% FBS (——) or 0% FBS (— —) for recording the enhanced and basal NHE1 activity (measured by the rate of intracellular pH [pHi] recovery). BIX 02565 was titrated to the cells with (—▲—) and without FBS (—●—). The 10% FBS-stimulated NHE1 activity was calibrated as 100%. The NHE1 activity under all the other treatments was calibrated as a ratio to the 10% FBS treatment. BIX 02565 inhibited serum-enhanced NHE1 activity in a dose-dependent manner while had no effect on basal NHE1 activity.
BIX02565 Improves Cardiac Function in NLC Mouse Hearts After I/R in a Dose-Dependent Manner
Although NHE1 inhibitors have been well documented to be beneficial in animal I/R models,5-7 clinical trials using NHE1 inhibitors showed mixed results.8-10 We suggest this may be due to the fact that the inhibitors studied inhibited both agonist-stimulated activity and basal NHE1 activity, while the latter is necessary for cell survival after exposure to I/R. Our previous study showed that overexpressing NHE1S703A in cardiac H9C2 cells decreased apoptosis in response to anoxia/reoxygenation.15 To further investigate whether inhibiting I/R activated NHE1 while maintaining the basal NHE1 activity is cardioprotective, we introduced ex vivo I/R with the Langendorff system using NLC mouse hearts pretreated with or without BIX02565 at different concentrations (Figure 3). The protocol of I/R is shown in Figure 3A. Before ischemia, there was no significant difference in left ventricular developed pressure (LVDP), dp/dt max, or rate pressure product (RPP) between BIX02565-and vehicle-treated groups (Figure 3B-D). After I/R, LVDP dramatically decreased to ∼8.7% of the basal level in NLC mouse hearts. In contrast, there was a significant recovery of LVDP in hearts treated with BIX02565. Treatment with BIX02565 (30 and 100 nmol/L) increased cardiac functional recovery in a dose-dependent manner (27.2% with 30 nmol/L and 44.6% with 100 nmol/L; Figure 3B). Similarly, a dose-dependent cardioprotection was observed in post-I/R dp/dt max and RPP (Figure 3C and D). However, no additional improvement was seen with further increase in BIX02565 to 300 nmol/L (Figure 3B-D). Therefore, 100 nmol/L BIX02565 was used for all subsequent experiments.
Figure 3.
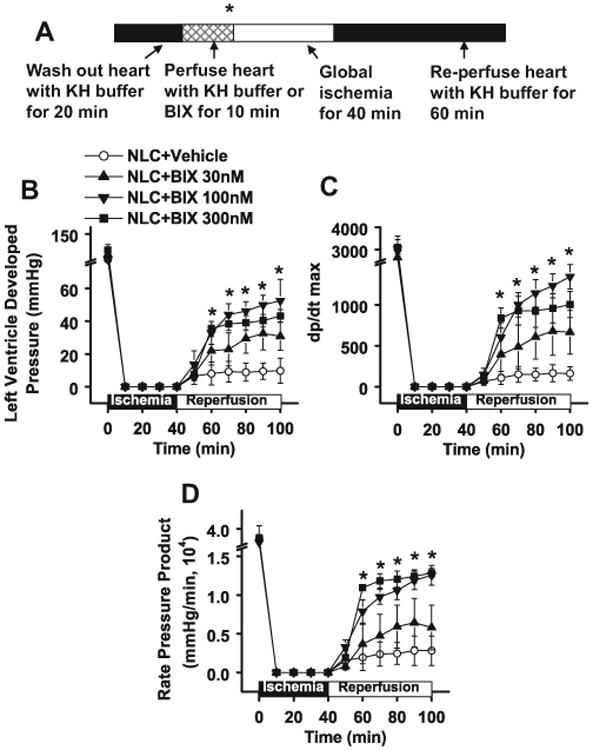
BIX02565 improves cardiac functional recovery in non-transgenic littermate control (NLC) mouse hearts in a dose-dependent manner. A, Diagram of ischemia/reperfusion (I/R) experimental protocol. In brief, hearts isolated from NLC mice were perfused with KH buffer for 20 minutes for stabilization. After this, hearts were perfused without or with BIX02565 (30, 100, and 300 nmol/L) for 10 minutes, followed by global ischemia for 40 minutes and reperfusion for 60 minutes. *Indicates the first data point in Figure 3B and D. B, Measurements of left ventricular–developed pressure (LVDP). C, dp/dt max. D, Rate pressure product (RPP) before and during I/R with vehicle or BIX02565 treatment in NLC mouse hearts. The RPP was calculated based on the equation: RPP = LVDP (mm Hg) × heart rate (bpm). Values are shown as mean ± standard error of the mean [SEM], n — 6, *P < .01.
The Cardioprotective Effect of BIX02565 is Mediated by the Inhibition of RSK
We previously showed that I/R-induced NHE1 activity was blunted in cardiac DN-RSK-Tg mouse hearts.15 To further understand the role of RSK in I/R injury, we evaluated cardiac functional recovery in BIX02565 (100 nmol/L)-treated and untreated NLC and compared the recovery to DN-RSK-Tg mouse hearts. Under basal conditions, there was no significant difference in LVDP or dp/dt between BIX02565 untreated and treated DN-RSK-Tg and NLC mouse hearts (first data point in Figure 4). After I/R, hearts from DN-RSK-Tg mice displayed a significant recovery of LVDP compared to the NLC (43% vs 8.7% of baseline, P < .01; Figure 4A). Similarly, post-I/R dp/dt max (35.4% vs 5.5%, P < .01; Figure 4B) and RPP in DN-RSK-Tg hearts were also significantly increased compared to NLC hearts (37.5% vs 8% of baseline, P < .01; Figure 4C). These data are consistent with our previous finding of enhanced cardiac function after in vivo I/R in DN-RSK-Tg mice.15
Figure 4.

The effect of BIX02565 is mediated by inhibition of ribosomal S6 Kinase (RSK). A, Hearts isolated from dominant-negative (DN)-RSK transgenic mice (DN-RSK-Tg) and non-transgenic littermate control (NLC) mice were perfused with KH buffer for 20 minutes for stabilization. Hearts were perfused with KH buffer or BIX02565 (100 nmol/L) for 10 minutes, followed by global ischemia for 40 minutes and reperfusion for 60 minutes. Both BIX-treated NLC hearts and untreated DN-RSK-Tg have significant increase in left ventricular–developed pressure (LVDP) after ischemia/reperfusion (I/R). A, Measurements of LVDP. B, dp/dt max. C, Rate pressure product (RPP) before and during I/R in NLC and DN-RSK-Tg mouse hearts (shown as mean ± standard error of the mean [SEM], n = 6, *P < .01).
BIX02565 treatment significantly increased LVDP in NLC hearts after I/R (8.7%-44.6%, P < .01; Figure 4A). Interestingly, this LVDP recovery was similar to what was seen in the untreated DN-RSK-Tg mouse hearts (44.6% vs 43%; not significant [ns]). The dp/dt max (5.5%-40.4%, P < .01) and RPP (8%-38.6%, P < .01) after I/R in NLC mouse hearts were also significantly increased by BIX02565 treatment.
It is interesting to notice that BIX02565 did not further improve any measurements of cardiac function in the DN-RSK-Tg mouse hearts (LVDP: 50.2% vs 43%, ns; dp/dt: 37.5% vs 41.3%, ns; and RPP: 49% vs 35.4%, ns; Figure 4). We interpret this result to show that the cardioprotective mechanism of BIX02565 is primarily through inhibiting RSK and NHE1. To further prove our hypothesis, we investigated the effect of BIX02565 on cardiac-specific WT-RSK-Tg mouse hearts after I/R. BIX02565 significantly improved LVDP in WT-RSK-Tg mouse hearts after I/R (7.4%-40.9%, P < .01; Figure S3). These results indicate inhibiting RSK is protective in cardiac I/R injury.
BIX02565 Reduces Infarction in NLC Hearts After I/R Injury
We next analyzed the infarct size after I/R. As shown in Figure 5, I/R resulted in a 40.1% infarct area in NLC mouse hearts. Treatment with 30 nmol/L BIX02565 had no significant change in infarct area (data not shown). However, treatment with 100 nmol/L BIX02565 dramatically reduced infarct size in NLC hearts (to 22.5%, P < .01). No additional protection was seen with 300 nmol/L BIX02565-treated mouse hearts (infarct to 25%) compared to 100 nmol/L BIX02565 (data not shown), which is consistent with the data shown in Figure 3. Similar to our previous findings,15 vehicle-treated DN-RSK-Tg mouse hearts had a significant decrease in infarct area after I/R compared to NLC (22.5% vs 40.1%, P < .01; Figure 5). However, no additional decrease in infarct size was seen in 100 nmol/L BIX02565-treated DN-RSK-Tg compared to untreated DN-RSK-Tg mouse hearts (22.5% vs 23.2%, ns; Figure 5).
Figure 5.

Both BIX02565 and dominant-negative (DN)-RSK transgenic mice (DN-RSK-Tg) reduce infarction in mouse hearts after ischemia/reperfusion (I/R) injury. Quantified infarct area from 3,5-triphenyltetrazolium chloride (TTC)-stained heart sections collected from non-transgenic littermate control (NLC) and DN-RSK-Tg mouse hearts treated without or with 100 nmol/L BIX02565. Both BIX-treated NLC hearts and untreated DN-RSK-Tg have significantly decreased infarct area after I/R. However, no significant change is seen in BIX untreated and treated DN-RSK-Tg mouse hearts. Values are shown as mean ± standard error of the mean [SEM], n = 6, *P < .01.
Inhibiting RSK Blunts I/R-induced pS703-NHE1
To determine whether BIX02565 inhibits pS703-NHE1 in the heart tissue, we collected mouse heart samples from different time points of I/R. In preliminary experiments, we found pS703-NHE1 started after reperfusion and peaked at 10 minutes after reperfusion (data not shown).15 Therefore, we compared I/R-induced pS703-NHE1 in NLC mouse hearts treated with vehicle or BIX02565 (100 nmol/L) as well as DN- RSK-Tg mouse hearts. As shown in Figure 6, there was no phosphorylation of NHE1 S703 detected at the end of 40 minutes ischemia (lane 1, NLC + vehicle “I” only). However, after 10 minutes reperfusion (lanes 2-5, NLC + vehicle “I/R”), there was a 4-fold increase in pS703-NHE1 in NLC mouse hearts (Figure 6B). In contrast, pS703-NHE1 was significantly decreased with the addition of BIX02565 (lanes 6-9, NLC + BIX 100 nmol/L “I/R”). As expected, I/R could not induce pS703-NHE1 in DN-RSK-Tg mouse hearts (lanes 10-13, DN-RSK-Tg + vehicle “I/R”), indicating RSK is responsible for I/R-induced pS703-NHE1.
Figure 6.
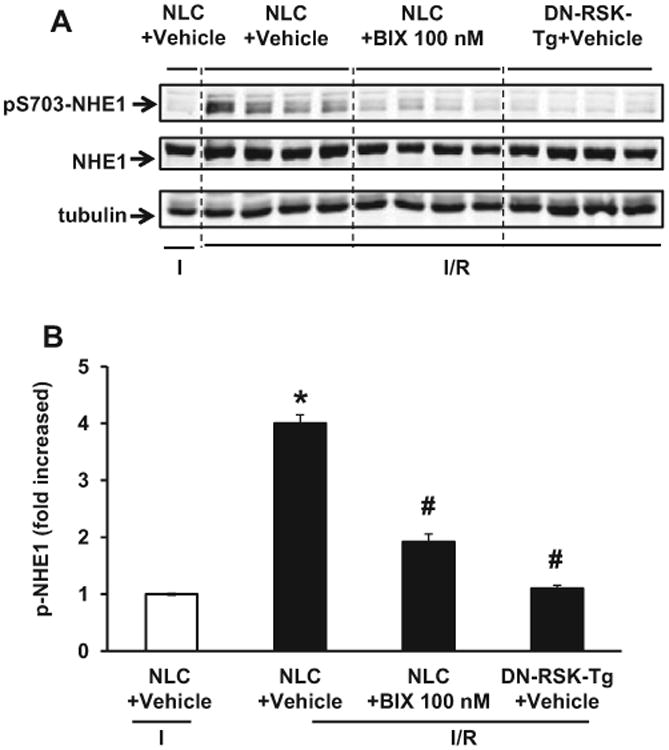
Inhibiting ribosomal S6 kinase (RSK) blunted ischemia/reperfusion (I/R)-induced Na+/H+ exchanger 1 (NHE1) S703 phosphorylation. A, Mouse heart protein samples were collected from (1) non-transgenic littermate control (NLC) + Vehicle harvested at the end of 40 minutes ischemia (I), (2) NLC + BIX02565 (100 nmol/L), and (3) dominant-negative (DN)-RSK-Tg + vehicle groups harvested at 10 minutes of reperfusion (I/R). Protein lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrphoresis (SDS-PAGE) and immunoblotted with anti-phospho S703-NHE1 antibody. Anti-NHE1 and anti-tubulin antibodies were used for the loading control. B, Quantified result of pS703-NHE1 normalized to total NHE1 protein levels. Reperfusion significantly increases NHE1 phosphorylation at S703 (*P < .01). The phosphorylation of NHE1 is significantly reduced in NLC mouse heart treated with BIX and DN-RSK-Tg mouse hearts (#P < .01). Values are shown as mean ± standard deviation [SD], n = 4.
Discussion
The major findings of the present study are that the novel RSK inhibitor BIX02565, which specifically targets the NTKD of RSK, inhibits pS703-NHE1/activation and improves cardiac function after I/R. Data to support this conclusion include (1) BIX02565 is highly selective for RSK compared to other mammalian kinases,31 (2) BIX02565 inhibits serum-enhanced NHE1 activity without affecting the basal NHE1, (3) cardiac-specific DN-RSK-Tg significantly improves cardiac functional recovery and decreases infarction size after I/R, (4) BIX02565 provides similar cardioprotective effect in NLC and WT-RSK-Tg compared to DN-RSK-Tg, and (5) both BIX02565 and DN-RSK-Tg inhibit I/R-induced pS703-NHE1 in mouse hearts.
We have previously shown that in response to extracellular stimuli (eg, Ang II, H2O2, and anoxia/reoxygenation), RSK phosphorylates NHE1 at S703, recruits 14-3-3 binding to NHE1, and maintains high activity of NHE1.13-15,26 Those discoveries were based on in vitro kinase assays and NHE1 activity measurements (pHi recovery assay) with NHE1 S703A and DN-RSK.13-15,26 We now extend those findings through directly detecting phosphorylation of NHE1 S703 with a phospho-specific antibody.
The rationale for inhibiting NHE1 for cardioprotection has been well documented and reviewed.32-36 Pharmacologic or genetic inhibition of NHE1 activity has been proven to protect mouse hearts from I/R injury,4-7 possibly through inhibiting mitochondria permeability transition pore and reactive oxygen species generation.31,37,38 However, clinical trials using NHE1 inhibitors showed minimal positive effects.8-10 In addition, 2 recent studies by Imahashi et al39 and Cook et al29 reported that transgenic mice with cardiac overexpression of NHE1 were protected from I/R. The latter study interpreted this phenotype was due to the increased endoplasmic reticulum stress by overexpressing NHE1. Another study using transgenic mice overexpressing activated form of NHE1 showed higher glycolysis, fatty acid oxidation, adenosine triphosphate (ATP) production, and less cardiac I/R injury compared to NLC and WT-NHE1-Tg.40 Interestingly, NHE1 inhibition still provided beneficial effect in both NHE1-Tg and NLC mice upon I/R.38,39 These results indicate the complicated role of NHE1 in I/R injury and support our hypothesis that maintaining basal NHE1 activity is important for I/R protection.
One study done by Karki et al reported that sustained intracellular acidosis (SIA) increased NHE1 activity independent of pS703-NHE1.41 They also showed DN-RSK (K100A) did not block SIA-increased NHE1 activity. However, the same group reported RSK was phosphorylated and activated by SIA. This RSK together with ERK then activated NHE1.42 It has been reported that both K100 and K451 sites are important for RSK activation of its substrates.43,44 Therefore, it will be interesting to see whether RSK inhibitor can block SIA-increased NHE1 activity. Importantly, there are significant differences between activation of RSK and NHE1 by I/R compared to SIA, especially greater oxidative stress in I/R.
Recently, chemicals targeting CTKD and NTKD of RSK have been reported to inhibit its activity in vitro.18,28,45,46 However, all the known RSK inhibitors have to be used at relatively high concentrations from 3 μmol/L (fmk)27,28 to 10 μmol/L (BI-D1870),45 which raises the possibility of nonspecific effects. BIX02565 is designed to compete with ATP binding on NTKD of RSK. Our study showed that BIX02565 inhibited RSK downstream signaling at 1 μmol/L, indicating a higher potency of this novel inhibitor. The NTKD of RSK has a similar structure to the active domain of PKC.47 To exclude the possibility that NHE1 is phosphorylated by PKC, we showed that PKC inhibitor calphostin C had no effect on pS703-NHE1. This result is consistent with our previous report that Ang II-stimulated pS703-NHE1 in RASM cells was mediated by RSK but not PKC.26 It further supports the concept that BIX02565 specifically inhibits RSK activation.
As the first report of targeting RSK (a key regulator of NHE1) inhibition in cardiac I/R, we suspect that RSK inhibitor may work in similar patient population (the subgroup of patients about to undergo coronary bypass surgery in the GUARDIAN trial) to NHE1 inhibitors and has to be present through both ischemia and reperfusion stages.8,10,48,49 Intriguingly, in the current study, we showed that BIX02565 (1 μmol/L) inhibited agonist-induced NHE1 phosphorylation/activity without affecting the basal NHE1. Moreover, significant cardiac improvement after I/R was observed in 100 nmol/L BIX02565-treated mouse hearts. These results may be explained by several mechanisms: The in vivo potency of BIX02565 is greater than in vitro, pS703-NHE1 phosphorylation is a better indicator than pHi recovery of NHE1 agonist-mediated activity, and partial inhibition of NHE1 is protective for I/R. Further investigation of RSK inhibitors in a variety of disease models may provide greater understanding of the potential of RSK inhibition as an alternative approach.
There are several caveats to the interpretation of the data presented here. First, RSK is directly phosphorylated and activated by ERK1/2 in response to various extracellular stimuli, such as hormones, growth factors, and neurotransmitters. Upon activation, RSK phosphorylates IκB,50 which leads to the ubiquitination/degradation of IκB, activation of transcription factor NFκB, and consequent promotion of cell inflammation and proliferation. Activation of NFκB has been reported in many cardiac diseases such as atherosclerosis,51-53 chronic heart failure,54 and I/R.55 Therefore, the RSK inhibitor may act through pathways other than NHE1 to provide cardioprotection. Second, a recent report found that BIX02565 had >50% inhibition on ligand binding for adrenergic α, β, and imidazoline I2 receptors, with the IC50 values between 0.052 and 1.820 μmol/L in vitro. BIX02565 at 3 μmol/L ex vivo and 21.8 μmol/L in vivo, respectively, affected aortic ring relaxation, mean arterial pressure, and heart rate in rat.21 Although BIX02565 was used at 30 times lower dose (100 nmol/L) in our ex vivo studies in mouse, we cannot fully excluded the possibility of off-target effect. Third, it is possible that BIX02565 attenuated ischemia-induced myocardial stunning by preventing ischemia-stimulated NHE1 activation (which leads to operation of the Na+/Ca2+ exchanger in reverse mode) and Ca2+ overloading in the cells.
In conclusion, the present study found that a novel RSK inhibitor, BIX02565, targeting NTKD kinase of RSK, significantly inhibited pS703-NHE1 and improved cardiac function after I/R. The present findings suggest that BIX02565 and related compounds may provide new therapeutic options for patients with myocardial infarction, coronary artery bypass graft surgery, and other situations where I/R affects organ function. However, the chemical characteristics of current RSK inhibitors need to be improved for in vivo use. Most importantly, more rapid of inhibition of RSK with improved inhibitors would be beneficial so that they can be given during reperfusion.
Supplementary Material
Acknowledgments
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Author Contribution: Xi Shi contributed to conception or design and acquisition, analysis, or interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; agreed to be accountable for all aspects of work ensuring integrity and accuracy. Margaret M. O'Neill, Scott MacDonnell, and Paul S. Brookes contributed to acquisition, analysis, or interpretation and gave final approval. Chen Yan contributed to acquisition, analysis, or interpretation; critically revised the manuscript; and gave final approval. Bradford C. Berk contributed to conception or design and acquisition, analysis, or interpretation; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring integrity and accuracy. The authors had full access to the data and take full responsibility for their integrity. All authors have read and agreed to the manuscript as written.
Declaration of Conflicting Interests: The author(s) declared potential conflicts of interest with respect to the research, authorship, and/or publication of this article: (a) Boehringer-Inglehiem. (b) Boehringer-Inglehiem employs Margaret M. O'Neill, Scott MacDonnell. They have less than 1% ownership of the company. (c) time and effort of Margaret M. O'Neill, Scott MacDonnell was partly supported for this project and BIX02565 was provided by the company.
Supplemental Material: The online [appendices/data supplements/etc] are available at http://cpt.sagepub.com/supplemental
References
- 1.Cingolani HE, Ennis IL, Mosca SM. Nhe-1 and nhe-6 activities: ischemic and reperfusion injury. Circ Res. 2003;93(8):694–696. doi: 10.1161/01.RES.0000097926.29243.96. [DOI] [PubMed] [Google Scholar]
- 2.Levitsky J, Gurell D, Frishman WH. Sodium ion/hydrogen ion exchange inhibition: A new pharmacologic approach to myocardial ischemia and reperfusion injury. J Clin Pharmacol. 1998;38(10):887–897. doi: 10.1002/j.1552-4604.1998.tb04383.x. [DOI] [PubMed] [Google Scholar]
- 3.Bobulescu IA, Di Sole F, Moe OW. Na+/h+ exchangers: Physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens. 2005;14(5):485–494. doi: 10.1097/01.mnh.0000174146.52915.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Meyer JW, Ashraf M, Shull GE. Mice with a null mutation in the nhe1 na+-h+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. 2003;93(8):776–782. doi: 10.1161/01.RES.0000094746.24774.DC. [DOI] [PubMed] [Google Scholar]
- 5.Chakrabarti S, Hoque AN, Karmazyn M. A rapid ischemiainduced apoptosis in isolated rat hearts and its attenuation by the sodium-hydrogen exchange inhibitor hoe 642 (cariporide) J Mol Cell Cardiol. 1997;29(11):3169–3174. doi: 10.1006/jmcc.1997.0561. [DOI] [PubMed] [Google Scholar]
- 6.Karmazyn M. The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann N Y Acad Sci. 1999;874:326–334. doi: 10.1111/j.1749-6632.1999.tb09248.x. [DOI] [PubMed] [Google Scholar]
- 7.Scholz W, Albus U, Counillon L, et al. Protective effects of hoe642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29(2):260–268. [PubMed] [Google Scholar]
- 8.Theroux P, Chaitman BR, Danchin N, et al. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the guardian trial. Guard during ischemia against necrosis (guardian) investigators. Circulation. 2000;102(25):3032–3038. doi: 10.1161/01.cir.102.25.3032. [DOI] [PubMed] [Google Scholar]
- 9.Zeymer U, Suryapranata H, Monassier JP, et al. The na+/h+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (escami) trial. J Am Coll Cardiol. 2001;38(6):1644–1650. doi: 10.1016/s0735-1097(01)01608-4. [DOI] [PubMed] [Google Scholar]
- 10.Mentzer RM, Jr, Bartels C, Bolli R, et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the expedition study. Ann Thorac Surg. 2008;85(4):1261–1270. doi: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 11.Takeishi Y, Huang Q, Abe J, et al. Activation of mitogen-activated protein kinases and p90 ribosomal s6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc Res. 2002;53(1):131–137. doi: 10.1016/s0008-6363(01)00438-2. [DOI] [PubMed] [Google Scholar]
- 12.Itoh S, Ding B, Shishido T, et al. Role of p90 ribosomal s6 kinasemediated prorenin-converting enzyme in ischemic and diabetic myocardium. Circulation. 2006;113(14):1787–1798. doi: 10.1161/CIRCULATIONAHA.105.578278. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi E, Abe J, Gallis B, et al. P90rsk is a serum-stimulated nhe1 kinase: Regulatory phosphorylation of serine 703 of na+/h+ exchanger isoform-1. J Biol Chem. 1999;274(29):20206–20214. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- 14.Lehoux S, Abe J, Florian JA, Berk BC. 14-3-3 binding to na+/h+ exchanger isoform-1 is associated with serum- dependent activation of na+/h+ exchange. J Biol Chem. 2001;276(19):15794–15800. doi: 10.1074/jbc.M100410200. [DOI] [PubMed] [Google Scholar]
- 15.Maekawa N, Abe J, Shishido T, et al. Inhibiting p90 ribosomal s6 kinase prevents na+-h+ exchanger-mediated cardiac ischemia-reperfusion injury. Circulation. 2006;113(21):2516–2523. doi: 10.1161/CIRCULATIONAHA.105.563486. [DOI] [PubMed] [Google Scholar]
- 16.Frodin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase rsk2 that recruits and activates pdk1. EMBO J. 2000;19(12):2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anjum R, Blenis J. The rsk family of kinases: Emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9(10):747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen TL. Targeting rsk: an overview of small molecule inhibitors. Anticancer Agents Med Chem. 2008;8(7):710–716. doi: 10.2174/187152008785914770. [DOI] [PubMed] [Google Scholar]
- 19.Kirrane TM, Boyer SJ, Burke J, et al. Indole rsk inhibitors. Part 2: optimization of cell potency and kinase selectivity. Bioorg Med Chem Lett. 2012;22(1):738–742. doi: 10.1016/j.bmcl.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 20.Boyer SJ, Burke J, Guo X, et al. Indole rsk inhibitors. Part 1: Discovery and initial sar. Bioorg Med Chem Lett. 2012;22(1):733–737. doi: 10.1016/j.bmcl.2011.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Fryer RM, Muthukumarana A, Chen RR, et al. Mitigation of off-target adrenergic binding and effects on cardiovascular function in the discovery of novel ribosomal s6 kinase 2 inhibitors. J Pharmacol Exp Ther. 2012;340(3):492–500. doi: 10.1124/jpet.111.189365. [DOI] [PubMed] [Google Scholar]
- 22.Vallega GA, Canessa ML, Berk BC, Brock TA, Alexander RW. Vascular smooth muscle na+-h+ exchanger kinetics and its activation by angiotensin ii. Am J Physiol. 1988;254(6 pt 1):C751–C758. doi: 10.1152/ajpcell.1988.254.6.C751. [DOI] [PubMed] [Google Scholar]
- 23.Guzman-Perez A, Wester RT, Allen MC, et al. Discovery of zoniporide: A potent and selective sodium-hydrogen exchanger type 1 (nhe-1) inhibitor with high aqueous solubility. Bioorg Med Chem Lett. 2001;11(6):803–807. doi: 10.1016/s0960-894x(01)00059-2. [DOI] [PubMed] [Google Scholar]
- 24.Brookes PS, Digerness SB, Parks DA, Darley-Usmar V. Mitochondrial function in response to cardiac ischemia-reperfusion after oral treatment with quercetin. Free Radic Biol Med. 2002;32(11):1220–1228. doi: 10.1016/s0891-5849(02)00839-0. [DOI] [PubMed] [Google Scholar]
- 25.Shi X, Yan C, Nadtochiy SM, Abe J, Brookes PS, Berk BC. P90 ribosomal s6 kinase regulates activity of the renin-angiotensin system: a pathogenic mechanism for ischemia-reperfusion injury. JMol Cell Cardiol. 2011;51(2):272–275. doi: 10.1016/j.yjmcc.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi E, Abe J, Berk BC. Angiotensin ii stimulates p90rsk in vascular smooth muscle cells. A potential na+/h+ exchanger kinase. Circ Res. 1997;81(2):268–273. doi: 10.1161/01.res.81.2.268. [DOI] [PubMed] [Google Scholar]
- 27.Cuello F, Snabaitis AK, Cohen MS, Taunton J, Avkiran M. Evidence for direct regulation of myocardial na+/h+ exchanger isoform 1 phosphorylation and activity by 90-kda ribosomal s6 kinase (rsk): Effects of the novel and specific rsk inhibitor fmk on responses to {alpha} 1-adrenergic stimulation. Mol Pharmacol. 2007;71(3):799–806. doi: 10.1124/mol.106.029900. [DOI] [PubMed] [Google Scholar]
- 28.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308(5726):1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cook AR, Bardswell SC, Pretheshan S, et al. Paradoxical resistance to myocardial ischemia and age-related cardiomyopathy in nhe1 transgenic mice: a role for er stress? J Mol Cell Cardiol. 2009;46(2):225–233. doi: 10.1016/j.yjmcc.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Coccaro E, Karki P, Cojocaru C, Fliegel L. Phenylephrine and sustained acidosis activate the neonatal rat cardiomyocyte na+/h+ exchanger through phosphorylation of amino acids ser770 and ser771. Am J Physiol Heart Circ Physiol. 2009;297(2):H846–H858. doi: 10.1152/ajpheart.01231.2008. [DOI] [PubMed] [Google Scholar]
- 31.Garciarena CD, Caldiz CI, Correa MV, et al. Na+/h+ exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J Appl Physiol. 2008;105(6):1706–1713. doi: 10.1152/japplphysiol.90616.2008. [DOI] [PubMed] [Google Scholar]
- 32.Avkiran M. Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am J Cardiol. 1999;83(10A):10G–17G. doi: 10.1016/s0002-9149(99)00215-5. discussion 17G-18G. [DOI] [PubMed] [Google Scholar]
- 33.Clements-Jewery H, Sutherland FJ, Allen MC, Tracey WR, Avkiran M. Cardioprotective efficacy of zoniporide, a potent and selective inhibitor of na+/h+ exchanger isoform 1, in an experimental model of cardiopulmonary bypass. Br J Pharmacol. 2004;142(10A):57–66. doi: 10.1038/sj.bjp.0705749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avkiran M, Marber MS. Na(+)/h(+) exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Coll Cardiol. 2002;39(5):747–753. doi: 10.1016/s0735-1097(02)01693-5. [DOI] [PubMed] [Google Scholar]
- 35.Karmazyn M, Sawyer M, Fliegel L. The na(+)/h(+) exchanger: a target for cardiac therapeutic intervention. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5(4):323–335. doi: 10.2174/1568006054553417. [DOI] [PubMed] [Google Scholar]
- 36.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88(2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villa-Abrille MC, Cingolani E, Cingolani HE, Alvarez BV. Silencing of cardiac mitochondrial nhe1 prevents mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol. 2011;300(4):H1237–H1251. doi: 10.1152/ajpheart.00840.2010. [DOI] [PubMed] [Google Scholar]
- 38.Luo J, Chen H, Kintner DB, Shull GE, Sun D. Inhibition of na+/h+ exchanger isoform 1 attenuates mitochondrial cytochrome c release in cortical neurons following in vitro ischemia. Acta Neurochir Suppl. 2006;96:244–248. doi: 10.1007/3-211-30714-1_52. [DOI] [PubMed] [Google Scholar]
- 39.Imahashi K, Mraiche F, Steenbergen C, Murphy E, Fliegel L. Overexpression of the na+/h+ exchanger and ischemia-reperfusion injury in the myocardium. Am J Physiol Heart Circ Physiol. 2007;292(5):H2237–H2247. doi: 10.1152/ajpheart.00855.2006. [DOI] [PubMed] [Google Scholar]
- 40.Mraiche F, Wagg CS, Lopaschuk GD, Fliegel L. Elevated levels of activated nhe1 protect the myocardium and improve metabolism following ischemia/reperfusion injury. J Mol Cell Cardiol. 2011;50(1):157–164. doi: 10.1016/j.yjmcc.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 41.Karki P, Coccaro E, Fliegel L. Sustained intracellular acidosis activates the myocardial na(+)/h(+) exchanger independent of amino acid ser(703) and p90(rsk) Biochim Biophys Acta. 2010;1798(8):1565–1576. doi: 10.1016/j.bbamem.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Malo ME, Li L, Fliegel L. Mitogen-activated protein kinase-dependent activation of the na+/h+ exchanger is mediated through phosphorylation of amino acids ser770 and ser771. J Biol Chem. 2007;282(9):6292–6299. doi: 10.1074/jbc.M611073200. [DOI] [PubMed] [Google Scholar]
- 43.Chrestensen CA, Sturgill TW. Characterization of the p90 ribosomal s6 kinase 2 carboxyl-terminal domain as a protein kinase. J Biol Chem. 2002;277(31):27733–27741. doi: 10.1074/jbc.M202663200. [DOI] [PubMed] [Google Scholar]
- 44.Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal s6 kinase (rsk) regulates extracellular signal-regulated kinase docking and rsk activity. Mol Cell Biol. 2003;23(14):4796–4804. doi: 10.1128/MCB.23.14.4796-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sapkota GP, Cummings L, Newell FS, et al. Bi-d1870 is a specific inhibitor of the p90 rsk (ribosomal s6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401(1):29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal s6 kinase (rsk) reveals an unexpected role for rsk in cancer cell proliferation. Cancer Res. 2005;65(3):1027–1034. [PubMed] [Google Scholar]
- 47.Frodin M, Gammeltoft S. Role and regulation of 90 kda ribosomal s6 kinase (rsk) in signal transduction. Mol Cell Endocrinol. 1999;151(1-2):65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 48.Avkiran M, Cook AR, Cuello F. Targeting na(+)/h(+) exchanger regulation for cardiac protection: a RSKy approach? Curr Opin Pharmacol. 2008;8(2):133–140. doi: 10.1016/j.coph.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 49.Murphy E, Allen DG. Why did the nhe inhibitor clinical trials fail? J Mol Cell Cardiol. 2009;46(2):137–141. doi: 10.1016/j.yjmcc.2008.09.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghoda L, Lin X, Greene WC. The 90-kda ribosomal s6 kinase (pp90rsk) phosphorylates the n-terminal regulatory domain of ikappabalpha and stimulates its degradation in vitro. J Biol Chem. 1997;272(34):21281–21288. doi: 10.1074/jbc.272.34.21281. [DOI] [PubMed] [Google Scholar]
- 51.Baker RG, Hayden MS, Ghosh S. Nf-kappab, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dabek J, Kulach A, Gasior Z. Nuclear factor kappa-light-chain-enhancer of activated b cells (nf-kappab): A new potential therapeutic target in atherosclerosis? Pharmacol Rep. 2010;62(5):778–783. doi: 10.1016/s1734-1140(10)70338-8. [DOI] [PubMed] [Google Scholar]
- 53.Demer L, Tintut Y. The roles of lipid oxidation products and receptor activator of nuclear factor-kappab signaling in atherosclerotic calcification. Circ Res. 2011;108(12):1482–1493. doi: 10.1161/CIRCRESAHA.110.234245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of nf-kappab in the heart: to be or not to nf-kappab. Circ Res. 2011;108(9):1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 55.Valen G. Signal transduction through nuclear factor kappa b in ischemia-reperfusion and heart failure. Basic Res Cardiol. 2004;99(1):1–7. doi: 10.1007/s00395-003-0442-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.