

Abstract
Belatacept is used to prevent allograft rejection, but fails to do so in a sizable minority of patients due to inadequate control of costimulation-resistant T cells. We have reported control of costimulation-resistant rejection when belatacept is combined with perioperative alemtuzumab-mediated lymphocyte depletion and rapamycin (ABR). To assess the means by which the ABR regimen controls belatacept-resistant rejection, we studied 20 ABR-treated patients, characterizing peripheral lymphocyte phenotype and functional responses to donor, third-party, and viral antigens using flow cytometry, intracellular cytokine staining, and CFSE-based lymphocyte proliferation. Compared to conventional immunosuppression in 10 patients, lymphocyte depletion evoked substantial homeostatic lymphocyte activation balanced by regulatory T and B cell phenotypes. The reconstituted T cell repertoire was enriched for CD28+ naïve cells, notably diminished in belatacept-resistant CD28- memory subsets, depleted of polyfunctional donor-specific T cells, but able to respond to third-party and latent herpes viruses. B cell responses were similarly favorable, without alloantibody development, and a reduction in memory subsets—changes not seen in conventionally treated patients. ABR regimen uniquely alters the immune profile, producing a repertoire enriched for CD28+ T cells, hyporesponsive to donor-alloantigen, and competent in its protective immune capabilities. The resulting repertoire is permissive for control of rejection with belatacept monotherapy.
TRIAL REGISTRATION
ClinicalTrials.gov - NCT00565773
Introduction
Conventional immunosuppression for kidney transplantation is based on regimens using calcineurin inhibitors (CNIs) (1-2). These regimens nonspecifically inhibit T cell activation, effectively preventing acute T cell-mediated allograft rejection at the expense of impaired T cell mediated immunity to viral infections. CNIs also have direct nephrotoxicity. As such, efforts have been made to replace CNIs with agents that more selectively control alloimmunity and avoid off-target side effects.
Belatacept, a B7-specific fusion protein, has been approved as a CNI replacement for kidney transplantation. Belatacept directly blocks the interaction between B7-expressing antigen presenting cells and CD28-expressing naïve T cells without significant off-target side effects (3-5). However, recent clinical studies have observed that patients treated with non-depletional induction followed by a belatacept-based regimen without CNIs experienced substantially higher acute rejection rates than CNI-based standard maintenance regimen (5-6). The underlying mechanisms of this B7 blockade-resistant allograft rejection have been attributed to the activation of allo-specific effector memory T cells (TEM) lacking CD28 expression (7-10).
Lymphocyte depletion using the humanized CD52-specific monoclonal antibody alemtuzumab effectively reduces the risk early acute rejection in kidney transplantation (11-13). Rapamycin, a mechanistic target of rapamycin inhibitor, has been shown experimentally to prolong allograft survival in combination with B7 costimulation blockade when used with or without pre-transplant donor hematopoietic cell infusion (14-17). Recently, we performed a pilot clinical trial (18) investigating the use of a regimen combining alemtuzumab induction with belatacept/rapamycin maintenance therapy (the ABR regimen) without CNIs and steroids. We demonstrated that this novel regimen effectively prevents costimulation blockade–resistant acute allograft rejection. Indeed, many patients selected for their low immunological risk were successfully weaned from rapamycin to belatacept monotherapy without rejection. Additionally, patients in this cohort showed a lack of belatacept-resistant T cell-mediated rejection. These peripheral T cells consist of naïve, central memory, effector memory, and terminally differentiated effector memory subsets. Allo-specific T cells are typically characterized as memory cells based on the lack of surface expression of CD197 and CD45RA (10), and these cells are resistant to B-7 costimulation blockade as they typically lack the CD28 surface protein.
Herein, we report a series of studies designed to elucidate the underlying mechanisms contributing to these favorable clinical outcomes of the ABR regimen. Our studies examine the dynamics, phenotypes, activation, proliferation and antigen specificity of reconstituting T and B lymphocytes seen under the ABR regimen. We demonstrate that the favorable clinical performance of this regimen is associated with reconstitution of a repertoire that is hyporesponsive to donor antigen, competent to third party and viral antigen, and enriched for cells expressing CD28, the downstream target of belatacept-mediated blockade. These data provide a first look at the mechanisms defining the efficacy of this regimen and provide further insight for the use of belatacept in renal transplantation.
Methods
Patients, Protocol Therapy, and Follow-up
This pilot study included 20 patients (median 45 years, range 20–69; 12 male:8 female, 16C:4AA, all EBV seropositive) enrolled under an IRB-approved, Food and Drug Administration sponsored clinical trial following informed consent. Patients received a renal allograft from either living related or unrelated donors. Immunosuppression consisted of alemtuzumab induction (30 mg, intravenously prior to transplantation) followed by maintenance therapy with intravenous infusion of belatacept and oral sirolimus as previously reported (18). All patients were included in the analysis regardless of randomization to donor specific transfusion or rapamycin weaning status. Patients were monitored weekly for the first month, monthly until 6 months, and then every 6 months until 36 months post-transplantation. Fresh blood from patients was collected in BD Vacutainers containing EDTA (BD Biosciences) before and after transplantation, and during each visit for flow cytometric analyses. An additional 10 patients served as the comparator group and were treated with basiliximab induction and a maintenance immunosuppressive regimen consisting of tacrolimus (trough levels 5–10 ng/mL), MMF (500 mg, twice daily), and steroids. These patients were selected for similar freedom from rejection, clinical stability and were also enrolled under an IRB-approved immune monitoring protocol following informed consent.
Reagents and Monoclonal Antibodies
The fluorochrome labeled monoclonal antibodies (mAbs) anti-CD2-FITC, anti-CD3-Alexa 700, anti-CD3-PerCP, anti-CD4-V450, anti-CD4-PE, anti-CD8-APC Fluor 780, anti-CD8-PacBlue, anti-CD16-FITC, anti-CD20 PECy7, anti-CD19-V450, anti-CD20-APCCy7, anti-CD25-PEcy7, anti- CD28-PE, anti-CD28-FITC, anti-CD38-PE, anti-CD39-FITC, anti-CD45-PerCP, anti-CD56-APC, anti-CD57-FITC, anti-CD197-PECy7, anti-HLA-DR, anti-IgM, anti-IgD, anti-Ki67-FITC, anti-Bcl-2-PE, anti-TNF-α-PE, anti-TNF-α-APC, IFN-γ-PcpCy5.5, and IL-2-PECy7 were purchased from BD Biosciences (Franklin Lakes, NJ). Anti-CD45RA-QDOT655 and anti-CD69-FITC mAb were obtained from Invitrogen (Carlsbad, CA). Anti-CD8-Alexa780, anti-CD27-Alexa Fluor 700, and anti-CD38-eFluor 650NC were obtained from E-bioscience (San Diego, CA). Anti-CD24-PEcy7 and anti-CD279-PE (PD-1) were purchased from Biolegend (San Diego, CA).
Human EBV protein and CMV peptide pool of pp65 sequence consisting of 138 peptides (15 mers with 11 amino acid overlaps) was purchased from JPT Peptide Technologies (Berlin, Germany).
All patients were DSA-free with a calculated panel reactive antibody (PRA) ≤20% at enrollment. Patient samples were assessed for donor-specific alloantibody post-transplantation as described previously (18).
Absolute Lymphocyte Subset Analysis
Absolute lymphocyte subsets were determined by BD Trucount analysis according to manufacturer’s protocol. Briefly, 50 μl of blood was added into BD Trucount tube, and incubated with mAb specific to CD3, CD4, CD8, CD16, CD20, CD45, and CD56 at room temperature for 15 minutes followed by incubation with 1 ml of High-Yield Lysing Solution (Invitrogen, Carlsbad, CA) at 37°C for 10 minutes. Cells were analyzed using LSR II polychromatic flow cytometry (BD Biosciences), and the data analysis was performed using FlowJo software (Tree Star, San Carlos, CA).
Cells and Flow Cytometry
Peripheral blood mononuclear cells (PBMCs) were isolated from fresh blood collected before and after transplantation by Ficoll density gradient centrifugation according to the manufacturer’s protocol (BD Biosciences). PBMCs were re-suspended in FACS buffer (PBS containing 1% fetal bovine serum), and at least 105 PBMCs were surface-stained with mAbs in the darkness at room temperature. Intracellular staining for Ki67 and Bcl-2 was carried out based on the protocol provided by manufacturer (BD Biosciences). Briefly, surface-stained cells were permeabilized/fixed with Perm/Fixation buffer (BD Biosciences) for 45 minutes on ice. Cells were stained with mAb specific for Ki67 and Bcl-2 at 4°C for 30 minutes. Cells were resuspended in FACS buffer and analyzed using LSR II polychromatic flow cytometry. Data analysis was performed using FlowJo software (Tree Star, San Carlos, CA).
Allogeneic Stimulation and Viral Peptide and Protein Stimulation
Blood samples were collected from patients prior to transplantation and at 12, 18, 24, and 36 months post-transplantation. PBMCs were isolated from fresh blood by Ficoll density gradient centrifugation according to the manufacturer’s protocol (BD Biosciences). Cells were diluted with frozen medium (FBS containing 10% DMSO) at 10 × 106 cells/ml followed by step-down frozen in Nalgene™ 1°C Frizzing container (Thermo Scientific, Rockford, IL) in -80°C freezer for 24 hours. Cells were then stored at -140°C. To ensure the quality of cryopreserved cells for functional assays, PBMCs from normal healthy volunteers or patients were randomly selected and assessed by trypan blue assay every three months, and the viability of cells after cryopreservation was 88 – 90 ± 2%.
T cell–mediated responses to specific allo-donors or HLA-mismatched third-party donors were evaluated using the method described previously (8, 10, 19). Briefly, recipient’s PBMCs were exposed to irradiated allo-stimulator PBMCs isolated from specific donors and HLA-mismatched third-party donors. Stimulators were depleted of CD3+ cells with magnetic CD3 MicroBeads and LS magnetic columns according to manufacturer’s instruction (Miltenyi Biotec, Auburn, CA). 2.5 × 105 responder PBMCs were incubated with 2.5 × 105 irradiated stimulators containing 1 μl/ml GolgiPlug at 37°C for 12 hours.
CMV- and EBV-specific memory T cell responses in patients before and after transplantation were investigated using the method described previously (10). Seropositivity for CMV and EBV was established before transplantation based on viral-IgG reactivity as determined by the Emory University clinical pathology lab. Frozen PBMCs from transplant patients were thawed and washed with RPMI-1640 medium containing 5% FBS, and then diluted with T cell culture medium followed by incubation at 37°C for 6 hours. Normal control PBMCs were isolated from CMV- and EBV-seropositive healthy volunteers for use as internal controls for the viral stimulation assay. Cells were washed and diluted to 106 cells/ml with RPMI-1640 medium containing 10% FBS. 2.5 × 105 cells were stimulated by 1.75 μg/ml of CMV pp65 peptides or EBV protein with 1 μl/ml of GolgiPlug at 37°C for 12 hours.
Intracellular Cytokine Staining (ICCS)
Cells were collected after 12-hour stimulation by allogeneic donor PBMCs and viral antigens, and then surface-stained with mAb at room temperature for 15 minutes followed by permeabilization and fixation with Cytofix/Cytoperm for 45 minutes on ice. ICCS with mAb directed to TNF-α, IFN-γ, and IL-2 was performed at 4°C for 30 minutes after final wash with Perm/Wash buffer. Cells were washed once with washing buffer and analyzed using polychromatic flow cytometry (BD Biosciences LSR II), and the analysis of data was performed using FlowJo software.
CFSE-Based Lymphocyte Proliferation Assay
To assess the allo-responding T cell proliferation before and after transplantation, a CFSE-based one-way mixed lymphocyte reaction was performed using the method described previously (8, 10, 20-21). Briefly, 2.5 × 105 CFSE-labeled responder PBMCs were incubated with 2.5 × 105 irradiated allo-stimulators in RPMI-1640 medium containing 10% FBS at 37°C for 5 days. Cells were collected and washed with FACS buffer, and then surface-stained with mAb directed to CD3, CD4, CD8, and CD28 at room temperature for 15 minutes. Cells were washed and analyzed acquisition with LSR II polychromatic flow cytometry.
Measurement of Serum BAFF and APRIL
To determine the serum levels of BAFF and APRIL during B cell repopulation after alemtuzumab induction, a standard ELISA was performed on serial serum samples in duplicate using human BAFF ELISA kit (R&D Systems, Minneapolis, MN), and human APRIL ELISA kit (Biolegend, San Diego, CA) according to the protocol provided by manufacturers.
Statistical Analysis
One-way ANOVA with post testing for linear trend was performed to compare variables between pre- and post-transplantation. Unpaired t test was performed to determine the statistical significance between standard care patients and patients treated with alemtuzumab induction and belatacept/rapamycin maintenance regimen. A p value of less than 0.05 was considered as statistically significant.
Results
Profound depletion of lymphocytes by alemtuzumab induction followed by rapid B cell and delayed T cell repopulation
T cell reconstitution following alemtuzumab-mediated depletion under the cover of belatacept-based therapy has not been previously described. We therefore longitudinally analyzed alemtuzumab-induced peripheral blood lymphocyte depletion and repopulation in the context of belatacept and sirolimus As shown in Figure 1, peripheral T (CD3+) and B (CD20+) lymphocytes were substantially depleted immediately after alemtuzumab induction consistent with previous reports (11-13,18). Absolute CD8+ cell counts returned slowly to baseline at 18 months post-depletion. CD4+ cells failed to return to baseline within three years. In contrast to T cells, a B cell reconstitution was relatively rapid, with absolute B cell counts reaching and then surpassing baseline counts at 6 months, remaining so through month 36. Absolute counts for granulocytes were not markedly decreased by depletional induction. Significant depletion of CD16+CD56+ cells was noted with reconstitution by 6 months post-depletion. Monocyte absolute counts were only transiently reduced in the first month post-depletion.
Figure 1. Repopulation of peripheral blood mononuclear cells after renal allograft transplantation.
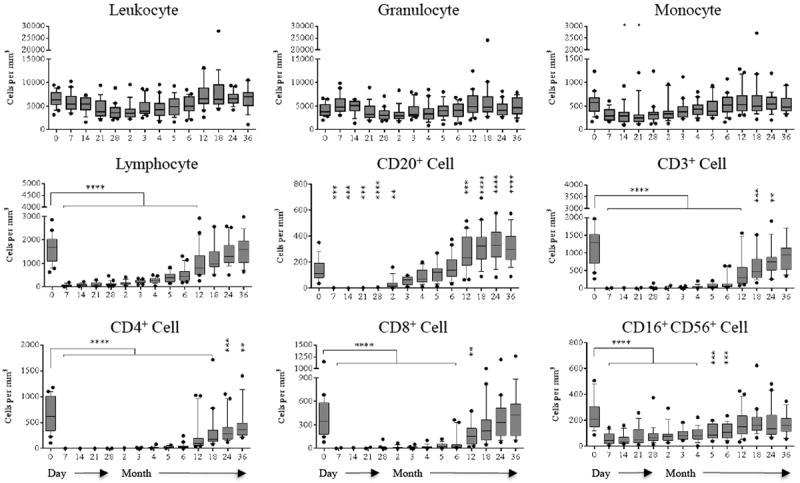
The absolute cell counts and subsets were analyzed by polychromatic flow cytometry. Leukocytes and granulocyte absolute numbers remain unchanged post-transplantation. Monocyte absolute counts decrease transiently within first month post-transplantation. In contrast, profound T cell (CD3+) and B cell (CD3-CD20+) depletion is achieved following alemtuzumab induction. T cell repopulation remained lethargic; however, B cells show rapid repopulation, with absolute numbers exceeding baseline values at 6 months post-transplantation. (*p ≤ 0.05; ** p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001)
Homeostatic proliferation and activation of both CD4+ and CD8+ TNaïve cells versus TEM cells
Lymphocyte activation status was markedly altered during repopulation. CD4+ and CD8+ T cell proliferation was measured longitudinally by assessing intracellular Ki67 expression, a nuclear protein involved in cell proliferation (22). As shown in Figure 2A, homeostatic T cell proliferation (both CD4+ and CD8+ cells) persisted through and beyond 6 months post-depletion. Patients treated with the study regimen were found to have a significantly higher frequency of Ki67-expressing T cells than patients treated with nondepleting standard care immunosuppression at similar time points. The lymphoproliferative burst post-depletion occurred predominantly in the TNaïve subset and waned as lymphocyte counts approached baseline. As such, Ki67 defined the period of homeostatic T cell proliferation.
Figure 2. Post-depletional homeostatic proliferation and activation of CD4+ and CD8+ cells after renal allograft transplantation.
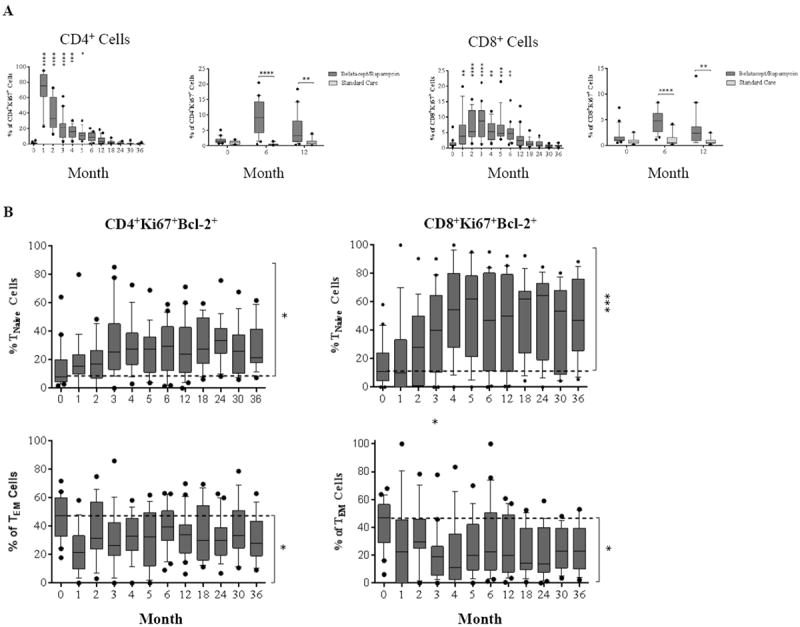
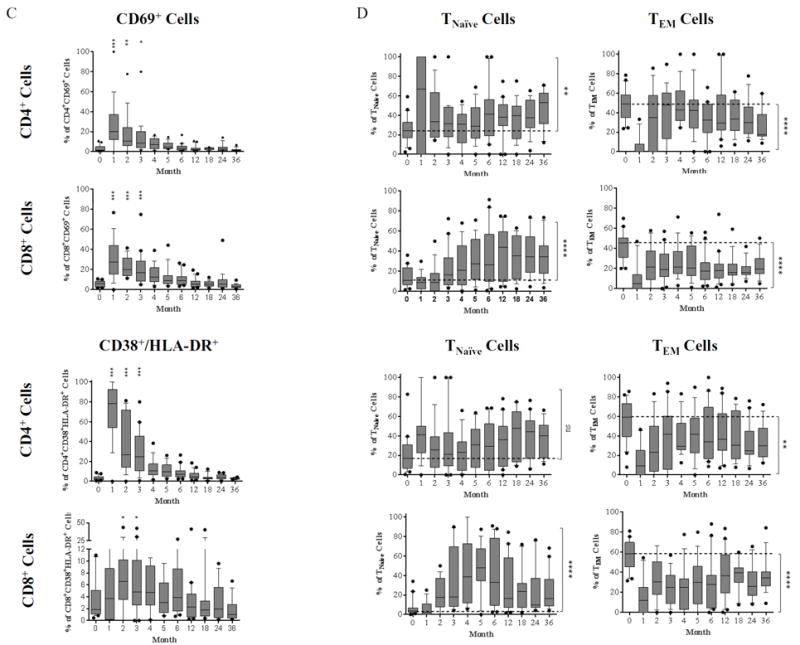
(A) The dynamics of homeostatic proliferation for both CD4+ and CD8+ cells post–alemtuzumab induction. Significantly higher percentages of Ki-67-expressing cells are seen in belatacept/rapamycin maintenance when compared with the standard care controls. (B) CD4+ Ki-67+ and CD8+ Ki-67+ cells which express the anti-apoptosis marker Bcl-2 are segregated by CD45RA and CD197 expression to determine naïve versus memory phenotypes. After depletion, both CD4+ and CD8+ Ki-67+Bcl-2+ double positive cells are more pronounced in the TNaïve population compared to baseline, with the reverse phenomenon observed in the TEM population. (C) The activation of T cells post-depletion is detected by up-regulation of CD69 and CD38/HLA-DR expression. Significant up-regulation for CD69 and CD38/HLA-DR is seen during early phase of both CD4+ and CD8+ T cell repopulation when compared with baseline. (D) CD69 and CD38/HLA-DR expressing cells are subdivided to memory and naïve subsets based on CD45RA and CD197 expression. TNaïve cells demonstrated a higher frequency of activation post-depletion, with decreased activation frequency in TEM cells. (*p ≤ 0.05; ** p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001)
Human T cells can be segregated into four distinct subsets based on the surface expression of CD197 (CCR7) and CD45RA (23): central memory (TCM, CD45RA-CD197+), naïve (TNaïve, CD45RA+CD197+), terminally differential effector memory (TEMRA, CD45RA+CD197-), and effector memory (TEM, CD45RA-CD197-). The memory phenotype of Ki67+Bcl-2+ expressing T cells was assessed based on the expression of CD45RA and CD197. In both CD4+ and CD8+ subsets, TNaïve populations expressed both Ki67 and the anti-apoptotic Bcl-2 (indicative of both proliferation and a survival advantage) to a significantly higher degree compared to TEM cells (Figure 2B). Contrary to prior evidence from murine models, T cell homeostatic proliferation did not derive solely from cells exhibiting a memory T cell phenotype (CCR7 negative, CD45RA negative), which has been described as an effector pseudomemory phenotype (24-25), but rather activation, as determined by upregulation of surface CD69 and CD38/HLA-DR expression, was disproportionately evident in the naïve compartment. Indeed, the effector pseudomemory phenotype is resistant to depletion and costimulation blockade, and the ABR may prevent this phenotype from prevailing after depletional induction.
An increased frequency of CD4+ and CD8+ cell activation was observed for approximately 3 months post-depletion (Figure 2C). Memory subset characterization demonstrated a significant increase in activation of the TNaïve compartment with concomitant reduction in the TEM compartment throughout the study period. At the end of the study period, this distribution remained predominated by TNaïve cells as the overall activation markers return to baseline (Figure 2D).
Homeostatic reconstitution with belatacept and rapamycin therapy produces CD8+ TEM and TNaïve frequencies favorable to costimulation blockade
Memory and naïve CD4+ cells ratios did not change significantly post-transplantation and throughout the study period (p ≥ 0.05, Figure 3A), although this observation may be due to low overall CD4+ counts. In contrast, repopulating CD8+ cells demonstrated a significant increase of TNaïve cell frequency (p ≤ 0.0001) with a significant decrease of TEM and TEMRA cell frequency (p ≤ 0.05) when compared with baseline proportions (Figure 3A). These reconstituting T cells, particularly CD8+ T cells, contained a significantly higher frequency of naïve cells and a significantly lower frequency of effector memory cells when compared with patients treated with a standard regimen (Figure 3B).
Figure 3. Dynamics of repopulating T cell subsets post-transplantation.
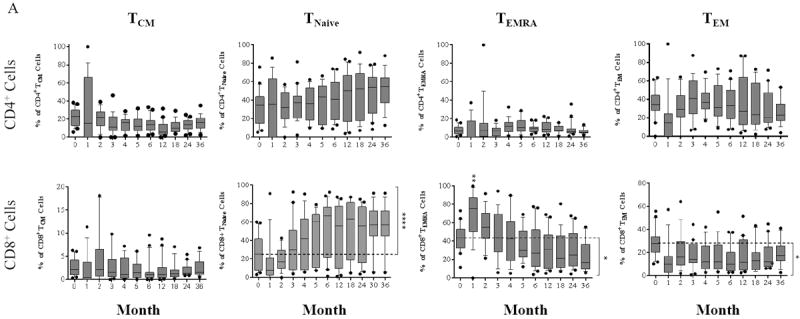
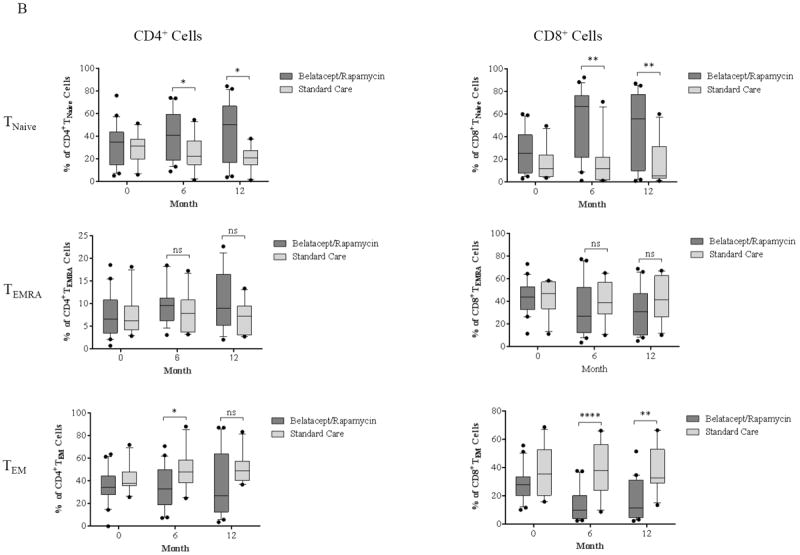
(A) CD4+ T cells to increase frequency of TNaïve cells and reduce the frequency of TEM cells during repopulation; however, this did not reach statistical significance when compared with baseline proportions. In contrast, CD8+ reconstitution contained increased in TNaïve and decreased in TEM and TEMRA cell percentages. (B) Patients treated with alemtuzumab induction followed by belatacept/rapamycin maintenance regimen show significantly higher percentages of TNaïve cells in both CD4+ and CD8+ compartments, with a significantly lower frequency of CD8+ TEM cells when compared with a cohort treated with basiliximab induction and standard care maintenance regimen. (*p ≤ 0.05; ** p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001)
Enrichment of CD8+ T cells with a costimulation sensitive phenotype during reconstitution
Previous studies have established that CD2hiCD28- T cells are resistant to B7 costimulation blockade (8, 26). We therefore examined how this regimen modulated CD28 expression. As shown in Figure 4A, CD4+ cells expression of CD28 was not significantly changed over the course of the study. CD8+CD28- T cell percentages were substantially more dynamic, transiently increasing in the first 2 months and decreasing thereafter. We further characterized both CD4+ and CD8+ T cells into four distinct subsets by CD2 and CD28 expression: CD2hiCD28-, CD2hiCD28+, CD2loCD28+, and CD2loCD28- (Figure 4B); CD4+ cells were predominantly comprised of CD2loCD28+ cells prior to depletion and did not have appreciable change post-depletion. In contrast, reconstituting CD8+ cells demonstrated a significant inversion of the CD2loCD28+ to CD2hiCD28+ T cell ratios when compared to baseline (p ≤ 0.001) and standard regimen controls (Figure 4B, p ≤ 0.001). The CD2loCD28+ subset was further defined by memory phenotype based on surface CD45RA and CD197 expression (Figure 4C) which demonstrated that reconstitution of CD8+CD2loCD28+ cells significantly enriched the TNaïve cell compartment over time when compared with baseline (p ≤ 0.001). The frequency of CD2loCD28+ TEM cells increased transiently followed by a dramatic reduction to below baseline at 6 months post-depletion (p ≤ 0.001).
Figure 4. Dynamics and memory phenotypes of CD28 expressing T cells during post-depletion repopulation.
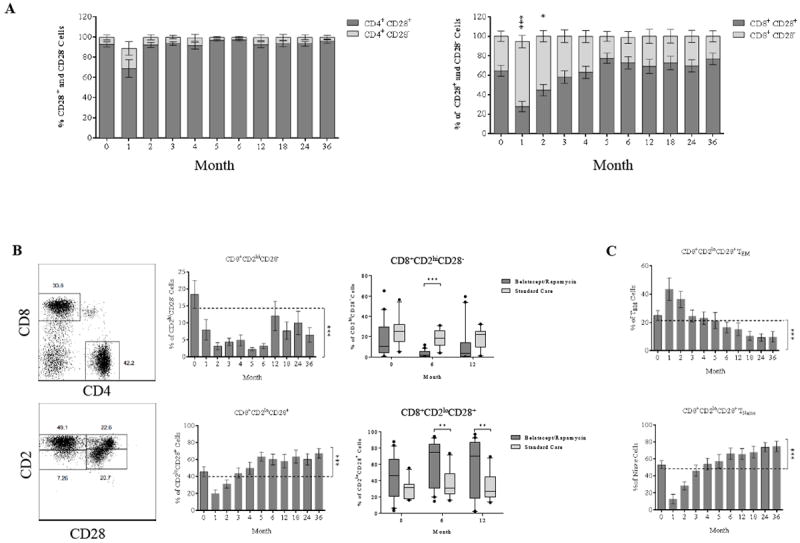
(A) Ratio of CD28 expression among repopulating CD4+ and CD8+ cells. The CD8+CD28- cells show a transient increase in frequency above the baseline within first two months. There is no change in frequency of CD28 expression observed in repopulating CD4+ cells. (B) CD8+ cells are segregated by CD2 and CD28 expression, demonstrating a significant reduction of CD8+CD2hiCD28- frequency with a concomitant increase in CD8+CD2loCD28+ frequency when compared and a standard care control group. (C) The CD8+CD2loCD28+ cells are subdivided based on CD45RA and CD197 expression, with a reduction in TEM frequency and increase in TNaïve frequency during reconstitution of the T cell population. (*p ≤ 0.05; ** p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001)
Rapid B cell reconstitution leading to a predominantly naïve and regulatory B cell phenotype
We observed a transient depletion and rapid repopulation of B cells following alemtuzumab induction without the development of donor-specific alloantibody in patients on the study regimen. To further study this phenomenon, we characterized repopulating B cells using both the CD19/CD27/IgD (27) and Bm1 through Bm5 classification systems (28-29) defining naïve and memory B cells.
Human CD19+ B cells were segregated into four subsets based on CD27 and IgD expression: switched memory (CD27+IgD-), un-switched memory (CD27+IgD+), naïve (CD27-IgD+), and exhausted memory (CD27-IgD-) (Figure 5A). The first month after depletion was marked by a significant but transient decrease in the frequency of naïve B cells, with memory subsets trending toward an increased frequency (Figure 5B). However, the frequencies of all memory subsets significantly decreased below baseline after the first month, with a reciprocal significant increase of naïve B cells that remained consistently higher than the pre-depletion baseline through the end of the study period. All memory B cell subsets were decrease relative to non-depletional controls, and naïve subsets were reciprocally increased (Figure 5C), indicating a unique B cell phenotype induced by this study regimen.
Figure 5. B cell repopulation and dynamic changes of phenotypes post-depletion.
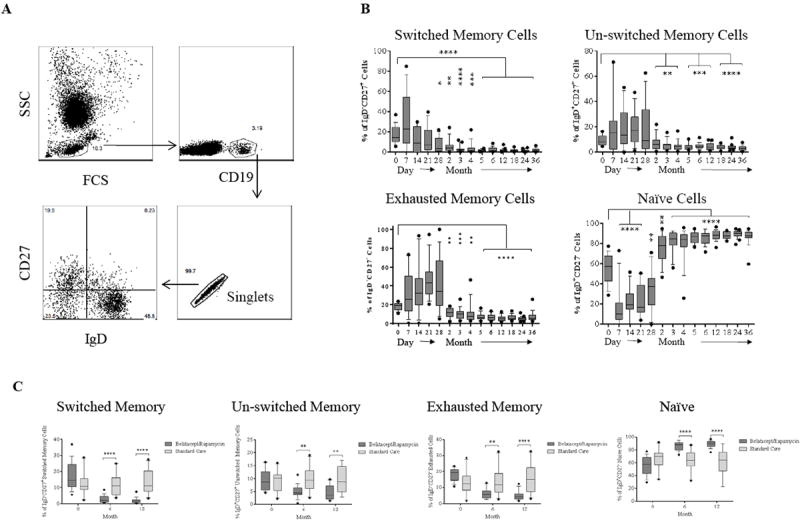
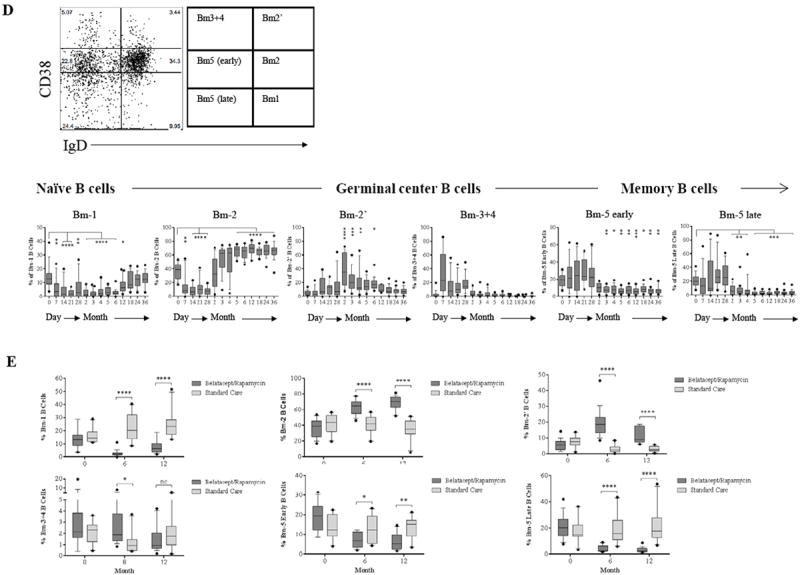
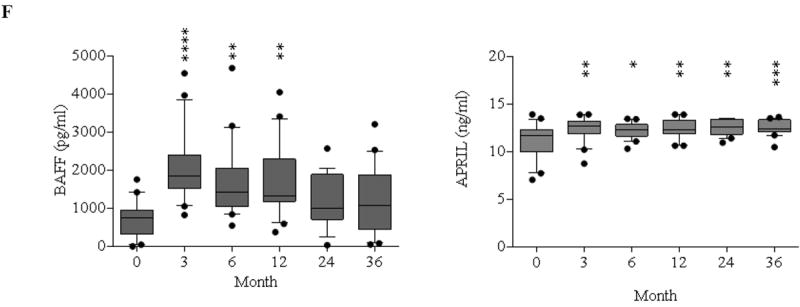
(A) Representative dot plots of gating on singlet of CD19+ B cells are segregated by the expression of CD27 and IgD into switched memory (CD27+IgD-), un-switched memory (CD27+IgD+), naïve (CD27-IgD+), and exhausted memory (CD27-IgD-) subsets. (B) Frequencies over time of memory and naïve B cells showing rapid repopulation of naïve cells compared to memory subsets post–alemtuzumab induction. (C) Comparisons of B cell subsets between patients receiving alemtuzumab induction followed by belatacept/rapamycin maintenance and patients with standard care immunosuppression. (D) CD19+ naïve and memory B cell subsets are defined by Bm1-Bm5 Classification based on CD38/IgD expression showing rapid repopulation of naïve Bm-2 subset. (E) Comparisons of B cell subsets defined by Bm1–Bm5 classification between study immunosuppression and standard care immunosuppression. (F) Serum BAFF and APRIL concentrations before and after transplantation measured by ELISA. (*p ≤ 0.05; ** p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001)
CD19+ B cells were divided into Bm1 to Bm5 subsets to further define peripheral blood B cell developmental stages. As shown in Figure 5D, patients treated with the ABR regimen displayed rapid repopulation of the naïve Bm2 subset within 2 months post-depletion, exceeding baseline from month 5 to the study endpoint. The frequency of the Bm1 subpopulation consisting of naïve and some memory cells (29) decreased transiently but returned to baseline 12 months after depletion. The presence of CD24hi and CD38hi regulatory B cells increased significantly between 2 and 6 months. This subset was negative for CD27 expression (data not shown). The germinal center Bm3+4 subsets demonstrated transient increases within the first month post-depletion with repopulation to baseline thereafter. B cell maturational arrest was evident as reconstitution of CD38+ IgD- (Bm5 early) and CD38- IgD- (Bm5 late) memory B cell subsets was essentially arrested 2 months post-depletion and did not return to baseline. Significantly higher percentages of Bm2 and Bm2’ subsets and significantly lower percentages of Bm1, Bm5 early, and Bm5 late subsets were observed in our study regimen patients compared to control patients (Figure 5E). A limitation of these studies is the access only to peripheral B cells, the development and differentiation of B cells within lymph node, spleen, and bone marrow is still unclear in these patients.
Previous studies have demonstrated that B cell activating factor (BAFF) and a proliferation-induced ligand (APRIL) play critical roles in regulating and enhancing B cell maturation, survival, activation, and proliferation (30). As shown in Figure 5F, serum BAFF concentrations were significantly elevated in the first year post-depletion, returning to baseline thereafter. Serum APRIL concentrations increased consistently post-depletion and did not return to baseline by 36 months.
Development of allo-specific hyporesponsiveness with intact viral-specific effector memory T cell responses
We have thus demonstrated that alemtuzumab depletion with belatacept costimulation blockade and rapamycin induces and maintains a predominantly naïve T and B cell phenotype. As this naiveté manifests in the activating and proliferating T cell populations, we therefore sought to assess the functional capacity of the persisting T cell repertoires toward allo- and viral antigens. PBMCs were stimulated with CD3-depleted PBMCs from either the original kidney donors, HLA-mismatched third-party allo-donors, CMV-pp65 peptides, or EBV proteins and interrogated by intracellular cytokine staining (ICCS). Previous studies have demonstrated that multi-cytokine producing T cells respond more vigorously to specific antigens when compared with single-cytokine producers (8, 10, 31-32). We therefore applied the definition of a TNF-α/IFN-γ/IL-2 triple cytokine producing T cell as a significant responder to antigen stimulation. To define memory triple-cytokine producers, activated CD4+ and CD8+ T cells were again delineated into memory and naïve subsets based on their surface expression of CD197 and CD45RA.
Prior to depletion and kidney transplantation, TNF-α/IFN-γ/IL-2 triple cytokine producers were found in both CD4+ and CD8+ cells after stimulation by donor specific and third-party cells (Figure 6A). Additionally, pre-depletion triple cytokine producers were predominantly CD4+ and CD8+ TEM cells prior to transplantation respectively (data not shown). The allo-specific CD4+ triple cytokine producers decreased after transplantation without reaching statistical significance; however, the frequency of allo-specific CD8+ triple cytokine producers was significantly reduced (p ≤ 0.01). In contrast, responders to HLA-mismatched third-party donors remained unchanged post-transplantation when compared with baseline, suggesting this study regimen induced highly allo-specific hyporesponsiveness without a generalized effect. Indeed, patients in this cohort also demonstrated the lack of allo-donor specific antibody formation during belatacept-based therapy, as recently reported (18).
Figure 6. Intracellular cytokine production and CD28 expression by allo- and viral-reactive TEM cells.
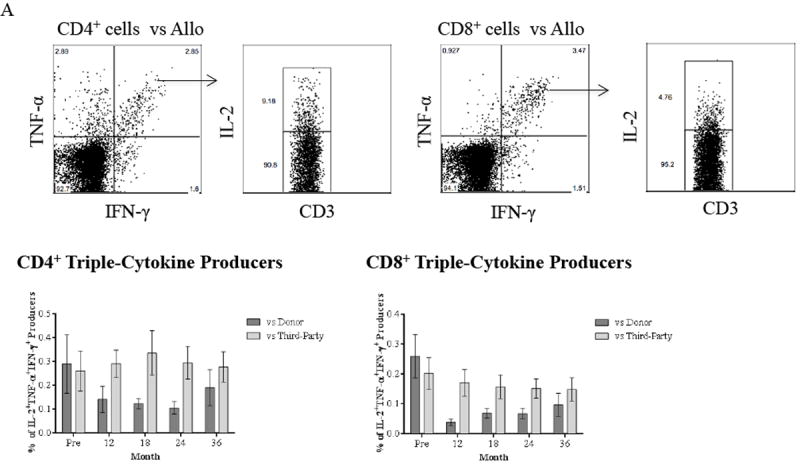
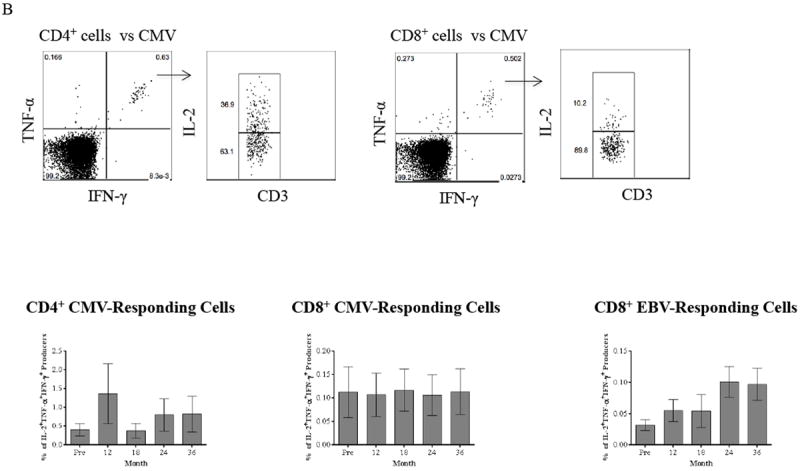
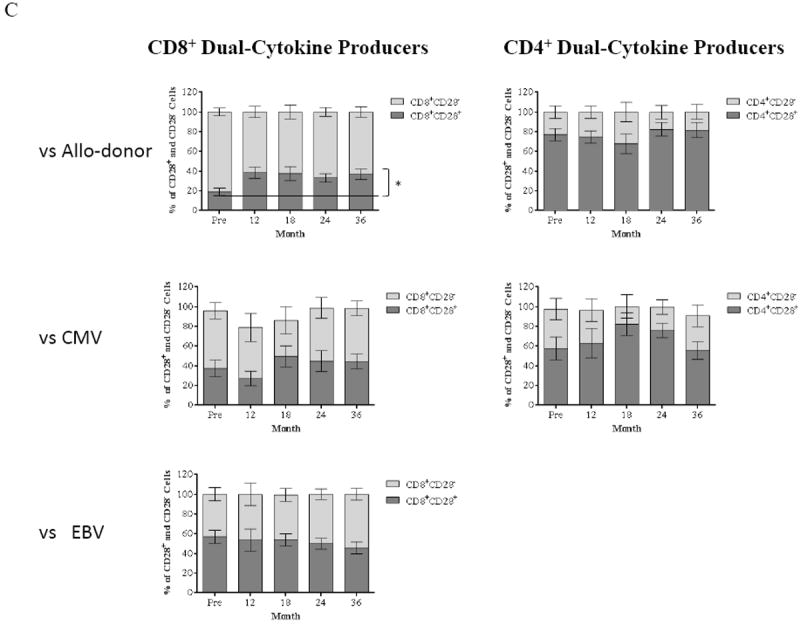
(A) PBMCs collected from patients are stimulated with CD3-depleted donor and third-party PBMCs followed by ICCS. Activating allo-specific CD4+ and CD8+ T cells are identified as TNF-α/IFN-γ dual producers, and the IL-2/TNF-α/IFN-γ triple producers are determined by gating on dual cytokine producers. Representative results from one individual are shown in top panel. The frequency of CD4+ and CD8+ triple producers in response to donor-specific antigens is decreased within 36 months after renal transplantation with this novel regimen. In contrast, T cell responses to HLA-mismatched third-party remain intact. (B) PBMCs from study patients are stimulated with CMV-pp65 peptides followed by ICCS to detect IL-2, TNF-α, and IFN-γ. Representative results from one individual are shown in top panel. Activation of CMV-specific is defined as triple cytokine producers, and both CMV-specific CD4+ and CD8+ T cell responses remain intact within 36 months post-transplantation. Activation of EBV-specific CD8+ T cells remains unchanged through the timeframe of the study with consistent phenotypes of IL-2/TNF-α/IFN-γ triple producers following stimulation with EBV proteins. (C) TNF-α and IFN-γ dual producers after allo- and viral-stimulation are segregated into CD28+ and CD28- subsets. The frequency of CD8+CD28- subset in allo-specific responders reduced significantly (p ≤ 0.0261) with an expected significant increase of the CD8+CD28+ subset (p ≤ 0.0261) within 36 months post-transplantation. In contrast, CD28 expression on viral-specific T cells after viral-stimulation remains unchanged post-depletion and transplant. The CD28 expression on CD4+ allo-responding cells remains unchanged post-depletion and transplant.
Our recently reported clinical observations have suggested that viral-specific protective immunity was not impaired, as readmissions for opportunistic infection were rare, with CMV and EBV viremia being absent or barely detectable with spontaneous resolution shortly after transplant (18). We therefore assessed PBMC responses to viral peptides or proteins to detect functional responses of viral-specific T cells. As shown in Figure 6B, patients treated with the study regimen demonstrated intact CMV-specific CD4+ and CD8+ T-cell responses as determined by the frequency of triple cytokine producers when compared to baseline. These CMV-reactive CD4+ and CD8+ triple cytokine producers were predominantly TEM cells (data not shown). Similarly, the EBV-responding CD8+ triple cytokine producers remained unchanged after depletion and transplantation with a predominantly TEM phenotype (data not shown). Thus, while the repertoire was skewed toward a naïve phenotype during repopulation, the pre-existing viral specific memory within the repertoire was not.
We have previously defined that the major subset responsible for rejection in the setting of costimulation blockade is the CD2hiCD28- T cell (8, 17). We therefore assessed CD28 expression on allo- and viral TNF-α/IFN-γ dual cytokine producers. As shown in Figure 6C, we observed a significant increase in the ratio of CD8+CD28+ to CD8+CD28- dual cytokine producers (p ≤ 0.026) post-transplantation. In contrast, CD28 expression on CD4+ dual cytokine producers after stimulation by donor cells remained unchanged. The CD28 expression on both CD4+ and CD8+ dual cytokine producers after CMV-pp65 peptide stimulation did not demonstrate significant changes. Furthermore, EBV-responding CD8+ dual cytokine producers were evenly divided between CD28+ and CD28-, and no significant changes were observed for CD28 expression on these EBV-specific T cells posttransplantation (Figure 6C). Thus, the ABR regimen induces an allo-specific modulation of CD28 expression, resulting in a favorable reduction of costimulation-resistant allo-specific CD8+CD28- T cells without affecting anti-viral responders, suggesting that the changes in the CD28 repertoire are alloantigen-specific.
Normal proliferative responses of naïve T cells post-depletion
The proliferative responses of both CD4+ and CD8+ T cells were assessed by a CFSE-based proliferation assay to observe the effects of antigen-specific memory T cells as well as de novo priming of a large fraction of naïve T cells. Strong proliferative responses of PBMCs from patients were detected after stimulation with positive control CD3CD28 beads. Proliferation of recipient PBMCs to donor specific and third-party antigens were not significantly altered post-transplantation when compared to baseline (Figure 7A). Furthermore, these proliferating CD4+ and CD8+ T cells were predominantly CD28 expressing cells, which are mechanistically favorable for belatacept’s efficacy (Figure 7B). Thus, although the effector functions of T cells, as measured by cytokine production, were impaired in a donor-specific manner, the proliferative responses were not. However, as all proliferating cells were all CD28+, they were controllable with belatacept. Therefore, the ABT regimen does not prevent the repopulation of cells with alloreactivity, but rather facilitates a situation in which repopulating cells can be controlled by CD28-B7 blockade.
Figure 7. Proliferative responses of allo-responding T cells posttransplantation.
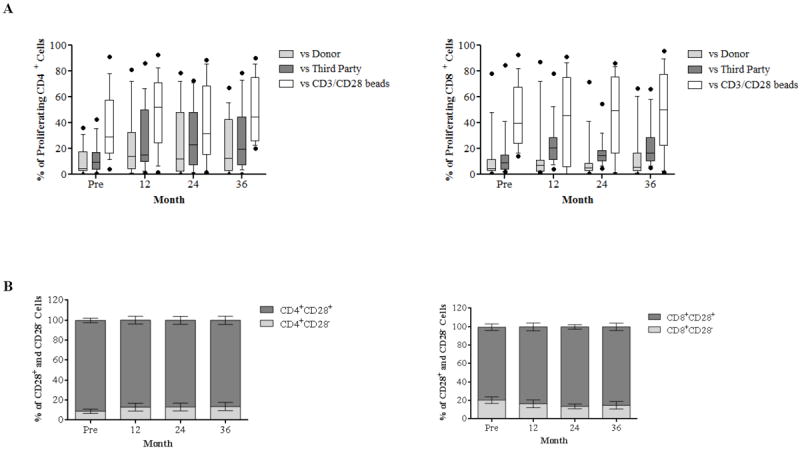
(A) CFSE-labeled responder PBMCs stimulated by donor and third-party cells and CD3/CD28 beads are analyzed for proliferation after 5 days through assessment of CFSE dilution. The proliferative responses to both specific donor and third-party allo-stimulation post-transplantation are comparable to baseline throughout the study period and predominated by CD28+ cells (B).
Discussion
We have recently reported that a regimen consisting of pretransplant alemtuzumab induction followed by a belatacept and rapamycin maintenance effectively prevents acute allograft rejection and enables some patients with low immunological risk to be weaned from rapamycin and maintain excellent graft function with belatacept monotherapy. (18) The success of this regimen prompted our investigation into the phenotypic and functional T and B cell profiles of these patients. In this study, we have examined the period of homeostatic repopulation, as this period has been suggested as a period ripe with opportunity to alter the immune repertoire (33), but also one in which homeostatic activation could foster costimulation resistance. We find that while standard non-depletional immunosuppressive regimens lead to a relatively static immune repertoire, that a depletional regimen combined with belatacept and rapamycin allows for substantial repertoire change, and that these changes appear to favor costimulation blockade-based therapy both mechanistically and clinically. Several aspects of this analysis deserve comment.
Ki67 is a surface marker long known to identify proliferating cells, including allograft-infiltrating memory T cells during acute and chronic allograft rejection (34-35), and human viral-specific memory T cells immunization (36). In this study we have shown this to be a reliable marker of homeostatic activation, as it rises during periods of lymphocyte accumulation, and coincides with markers of activation including CD69, HLA-DR and CD38. Ki67/Bcl-2 dual expressing cells are rare pre-depletion, and largely limited to activated memory T cells. However, following depletion we observed a dramatic shift of expression from memory to naïve that persisted throughout reconstitution. Furthermore, these repopulating naïve cells expressed CD28 and therefore were susceptible to costimulation blockade (8, 10), supporting this as a mechanism for the efficacy of this regimen. Practically, this introduces a method to formally assess the dynamic period of post-depletional repopulation, and we would suggest that Ki67 measurement be considered as a mechanistic adjunct to studies involving vigorous induction therapy, particularly when subsequent immunosuppressive minimization is concerned. As relates to this particular regimen, the increase in CD28 expression observed appears to derive from proliferating naïve T cells. While we have shown that terminally differentiated T cells are immediately resistant to antibody mediated T cell depletion (37), the resulting repertoire likely derives from cells without antigen experience. This clearly favors subsequent use of costimulation-based therapies. While it may be contradistinction to use the data of analyzing recipients treated with non-depletional induction immunotherapy as controls for alemtuzumab induction, the further investigation is needed to compare ABR approach with depletional induction followed by standard immunosuppression.
The antigen specificity of this effect is noteworthy. General alloreactivity was not decreased in the reconstituted repertoire, but a substantial (though incomplete) reduction in donor specific reactivity was seen, suggesting that the presence of donor antigen during repopulation is an important element in reducing the number of donor specific T cells. Whether this effect is unique to the ABR regimen, or a characteristic of post-depletional repopulation in general, will require specific comparative study. However, it is important to note that pre-existing viral-specific memory was left in tact. This is important to consider in that heterologous reactivity between viral and allo-specific T cells has been a major concern as a barrier to costimulation blockade (9, 38-40).
Previous experimental costimulation studies have showed that allospecific effector memory cells can be eliminated via activation induced cell death, particularly when mTOR inhibition is used as adjuvant therapy (14, 41). Based on recent studies, we have demonstrated that the vast majority of allospecific T cells, specifically those resulting from heterologous cross reactivity, mount low quality or incomplete responses relative to cells responding to cognate viral antigen. We thus propose that the effects described herein reflect a hierarchy of response to the regimen, with low affinity or poorly developed memory, that typified by cross-alloreactive cells, being poorly suited to survive or evade activation induced cell death, while more developed cells resulting from an explicit response to viral antigens have a greater capacity to avoid mechanisms of peripheral deletion. Indeed, mTOR inhibition has been shown to facilitate viral-responsive TEM cell differentiation (42-43). Teleological, this would be advantageous in periods of viral-induced lymphopenia, and as allospecific cross-reactivity lacks the affinity of a matured viral response, this difference in affinity may be exploited to prune allospecific clones without impairing viral memory. The current study only included non-sensitized recipients of living donor transplants and excluded transplants between donors with seropositive for CMV and seronegative recipients. In this vein, ABR approach would be expected to perform poorly in the face of an explicitly pre-sensitized individual, but appears to handle heterologous reactions or those derived from homeostatic activation well.
The role of B cells in allograft fate has extended beyond a discussion of antibody and increasingly been recognized to be relevant in T cell-mediated rejection, perhaps though antigen presenting functions. This has been accentuated by reports associating a signature of B cell maturational arrest and accumulation of transitional B cells, with tolerance. Certainly, the ABR regimen controlled alloantibody formation (18), distinguishing this from other alemtuzumab-based regimens.(44-45) Beyond this, and consistent with reports from previous investigators, we observed that recovery from transient B cell depletion promoted the accumulation of regulatory/transitional-like B cell subsets. This unique signature correlated with stable allograft function and immune tolerance in renal transplant patients (18, 46-47). B cell compartments dramatically skewed toward the naïve phenotype with significantly fewer differentiated memory B cell subsets, and the repopulating naïve subset arrested in the maturation process as they exceed the baseline over the 36-month course of the study with minimal repopulation of the Bm5 compartment. Other pro-regulatory effects were also seen, including increasing CD4+CD25+Foxp3+ regulatory T cells, CD3+Vδ1+ T cells, and CD27-CD38+IgDhi transitional/regulatory B cells (18). Indeed, these repopulating naïve B cells may enhance the expansion of FoxP3+regulatory T cells during early T cell reconstitution as suggested by previous studies (48-49), which may suggest an additional mechanistic benefit of this immunosuppression regimen.
BAFF and APRIL, as members of TNF ligand superfamily, are regulated by consumption, and play critical roles in regulating and enhancing B cell maturation, survival, activation and proliferation (30). Previous studies have demonstrated that an increased BAFF level directly correlates with the risk of alloantibody-mediated acute rejection in patients who received alemtuzumab induction and rapamycin monotherapy (50) as well as the patients who underwent antibody incompatible renal transplantation without depletional induction (51). However, we demonstrate that an elevated BAFF level with a rapid repopulation of naïve but not memory B cells in the context of belatacept therapy supports this agent’s effectiveness in abrogating the production of donor specific antibodies. These findings are supported by previous preclinical studies demonstrating the inhibitory effects of belatacept-based regimen in preventing de novo donor-specific antibody in nonhuman primate allotransplantation (19-20). We also find that APRIL levels remain elevated throughout the study timeframe. As this appears to correlate with the rapid and consistent elevation of naïve B cells, it may suggest that APRIL plays a role in promoting and/or maintaining naïve B cell expansion post-alemtuzumab induction.
Taken together, these data characterize the dynamics and functional responses of lymphocytes during homeostatic reconstitution in the context of kidney transplantation using a novel immunosuppressive regimen of alemtuzumab depletion with belatacept and rapamycin maintenance. These stark changes do not appear to occur under standard non-depletional conditions, but which aspects of the current regimen are vital for the effect remains undefined by this initial experience. Nevertheless, these findings speak to a mechanistic explanation for the salutary effects of the ABR regimen, and provide useful insights that will help guide the development of subsequent costimulation-blockade trials.
Acknowledgments
The authors would like to thank US/FDA, the Georgia Research Alliance, the National Institutes of Health, and Roche ROTRF for research funding, and Emory Transplant Center Biorepository for sample collection and storage.
FUNDING. This work was funded in part by grants from the US Food and Drug Administration (1R01 FD003539-01, ADK), the National Institutes of Health (R01 AI097423, ADK), the Georgia Research Alliance (ADK), and the Roche Organ Transplant Research Foundation grant (346678023, HX).
Abbreviations
- ABR
Alemtuzumab, Belatacept, Rapamycin
- CNIs
calcineurin inhibitors
- mAb
monoclonal antibody
- PBMCs
peripheral blood mononuclear cells
- TNaive
naïve T cells
- TC
central memory T cells
- TEM
effector memory T cells
- TEMRA
terminally differential effector memory T cells
- CMV
cytomegalovirus
- EBV
Epstein-Barr virus
- ICCS
intracellular cytokine staining
- TNF-α
tumor necrosis factor alpha
- IFN-γ
interferon gamma
- PBS
phosphate-buffered saline
- FBS
fetal bovine serum
- CFSE
carboxyfluorescein succinimidyl ester
- MLR
mixed lymphocyte reaction
- BAFF
B cell activating factor of the FNF family
- PBMCs, APRIL
a proliferation-inducing ligand
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Matas AJ, et al. OPTN/SRTR 2012 annual data report: Kidney. Am J Transplant. 2014;14(S1):11–44. doi: 10.1111/ajt.12579. [DOI] [PubMed] [Google Scholar]
- 2.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011;11(3):450–462. doi: 10.1111/j.1600-6143.2010.03283.x. [DOI] [PubMed] [Google Scholar]
- 3.Larsen C, Pearson T, Adams A, Tso P, Shirasugi N, Strobert E, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA-4Ig with potent immunosuppressive properties. Am J Transplant. 2005;5(3):445–453. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 4.Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, et al. Costimulation blockade with belatacept in renal transplantation. N Engl J Med. 2005;353(8):770–781. doi: 10.1056/NEJMoa050085. [DOI] [PubMed] [Google Scholar]
- 5.Vincenti F, Charpentier B, Vanrenterghem Y, Rostain L, Bresnahan B, Darji P, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study) Am J Transplant. 2010;10(3):535–546. doi: 10.1111/j.1600-6143.2009.03005.x. [DOI] [PubMed] [Google Scholar]
- 6.Vincenti F, Larsen C, Alberu J, Bresnahan B, Garcia V, Kohari J, et al. Three-year outcomes form BENEFIT, a randomized active-controlled, parallel-group study in adult kidney transplant recipient. Am J Transplant. 2012;12(1):201–217. doi: 10.1111/j.1600-6143.2011.03785.x. [DOI] [PubMed] [Google Scholar]
- 7.Trambley J, Bingaman A, Lin A, Elwood E, Waitze S, Ha J, et al. Asial GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J Clin Invest. 1999;104(12):1715–1722. doi: 10.1172/JCI8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo D, Weaver T, Stempora L, Mehta A, Ford M, Larsen C, et al. Selective targeting of human alloresponsive CD8+ effector memory T cells based on CD2 expression. Am J Transplant. 2011;11(1):22–33. doi: 10.1111/j.1600-6143.2010.03317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams A, Williams M, Jones T, Shirasugi N, Durham M, Kaech S, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111(12):1887–1889. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu H, Perez S, Cheeseman A, Mehta A, Kirk A. The allo- and viral-specific immunosuppressive effect of belatacept, but not tacrolimus, attenuates with progressive T cell maturation. Am J Transplant. 2014;14(2):319–332. doi: 10.1111/ajt.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calne R, Friend P, Moffatt S, Bradley A, Hale G, Firth J, et al. Prope tolerance, perioperative campath 1H, and low-dose cyclosporine monotherapy in renal allograft recipients. Lancet. 1998;351(9117):1701–1702. doi: 10.1016/S0140-6736(05)77739-4. [DOI] [PubMed] [Google Scholar]
- 12.Kirk A, Hale D, Mannon R, Kleiner D, Hoffmann S, Kampen R, et al. Results from a human renal allograft tolerance trial evaluating the humanized CD52-specific monoclonal antibody alemtuzumab (campath-1H) 2003;76(1):120–129. doi: 10.1097/01.TP.0000071362.99021.D9. [DOI] [PubMed] [Google Scholar]
- 13.Knechtle S, Pascual J, Bloom d, Torrealba J, Jankowska-Gan E, Burlingham W, et al. Early and limited use of tacrolimus to avoid rejection in an alemtuzumab and sirolimus regimen for kidney transplantation: clinical results and immune monitoring. Am J Transplant. 2009;9(5):1087–1098. doi: 10.1111/j.1600-6143.2009.02581.x. [DOI] [PubMed] [Google Scholar]
- 14.Wells A, Li X, Li Y, Walsh M, Zheng X, Wu Z, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5(11):1301–1307. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- 15.Preston E, Xu H, Dhanireddy K, Pearl J, Leopardi F, Starost M, et al. IDEC-131 (anti-CD154), sirolimus and donor-specific transfusion facilitate operational tolerance in non-human primates. Am J Transplant. 2005;3(11):1350–1354. doi: 10.1111/j.1600-6143.2005.00796.x. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Montgomery S, Preston E, Tadaki D, Hale A, Harlan D, et al. Studies investigating pretransplant donor-specific blood transfusion, rapamycin, and the CD154-specific antibody IDEC-131 in a nonhuman primate model of skin allotransplantation. J Immunol. 2003;170(5):2776–2782. doi: 10.4049/jimmunol.170.5.2776. [DOI] [PubMed] [Google Scholar]
- 17.Lo D, Anderson D, Weaver T, Leopardi F, Song M, Farris A, et al. Belatacept and sirolimus prolong nonhuman primate renal allograft survival without a requirement for memory T cell depletion. Am J Transplant. 2013;13(2):320–328. doi: 10.1111/j.1600-6143.2012.04342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirk A, Guasch A, Xu H, Cheeseman J, Mead S, Ghali A, et al. Renal transplantation using belatacept without maintenance steroids or calcineurin inhibitors. Am J Transplant. 2014;14(5):1142–1151. doi: 10.1111/ajt.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badell I, Russell M, Cardona K, Shaffer V, Turner A, Avila J, et al. CTLA4Ig prevents alloantibody formation following nonhuman primate islet transplantation using the CD40-specific antibody 3A8. Am J Transplant. 2012;12(7):1918–1923. doi: 10.1111/j.1600-6143.2012.04029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim e, Kwun J, Gibby A, Hong J, Farris A, Iwakoshi N, et al. Costimulation blockade alters germinal center responses and prevents antibody-mediated rejection. Am J Transplant. 2014;14(1):59–69. doi: 10.1111/ajt.12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bloom D, Hu H, Fechner J, Knechtle S. T-lymphocyte alloresponses of campath-1H-treated kidney transplant patients. Transplantation. 2006;81(1):81–87. doi: 10.1097/01.tp.0000191940.13473.59. [DOI] [PubMed] [Google Scholar]
- 22.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 23.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 24.Neujahr D, Chen C, Huang X, Markmann J, Cobbold S, Waldmann H, et al. Accelerated memory cell homeostasis during T cell depletion and approaches to overcome it. J Immunol. 2006;176(8):4632–4639. doi: 10.4049/jimmunol.176.8.4632. [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Bensinger S, Zhang J, Chen C, Yuan X, Huang X, et al. Homeostatic proliferation is a barrier to transplantation tolerance. Nat Med. 2004;10(1):87–92. doi: 10.1038/nm965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weaver T, Charafeddine A, Agarwal A, Turner A, Russell M, Leopardi F, et al. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat Med. 2009;15(7):746–749. doi: 10.1038/nm.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188(9):1679–1689. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pascual V, Liu Y, Magalski A, Bouteiller O, Banchereau J. Analysis of somatic mutation in five B cell subsets of human tonsil. J Exp Med. 1994;180(1):329–339. doi: 10.1084/jem.180.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohnhorst J, Bjorgan M, Thoen J, Natvig J, Thompson K. Bm1-Bm5 classification of peripheral B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjogren’s syndrome. J Immunol. 2001;167(7):3610–3618. doi: 10.4049/jimmunol.167.7.3610. [DOI] [PubMed] [Google Scholar]
- 30.Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 31.Pantaleo G, Harari A. Functional signatures in antiviral T-cell immunity for monitoring virus-associated diseases. Nat Rev Immunol. 2006;6(5):417–423. doi: 10.1038/nri1840. [DOI] [PubMed] [Google Scholar]
- 32.Kannanganat S, Ibegbu C, Chennareddi L, Robinson H, Amara R. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol. 2007;8(16):8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stock P, Kirk AD. The risk and opportunity of homeostatic repopulation. Am J Transplant. 2011;11(7):1349–1350. doi: 10.1111/j.1600-6143.2011.03543.x. [DOI] [PubMed] [Google Scholar]
- 34.Kerjaschki D, Regele H, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol. 2004;15:603–612. doi: 10.1097/01.asn.0000113316.52371.2e. [DOI] [PubMed] [Google Scholar]
- 35.Dollinger M, Howie S, Plevris J, Graham L, Hayes P, Harrison D. Intrahepatic proliferation of naïve and memory T cells during liver allograft rejection: primary immune response within the allograft. FASEB. 1998;12:939–947. doi: 10.1096/fasebj.12.11.939. [DOI] [PubMed] [Google Scholar]
- 36.Stubbe M, Vanderheyde N, Goldman M, Marchant A. Antigen-specific central memory CD4+ T lymphocytes produce multiple cytokines and proliferate in vivo in humans. J Immunol. 2006;177(11):8185–8190. doi: 10.4049/jimmunol.177.11.8185. [DOI] [PubMed] [Google Scholar]
- 37.Pearl JP, Parris J, Hale DA, Hoffmann SC, Bernstein WB, McCoy KL, et al. Immunocompetent T-cells with a memory-like phenotype are the dominant cell type following antibody-mediated T-cell depletion. Am J Transplant. 2005;5(3):465–474. doi: 10.1111/j.1600-6143.2005.00759.x. [DOI] [PubMed] [Google Scholar]
- 38.Selin L, Varga S, Wong I, Welsh R. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J Exp Med. 1998;188(9):1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welsh R, Selin L. No one is naïve: The significance of heterologous T-cell immunity. Nat Rev Immunol. 2002;2(6):417–426. doi: 10.1038/nri820. [DOI] [PubMed] [Google Scholar]
- 40.Amir A, D’Orsongna L, Roelen D, van Loenen M, Hagedoom R, de Boer R, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood. 2010;115(15):3146–3157. doi: 10.1182/blood-2009-07-234906. [DOI] [PubMed] [Google Scholar]
- 41.Gao W, Lu Y, Ei Essawy B, Oukka M, Kuchroo V, Strom T. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant. 2007;7(7):1722–1733. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Araki K, Turner A, Shaffer V, Gangappa S, Seller S, Bachmann M, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner A, Shaffer V, Araki K, Martens C, Turner P, Gangappa S, et al. Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant. 2011;11(3):613–618. doi: 10.1111/j.1600-6143.2010.03407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knechtle S, Pirsch J, Fechner J, Necker B, Friedl A, Colvin R, et al. Campath-1H induction rapamycin monotherapy for renal transplantation: results of a pilot study. Am J Transplant. 2003;3(6):722–730. doi: 10.1034/j.1600-6143.2003.00120.x. [DOI] [PubMed] [Google Scholar]
- 45.Heidt S, Hester J, Shankar S, Friend P, Wood K. B cell population after alemtuzumab induction-transient increase in transitional B cells and long-term dominance of naïve B cells. Am J Transplant. 2012;12(7):1784–1792. doi: 10.1111/j.1600-6143.2012.04012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newell K, Ssare A, Kirk A, Gisler T, Bourcier K, Suthanthiran M, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest. 2010;120(6):1836–1847. doi: 10.1172/JCI39933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leibler C, Matignon M, Pilon c, Montespan F, Bigot J, Lang P, et al. Kidney transplant recipients treated with belatacept exhibit increased naïve and transitional B cells. Am J Transplant. 2014;14(5):1173–1182. doi: 10.1111/ajt.12721. [DOI] [PubMed] [Google Scholar]
- 48.Chen X, Jensen P. Direct expansion of human allospecific FoxP3+CD4+ regulatory T cells with allogeneic B cells for therapeutic application. J Immunol. 2009;183(6):4094–4102. doi: 10.4049/jimmunol.0901081. [DOI] [PubMed] [Google Scholar]
- 49.Reichardt P, Reichardt P, Dombach B, Rong S, Beissert S, Gueler F, et al. Naïve B cells generate regulatory T cells in the presence of a mature immunologic synapse. Blood. 2007;110(5):1519–1529. doi: 10.1182/blood-2006-10-053793. [DOI] [PubMed] [Google Scholar]
- 50.Bloom D, Chang Z, Pauly K, Kwun J, Fechner J, Hayes C, et al. BAFF is increased in renal transplant patients following treatment with alemtuzumab. Am J Transplant. 2009;9(8):1835–1845. doi: 10.1111/j.1600-6143.2009.02710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banham G, Prezzi D, Harford S, Taylor C, Hamer R, Higgins R, et al. Elevated pre-transplantation soluble BAFF is associated with an increased risk of acute antibody-mediated rejection. Transplantation. 2013;96(4):412–420. doi: 10.1097/TP.0b013e318298dd65. [DOI] [PMC free article] [PubMed] [Google Scholar]